

Abstract
Niemann-Pick disease type C1 (NPC1) is a rare genetic cholesterol storage disorder caused by mutations in the NPC1 gene. Mutations in this transmembrane late endosome protein lead to loss of normal cholesterol efflux from late endosomes and lysosomes. It has been shown that broad spectrum histone deacetylase inhibitors (HDACi's) such as Vorinostat correct the cholesterol accumulation phenotype in the majority of NPC1 mutants tested in cultured cells. In order to determine the optimal specificity for HDACi correction of the mutant NPC1s, we screened 76 HDACi's of varying specificity. We tested the ability of these HDACi's to correct the excess accumulation of cholesterol in patient fibroblast cells that homozygously express NPC1I1061T, the most common mutation. We determined that inhibition of HDACs 1, 2, and 3 is important for correcting the defect, and combined inhibition of all three is needed to achieve the greatest effect, suggesting a need for multiple effects of the HDACi treatments. Identifying the specific HDACs involved in the process of regulating cholesterol trafficking in NPC1 will help to focus the search for more specific druggable targets.
Keywords: Niemann-Pick Type C, HDAC inhibition, cholesterol
Introduction
Niemann-Pick disease type C (NPC) is a rare autosomal recessive lysosomal storage disorder. Analysis of multiple exosome sequencing databases predicts an incidence rate of 1:15 000 to 1:20 000 when including late-onset forms of the disease.1 New screening initiatives and assays will soon improve our understanding of the prevalence of this disease.2 The characteristics of NPC disease include accumulation of unesterified cholesterol and other lipids in the late endosomes and lysosomes (LE/Ly) of several tissues, with the most serious effects in the central nervous system (CNS). Abnormal lipid accumulation in peripheral organs also contributes to the pathology of this disease.3 A large fraction of patients experience progressive neurological degeneration and often die before the age of 20.4−7 Mutations in two genes, NPC1 and NPC2, are the cause of NPC disease, and NPC1 mutations are responsible for approximately 95% of cases.8−10
NPC1 is a multipass transmembrane protein in the limiting membrane of the LE/Ly. NPC1 has an N-terminal cholesterol-binding domain and a sterol-sensing domain associated with its transmembrane domains.11−15 NPC2 is a small, soluble LE/Ly protein that binds cholesterol.16 NPC2 binds to NPC1 and delivers cholesterol to the N-terminal cholesterol binding domain of NPC1.13,14,17−19 Recent studies have indicated that NPC1 functions as a sterol tunnel, moving cholesterol past the glycocalyx of the lysosome to the limiting membrane for extraction by sterol transport proteins.20−22
More than 650 variants of NPC1 have been described, with over 250 of these variants having a known or likely pathogenic nature (Niemann-Pick Type C Database: https://medgen.medizin.uni-tuebingen.de/NPC-db2/index.php).3,23−25 The most common mutant allele found in patients is NPC1I1061T (14–18% of disease alleles26,27). Cells with this mutation express low levels of the mature protein due to poor folding in the endoplasmic reticulum (ER), followed by ER associated degradation (ERAD).28 High levels of overexpression of the NPC1I1061T protein rescue the cholesterol storage phenotype,28 suggesting that the mutant protein is functional if a sufficient amount is delivered to the LE/Ly.
Treatment of NPC patients with Miglustat (Zavesca), an approved treatment in many countries but not in the US,29,30 slows disease progression by inhibiting glycosphingolipid synthesis. This reduces the production of some lipids that accumulate along with cholesterol in LE/Ly of NPC mutant cells. A clinical trial treatment for NPC has been based on intrathecal injection of VTS-270, a formulation of 2-hydroxypropyl β-cyclodextrin (https://clinicaltrials.gov/ct2/show/NCT02534844).31 β-Cyclodextrins act by binding cholesterol in LE/Ly, bypassing the requirement for NPC1 and NPC2, to shuttle cholesterol out of LE/Ly.32−35 Hydroxypropyl β-cyclodextrin treatment has been effective in animal studies,36−42 and initial human data were promising.43,44 However, hydroxypropyl β-cyclodextrin does not cross the blood brain barrier (BBB),45 requiring direct infusion into the CNS. Clinical studies using intrathecal injection of VTS-270, a pharmaceutical hydroxypropyl β-cyclodextrin, have indicated slowing of disease progression.46,47
There remains a need for effective NPC disease drug therapies, preferably with an easy and safe route of administration. Valproic acid, a weak histone deacetylase inhibitor (HDACi), had been shown to have some benefit in NPC1–/– neural stem cells.48 We identified more potent and specific HDAC inhibitors as a class of molecules that reversed the NPC1 mutant phenotype in fibroblasts from NPC1 patients.49 Treatment with broad spectrum HDACi's led to increased expression of the mutant NPC1 protein in patient fibroblasts expressing one or two NPC1I1061T alleles. Specifically, treatment with a broad spectrum HDACi, Vorinostat (SAHA), reduced ERAD of the NPC1I1061T protein, which was correctly delivered to LE/Ly.50 We also tested the effectiveness of Vorinostat and Panobinostat (Farydak, LBH589) in U2OS-SRA cells engineered to express 1 of 60 different NPC1 mutations found in patients. In these cells, treatment with the HDACi corrected the phenotype for over 80% of the tested mutations.50 However, mutations in regions required for NPC1 function, such as the N-terminal cholesterol binding domain or the lumenal domain to which NPC2 binds, remained uncorrected with HDACi treatment.
Some studies of HDACi's in mouse models expressing mutant Npc1 have used SAHA (Vorinostat), a HDACi that has relatively poor BBB penetration.51−53 One study in Npc1nmf164 mice, which express a missense D1005G mutation in the Npc1 gene, showed improved liver function and reduced liver pathology. Additionally, there were changes in transcriptional regulation of cholesterol and apolipoprotein B homeostasis consistent with improved cholesterol trafficking. However, SAHA treatment of Npc1nmf164 mice at a maximum tolerated dose did not ameliorate disease progression, weight loss or animal morbidity. The very low levels of SAHA found in brain tissue may explain this lack of effect.53 A study in Npc1nmf164 mice reported that a combination therapy with Vorinostat, hydroxypropyl β-cyclodextrin, and polyethylene glycol 400 led to slowed neuronal degeneration and improved lifespan in Npc1nmf164 mutant animals.54 However, in a very similar study using NPC1I1061T mice, the hydroxypropyl β-cyclodextrin component alone proved effective,55 suggesting that this may have been the case in the earlier study as well. Thus, although HDACi's have been able to rescue most NPC1 mutants in cell culture, there is not yet evidence of a benefit in animal models. It remains possible that other HDACi's would provide a benefit.
Other recent papers have shown effectiveness of HDACi treatments in cells.56,57 HDACi treatment has also been shown to correct the lysosomal enzyme defect in fibroblasts expressing a common mutation found in Gaucher disease, which is also associated with ERAD of the mutant protein and alterations of molecular chaperone expression.58 HDACi treatment also has been reported to rescue several CFTR mutations associated with cystic fibrosis59 as well as Huntington’s and Alzheimer’s disease.60,61 These results suggest that improving proper folding and exit of mutants out of the endoplasmic reticulum can provide benefit for several inherited diseases.
The most common function of a HDAC enzyme is to catalyze the deacetylation of acetyl-l-lysine side chains of histones and other proteins. HDACs are broadly divided into five classes: Class I (HDACs 1, 2, 3, and 8), IIa (HDACs 4, 5, 7, and 9), IIb (HDACs 6 and 10, which have two HDAC domains), III (sirtuins), and IV (HDAC 11, which shares properties with classes I and II).62 For each class, the region of homology is the conserved deacetylase domain (reviewed in ref (63)). However, HDACs 4, 5, 7, and 9 have little or no deacetylase activity.64 The inhibition of HDACs affect NPC disease because they modulate multiple biological processes.65 HDACs interact with their targeted acetylated proteins and, as members of multiprotein complexes, can alter gene expression and change chromatin structure. Many of the complexes formed include multiple HDACs, indicating that more than one member of an HDAC class may be needed to achieve the desired genetic response. They also influence protein folding through their effect on chaperones and the proteasome by controlling the ubiquitination of lysines.63
Our previous work indicated that a series of broad spectrum HDAC inhibitors that are either U.S. Food and Drug Administration (FDA) approved or have been investigated in clinical studies (SAHA, TSA, CI-994, and LBH589) were effective in reducing cholesterol in NPC1 mutant fibroblasts while a HDAC 8 selective inhibitor did not have an effect.49 Treatment with valproic acid, a weak Class I HDAC inhibitor,66 corrected cholesterol accumulation in NPC1 fibroblasts and improved lysosomal targeting of NPC1.56 The dose-limiting toxicity of these HDACi's is thought to be due to the broad-spectrum inhibition, and more selective HDACi's have been proposed to be less toxic.67 Selective HDACi's that discriminate among these HDACs are now available. We sought to determine if any of the more selective HDACi compounds available to us would optimize restoration of cholesterol trafficking in NPC1 mutant fibroblasts. A more selective compound might not only reduce the adverse side effects associated with HDACi therapy67,68 but also provide insights into the mechanism of action of HDACi's in NPC by clarifying which HDAC isoform or combination thereof is responsible for the observed effect. We thus examined the effect of 76 known and novel HDACi's with different inhibitor profiles on NPC1I1061T patient-derived fibroblasts. As we will show, inhibition of the combination of HDAC 1, 2, and 3 rather than a narrower subset leads to the best reduction of cholesterol storage in NPC1 mutant fibroblasts. We also confirm that inhibition of HDACs 6 and 8 is not required.
