

Abstract
Antibody class switch recombination (CSR) is a locus‐specific genomic rearrangement mediated by switch (S) region transcription, activation‐induced cytidine deaminase (AID)‐induced DNA breaks, and their resolution by non‐homologous end joining (NHEJ)‐mediated DNA repair. Due to the complex nature of the recombination process, numerous cofactors are intimately involved, making it important to identify rate‐limiting factors that impact on DNA breaking and/or repair. Using an siRNA‐based loss‐of‐function screen of genes predicted to encode PHD zinc‐finger‐motif proteins, we identify the splicing factor Phf5a/Sf3b14b as a novel modulator of the DNA repair step of CSR. Loss of Phf5a severely impairs AID‐induced recombination, but does not perturb DNA breaks and somatic hypermutation. Phf5a regulates NHEJ‐dependent DNA repair by preserving chromatin integrity to elicit optimal DNA damage response and subsequent recruitment of NHEJ factors at the S region. Phf5a stabilizes the p400 histone chaperone complex at the locus, which in turn promotes deposition of H2A variant such as H2AX and H2A.Z that are critical for the early DNA damage response and NHEJ, respectively. Depletion of Phf5a or p400 blocks the repair of both AID‐ and I‐SceI‐induced DNA double‐strand breaks, supporting an important contribution of this axis to programmed as well as aberrant recombination.
Keywords: CSR, genomic instability, H2A.Z, NHEJ, Phf5a
Subject Categories: DNA Replication, Repair & Recombination; Immunology
Regulation of DNA damage responses and NHEJ repair factor recruitment represents a new, splicing‐independent role for spliceosome‐linked PHD finger protein Phf5a/Sf3b14b.
Introduction
In mature B cells, activation‐induced cytidine deaminase (AID) induces DNA breaks at the IgH locus in order to diversify the antibody gene locus via somatic hypermutation (SHM) and class switch recombination (CSR) (Muramatsu et al, 2000; Muramatsu et al, 2007). The cellular DNA repair system plays a key role during these events as the DNA breaks at the variable and the switch (S) regions are processed distinctly, leading to SHM and CSR, respectively. In particular, CSR requires an elaborate DNA repair process to join the two S region DNA double‐strand break (DSB) ends. The acceptor and the donor S regions, which are located several kilobases apart, are brought into proximity and undergo ligation predominantly mediated by non‐homologous end joining (NHEJ) (Stavnezer et al, 2010; Boboila et al, 2012a). However, genomic loci other than IgH are often mis‐targeted by AID, and the repair of these DNA breaks by NHEJ is a potential source of oncogenic mutations and chromosomal translocations. Therefore, CSR and the associated genomic instability provide a unique opportunity to investigate DNA break‐repair pathway and its regulation.
Active chromatin marks and their combinatorial histone codes are known to be involved in AID‐induced genomic instability (Daniel et al, 2010; Begum & Honjo, 2012; Li et al, 2013; Sheppard et al, 2018). Specific histone chaperones and transcription elongation complexes play important roles in the chromatin organization and histone modification regulation at the IgH locus (Pavri et al, 2010; Stanlie et al, 2010; Stanlie et al, 2012). Moreover, several chromatin reader proteins, such as 53BP1, Brd4, and PTIP, are also known to promote DNA repair step of CSR (Reina‐San‐Martin et al, 2007; Daniel et al, 2010; Stanlie et al, 2014). Thus, the chromatin‐associated proteins involved in recognizing and/or remodeling the histone codes at the S regions can greatly impact CSR efficiency by influencing the DSB formation and/or recombination. For example, CSR requires histone post‐translational modification H3K4me3, which promotes AID‐induced DNA break in the recombining S regions. On the other hand, transcription elongation‐associated FACT, SPT6, and DSIF complexes play a critical role in regulating H3K4me3 in the S regions. Therefore, deficiencies not only in FACT or SPT6, but also in any proteins involved in H3K4me3 formation (Ash2, CxxC1, Wdr82, Set1, Mll1, or PTIP) can impair CSR by blocking the S region DSB formation (Daniel et al, 2010; Stanlie et al, 2010; Begum et al, 2012; Stanlie et al, 2012). A number of chromatin‐associated proteins with a PHD zinc‐finger module are known to bind H3K4me3 (Bienz, 2006; Sanchez & Zhou, 2011). In V(D)J recombination, locus‐specific H3K4me3 marks are recognized by the RAG2 PHD motif, which promotes RAG1 recruitment and subsequently DNA cleavage (Matthews et al, 2007; Bettridge et al, 2017).
Recently, PHD motif‐containing Phf5a protein has been reported to be involved in embryonic pluripotency being associated with Paf1/Ski8 transcription elongation complex (Trappe et al, 2002; Strikoudis et al, 2016; Zheng et al, 2018). It has also been identified as part of a splicing complex, in which a novel Phf5a mutant mitigated the action of splicing inhibitors used in cancer therapy (Teng et al, 2017). Similarly, the function of Phf5a in splicing was found to be important for the survival of tumor but not normal cells (Hubert et al, 2013). In mouse spermatocyte, Phf5a has been reported to interact with splicing factor and helicases, but its function in meiosis remains undefined (Rzymski et al, 2008).
During a systematic screening of PHD finger‐containing proteins, we identified Phf5a as a novel CSR regulatory protein. Due to its reported association with transcription and splicing complexes, we initially hypothesized for the role of Phf5a in regulating switch germline transcripts that are important for CSR (Stavnezer et al, 2008). However, through a series of genetic and biochemical analysis, we revealed that Phf5a is involved in S region variant H2A chromatin remodeling, in particular H2AX and H2A.Z, which are required for optimal DNA damage response (DDR) and efficient DNA repair by NHEJ, respectively. Mechanistically, the structural integrity of the Phf5a zinc fingers is essential in stabilizing the p400‐H2A.Z histone chaperone complex, which is central to the regulation of AID‐induced CSR and genomic instability.
Results
Depletion of Phf5a strongly inhibits CSR and IgH/cMyc translocation
We applied siRNA‐mediated gene knockdown (KD) approach to screen a panel of candidate genes that are known or predicted to have a PHD zinc‐finger motif (Appendix Fig S1). This includes Phf5a, a small 110 amino acid protein with a central PHD zinc‐finger domain (Trappe et al, 2002; Sanchez & Zhou, 2011). To confirm the requirement of Phf5a in CSR, we depleted Phf5a in a mouse B‐cell line (CH12F3‐2A) that undergoes CSR with high efficiency when stimulated with CD40 ligand, IL‐4, and TGF‐β(CIT). Phf5a KD by small interfering RNAs (siRNAs) strongly impaired the IgM to IgA switching induced by CIT for 24 or 48 h (Fig 1A). We confirmed that the Phf5a protein was significantly depleted by the three siRNAs tested (Fig 1B). To verify the specificity of this effect, we designed a Phf5a construct whose transcripts were resistant to degradation by siPhf5a#3. In addition, to distinguish its expression from the endogenous Phf5a expression, we fused a Myc‐Flag epitope to the C‐terminus (Phf5aR‐MF). The inhibition of CSR by siPhf5a#3 was observed when the cells were co‐transfected with empty vector but not with Phf5aR‐MF (Fig 1C). As expected, Phf5aR‐MF was well expressed, while the endogenous Phf5a targeted by siPhf5a was significantly depleted (Fig 1D). Moreover, primary B cells stimulated by LPS or in combination with LPS and IL4 showed not only gradual increase in the expression of Phf5a as compared to non‐stimulated naïve B cells in a time‐dependent manner, but the timing also coincides with that of AID (Appendix Fig S2A). As expected, depletion of Phf5a in primary B cells impaired IgG1 as well as IgG3 switching without perturbing AID expression and switch germline transcription (Appendix Fig S2B and C). Taken together, these results validate an essential requirement of Phf5a in CSR.
Figure 1. Knockdown of Phf5a inhibits CSR.
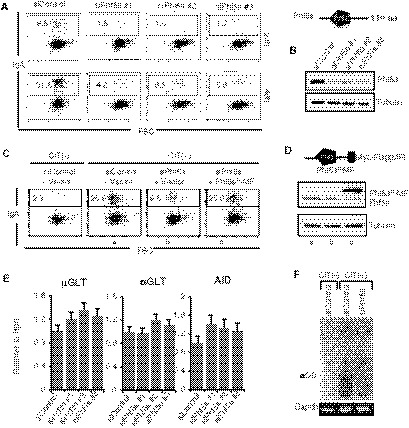
- Representative FACS profiles of IgA switching in CH12F3‐2A cells. The control and Phf5a siRNAs are indicated at the top. The number in each FACS profile indicates the percent (%) IgA+ cells at 24 and 48 h after CIT stimulation.
- Top: Illustration of Phf5a with a central PHD domain. Bottom: Confirmation of Phf5a KD by Western blot.
- CSR complementation assay using siRNA‐resistant Phf5a with Myc and Flag (MF) epitopes tagged at the C‐terminus.
- Schematic of MF‐tagged siRNA‐resistant Phf5a (Phf5aR‐MF). Asterisks (*) indicate the approximate locations of mutations in the cDNA made to generate the siRNA‐resistant version. Western blot confirmed the KD efficiency of the endogenous Phf5a, and the expression of Phf5aR‐MF.
- Quantitative RT–PCR (qRT–PCR) of µGLT, αGLT, and AID from siControl‐ and siPhf5a‐treated samples. The values are presented as mean ± SD (n = 3).
- Ethidium Bromide‐stained gels showing the PCR detection of excision circular DNA (αCD) and Gapdh as a control in the genomic DNA samples indicated.
Since the expression of AID and germline switch transcripts (µGLT and αGLT) were unperturbed in Phf5a‐depleted CH12F3‐2A cells (Appendix Fig 1E), we examined alpha circular DNA (αCD) production, a hallmark of the completion of IgM to IgA switching through looping out recombination mechanism at the genomic DNA level (Kinoshita et al, 2001; Dong et al, 2015). PCR‐amplified product of the Sµ and Sα junctions present in αCDs is normally detected as smear on a gel as the joining sites vary widely. Consistent with the impairment of IgA expression on the cell surface, αCD products were barely detectable in the samples from Phf5a‐depleted cells, whereas a thick smear of DNA derived from αCD PCR product was readily detectable in the control sample (Fig 1F). As expected, no αCD product can be detected in the absence of CIT stimulation.
We further confirmed the lack of productive or deletional recombination in the Phf5a KD cells by examining the recombined locus through the well‐documented Digestion Circularization PCR assay (Chu et al, 1992; Dong et al, 2015). Since CSR predominantly occurs through deletional recombination pathway, we observed a strong signal of direct Sµ‐Sα joining in the sample from CIT‐stimulated control cells, but not in the sample from CIT‐stimulated Phf5a‐depleted cells (Appendix Fig S2E and F). Inversional or non‐productive recombination also occurs, but at a low frequency, which does not produce excision circle DNA (Dong et al, 2015). Consistent with the low level of inversional joining, a weak signal was detected only in the control sample, suggesting that S‐S recombination involving deletion as well as inversion is severely impaired by Phf5a deficiency.
Since the Sµ–Sα recombination was strongly impaired in both orientations, we subsequently asked whether Phf5a depletion also impairs trans‐chromosomal recombination between IgH and other loci. To assess this possibility, we examined the frequency of chromosomal translocation between IgH and c‐Myc loci (Fig 2A), which routinely occurs during CSR activation (Ramiro et al, 2004; Nussenzweig & Nussenzweig, 2010). Following established protocol, translocated junctions were PCR‐amplified, followed by Southern hybridization with a locus‐specific probe. Similar to CSR impairment, the IgH/c‐Myc translocation was severely impaired by Phf5a KD (Fig 2B and C). Our group has previously showed that the depletion of Smarca4 and Top1 dramatically increases AID‐induced chromosomal translocation (Husain et al, 2016). Remarkably, the high frequency of IgH/c‐Myc translocation induced by Smarca4 or Top1 depletion was also counteracted significantly by Phf5a depletion (Fig 2B and C). Similar inhibitory effect was also evident in the case of two other chromatin remodelers, Bptf1 and Smarca2, whose KD led to an elevated level of both CSR and IgH/c‐Myc translocation frequency (Fig 2B and C). Therefore, we concluded that Phf5a potentially is a key regulator of both cis‐ and trans‐recombination.
Figure 2. Knockdown of Phf5a suppresses IgH/c‐Myc translocation.
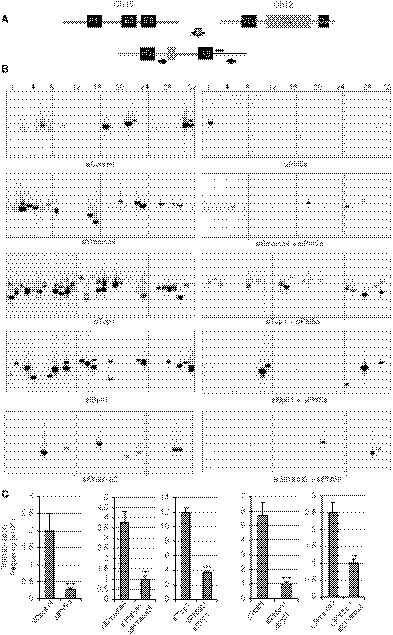
- Schematic representation of the IgH and c‐Myc chromosomal loci before and after translocation. Arrows and black bar indicate the positions of the PCR primers used to amplify the rearranged region and the probe for Southern hybridization, respectively.