Results and Discussion
As a model for NPC1 disease, we use NPC1I1061T patient-derived fibroblast cells (Coriell, GM18453). These cells have unesterified (free) cholesterol accumulated in lysosome-like storage organelles (LSOs) that accumulate near the center of the cells. To label free cholesterol in the cells, we use filipin, a fluorescent dye that binds unesterified cholesterol.69 As we have shown previously,50 treatment of GM18453 cells with 10 μM SAHA for 3 days greatly reduces the filipin-labeled cholesterol in the cells (Supplemental Figure 1A). To quantify this, we measure the fluorescence power in the bright areas using a threshold and then divide this fluorescence power by the total area occupied by cells in each field. We call this the LSO ratio, and we normalize it for each experiment to the ratio value for DMSO-treated cells (solvent control). We found that 10 μM SAHA, which we use herein as a reference treatment, was nearly as effective as 100 μM hydroxypropyl β-cyclodextrin with 3 days treatment for each.32,49
Broad Spectrum HDAC Inhibitors Correct NPC1 Cholesterol Phenotype
Using this filipin assay, we have shown that broad spectrum HDACi's such as SAHA, trichostatin A (TSA), or LBH589 correct the cholesterol accumulation phenotype of several fibroblast cell lines containing a variety of NPC1 mutations.49 In agreement with previous results,49 several of the broad spectrum HDACi's tested corrected the cholesterol accumulation phenotype of NPC1I1061T fibroblast cells (Supplemental Figure 1). A 3 day treatment with SAHA (Vorinostat), Chidamide,70 or AR4271 efficiently corrected the cholesterol accumulation phenotype of NPC1I1061T fibroblasts (Supplemental Figure 1B). Broad spectrum HDACi's such as LBH589 increase cholesterol esterification as a consequence of release of cholesterol from LSOs,49 and CI-994 also increases lipid droplet formation in treated cells (Supplemental Figure 1D).
We used SAHA as the reference compound in all subsequent compound screens. SAHA is an effective inhibitor of HDACs 1, 2, 3, and 6 and, to a lesser extent, HDAC 8 (Table 2).49 To determine which HDAC isoform plays the most important role in the correction of cholesterol accumulation in NPC1 fibroblast cells, we tested 76 compounds with a range of potency and selectivity for individual HDACs or groups of HDACs.
Table 2. IC50’s and ED50’s for HDAC 1, 2, and 3 Specific Inhibitors (Figure 2A Compounds)a.

Color coded boxes indicate the specificity of the compound. Green borders, low IC50 values (μM); and red, no specific inhibitory activity. Superscripts indicate the method for IC50 value determination. IC50 values provided by Reaction Biology Corporation (method a) or KDAc Therapeutics (method c). Mean ED50’s in μM (and ranges) for reduction of cholesterol accumulation for each compound.
The broad spectrum HDAC inhibitor TSA has been shown to correct the cholesterol accumulation phenotype in NPC1 patient fibroblasts.49 The R-enantiomer of TSA is a broad range inhibitor that, like SAHA, inhibits multiple HDACs. The S-enantiomer of TSA is much more specific for HDAC 6 (Table 1).72 Class I HDACs HDAC 1–3 and class II HDAC 10 were all strongly inhibited by (R)-TSA but only weakly by (S)-TSA, whereas other HDACs, including HDAC 6, were inhibited about equally or with only small differences (Table 1). Thus, although (R)-TSA is a broad spectrum HDAC inhibitor, (S)-TSA, the unnatural enantiomer, had in vitro moderate selectivity for HDAC 6 (∼20-fold lower IC50 compared to the next closest isoform, HDAC 1). Treating NPC1I1061T patient-derived fibroblasts with the separate enantiomers demonstrated that (R)-TSA corrected the cholesterol accumulation phenotype much more effectively than (S)-TSA (Welch’s ANOVA, P < 0.0001 and P = 0.23, respectively) (Figure 1). The racemic TSA is less potent than (R)-TSA at lower doses but still significantly increases cholesterol clearance with the maximal dosage (P < 0.0001, Figure 1).
Table 1. IC50’s and ED50’s of TSA Enantiomersa.

Broad spectrum HDAC inhibitors have varying specificity for individual HDACs. IC50 values (μM) for each HDAC indicated are shown in color-coded boxes. Green border boxes indicate low IC50 values and high efficacy for the indicated HDAC. The IC50’s for these compounds were measured by Reaction Biology Corporation (method a). Mean ED50s in μM (and ranges) for reduction of cholesterol accumulation for each compound.
Figure 1.
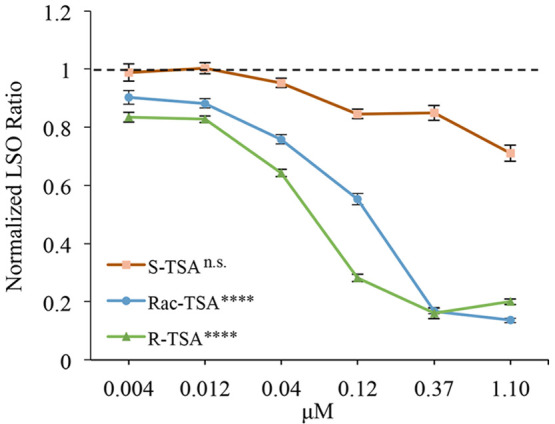
Enantiomers of TSA demonstrate the importance of HDAC 1, 2, and 3 inhibition for cholesterol clearance and that inhibition of HDACs 6 and 8 is not necessary. Racemic TSA was separated into R- and S-enantiomers. NPC1I1061T fibroblasts were treated with these enantiomers for 72 h at concentrations of 4 nM to 1.10 μM. R-TSA produced by University of Notre Dame (UND; green line) or racemic TSA (produced by UND) (blue line) specifically inhibits HDACs 1, 2, 3, 6, and 8 (see Table 1). Both dose-dependently increased cholesterol clearance from treated NPC1I1061T fibroblasts. S-TSA produced by UND (red line) more specifically inhibits HDACs 6 and 8 (see Table 1) and did not increase cholesterol clearance in treated NPC1I1061T fibroblasts. N = 3 independent cell culture experiments. Bars are SEM ****, P < 0.0001; n.s., not significant.
To categorize the compounds studied, they were binned in three classes according to their IC50’s. An IC50 value below 2 μM was considered to be an effective inhibitor of the target HDAC (green borders in Tables 1–4). An IC50 between 2 and 10 μM (yellow) was considered low inhibitory activity, while a compound was considered inactive if the IC50 was above 10 μM (red). The IC50 values are from published sources when available or the provider of the compounds. (See Methods section for details on compound providers a–c as indicated by superscripts in the tables.)
Table 4. IC50’s for Inhibitors of HDACs 1, 2, or 3 (Figure 3 Compounds)a.

Color coded box borders indicate the specificity of the compound. Green, low IC50 values (μM); yellow, residual inhibitory activity; and red, no specific inhibitory activity. IC50 values provided by Dana-Farber for UND (method b) or KDAc therapeutics (method c).
Testing HDAC Inhibitor Isoform Specificity For Correction of NPC1 Cholesterol Phenotype
To test the hypothesis that inhibition of only HDACs 1–3 was responsible for increased cholesterol clearance, we used two different approaches. In the first approach, we investigated inhibitors that were specific for HDACs 1, 2, and 3 (Table 2) but not HDAC 6 or 8. Most of these HDAC 1, 2, and 3 inhibitors corrected the cholesterol accumulation phenotype, and the dose–response curves of the compounds are shown in Figure 2. Several of the compounds tested that have low IC50 values for HDAC 1, 2, and 3 led to cholesterol clearance comparable that for to SAHA (CI-994, P = 0.0007; K-1,2,3-S, P = 0.0042; K-1,2,3-V, P = 0.0033; Figure 2A and Supplemental Figure 2).
Figure 2.

Inhibitors specific to HDACs 1, 2, and 3 increased cholesterol clearance in NPC1I1061T fibroblasts. (A) Inhibitors of HDACs 1, 2, and 3 dose-dependently increase cholesterol clearance similar to SAHA. N = 3 independent cell culture experiments. Bars are SEM. ***, P < 0.001; **, P < 0.005. (B) Compounds that do not inhibit HDACs (negative controls) do not increase cholesterol clearance in treated cells. N = 3 independent cell culture experiments. Bars are SEM. (C) Structures of HDAC inhibitors and negative controls.
Metal Chelation is Required for HDACi Cholesterol Clearance
Several HDAC inhibitors are also active against a variety of other targets. For example, valproic acid is a HDACi that was found to correct NPC1 defects in cells,48,56 but it also inhibits a number of other targets.73 Furthermore, nonmetal chelating binding modes of TSA have been found in crystal structures.74 In our second approach, we tested the importance of metal chelation in the HDAC active site for increased cholesterol clearance. We designed and tested three different HDACi analogues that do not contain a metal-chelating moiety and therefore do not inhibit any HDACs (Table 3), but are otherwise structurally very similar, as negative controls. For the first, the hydroxamic acid, which binds zinc in the HDAC catalytic site,71 is replaced by a much weaker metal binder in suberoylanilide carboxylic acid (SACA, see structures in Figure 2C). As a complementary negative control of benzamide-type HDAC inhibitors, we designed N-NC-A, which is a protected precursor of N-1,2-C (Figure 3A,C) in which the benzamide group required for zinc binding is blocked. To exclude the possibility that modification of the zinc binding moiety would lead to a negative result for reasons other than the disruption of metal binding, we designed BRD4097,75 a modified form of CI-994 that leaves the zinc binding moiety intact but disrupts binding through steric repulsion via an additional methyl group. None of these compounds led to cholesterol clearance in treated NPC1I1061T fibroblasts (Figure 2B), strongly suggesting that metal chelation by HDACi's is required for increased cholesterol clearance.
Table 3. Specificity of Negative Control Compounds for HDAC Inhibitors (Figure 2B Compounds)a.

IC50 values (μM) for each HDAC indicated are shown in color-coded boxes (red for no inhibitory activity against listed HDAC). IC50 values provided by KDAc Therapeutics (method c) and Reaction Biology Corporation (method a).
Figure 3.

Inhibitors specific for individual HDACs (1, 2, or 3) do not substantially increase cholesterol clearance from NPC1I1061T fibroblasts. (A) Compounds that more specifically inhibit HDAC1 do not increase cholesterol clearance in treated NPC1I1061T fibroblasts. N = 3 independent cell culture experiments. Bars are SEM. *P < 0.05; n.s., not significant. (B) Compounds that inhibit HDACs 1 and 2 only partially increase cholesterol clearance in treated NPC1I1061T fibroblasts. N = 3 independent cell culture experiments. Bars are SEM. (C) Compounds that inhibit HDAC3 alone do not significantly increase cholesterol clearance in treated NPC1I1061T fibroblasts. N = 3 independent cell culture experiments. Bars are SEM. (D) Structures of HDACi compounds in (A)–(C).
Testing Individual HDAC Isoforms
We studied the question of whether one HDAC isoform was solely responsible for the observed effect. While there are isoform selective inhibitors available that inhibit HDAC 1 and 2 over HDAC 3, HDACs 1 and 2 are 83% identical76 and the sequence differences are far removed from the active site. Therefore, compounds that inhibit HDAC 1 also inhibit HDAC 2 with some level of potency (e.g., Tables 2 and 4). We therefore used an approach in which we tested compounds with varying specificity for the individual HDACs. The results were correlated with the overall effect on cholesterol clearance to determine if inhibition of one HDAC alone could result in the cholesterol clearance response. The HDAC inhibitor that most specifically targeted HDAC 1 (BRD3227,77 see Table 4) did not correct the cholesterol accumulation phenotype in NPC1I1061T fibroblast cells (Figure 3A). Compound N-1,2-C, which had a lower IC50 for inhibiting HDAC3 (Table 4) was also less effective at reducing cholesterol storage in the NPC1I1061T fibroblasts (Figure 3A). We included cells treated with SAHA to confirm that the cells were responding to treatment (Figure 3B).
Compounds (N-1,2-A and N-1,2-D) with very weak activity against HDAC 3 compared to HDAC 1 and 2 did not lead to cholesterol clearance. These compounds arose from a computationally designed modification of a previously reported HDAC 1 and 2 inhibitor that exploits the mutation of a serine at the bottom of the 14 Å side pocket adjacent to the active site in HDAC 1/2 to a tyrosine in HDAC 3 (Supplemental Figure 3).78,79 The interactions of BRD3227 in the active site of HDAC1/2 vs 3 have been discussed elsewhere80 and served as the inspiration for the computational design of N-1,2,-C and related compounds. The basic design idea for these compounds is to position hydrogen bond donors and acceptors into the 14 Å pocket78 that is different between HDAC1/2 and 3 to achieve selectivity. Suitable modification of the benzamide was predicted to alter the HDAC 3 activity while maintaining the HDAC 1/2 activity in closely related analogs. If only HDAC 1/2 activity would be relevant for the observed phenotype, then these two compounds should be very similar in their efficacy. Conversely, significant differences in the observed phenotype correction should be due to the differences in HDAC 3 activity. Treatment with the inhibitors more specific to HDAC 3 (Figure 3C), K-3-A and K-3-B, resulted in minimal cholesterol clearance. Overall, these compounds failed to show the robust correction that we observed with inhibition of HDACs 1, 2, and 3.
NPC1 Cholesterol Phenotype Correction Requires Inhibition of HDACs 1, 2, and 3
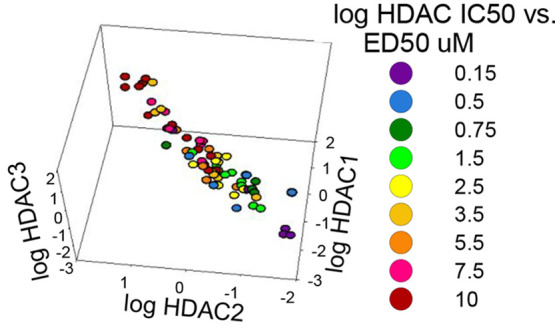
The effects of specificity when inhibiting HDACs 1–3 for reversal of the cholesterol storage phenotype of NPC1 fibroblasts are illustrated in a 3D graph in which the ED50 for lowering cholesterol (i.e., the dose at which it led to 50% clearance of cholesterol in NPC1I1061T fibroblasts) is color coded and plotted versus the log of the IC50 value for each of the three HDACs (Figure 4). Violet represents the lowest ED50 values (0.15 μM), and red represents the highest ED50 values (10 μM). In general, the compounds with the lowest ED50 values (blue and violet) are clustered at the lowest IC50 values for all three HDACs. This 3D correlation plot is rotated to focus on each HDAC individually and then oriented to show all three with equal emphasis. This plot demonstrates the relationship between specificity for the three HDACs and the ability of each compound to exert a corrective effect on cholesterol clearance within NPC1I1061T fibroblasts. The very high similarity of HDACs 1 and 2 is reflected in the better correlation between the specificities for these two enzymes and the concentrations at which cholesterol is cleared from the cells. Although the correlation is less clear for HDAC 3 than for HDACs 1 and 2, the clearance of cholesterol depends on inhibition of all three enzymes. HDAC 3 is a member of the same family as HDACs 1 and 2 (Class I), but it has less similarity to 1 and 2 as they do to each other.62
Figure 4.

Correlation between HDACi specificity (IC50) and dosage (ED50) for cholesterol clearance with compounds that inhibit HDACs 1, 2, and 3. (A) HDAC1 focused view. The (log) IC50 values for HDAC1 on the front facing axis show that points of similar ED50’s (color-coded) cluster in separate halves (reds to left, blue/purple to right). The ED50 values range from 0.15 μM (purple) to 10 μM (red). (B) HDAC2 focused view. The (log) IC50 values for HDAC2 on the front facing axis show a pattern that is a mirror image of the HDAC1 focused view. (C) HDAC3 focused view. The (log) IC50 values for HDAC3 on the front facing axis show an ED50 cluster pattern that is less distinct with red/pink clustered at the top while blue/purple is on the bottom. (D) Equal weighted plot for all three HDACs. The specificity of the inhibitor for HDACs 1, 2, and 3 is correlated with the experimentally determined ED50.