- Southern blots of the IgH/c‐Myc translocation assay using genomic DNA extracted from CIT‐stimulated CH12F3‐2A cells treated with the siRNAs shown at the bottom of each panel. Numbers on the top indicate gel lanes.
- Frequency of IgH/c‐Myc chromosomal translocations derived from the number of translocations detected in the total number of PCRs per sample (Boboila et al, 2012b). The values are presented as mean ± SD (n = 3). Statistical significance was assessed by two‐tailed unpaired Student’s t‐test (**P ≤ 0.01 and ***P ≤ 0.001).
Phf5a is dispensable for SHM and DNA breaks at the IgH locus
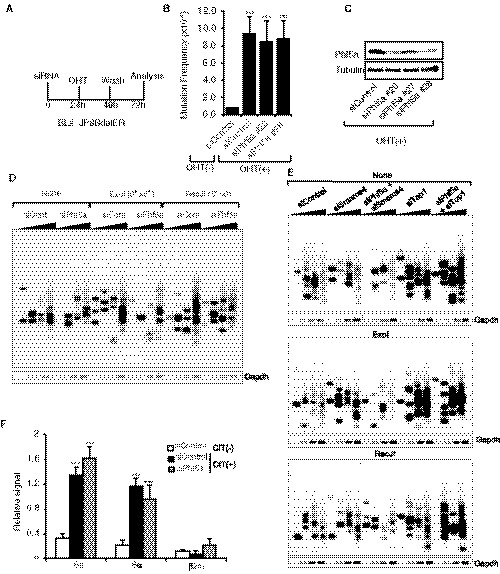
To clarify whether Phf5a also impacts AID‐induced SHM, a human B‐cell line (BL2) expressing a mutant AID fused to the estrogen receptor (JP8Bdel‐ER) was utilized (Nagaoka et al, 2005). In this cell line, activation of JP8Bdel‐ER by 4‐hydroxytamoxifen (OHT) induces high frequency of mutations in the recombined V(D)J region. Two (#20 and #28) out of the three siRNAs tested significantly reduced the Phf5a protein in BL2 line. However, neither of them showed a marked effect on the SHM frequency or mutational base bias (Fig 3A–C; Appendix Fig S3A and B). As CH12F3‐2A cells do not undergo SHM at the recombined V(D)J region, we instead initiated S region SHM analysis by sequencing the 5′ region of Sµ following 48 h of CIT stimulation (Appendix Fig S3C and D). The AID‐induced mutation frequency was found to be comparable between siControl‐ and siPhf5a‐treated samples, indicating the dispensability of Phf5a for both V and S region SHM.
Figure 3. Phf5a is dispensable for AID‐induced SHM and DNA breaks.
-
A–CSHM analysis of the variable (V) region of the human BL2 cell line expressing an AID C‐terminal mutant (JP8Bdel) fused to ER. Scheme shows the time points of siRNA transfection, the activation of JP8Bdel‐ER by OHT, and the genomic DNA isolation for mutation analysis (A). Mutation frequency at the V region after 48 h of AID (JP8Bdel) activation; plot represents the summary of three independent experiments (B). The values are presented as mean ± SD (n = 3). Statistical significance was assessed by two‐tailed unpaired Student’s t‐test (***P ≤ 0.001). Western blot shows the knockdown efficiency by individual siRNAs (C).
-
D, EDetection of Sµ DNA double‐strand breaks by LM‐PCR in CH12F3‐2A cells transfected with the indicated siRNA combinations. The DNA samples were either left untreated (none) or treated with indicated end‐processing enzymes prior to linker ligation.
-
FDetection of ssDNA breaks by DNA break end labeling. The DNA breaks were end‐labeled in situ by incorporating biotin‐16‐dUTP and TdT. The DNA was fragmented and labeled fragments were enriched by streptavidin pull‐down, followed by quantification by qPCR. The signal from the β₂‐Macroglobulin (β₂m) locus served as a negative control. The values are presented as mean ± SD (n = 3). Statistical significance was assessed by two‐tailed unpaired Student’s t‐test (***P ≤ 0.001). The two DNA break assays were illustrated in (Appendix Fig S3E, B and F).
Since both SHM and CSR are initiated by DNA breaks at the IgH locus, we directly examined the requirement of Phf5a for AID‐induced DNA breaks in CSR. We evaluated the S region DNA DSB formation by performing the well‐documented LM‐PCR assay (Appendix Fig S3E) (Schrader et al, 2005; Husain et al, 2016). However, LM‐PCR‐generated break signals were comparable between control and Phf5a‐depleted samples (Fig 3D, panel of none). We also treated genomic DNA with ssDNA‐digesting enzyme prior to linker ligation for additional sensitivity, but it also did not show any striking difference between siControl‐ and siPhf5a‐treated samples (Fig 3D, panels of ExoI and RecJ). LM‐PCR assay was also conducted for siSmarca4‐ or siTop1‐treated samples in the presence or absence of Phf5a siRNA (Fig 3E). Phf5a KD had little effect on the DSB signals produced in Smarca4‐depleted cells. A robust DSB signal was detected after Top1 KD, which was only minimally affected by Phf5a KD. Finally, we also assessed the single‐strand (ss) DNA break events using DNA end‐labeling assay (Doi et al, 2009) in which biotin‐dUTP is incorporated at the DNA break ends (5′) (Appendix Fig S3F). Streptavidin pull‐down of the biotin‐dUTP‐labeled DNA break ends, followed by PCR of the target region in the Sµ, enables ssDNA break incidence estimation. Similar to the DSB detection data, no alteration in the ssDNA break signal was observed after Phf5a KD (Fig 3F). Taken together, these data unequivocally exclude the involvement of Phf5a in the generation of AID‐induced DSBs at the IgH locus.