We found that the specificity of the inhibitor for HDACs 1, 2, and 3 is correlated with the concentration at which it leads to correction of the cholesterol accumulation phenotype in NPC1I1061T fibroblasts. This finding overlaps with findings in other disease models including Alzheimer’s disease and cystic fibrosis.60,61,80,81
Broad spectrum HDAC inhibitors and compounds that inhibit HDACs 1, 2, and 3 increase cholesterol clearance in NPC1I1061T patient derived fibroblasts (Coriell, GM18453). Although broad spectrum HDACi's such as SAHA have been FDA approved for a range of diseases, and SAHA has been evaluated in a clinical trial as a potential treatment for NPC, it has undesirable side effects that have been attributed to its limited selectivity.82−84 The therapeutic and/or toxic effects seen in clinical trials are thought to be due to a lack of HDAC isoform-specific inhibition, and the development of more selective inhibitors is predicted to result in better clinical outcomes.67,68 Determining which HDACs are important for this process would avoid unnecessary side effects resulting from the inhibition of HDACs not involved in cholesterol clearance. Furthermore, the mechanism by which SAHA leads to improved cholesterol clearance is not well understood. This is similar to many other epigenetic treatments where the downstream effects of the inhibition of the epigenetic target are unclear. We did find that the broad spectrum HDAC inhibitory activity of SAHA was necessary for improving the cholesterol clearance in NPC1 fibroblasts, similar to results seen with valproic acid.56 It is therefore important to determine which HDAC isoform(s) are responsible for the effect observed in the cellular assay.
While the broad spectrum HDAC inhibitors SAHA, Chidamide, AR42, and TSA all show correction of the cholesterol phenotype in NPC1I1061T fibroblast cells, we aimed to identify specific HDACs as the necessary targets for inhibition. By testing a library of compounds with different HDAC isoform selectivity profiles, we have identified that it is necessary to simultaneously inhibit HDACs 1, 2, and 3 together to increase cholesterol clearance from NPC1I1061T fibroblasts. The combined inhibition of 1, 2, and 3 explains all the observed effects of broad spectrum HDAC inhibitors.
It was not surprising that inhibition of both HDACs 1 and 2 was necessary in our studies. They appear to have overlapping functions in complexes and are 83% identical.76 Interestingly, we found that HDAC 3, which has some significant differences to HDAC 1/2 that can be exploited for isoform selectivity,85 must also be inhibited to see the optimal effect on cholesterol clearance. This result can be rationalized by the fact that HDACs, like many epigenetic modulators, exert their biological effect in a variety of protein–protein complexes. For example, HDACs 1 and 2 can exist as homodimers or heterodimers within different complexes.76 Inhibition of HDACs 1–3 has recently been shown to downregulate transcription of core regulatory transcription factors and lead to more open chromatin architecture,80 potentially allowing for transcription of other genes involved in the trafficking of cholesterol. HDACi's selected for cancer treatments reflect changes in transcription within rapidly growing cells.86 In some diseases, HDACi treatment alters proteostasis due to activation of the chaperone-mediated refolding of pathway specific proteins.87 However, the cellular basis by which HDACi's rescue the activity of mutated NPC1 is not yet known. Identifying which HDACs play the largest part in the process is a crucial step forward.
We have identified the inhibition of HDACs 1, 2, and 3 as necessary for the clearance of excess cholesterol accumulation in the lysosomes of NPC cells. In contrast, we found that it was not necessary to inhibit HDACs 6, 8, and 11, which are also targeted by the broad spectrum HDAC inhibitors previously shown to correct the phenotype. Based on a recent study in NPC1I1061T mice,55 the HDAC inhibitors tested have not been effective treatments for NPC1 disease in mouse models. However, the identification of newer HDACi's and the pathways and genes regulated by HDACs 1, 2, and 3 that yield phenotypic improvements in NPC1 mutant cells may offer new targets for improving cholesterol trafficking in NPC1 patients. Effective treatments will also require efficient entry into the CNS, and only some of the HDACi's we used have been tested for this action.66
Materials and Methods
GM18453 cell line, NPC1I1061T patient-derived fibroblast cells from Coriell Institute for Medical Research (RRID:CVCL_DA78). 384 well plates, Corning #3712 (2018). Chidamide, Cayman #13686 (RRID:SCR_008945). FBS, LifeTechnologies #10437028 (2018). DMSO, Sigma #34869 (2018), dried with Fluka Chemika, Molecular Sieves #69839 (2018). Filipin, Sigma #F9765 (2018). DRAQ5, ThermoFisher #DR51000/62252 (2018). Modified Eagle Medium (MEM), ThermoFisher #11095098 (2018). LipidTox Red, Life Technologies #H34476 (2021).
Compound Synthesis
Procedures for synthesis of the Notre Dame compounds are provided in the Supporting Information. The compounds provided for screening by KDAc are extensively described in US patent US9365498B2.88 Custom made materials will be shared upon reasonable request.
Software
ChemDraw 16.0, PerkinElmer, Waltham MA.
Microsoft Excel, Office 365, Redmond WA.
MetaMorph and MetaXpress software, Molecular Devices LLC, San Jose, CA.
Prism 8.0, GraphPad Software, San Diego, CA.
PyMOL Molecular Graphics System, ver. 2.0, Schrödinger, LLC.
SigmaPlot 14.0, Systat Software, San Jose, CA.
Equipment
AiryScan Confocal microscope, Zeiss.
ImageXpress Micro Imager (automated wide-field fluorescence microscope), MultiDrop384 Dispenser, and BioTek Plate Washer.
Methods
No ethical approval was required for this study. To test HDAC inhibitors for their ability to correct the cholesterol accumulation phenotype of NPC1 patient derived fibroblast cells (GM18453), we used a high-throughput filipin assay, previously described in detail.89 Briefly, GM18453 fibroblast cells homozygously express the I1061T mutant form for the NPC1 protein. These cells were grown in Modified Eagle Medium with 5.5% FBS on a 384-well plate and maintained at 37 °C in a humidified 5% CO2 incubator. On the second day of plating, the cells were treated with (dry) DMSO (control) or with a HDAC inhibitor compound dissolved in (dry) DMSO at concentrations ranging from 40 nM to 10 μM for 72 h. Following treatment, the cells were fixed with 3% paraformaldehyde (PFA) for 20 min, stained with 50 μg/mL filipin for 45 min, followed by a nuclear stain of 2 μM DRAQ5. All staining was done at room temperature. Images of the cells were acquired on an ImageXpress automated wide field microscope at 10× magnification using UV and CY5 filters. Each compound concentration was tested in triplicate on each plate. The results plotted are the average of three separate cell culture experiments. Filipin stains the outer membrane of the cell and the “lysosomal storage organelles” (LSOs). The LSO ratio value is the filipin intensity in the lysosomal storage organelles above the intensity of the cell membrane. DMSO-treated control cells are included in each plate, and the average value from these cells is used to set the “normal value” (1.0) for each plate. The normalized LSO ratio plotted for each treatment condition represents the LSO ratio value for the treated cells divided by the DMSO-treated cells in the same plate, allowing a comparison to be made for experiments performed on separate days. All data are plotted as mean ± standard error measurement (SEM). GM18453 fibroblast cells were treated with DMSO or 10 μM CI-994 for 48 h. After treatment, cells were stained with LipidTox Red for 30 min (1:2000). Cells were imaged with an AiryScan confocal microscope using a 40× oil immersion objective (1.3 NA) and a 561 laser line. Total fluorescence intensities in lipid droplets per image were measured with the granularity feature in the MetaXpress software. Data are shown from three independent experiments; error bars are ± SEM.
Methods for IC50 Value Determination
Method a: Reaction Biology Corp. IC50 Profiling Service (reactionbiology.com)
Compounds were tested by Reaction Biology Corp. in singlet 10 dose IC50 mode with 3-fold serial dilution starting at 100 μM against HDACs 1, 2, 3, 6, and 8. The substrate for HDACs 1, 2, 3, and 6 was a fluorogenic peptide from p53 residues 379–382 (RHKK(Ac)AMC). The substrate for HDAC8 was a fluorogenic peptide from p53 residues 379–382 (RHK(Ac)K(Ac)AMC). The IC50 values were calculated using the GraphPad Prism 4 program based on the sigmoidal dose–response equation. The blank (DMSO) value was entered as 1.00 × 10–12 of concentration for curve fitting.
Method b: Biochemical HDAC assay for UND Produced Compounds (Performed at Dana-Farber Cancer Institute)
HDAC activity was determined using an in vitro optimized assay performed in 384-well plates as previously described.90−92 Briefly, each compound was incubated with recombinant full-length HDAC protein (BPS Biosciences, San Diego, CA) for 3 h prior to enzymatic reactions. Reactions were initiated by the addition of fluorophore-conjugated substrates, MAZ1600 and MAZ1675, at a concentration equal to the substrate KM for each individual HDAC (MAZ1600: 9 μM for HDAC 1, 10 μM for HDAC 2, 8 μM for HDAC 3; MAZ1675: 202 μM for HDAC 8). The assay was carried out in assay buffer (50 mM HEPES, 100 mM KCl, 0.001% (v/v) Tween 20, 0.05 (w/v) bovine serum albumin, pH 7.4) and followed for fluorogenic release of 7-amino-4-methylcoumarin from substrate upon deacetylase and trypsin enzymatic activity. Fluorescent measurements were obtained every 5 min using a multilabel plate reader and plate stacker (Envision; PerkinElmer, Waltham, MA). The first derivative of the fluorescent intensity over time within the linear range of the data was taken (Spotfire DecisionSite) and imported into analytical software (GraphPad Prism) for analysis. Replicate experimental data (n = 4) from incubations with inhibitor were normalized to DMSO ([DMSO] < 5%). IC50 values were determined by logistic regression.
Method c: KDAc/BRD
Purified HDACs were incubated with a FAM-labeled fluorescent substrate. For HDACs 1–3, compounds were preincubated for 3 h; all other HDACs had no preincubation and were tested at room temperature for 1 h. The fluorescence intensity of electrophoretically separated substrate and product was measured, and the percent inhibition was plotted against the compound concentration. The IC50 values were determined by curve fitting with Origin 8.0 software.77
Racemic TSA Synthesis and Chiral Separation of (R)-TSA and (S)-TSA
Racemic TSA was obtained via synthetic methods described in.93 Racemic TSA was purified using a Waters XBridge Prep C18 5 μm OBD column (19 × 50 mm) and a water/acetonitrile gradient (10–90% ACN in water in 10 min, pH = 7; 14 min run, flow rate = 20 mL/min; racemic TSA RT = 7.0 min). With analytically pure racemic TSA in hand, a method was developed for the chiral separation. Racemic TSA was determined to be separable using a Daicel ChiralPAK AD-H 5 μm column (amylose tris(3,5-dimethylphenylcarbamate)-coated 5 μm silica gel, 4.6 × 250 mm) and heptane/isopropanol gradient (10–90% IPA in heptane over 15 min, no additives; total 25 min run, flow rate = 1.0 mL/min). (S)-TSA was eluted at 14.64 min. (R)-TSA was eluted at RT = 15.77 min. These samples were collected, and comparison of (R)-TSA to the commercially available (R)-TSA was performed on the same chromatography column. Commercially available (R)-TSA was confirmed to have >95% enantiomeric purity under the same mobile phase conditions ((S)-TSA is not commercially available at the time of this report). Co-injection during chiral HPLC of commercially available (R)-TSA and the isolated (R)-TSA sample derived from the separated racemic mixture confirmed that a single enantiomer was isolated and matched known material. 1H NMR and LCMS data conform to previously reported structural data in ref (66). Commercially available (R)-TSA was purchased from TCI AMERICA (Lot No. CZU7I-GC) for use in HPLC analysis.
Statistics
One-way Welch’s ANOVA or Mann–Whitney, as indicated in figures, using GraphPad Prism 8.0 software.
Acknowledgments
Justin M. Roberts of Dana-Farber Cancer Institute for the HDAC specificity assay of UND compounds. Harold Ralph of Weill Cornell Medicine Microscopy & Image Analysis Core Facility. This work was supported by NIH Grant R01 NS092653 (F.R.M., P.H., O.W.), the Ara Parseghian Medical Research Foundation, Peter G. Pentchev Research Fellowship from National Niemann-Pick Type-C Disease Foundation (Fellowship #11, D.G.), and a CBBI fellowship (T.R.Q.) T32GM075762.
Glossary
Nonstandard Abbreviations
- UND
University of Notre Dame
- TSA
trichostatin A
- HDACi
histone deacetylase inhibitor
- SACA
suberoylanilide carboxylic acid (inactive SAHA)
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00033.
Synthetic procedures for compounds and components described; design and synthesis of modified HDAC 1, 2, and 3 inhibitors (computational modeling and design); synthetic procedures for additional compounds and components tested, but not described in main text; additional broad spectrum HDACi’s that clear cholesterol from NPC mutant fibroblasts and images (PDF)
Author Contributions
Cell culture experimental design and work, data analysis, manuscript, and figure preparation (D.L.C.); cell culture experimental design and work, data analysis (N.P., S.M, D.G.); experimental design, manuscript, and figure editing (F.R.M.); design and synthesis of HDAC inhibitors, UND (G.L., M.G., M.O., T.R.Q., A.K., A.E., A.D., C.G., B.J.M, P.H., O.W.); Supporting Information preparation, manuscript, and figure editing (M.O., T.R.Q., P.H., O.W.); design and synthesis of HDAC inhibitors, KDAc (E.H., F.F.W.).
The authors declare the following competing financial interest(s): E.H. and F.F.W. are shareholders in KDAc Therapeutics. A.E. is employed by Poolia and contracted out to AstraZeneca.
Supplementary Material
References
- Patterson M. C.; Clayton P.; Gissen P.; Anheim M.; Bauer P.; Bonnot O.; Dardis A.; Dionisi-Vici C.; Klünemann H. H.; Latour P.; Lourenço C. M.; Ory D. S.; Parker A.; Pocoví M.; Strupp M.; Vanier M. T.; Walterfang M.; Marquardt T. (2017) Recommendations for the detection and diagnosis of Niemann-Pick disease type C: An update. Neurol. Clin. Pract. 7 (6), 499–511. 10.1212/CPJ.0000000000000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrido M. J.; Bauer P.; de Koning T.; Klopstock T.; Nadjar Y.; Patterson M. C.; Synofzik M.; Hendriksz C. J. (2019) Recommendations for patient screening in ultra-rare inherited metabolic diseases: what have we learned from Niemann-Pick disease type C?. Orphanet J. Rare Dis. 14 (1), 20. 10.1186/s13023-018-0985-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier M. T. (2010) Niemann-Pick disease type C. Orphanet J. Rare Dis. 5, 16. 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier M. T.; Millat G. (2003) Niemann-Pick disease type C. Clin. Genet. 64 (4), 269–81. 10.1034/j.1399-0004.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- Wraith J. E.; Guffon N.; Rohrbach M.; Hwu W. L.; Korenke G. C.; Bembi B.; Luzy C.; Giorgino R.; Sedel F. (2009) Natural history of Niemann-Pick disease type C in a multicentre observational retrospective cohort study. Mol. Genet. Metab. 98 (3), 250–4. 10.1016/j.ymgme.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Spiegel R.; Raas-Rothschild A.; Reish O.; Regev M.; Meiner V.; Bargal R.; Sury V.; Meir K.; Nadjari M.; Hermann G.; Iancu T. C.; Shalev S. A.; Zeigler M. (2009) The clinical spectrum of fetal Niemann-Pick type C. Am. J. Med. Genet., Part A 149A (3), 446–50. 10.1002/ajmg.a.32642. [DOI] [PubMed] [Google Scholar]
- Vanier M. T.; Wenger D. A.; Comly M. E.; Rousson R.; Brady R. O.; Pentchev P. G. (1988) Niemann-Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification. A collaborative study on 70 patients. Clin. Genet. 33 (5), 331–48. 10.1111/j.1399-0004.1988.tb03460.x. [DOI] [PubMed] [Google Scholar]
- Vanier M. T.; Duthel S.; Rodriguez-Lafrasse C.; Pentchev P.; Carstea E. D. (1996) Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. Am. J. Hum. Genet. 58 (1), 118–25. [PMC free article] [PubMed] [Google Scholar]
- Carstea E. D.; Morris J. A.; Coleman K. G.; Loftus S. K.; Zhang D.; Cummings C.; Gu J.; Rosenfeld M. A.; Pavan W. J.; Krizman D. B.; Nagle J.; Polymeropoulos M. H.; Sturley S. L.; Ioannou Y. A.; Higgins M. E.; Comly M.; Cooney A.; Brown A.; Kaneski C. R.; Blanchette-Mackie E. J.; Dwyer N. K.; Neufeld E. B.; Chang T. Y.; Liscum L.; Strauss J. F.; Ohno K.; Zeigler M.; Carmi R.; Sokol J.; Markie D.; O’Neill R. R.; van Diggelen O. P.; Elleder M.; Patterson M. C.; Brady R. O.; Vanier M. T.; Pentchev P. G.; Tagle D. A. (1997) Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 277 (5323), 228–31. 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Naureckiene S.; Sleat D. E.; Lackland H.; Fensom A.; Vanier M. T.; Wattiaux R.; Jadot M.; Lobel P. (2000) Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290 (5500), 2298–301. 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- Infante R. E.; Abi-Mosleh L.; Radhakrishnan A.; Dale J. D.; Brown M. S.; Goldstein J. L. (2008) Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 283 (2), 1052–63. 10.1074/jbc.M707943200. [DOI] [PubMed] [Google Scholar]
- Infante R. E.; Radhakrishnan A.; Abi-Mosleh L.; Kinch L. N.; Wang M. L.; Grishin N. V.; Goldstein J. L.; Brown M. S. (2008) Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem. 283 (2), 1064–75. 10.1074/jbc.M707944200. [DOI] [PubMed] [Google Scholar]