Phf5a is critical for the NHEJ‐dependent DNA repair in CSR
Since AID‐induced DSBs in CSR are mainly resolved through NHEJ‐mediated DNA repair, we embarked on exploring the role of Phf5a in NHEJ. Utilizing a well‐documented NHEJ assay coupled to I‐SceI‐mediated DNA breaks (Ogiwara et al, 2011; Stanlie et al, 2014), the effect of Phf5a KD on this specific DNA repair pathway was evaluated. In this assay, a GFP reporter gene is expressed only after a successful joining event between the two cleaved I‐SceI sites, which removes an intervening thymidine kinase (TK) gene that initially prevents the GFP expression (Fig 4A). The siRNA against Phf5a was introduced into the reporter cell line H1299dA3‐1 (Ogiwara et al, 2011) along with I‐SceI‐expressing plasmid. The number of GFP‐positive cells was dramatically reduced in Phf5a‐treated cells compared to control, indicating that Phf5a plays a key role during NHEJ (Fig 4B). Consistent with this finding, PCR product of the repaired I‐SceI break points (“Joined”) was barely observed in the genomic DNA isolated from Phf5a‐depleted cells (Fig 4C), which further confirmed that the NHEJ was severely defective. The KD efficiency of Phf5a by the individual siRNAs was well correlated with their inhibitory effect on NHEJ (Fig 4D). Utilizing a similar assay strategy, but with a homologous recombination (HR)‐dependent GFP reporter construct (Sakamoto et al, 2007; Stanlie et al, 2012), we did not detect any reduction in GFP expression after Phf5a depletion (Appendix Fig S4A–C). This indicates that Phf5a functions exclusively in NHEJ. Consistent with Phf5a’s role in NHEJ, a higher incidence of nucleotide insertions was found in the I‐SceI‐cleaved junctions derived from Phf5a KD cells (Fig 4E; Appendix Fig S5).
Figure 4. Phf5a depletion impairs NHEJ.
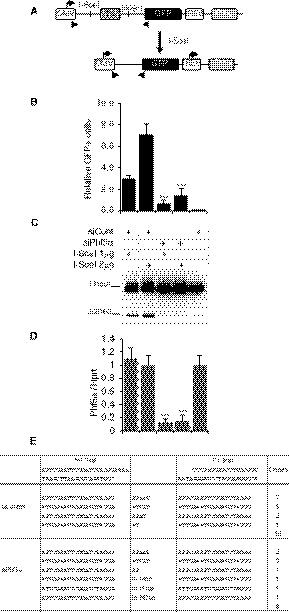
- Schematic diagram of the reporter construct for analyzing the DNA DSB repair by NHEJ. Incompatible DNA ends produced by I‐SceI‐induced DNA cleavage are repaired by NHEJ, which removes the intervening TK, resulting in GFP expression. Triangles indicate the positions of primers used to amplify a fragment encompassing the repaired junction after I‐SceI cleavage.
- Effect of Phf5a knockdown on I‐SceI‐induced NHEJ. Plot represents the GFP‐positive cells obtained by FACS analysis after co‐transfection of the I‐SceI expression plasmid and the indicated siRNAs into a cell line harboring the NHEJ reporter construct. The values are presented as mean ± SD (n = 3). Statistical significance was evaluated against corresponding siControl by two‐tailed unpaired Student’s t‐test (***P ≤ 0.001).
- Genomic DNA was isolated from each transfectant and subjected to PCR using the primer pair indicated in A.
- Confirmation of Phf5a KD in the samples by qRT–PCR. The values are presented as mean ± SD (n = 3). Statistical significance was evaluated against corresponding siControl by two‐tailed unpaired Student’s t‐test (***P ≤ 0.001).
- Representative NHEJ junctions obtained from siControl‐ and siPhf5a‐treated samples.
To confirm that Phf5a KD causes NHEJ defect in CSR, we examined the Sµ and Sα junction sequences of CH12F3‐2A cells stimulated for IgM to IgA switching. The CSR junctions were PCR‐amplified from the genomic DNA obtained from switched (IgA+) siControl‐ and siPhf5a‐treated cells. Many clones with a variety of unique junctional recombinations were readily obtained from the control sample (Fig 5A and B). However, multiple rounds of experiments were necessary to accumulate 60 of such junctions from Phf5a KD sample. As expected, the CSR junctions of the Phf5a‐depleted samples showed less direct joining (0–2 bp) and more microhomology (> 3 bp) and insertions (Boboila et al, 2012a; Chang et al, 2017). Moreover, our results derived from an adaptation of a DNA end‐resection assay (Gao et al, 2020) within a defined region of Sµ also suggest that there is an increase of DSB end resection in Phf5a‐depleted cells (Appendix Fig S4D and E). We therefore speculated that in the absence of Phf5a, the recruitment of NHEJ DNA repair factors might be defective during S‐S recombination. In support of this idea, chromatin immunoprecipitation (ChIP) analysis (Fig 5C) revealed a reduction in the recruitment of a key NHEJ factor, Ku80, which is known to protect DSB ends and promote Ligase 4‐mediated classical NHEJ (c‐NHEJ). The recruitment of DNA end‐processing enzymes Exo1 and Mre11 to the S region was compromised, but not completely depleted in Phf5a KD cells. However, upon Phf5a depletion, we did not observe significant difference in the recruitment of CtIP (Fig 5C), which is known to have both catalytic and noncatalytic roles in DSB end resection (Makharashvili et al, 2014). Non‐catalytically, CtIP enhances short‐ and long‐range resections mediated by MRN and helicase helicase–DNA2 complex, respectively, leading to diminished C‐NHEJ efficiency (Lee‐Theilen et al, 2011; Daley et al, 2017; Ceppi et al, 2020).
Figure 5. Impaired recruitment of NHEJ factors to the IgH locus upon Phf5a deficiency.
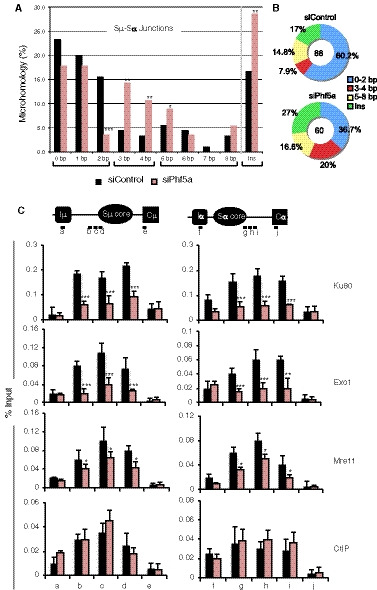
- Analysis of Sµ‐Sα recombination junctions in the genomic DNA isolated from siControl‐ and siPhf5a‐transfected CH12F3‐2A cells stimulated by CIT for 48 h. The values are presented as mean ± SD (n = 3). Statistical significance was evaluated in reference to siControl by two‐tailed unpaired Student’s t‐test (*P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001).