- Deffieu M. S.; Pfeffer S. R. (2011) Niemann-Pick type C 1 function requires lumenal domain residues that mediate cholesterol-dependent NPC2 binding. Proc. Natl. Acad. Sci. U. S. A. 108 (47), 18932–6. 10.1073/pnas.1110439108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Saha P.; Li J.; Blobel G.; Pfeffer S. R. (2016) Clues to the mechanism of cholesterol transfer from the structure of NPC1 middle lumenal domain bound to NPC2. Proc. Natl. Acad. Sci. U. S. A. 113 (36), 10079–84. 10.1073/pnas.1611956113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Wang J.; Coutavas E.; Shi H.; Hao Q.; Blobel G. (2016) Structure of human Niemann-Pick C1 protein. Proc. Natl. Acad. Sci. U. S. A. 113 (29), 8212–7. 10.1073/pnas.1607795113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland N.; Liou H. L.; Lobel P.; Stock A. M. (2003) Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proc. Natl. Acad. Sci. U. S. A. 100 (5), 2512–7. 10.1073/pnas.0437840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante R. E.; Wang M. L.; Radhakrishnan A.; Kwon H. J.; Brown M. S.; Goldstein J. L. (2008) NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. U. S. A. 105 (40), 15287–92. 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.; Farver W.; Kodukula S.; Storch J. (2008) Regulation of sterol transport between membranes and NPC2. Biochemistry 47 (42), 11134–43. 10.1021/bi801328u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H.; Wu X.; Du X.; Yao X.; Zhao X.; Lee J.; Yang H.; Yan N. (2020) Structural Basis of Low-pH-Dependent Lysosomal Cholesterol Egress by NPC1 and NPC2. Cell 182 (1), 98–111.e18. 10.1016/j.cell.2020.05.020. [DOI] [PubMed] [Google Scholar]
- Winkler M. B. L.; Kidmose R. T.; Szomek M.; Thaysen K.; Rawson S.; Muench S. P.; Wüstner D.; Pedersen B. P. (2019) Structural Insight into Eukaryotic Sterol Transport through Niemann-Pick Type C Proteins. Cell 179 (2), 485–497.e18. 10.1016/j.cell.2019.08.038. [DOI] [PubMed] [Google Scholar]
- Pfeffer S. R. (2019) NPC intracellular cholesterol transporter 1 (NPC1)-mediated cholesterol export from lysosomes. J. Biol. Chem. 294 (5), 1706–1709. 10.1074/jbc.TM118.004165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha P.; Shumate J. L.; Caldwell J. G.; Elghobashi-Meinhardt N.; Lu A.; Zhang L.; Olsson N. E.; Elias J. E.; Pfeffer S. R. (2020) Inter-domain dynamics drive cholesterol transport by NPC1 and NPC1L1 proteins. eLife 9, e57089. 10.7554/eLife.57089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Valero E. M.; Ballart A.; Iturriaga C.; Lluch M.; Macias J.; Vanier M. T.; Pineda M.; Coll M. J. (2005) Identification of 25 new mutations in 40 unrelated Spanish Niemann-Pick type C patients: genotype-phenotype correlations. Clin. Genet. 68 (3), 245–54. 10.1111/j.1399-0004.2005.00490.x. [DOI] [PubMed] [Google Scholar]
- Fancello T.; Dardis A.; Rosano C.; Tarugi P.; Tappino B.; Zampieri S.; Pinotti E.; Corsolini F.; Fecarotta S.; D’Amico A.; Di Rocco M.; Uziel G.; Calandra S.; Bembi B.; Filocamo M. (2009) Molecular analysis of NPC1 and NPC2 gene in 34 Niemann-Pick C Italian patients: identification and structural modeling of novel mutations. Neurogenetics 10 (3), 229–39. 10.1007/s10048-009-0175-3. [DOI] [PubMed] [Google Scholar]
- Bauer P.; Knoblich R.; Bauer C.; Finckh U.; Hufen A.; Kropp J.; Braun S.; Kustermann-Kuhn B.; Schmidt D.; Harzer K.; Rolfs A. (2002) NPC1: Complete genomic sequence, mutation analysis, and characterization of haplotypes. Hum. Mutat. 19 (1), 30–8. 10.1002/humu.10016. [DOI] [PubMed] [Google Scholar]
- Millat G.; Marçais C.; Rafi M. A.; Yamamoto T.; Morris J. A.; Pentchev P. G.; Ohno K.; Wenger D. A.; Vanier M. T. (1999) Niemann-Pick C1 disease: the I1061T substitution is a frequent mutant allele in patients of Western European descent and correlates with a classic juvenile phenotype. Am. J. Hum. Genet. 65 (5), 1321–9. 10.1086/302626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park W. D.; O’Brien J. F.; Lundquist P. A.; Kraft D. L.; Vockley C. W.; Karnes P. S.; Patterson M. C.; Snow K. (2003) Identification of 58 novel mutations in Niemann-Pick disease type C: correlation with biochemical phenotype and importance of PTC1-like domains in NPC1. Hum. Mutat. 22 (4), 313–25. 10.1002/humu.10255. [DOI] [PubMed] [Google Scholar]
- Gelsthorpe M. E.; Baumann N.; Millard E.; Gale S. E.; Langmade S. J.; Schaffer J. E.; Ory D. S. (2008) Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 283 (13), 8229–36. 10.1074/jbc.M708735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyseng-Williamson K. A. (2014) Miglustat: a review of its use in Niemann-Pick disease type C. Drugs 74 (1), 61–74. 10.1007/s40265-013-0164-6. [DOI] [PubMed] [Google Scholar]
- Santos-Lozano A.; Villamandos García D.; Sanchis-Gomar F.; Fiuza-Luces C.; Pareja-Galeano H.; Garatachea N.; Nogales Gadea G.; Lucia A. (2015) Niemann-Pick disease treatment: a systematic review of clinical trials. Ann. Transl. Med. 3 (22), 360. 10.3978/j.issn.2305-5839.2015.12.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottinger E. A.; Kao M. L.; Carrillo-Carrasco N.; Yanjanin N.; Shankar R. K.; Janssen M.; Brewster M.; Scott I.; Xu X.; Cradock J.; Terse P.; Dehdashti S. J.; Marugan J.; Zheng W.; Portilla L.; Hubbs A.; Pavan W. J.; Heiss J.; Vite C. H.; Walkley S. U.; Ory D. S.; Silber S. A.; Porter F. D.; Austin C. P.; McKew J. C. (2014) Collaborative development of 2-hydroxypropyl-β-cyclodextrin for the treatment of Niemann-Pick type C1 disease. Curr. Top. Med. Chem. 14 (3), 330–9. 10.2174/1568026613666131127160118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum A. I.; Zhang G.; Warren J. D.; Maxfield F. R. (2010) Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc. Natl. Acad. Sci. U. S. A. 107 (12), 5477–82. 10.1073/pnas.0914309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum A. I.; Maxfield F. R. (2011) Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J. Neurochem. 116 (5), 789–95. 10.1111/j.1471-4159.2010.06976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi-Mosleh L.; Infante R. E.; Radhakrishnan A.; Goldstein J. L.; Brown M. S. (2009) Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. U. S. A. 106 (46), 19316–21. 10.1073/pnas.0910916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez C. M.; Liu B.; Taylor A. M.; Repa J. J.; Burns D. K.; Weinberg A. G.; Turley S. D.; Dietschy J. M. (2010) Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr. Res. 68 (4), 309–15. 10.1203/PDR.0b013e3181ee4dd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo F.; Erickson R. P.; Garver W. S.; Hossain G. S.; Carbone P. N.; Heidenreich R. A.; Blanchard J. (2001) Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 70 (2), 131–42. 10.1016/S0024-3205(01)01384-4. [DOI] [PubMed] [Google Scholar]
- Davidson C. D.; Ali N. F.; Micsenyi M. C.; Stephney G.; Renault S.; Dobrenis K.; Ory D. S.; Vanier M. T.; Walkley S. U. (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One 4 (9), e6951. 10.1371/journal.pone.0006951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Ramirez C. M.; Miller A. M.; Repa J. J.; Turley S. D.; Dietschy J. M. (2010) Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 51 (5), 933–44. 10.1194/jlr.M000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peake K. B.; Vance J. E. (2012) Normalization of cholesterol homeostasis by 2-hydroxypropyl-β-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J. Biol. Chem. 287 (12), 9290–8. 10.1074/jbc.M111.326405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A. M.; Liu B.; Mari Y.; Liu B.; Repa J. J. (2012) Cyclodextrin mediates rapid changes in lipid balance in Npc1–/– mice without carrying cholesterol through the bloodstream. J. Lipid Res. 53 (11), 2331–42. 10.1194/jlr.M028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez A. M.; Terpack S. J.; Posey K. S.; Liu B.; Ramirez C. M.; Turley S. D. (2014) Systemic administration of 2-hydroxypropyl-β-cyclodextrin to symptomatic Npc1-deficient mice slows cholesterol sequestration in the major organs and improves liver function. Clin. Exp. Pharmacol. Physiol. 41 (10), 780–7. 10.1111/1440-1681.12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vite C. H.; Bagel J. H.; Swain G. P.; Prociuk M.; Sikora T. U.; Stein V. M.; O’Donnell P.; Ruane T.; Ward S.; Crooks A.; Li S.; Mauldin E.; Stellar S.; De Meulder M.; Kao M. L.; Ory D. S.; Davidson C.; Vanier M. T.; Walkley S. U. (2015) Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci. Transl. Med. 7 (276), 276ra26. 10.1126/scitranslmed.3010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter F. D.; Farhat N. Y.; Ottinger E. A.; McKew J. C.; Weissfeld L.; Machielse B.; Berry-Kavis E. M.; Vite C. H.; Walkley S. U.; Ory D. S. (2016) Phase 1/2 evaluation of intrathecal 2-hydroxypropyl-β-cyclodextrin for the treatment of Niemann-Pick disease type C1. Mol. Genet. Metab. 117 (2), S97. 10.1016/j.ymgme.2015.12.411. [DOI] [Google Scholar]