- The proportion of insertions versus microhomology regions of various lengths in the Sµ‐Sα junctions presented as a pie chart. The number of clones analyzed from control and Phf5a‐depleted samples is indicated at the center.
- Top: Schematic diagram of the position of the ChIP assay PCR products specific to the S region. Bottom: ChIP assay of the indicated DNA repair proteins using CIT‐stimulated CH12F3‐2A cells transfected with siControl or siPhf5a. The values are presented as mean ± SD (n = 3). Statistical significance was evaluated in reference to siControl by two‐tailed unpaired Student’s t‐test (*P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001).
Loss of Phf5a perturbed the H2A variant stoichiometry in the S region
To further investigate the cause of the NHEJ defect by Phf5a KD, we analyzed the early phase of DDR. Phosphorylation of H2AX (γH2AX) is a key step in DDR, which is required for the initial assembly of DNA repair proteins at the site of damage, including AID‐induced breaks in the S regions (Rogakou et al, 1999; Reina‐San‐Martin et al, 2003). In CH12F3‐2A cells, Phf5a KD decreased not only the γH2AX formation in the S regions but also H2AX deposition (Fig 6A and B). This finding prompted us to examine whether there was any anomaly in other histone H2A variant recruitment to the S regions after Phf5a depletion. We first examined the deposition of H2A.Z, which is known to be critical for NHEJ (Xu et al, 2012). Interestingly, the H2A.Z level was reduced at both Sµ and Sα, but not at the promoter regions upon Phf5a KD. Another H2A variant, H2A.Bbd, which is known to associate with actively transcribing loci (Tolstorukov et al, 2012), was also reduced. On the other hand, the recruitment of mH2A (macroH2A), which is associated with transcriptionally active loci (Gaspar‐Maia et al, 2013), was increased, further indicating that the overall deposition of H2A variants was perturbed upon Phf5a depletion. We confirmed that none of the H2A variants or core histones were decreased at the protein level in the Phf5a‐depleted cells (Fig 6C; Appendix Fig S6A). Similar observation was also made for several histone post‐translational modifications examined at both the overall expression level and their depositions in Sµ and Sα loci (Fig 6C; Appendix Fig S6A and B).
Figure 6. Knockdown of Phf5a alters the H2A variant deposition at the S region.
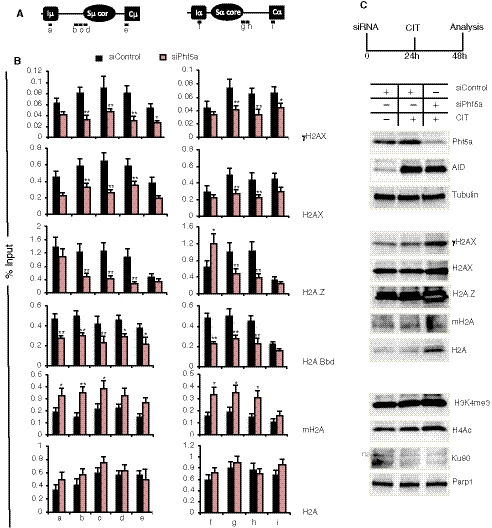
- Schematic illustration of the Sµ and Sα regions showing the position of the ChIP PCR products.
- ChIP analysis of H2A and its variants using CIT‐stimulated CH12F3‐2A cells transfected with siControl or siPhf5a. The antibodies used for each ChIP set are indicated at right. The values are presented as mean ± SD (n = 3). Statistical significance was evaluated in reference to the siControl by two‐tailed unpaired Student’s t‐test (*P ≤ 0.05 and **P ≤ 0.01).
- Western blot analysis of the whole‐cell extract from CIT‐stimulated CH12F3‐2A cells transfected with the indicated siRNAs. Expression of various proteins including AID, variant H2As, and Ku80 was not decreased by Phf5a KD (ns, non‐specific).
The NHEJ defect in Phf5a‐deficient cells corresponds to the H2A.Z deficiency
We hypothesized that, in addition to the defective DDR due to reduced γH2AX formation, the loss of H2A variants like H2A.Z caused a severe DNA repair defect, which led to impaired CSR and IgH/cMyc translocation. To test this scenario, we first verified the effect of H2A.Z KD on NHEJ using the same GFP reporter cell line used for Phf5a KD (Fig 4). Under identical conditions, the I‐SceI expression plasmid was co‐transfected with either siControl or siH2A.Z. The I‐SceI‐dependent increase in GFP‐positive cells was readily observed in the siControl‐treated samples, whereas GFP‐positive cells were hardly detectable in the siH2A.Z‐treated cells (Appendix Fig S4F). There was barely any detectable PCR product of the NHEJ‐mediated recombination from the isolated genomic DNA, in agreement with the strong blockage of NHEJ upon H2A.Z depletion (Appendix Fig S4G). The KD efficiency of H2A.Z also correlated well with these results (Appendix Fig S4H).
A co‐immunoprecipitation (co‐IP) analysis using Myc‐Flag epitope‐tagged H2A.Z (H2A.Z‐MF) confirmed that H2A.Z interacts with various DNA repair proteins (Appendix Fig S4I). The H2A.Z co‐IP not only pulled down Ku80, a bona fide NHEJ repair factor, but also other DNA repair‐associated factors like Mre11, CtIP, and Exo1, all of which are known to be involved in DNA end processing prior to end joining and in CSR (Dinkelmann et al, 2009; Eccleston et al, 2011; Lee‐Theilen et al, 2011). However, Msh2, a mismatch DNA repair protein involved in CSR, was not detected in the co‐IP under identical conditions, indicating that H2A.Z specifically associates with specific DNA repair factors, which is consistent with an earlier report (Obri et al, 2014).
Finally, we analyzed the repaired junctions derived from the I‐SceI‐cleaved NHEJ. Both Phf5a KD and H2A.Z KD samples showed an increased proportion of deletions and longer insertions in the junctions (Appendix Fig S5) as compared to the control sample. Taken together, these findings suggest that Phf5A is functionally linked with H2A.Z and NHEJ.