- Machielse B. N.; Porter F. D.; Yergey A. L.; Berry-Kravis E. M.; Darling A.; Rao R. (2017) VTS-270 for the treatment of Niemann-Pick disease type C. Mol. Genet. Metab. 120 (1–2), S89–S90. 10.1016/j.ymgme.2016.11.219. [DOI] [Google Scholar]
- Pontikis C. C.; Davidson C. D.; Walkley S. U.; Platt F. M.; Begley D. J. (2013) Cyclodextrin alleviates neuronal storage of cholesterol in Niemann-Pick C disease without evidence of detectable blood-brain barrier permeability. J. Inherited Metab. Dis. 36 (3), 491–8. 10.1007/s10545-012-9583-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory D. S.; Ottinger E. A.; Farhat N. Y.; King K. A.; Jiang X.; Weissfeld L.; Berry-Kravis E.; Davidson C. D.; Bianconi S.; Keener L. A.; Rao R.; Soldatos A.; Sidhu R.; Walters K. A.; Xu X.; Thurm A.; Solomon B.; Pavan W. J.; Machielse B. N.; Kao M.; Silber S. A.; McKew J. C.; Brewer C. C.; Vite C. H.; Walkley S. U.; Austin C. P.; Porter F. D. (2017) Intrathecal 2-hydroxypropyl-β-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial. Lancet 390 (10104), 1758–1768. 10.1016/S0140-6736(17)31465-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer C. A.; Thurm A.; Farhat N.; Bianconi S.; Keener L. A.; Porter F. D. (2019) Long-Term Neuropsychological Outcomes from an Open-Label Phase I/IIa Trial of 2-Hydroxypropyl-β-Cyclodextrins (VTS-270) in Niemann-Pick Disease, Type C1. CNS Drugs 33 (7), 677–683. 10.1007/s40263-019-00642-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. J.; Lee B. H.; Lee Y. S.; Kang K. S. (2007) Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 360 (3), 593–9. 10.1016/j.bbrc.2007.06.116. [DOI] [PubMed] [Google Scholar]
- Pipalia N. H.; Cosner C. C.; Huang A.; Chatterjee A.; Bourbon P.; Farley N.; Helquist P.; Wiest O.; Maxfield F. R. (2011) Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. U. S. A. 108 (14), 5620–5. 10.1073/pnas.1014890108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipalia N. H.; Subramanian K.; Mao S.; Ralph H.; Hutt D. M.; Scott S. M.; Balch W. E.; Maxfield F. R. (2017) Histone deacetylase inhibitors correct the cholesterol storage defect in most Niemann-Pick C1 mutant cells. J. Lipid Res. 58 (4), 695–708. 10.1194/jlr.M072140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M.; Benn C. L.; Franklin S. A.; Smith D. L.; Woodman B.; Marks P. A.; Bates G. P. (2011) SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS One 6 (11), e27746. 10.1371/journal.pone.0027746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson J. E.; La H.; Plise E.; Chen Y. H.; Ding X.; Hanania T.; Sabath E. V.; Alexandrov V.; Brunner D.; Leahy E.; Steiner P.; Liu L.; Scearce-Levie K.; Zhou Q. (2013) SAHA enhances synaptic function and plasticity in vitro but has limited brain availability in vivo and does not impact cognition. PLoS One 8 (7), e69964. 10.1371/journal.pone.0069964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkacsi A. B.; Hammond N.; Schneider R. T.; Senanayake D. S.; Higaki K.; Lagutin K.; Bloor S. J.; Ory D. S.; Maue R. A.; Chen F. W.; Hernandez-Ono A.; Dahlson N.; Repa J. J.; Ginsberg H. N.; Ioannou Y. A.; Sturley S. L. (2017) Normalization of Hepatic Homeostasis in the Npc1(nmf164) Mouse Model of Niemann-Pick Type C Disease Treated with the Histone Deacetylase Inhibitor Vorinostat. J. Biol. Chem. 292 (11), 4395–4410. 10.1074/jbc.M116.770578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M. S.; Getz M.; Haldar K. (2016) Chronic administration of an HDAC inhibitor treats both neurological and systemic Niemann-Pick type C disease in a mouse model. Sci. Transl. Med. 8 (326), 326ra23. 10.1126/scitranslmed.aad9407. [DOI] [PubMed] [Google Scholar]
- Davidson J.; Molitor E.; Moores S.; Gale S. E.; Subramanian K.; Jiang X.; Sidhu R.; Kell P.; Zhang J.; Fujiwara H.; Davidson C.; Helquist P.; Melancon B. J.; Grigalunas M.; Liu G.; Salahi F.; Wiest O.; Xu X.; Porter F. D.; Pipalia N. H.; Cruz D. L.; Holson E. B.; Schaffer J. E.; Walkley S. U.; Maxfield F. R.; Ory D. S. (2019) 2-Hydroxypropyl-β-cyclodextrin is the active component in a triple combination formulation for treatment of Niemann-Pick C1 disease. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1864 (10), 1545–1561. 10.1016/j.bbalip.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian K.; Hutt D. M.; Scott S. M.; Gupta V.; Mao S.; Balch W. E. (2020) Correction of Niemann-Pick type C1 trafficking and activity with the histone deacetylase inhibitor valproic acid. J. Biol. Chem. 295 (23), 8017–8035. 10.1074/jbc.RA119.010524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton J.; Hait N. C.; Maceyka M.; Colaco A.; Maczis M.; Wassif C. A.; Cougnoux A.; Porter F. D.; Milstien S.; Platt N.; Platt F. M.; Spiegel S. (2017) FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibroblasts. FASEB J. 31 (4), 1719–1730. 10.1096/fj.201601041R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.; Rahimpour S.; Lu J.; Pacak K.; Ikejiri B.; Brady R. O.; Zhuang Z. (2013) Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc. Natl. Acad. Sci. U. S. A. 110 (3), 966–71. 10.1073/pnas.1221046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Li P.; Merz K. M. (2018) Extended Zinc AMBER Force Field (EZAFF). J. Chem. Theory Comput. 14 (1), 242–254. 10.1021/acs.jctc.7b00773. [DOI] [PubMed] [Google Scholar]
- Yang S. S.; Zhang R.; Wang G.; Zhang Y. F. (2017) The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 6, 19. 10.1186/s40035-017-0089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G.; Cha J. H. (2006) Histone deacetylase inhibitors: a novel therapeutic approach to Huntington’s disease (complex mechanism of neuronal death). Curr. Alzheimer Res. 3 (4), 403–8. 10.2174/156720506778249407. [DOI] [PubMed] [Google Scholar]
- Gregoretti I. V.; Lee Y. M.; Goodson H. V. (2004) Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338 (1), 17–31. 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Seto E.; Yoshida M. (2014) Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harbor Perspect. Biol. 6 (4), a018713. 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra M. (2015) Class IIa HDACs - new insights into their functions in physiology and pathology. FEBS J. 282 (9), 1736–44. 10.1111/febs.13061. [DOI] [PubMed] [Google Scholar]
- Helquist P.; Maxfield F. R.; Wiech N. L.; Wiest O. (2013) Treatment of Niemann--pick type C disease by histone deacetylase inhibitors. Neurotherapeutics 10 (4), 688–97. 10.1007/s13311-013-0217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weïwer M.; Lewis M. C.; Wagner F. F.; Holson E. B. (2013) Therapeutic potential of isoform selective HDAC inhibitors for the treatment of schizophrenia. Future Med. Chem. 5 (13), 1491–508. 10.4155/fmc.13.141. [DOI] [PubMed] [Google Scholar]
- Witt O.; Lindemann R. (2009) HDAC inhibitors: magic bullets, dirty drugs or just another targeted therapy. Cancer Lett. 280 (2), 123–4. 10.1016/j.canlet.2009.02.038. [DOI] [PubMed] [Google Scholar]
- Wagner J. M.; Hackanson B.; Lübbert M.; Jung M. (2010) Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenet. 1 (3–4), 117–136. 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpl G.; Gehrig-Burger K. (2011) Probes for studying cholesterol binding and cell biology. Steroids 76 (3), 216–31. 10.1016/j.steroids.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Ning Z. Q.; Li Z. B.; Newman M. J.; Shan S.; Wang X. H.; Pan D. S.; Zhang J.; Dong M.; Du X.; Lu X. P. (2012) Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 69 (4), 901–9. 10.1007/s00280-011-1766-x. [DOI] [PubMed] [Google Scholar]
- Lu Q.; Wang D.-S.; Chen C.-S.; Hu Y.-D.; Chen C.-S. (2005) Structure-based optimization of phenylbutyrate-derived histone deacetylase inhibitors. J. Med. Chem. 48 (17), 5530–5. 10.1021/jm0503749. [DOI] [PubMed] [Google Scholar]
- Miyake Y.; Keusch J. J.; Wang L.; Saito M.; Hess D.; Wang X.; Melancon B. J.; Helquist P.; Gut H.; Matthias P. (2016) Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 12 (9), 748–54. 10.1038/nchembio.2140. [DOI] [PubMed] [Google Scholar]
- Kazantsev A. G.; Thompson L. M. (2008) Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discovery 7 (10), 854–68. 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- Somoza J. R.; Skene R. J.; Katz B. A.; Mol C.; Ho J. D.; Jennings A. J.; Luong C.; Arvai A.; Buggy J. J.; Chi E.; Tang J.; Sang B. C.; Verner E.; Wynands R.; Leahy E. M.; Dougan D. R.; Snell G.; Navre M.; Knuth M. W.; Swanson R. V.; McRee D. E.; Tari L. W. (2004) Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12 (7), 1325–34. 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Wagner F. F.; Lundh M.; Kaya T.; McCarren P.; Zhang Y. L.; Chattopadhyay S.; Gale J. P.; Galbo T.; Fisher S. L.; Meier B. C.; Vetere A.; Richardson S.; Morgan N. G.; Christensen D. P.; Gilbert T. J.; Hooker J. M.; Leroy M.; Walpita D.; Mandrup-Poulsen T.; Wagner B. K.; Holson E. B. (2016) An Isochemogenic Set of Inhibitors To Define the Therapeutic Potential of Histone Deacetylases in β-Cell Protection. ACS Chem. Biol. 11 (2), 363–74. 10.1021/acschembio.5b00640. [DOI] [PubMed] [Google Scholar]
- Kelly R. D.; Cowley S. M. (2013) The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem. Soc. Trans. 41 (3), 741–9. 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- Wagner F. F.; Zhang Y. L.; Fass D. M.; Joseph N.; Gale J. P.; Weïwer M.; McCarren P.; Fisher S. L.; Kaya T.; Zhao W. N.; Reis S. A.; Hennig K. M.; Thomas M.; Lemercier B. C.; Lewis M. C.; Guan J. S.; Moyer M. P.; Scolnick E.; Haggarty S. J.; Tsai L. H.; Holson E. B. (2015) Kinetically Selective Inhibitors of Histone Deacetylase 2 (HDAC2) as Cognition Enhancers. Chem. Sci. 6 (1), 804–815. 10.1039/C4SC02130D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. F.; Wiest O.; Helquist P.; Lan-Hargest H. Y.; Wiech N. L. (2004) On the function of the 14 A long internal cavity of histone deacetylase-like protein: implications for the design of histone deacetylase inhibitors. J. Med. Chem. 47 (13), 3409–17. 10.1021/jm0498497. [DOI] [PubMed] [Google Scholar]
- Wang D. F.; Helquist P.; Wiech N. L.; Wiest O. (2005) Toward selective histone deacetylase inhibitor design: homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J. Med. Chem. 48 (22), 6936–47. 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]
- Gryder B. E.; Wu L.; Woldemichael G. M.; Pomella S.; Quinn T. R.; Park P. M. C.; Cleveland A.; Stanton B. Z.; Song Y.; Rota R.; Wiest O.; Yohe M. E.; Shern J. F.; Qi J.; Khan J. (2019) Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 10 (1), 3004. 10.1038/s41467-019-11046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglès F.; Hutt D. M.; Balch W. E. (2019) HDAC inhibitors rescue multiple disease-causing CFTR variants. Hum. Mol. Genet. 28 (12), 1982–2000. 10.1093/hmg/ddz026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvic M.; Talpur R.; Ni X.; Zhang C.; Hazarika P.; Kelly C.; Chiao J. H.; Reilly J. F.; Ricker J. L.; Richon V. M.; Frankel S. R. (2007) Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109 (1), 31–9. 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruserud Ø.; Stapnes C.; Ersvaer E.; Gjertsen B. T.; Ryningen A. (2007) Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cell. Curr. Pharm. Biotechnol. 8 (6), 388–400. 10.2174/138920107783018417. [DOI] [PubMed] [Google Scholar]
- Bertino E. M.; Otterson G. A. (2011) Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin. Invest. Drugs 20 (8), 1151–8. 10.1517/13543784.2011.594437. [DOI] [PubMed] [Google Scholar]
- Methot J. L.; Chakravarty P. K.; Chenard M.; Close J.; Cruz J. C.; Dahlberg W. K.; Fleming J.; Hamblett C. L.; Hamill J. E.; Harrington P.; Harsch A.; Heidebrecht R.; Hughes B.; Jung J.; Kenific C. M.; Kral A. M.; Meinke P. T.; Middleton R. E.; Ozerova N.; Sloman D. L.; Stanton M. G.; Szewczak A. A.; Tyagarajan S.; Witter D. J.; Secrist J. P.; Miller T. A. (2008) Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 18 (3), 973–8. 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- Mitsiades C. S.; Mitsiades N. S.; McMullan C. J.; Poulaki V.; Shringarpure R.; Hideshima T.; Akiyama M.; Chauhan D.; Munshi N.; Gu X.; Bailey C.; Joseph M.; Libermann T. A.; Richon V. M.; Marks P. A.; Anderson K. C. (2004) Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc. Natl. Acad. Sci. U. S. A. 101 (2), 540–5. 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutt D. M.; Herman D.; Rodrigues A. P.; Noel S.; Pilewski J. M.; Matteson J.; Hoch B.; Kellner W.; Kelly J. W.; Schmidt A.; Thomas P. J.; Matsumura Y.; Skach W. R.; Gentzsch M.; Riordan J. R.; Sorscher E. J.; Okiyoneda T.; Yates J. R.; Lukacs G. L.; Frizzell R. A.; Manning G.; Gottesfeld J. M.; Balch W. E. (2010) Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol. 6 (1), 25–33. 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holson E., Wagner F. F., Weiwer M., Tsai L.-H., Haggarty S., and Zhang Y.-L. (2016) Inhibitors of histone deacetylase. US9365498B2.
- Pipalia N. H.; Huang A.; Ralph H.; Rujoi M.; Maxfield F. R. (2006) Automated microscopy screening for compounds that partially revert cholesterol accumulation in Niemann-Pick C cells. J. Lipid Res. 47 (2), 284–301. 10.1194/jlr.M500388-JLR200. [DOI] [PubMed] [Google Scholar]
- Bradner J. E.; West N.; Grachan M. L.; Greenberg E. F.; Haggarty S. J.; Warnow T.; Mazitschek R. (2010) Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6 (3), 238–243. 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers A. A.; Greshock T. J.; West N.; Estiu G.; Schreiber S. L.; Wiest O.; Williams R. M.; Bradner J. E. (2009) Synthesis and conformation-activity relationships of the peptide isosteres of FK228 and largazole. J. Am. Chem. Soc. 131 (8), 2900–5. 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers A.; West N.; Taunton J.; Schreiber S. L.; Bradner J. E.; Williams R. M. (2008) Total synthesis and biological mode of action of largazole: a potent class I histone deacetylase inhibitor. J. Am. Chem. Soc. 130 (33), 11219–22. 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosner C. C.; Bhaskara Reddy Iska V.; Chatterjee A.; Markiewicz J. T.; Corden S. J.; Löfstedt J.; Ankner T.; Richer J.; Hulett T.; Schauer D. J.; Wiest O.; Helquist P. (2013) Evolution of Concise and Flexible Synthetic Strategies for Trichostatic Acid and the Potent Histone Deacetylase Inhibitor Trichostatin A. Eur. J. Org. Chem. 2013 (1), 162–172. 10.1002/ejoc.201201233. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.