P400 deficiency phenocopied the Phf5a deficiency
To deplete H2A.Z from the IgH locus, we performed siRNA‐mediated KD of histone chaperone p400, which is responsible for H2A.Z deposition on chromatin in mammalian cells (Mizuguchi et al, 2004; Gevry et al, 2009). Knockdown of p400 impaired CSR in both CH12F3‐2A (CSR to IgA) and primary B cells (CSR to IgG1 and IgG3) without any significant reduction in AID expression or GLTs (Appendix Fig S7A‐D). Strikingly, the depletion of p400 strongly impaired IgH/c‐Myc translocation, similar to Phf5a KD (Appendix Fig S7E and F). Moreover, there was no reduction in the S region DNA breaks, reminiscent of our earlier observation with Phf5a KD (Appendix Fig S7G). Furthermore, p400 KD reduced γH2AX signal and also deposition of H2AX, H2A.Z, H2A. Bd, Ku80, and Exo1, but not CtIP at the S region (Appendix Fig S7H). Interestingly, just as observed in Phf5a deficiency, mH2A level was also elevated in p400‐depleted condition. In summary, p400 KD recapitulates the recruitment defect of H2A variants and DNA repair proteins as observed in the case of Phf5a deficiency.
The overwhelming and numerous similarities observed in depleting Phf5a or p400 prompted us to examine whether Phf5a KD altered p400 accumulation at the IgH locus. Remarkably, ChIP analysis revealed that Phf5a loss led to a decrease in the level of p400 in the S region, suggesting that Phf5a contributes to the stability of p400 in this region (Appendix Fig S7I). On the other hand, p400 depletion has no impact on the status of Phf5a in both Sµ and Sα.
Therefore, it can be concluded that the effect of Phf5a KD in AID‐induced genomic instability was manifested through the impairment of the p400‐H2A.Z pathway. The presence of Phf5a in the IgH locus is critical for p400‐mediated H2A.Z deposition that would in turn promote NHEJ and subsequently CSR.
Identification of loss‐of‐CSR‐function mutants of Phf5a
To gain insight into how Phf5a functions to stabilize a large complex like p400, we hypothesized that the non‐canonical PHD zinc‐finger structure of Phf5a may be required for the stable chromatin complex formation. If this is indeed the case, it should be feasible to generate loss‐of‐CSR‐function mutants by mutating the PHD domain of Phf5a. While it is common for a PHD motif to contain 2 zinc fingers consisting of 4 CxxC motifs in a cross‐braced conformation and spanning 50–80 amino acid residues (Sanchez & Zhou, 2011), Phf5a, which is only 110 amino acids long, uniquely possesses 13 cysteine (Cys) residues including five CxxC motifs (Fig 7A; Appendix Fig S8A–C). Therefore, the predicted PHD motif of Phf5a contains 3 zinc fingers (ZF1‐ZF3) and forms a closed ring structure involving all of the CxxC motifs and 2 additional Cys residues (Fig 7A; Appendix Fig S8C). We mutated each of the CxxC motifs and every Cys residue individually by substituting it to alanine (Ala) (Appendix Fig S8B). Unlike the wild‐type Phf5a, none of the CxxC motif mutants showed any CSR complementation, despite their sufficient expression level (Fig 7B; Appendix Fig S8D and E). On the other hand, the single Cys mutants showed a spectrum of CSR complementation, including complete CSR loss in the case of C23A (ZF1), C30A (ZF2), and C72A (ZF2) mutants (Fig 7B; Appendix Fig S8F and G). The C72xxC75 motif of ZF2 showed an interesting phenomenon—while the C72A mutant failed to complement CSR, the C75A mutant fully complemented CSR, indicating that the loss of CSR activity of the C72xxC75 mutant was due to C72A mutation. These mutants also showed better expression stability compared to the C23A and C30A mutants or their CxxC motif double mutants (C23A/26A, C30A/C33A). Therefore, the single C72A mutant in ZF2 was detrimental for CSR without having an adverse effect on the stability of the protein. We considered C72A mutation as the important CSR loss‐of‐function mutant which will be analyzed further for functional analysis.
Figure 7. The Phf5a mutants with a loss of CSR function show defects in protein–protein interactions.
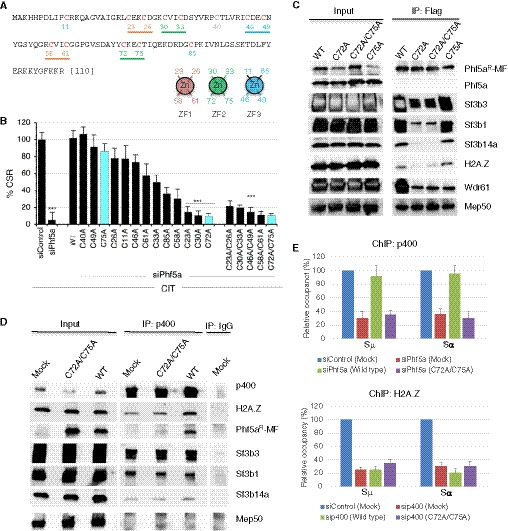
- Amino acid sequence of mouse Phf5a protein. The 12 Cys residues indicated in red are part of the PHD finger‐like domain. Each CxxC motif is underlined and color coded according to their pairing with another CxxC motif to form a zinc finger (ZF). A pair of CxxC motifs holds the zinc atom in each ZF. Arrangement of 4 Cys residues in ZF1‐3 is schematically shown below the Phf5a sequence.
- CH12F3‐2A cells were transfected with siRNAs along with empty vectors or vectors expressing various zinc‐finger mutants of Phf5a (Phf5aR‐MF). Twenty‐four hours after transfection, the cells were stimulated by CIT for another 24 h and then examined for CSR. The efficiency of IgA switching was calculated relative to the value obtained for siControl and empty vector‐transfected cells. The data are compiled from three independent experiments. The values are presented as mean ± SD (n = 3). Statistical significance was performed in reference to siControl by two‐tailed unpaired Student’s t‐test (***P ≤ 0.001). See Appendix Fig S8 for a representative data set.
- CH12F3‐2A cells were transfected with WT Phf5a or its mutants (Phf5aR‐MF) along with siRNA against Phf5a. An anti‐Flag antibody was used to immunoprecipitate and immunodetect Phf5aR‐MF. An anti‐Phf5a immunoblot confirmed the knockdown efficiency of the endogenous Phf5a. The antibodies used to detect the co‐associated proteins are indicated next to the respective blot.
- CH12F3‐2A cells transfected with WT or a mutant Phf5a (Phf5aR‐MF) were subjected to anti‐p400 immunoprecipitation. Associations with relevant interacting proteins were examined by immunoblot analysis and are indicated next to each blot.
- ChIP assay derived from CH12F3‐2A cells transfected with the indicated siRNA and Phf5a (Phf5aR‐MF) expression constructs. Antibodies used for ChIP are indicated on the top of the respective plot. The data are summarized from 2 independent experiments and presented relative to siControl‐treated sample, which was set as 100. The error bars represent the mean ± SD.
To verify mutations outside the CxxC frame work, we performed serial truncations at the C‐ or N‐terminus of Phf5a. The C‐terminus truncation mutants that did not encompass any Cys residue were fully functional in CSR. A similar observation was made with a mutant truncated at the N‐terminus (ΔN5); however, longer deletions including Cys11 dramatically affected protein expression, which correlates with the progressive loss of CSR efficiency (Appendix Fig S8A and B).
In order to avoid deleterious effect due to truncations, we examined several point mutants that destroyed potential phosphorylation and acetylation sites in Phf5a. All of the phospho‐site mutants were CSR proficient (Appendix Fig S9A and B), including Y26A and Y26C, which are frequently found in cancer cells, making them resistance to the splicing inhibitors (Teng et al, 2017).
To gain insight into the effect of Phf5a acetylation, we specifically investigated mutation at position K29 (Appendix Fig S9A), which has been reported to impair histone demethylase Kdm3a expression and subsequently deregulate stress‐induced gene expression (Wang et al, 2019). Although we did not observe any change in Kdm3a gene transcription after Phf5a KD in CH12F3‐2A cells (Appendix Fig S15), we still verified the impact of K29 mutation in CSR. The two mutants, K29R and K29Q, corresponding to acetylation defective and mimetic, respectively, were found be CSR proficient (Appendix Fig S10A). We also confirmed by immunoprecipitation (IP) that Phf5a does not undergo acetylation during CSR in CH12F3‐2A cells (Appendix Fig S10B and C).
Interestingly, when we introduced a Cys mutation such as C23A, both the mutants (K29R/Q) lost CSR complementation ability (Appendix Fig S10A), suggesting the importance of CxxC domain in CSR. Since mutation in any of the five CxxC motifs led to complete failure in complementing CSR, we speculated that the trefoil knot (van Roon et al, 2008; Teng et al, 2017) structure formed by the three ZFs is critical for a stable protein complex formation that is necessary for CSR.
Phf5a forms a chromatin complex with p400 and Sf3b components
Next, we sought to identify the Phf5a‐associated protein complex and to understand its association with chromatin and the p400 complex. To perform efficient IP, we expressed Phf5aR‐MF in CH12F3‐2A cells and performed Flag‐IP using nuclear and chromatin‐bound fractions (Appendix Fig S11A). Mass spectrometry (MS) analysis of IPed proteins identified spliceosomal subunits and additional proteins including chromatin proteins and histones (Appendix Table S1). Careful examination of the interacting proteins indicated that they were components of various large complexes such as major and minor U2snRNPs, RNA methylation, p300, Paf1, and senataxin complexes (Appendix Fig S11B). This observation is consistent with previous reports that found the association of Phf5a with functionally different protein complexes (Strikoudis et al, 2016; Teng et al, 2017; Wang et al, 2019).
In order to further dissect the relevance of the identified Phf5a complexes in the context of CSR, we selected notable Phf5a‐interacting partners from each protein complex, such as Sf3b1, Sf3b3, and Sf3b14a from the U2snRNP complex, Mep50 from the RNA methylation complex, Wdr61 from the Paf1/Ski8 transcriptional complex, and H2A.Z from the p400 complex, and subsequently compared their interaction with wild‐type Phf5a and the loss‐of‐CSR‐function mutant. We selected C72A and C72A/C75A mutants that were unable to support CSR and C75A mutant that retained the CSR activity that is comparable to that of wild‐type Phf5a (Fig 7A and B). Co‐IP analysis revealed that both loss‐of‐function mutants had significantly reduced interaction with the Sf3b subunits and with H2A.Z (Fig 7C). In contrast, these mutants interacted well with Mep50, suggesting that Mep50 or its associated RNA methylation complex is not involved in Phf5a‐dependent CSR. Similarly, the involvement of Wdr61 in CSR was also excluded, as all of the mutants regardless of their CSR complementation efficiency showed a similar level of interaction with this protein (Fig 7C). In addition, we did not detect Ctr9, a key component of the Paf1 complex (Xie et al, 2018), in our Phf5a co‐IPed proteins (Appendix Table S1). Therefore, the possible involvement of Wdr61 or Ctr9 (Paf1/ski8 complex) in Phf5a‐associated CSR was nullified. Consistent with these findings, the KD of Mep50 or Wdr61 did not significantly affect CSR.
The loss of interaction of either C72A or C72A/C75A mutant with the Sf3b subunits and H2A.Z correlates well with the failure to complement CSR by these mutants, thus suggesting a link between the H2A.Z‐p400 complex and the U2snRNP complex. To examine this possibility, we conducted co‐IP analyses of p400. The data confirm that p400 indeed interacts with wild‐type Phf5a, but not with its loss‐of‐function mutant counterpart (Fig 7D). Importantly, p400 IP also pulled down Sf3b1, Sf3b3, and Sf3b14a, indicating that the p400‐H2A.Z complex is associated with Phf5a‐U2snRNP complex. Phf5a depletion therefore diminishes the interaction of p400 with the Sf3b subunits but not with H2A.Z or H2AX (Appendix Fig S12). On the other hand, p400 complex did not interact with Mep50, which was confirmed to be dispensable for CSR.
Finally, since we observed that the stability of p400 in the S region depends on Phf5a (Appendix Fig S7I), we examined whether the loss‐of‐CSR‐function mutant (C72A/C75A) fails to stabilize the p400‐H2A.Z complex in the IgH locus. We performed p400 ChIP analysis in Phf5a‐depleted condition (siPhf5a) co‐transfected with siRNA‐resistant wild‐type and mutant Phf5a. As expected and consistent with the IP result, p400 enrichment at the switch regions was observed only in the presence of wild type but not in loss‐of‐function Phf5a mutant (Fig 7E). No enrichment of H2A.Z was observed when p400 was depleted. A similar CSR complementation experiment was also conducted through the expression of C72A/C75A and C75 mutants, which are CSR deficient and proficient, respectively (Fig 8A and B). The C72/C75 mutant was unable to restore CSR and the occupancy of H2A.Z at the IgH locus in endogenous Phf5a‐depleted condition. The C75 mutant, however, was capable to fully restore both CSR and H2A.Z status under identical experimental setup, indicating a critical requirement of Phf5a ZF2 in CSR and epigenomic regulation (Fig 8C and D). Taken together, we conclude that Phf5a‐p400 crosstalk contributes to not only H2A.Z and H2AX regulation, but also DNA repair factor deposition in the S region. These concerted modulations are in turn required to elicit optimal DDR, promote NHEJ, and eventually drive CSR to its completion (Fig 8E).
Figure 8. Phf5a zinc finger‐2 (ZF2) plays an important role in CSR and epigenomic regulation required for DNA repair.
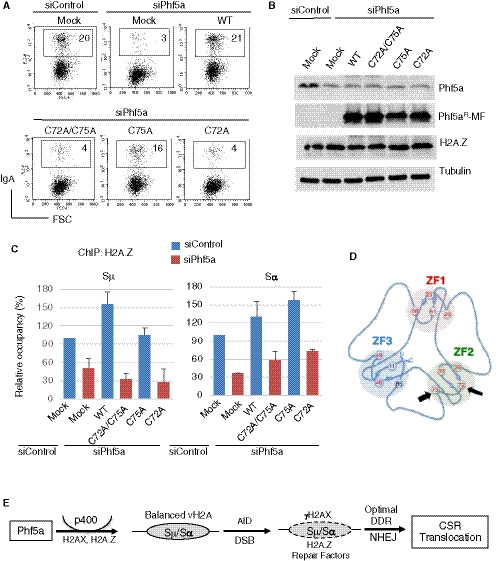
- A representative FACS profile of CSR complementation assay using WT and mutant Phf5 as indicated and describe in Fig 7B.
- Western blot showing expression of endogenous and exogenously introduced Phf5a.
- H2A.Z ChIP assay was performed using CH12F3‐2A cells transfected with indicated siRNA and the Phf5a expression constructs. The data are compiled from 2 experiments and presented relative to siControl‐treated sample, which was set as 100. The error bars represent the mean ± SD.
- The schematic illustration of Phf5 zinc fingers (ZF1‐3) was adapted from van Roon et al, 2008. The Cys75 and Cys72 are indicated by thick and thin arrows, respectively.
- Illustration depicting AID‐induced DNA breaks and repair in the S region, which leads to CSR and IgH/c‐Myc chromosomal translocation. The scheme also highlights the involvement of the Phf5a‐p400 pathway, which promotes H2A.Z and H2AX deposition in the recombining S regions and facilitates NHEJ‐mediated DNA repair.
Discussion
The DNA damage response and the subsequent choice of DNA repair pathway shape the genomic integrity of a cell type. In antigen‐activated B cells, the IgH locus undergoes AID‐induced programmed genomic alterations to diversify the antibody gene locus, which requires extensive involvement of DNA repair and recombination mechanism (Stavnezer et al, 2008). Although AID‐induced DNA breaks predominantly target the IgH locus, considerable DNA breaks in non‐IgH loci can also occur (Ramiro et al, 2004; Nussenzweig & Nussenzweig, 2010). NHEJ is the key DNA repair pathway that plays a central role in resolving these breaks, leading to either programmed recombination such as CSR or deleterious joining with the potential to drive oncogenic rearrangements (Ramiro et al, 2004; Nussenzweig & Nussenzweig, 2010). In this process, the chromatin structure at the target locus serves as a docking platform for the recombination‐repair complex and any perturbation of the local chromatin structure or epigenomic state may cause inefficient DNA repair and subsequently impair CSR.
In the present study, we demonstrate that Phf5a is a novel chromatin regulator, which promotes p400‐mediated IgH locus chromatin remodeling and impacts cis‐ and trans‐recombination during CSR and IgH/cMyc translocation, respectively. Phf5a is not involved in SHM or AID‐induced DNA breaks, but is critical for NHEJ‐mediated DNA repair during CSR. Surprisingly, we found that Phf5a affects NHEJ by regulating variant H2A deposition, and consequently other repair proteins, at the target locus. The loss of Phf5a down‐regulates the recruitment of two critical H2A variants, H2AX and H2A.Z, which are intimately linked to the initiation of DNA damage repair (Foster & Downs, 2005; Bonisch & Hake, 2012). Immediately after DSB formation, ATM‐dependent phosphorylation of H2AX spreads rapidly over a large area surrounding the DSB. γH2AX formation is critical for activating a series of downstream repair cascades to promote DNA repair via homologous or non‐homologous recombination. On the other hand, H2A.Z, which plays a role downstream of H2AX phosphorylation, contributes exclusively to NHEJ. The presence of H2A.Z at the breakpoint is known to promote DNA repair in multiple ways—firstly, H2A.Z facilitates the recruitment of Ku complex to the site of DSBs on chromatin, which further promotes the recruitment of other NHEJ factors, such as DNA‐PKcs and the Xrcc4–Xlf–DNA ligase 4 complex, to complete the DNA end joining process (Xu et al, 2012; Jiang et al, 2015). Secondly, H2A.Z increases the chromatin accessibility to DNA end‐processing enzymes such as Exo1 (Adkins et al, 2013). Thirdly, the presence of H2A.Z flanking DSB has been suggested to determine the upstream and downstream boundaries of the DNA break, which in turn may protect DNA ends from excessive resection to promote NHEJ.
Consistent with the DSB resolution defect, increased insertions and deletions (indels) were evident in both CSR and I‐SceI‐cleaved junctions upon Phf5a depletion. Indeed, KD of either Phf5a or H2A.Z reduced the NHEJ efficiency dramatically (Fig 4), and the frequency of indels at the DNA repaired junctions was significantly increased in either Phf5a‐ or H2A.Z‐deficient cells (Fig 4; Appendix Fig S5), suggesting their involvement in a common pathway during NHEJ (Ogiwara & Kohno, 2011). The high incidence of long insertions and deletions may reflect an alternative mode of DNA repair such as homology‐directed or copy‐paste‐mediated mechanism while attempting to “rescue” the resected DNA break ends (Onozawa et al, 2014; Iliakis et al, 2015). We envisage that the loss of Phf5a/p400 leads to impaired DDR and consequentially the recruitment defect of crucial DSB end shielding factors like Ku80, 53BP1, and others (Fig 5 and unpublished). Moreover, CtIP level remains unchanged, which might promote DNA end resection directly or indirectly with the available MRN/DNA2 complex, especially when the S region DSBs are no longer flanked by sufficient H2A.Z (Sartori et al, 2007; Xu et al, 2012).
Activation of AIDER, which induces DNA breaks in the S regions, led to the accumulation of γH2AX, Ku80, and Exo1, but not of H2AX or H2A.Z at these sites (Appendix Fig S13A). Using AID‐deficient CH12F3‐2A cells, we further confirmed that Phf5a, but not AID activation, regulates H2A.Z and p400 level in the IgH locus (Appendix Fig S13B). The finding may suggest that the S regions are preloaded and/or continuously supplied with histone H2A variants. Indeed, CSR is also strongly affected by the KD of H2A.Z chaperone p400. Unexpectedly, H2AX was found to interact with p400, and its recruitment was reduced upon p400 or Phf5a KD, suggesting that p400 is also involved in H2AX deposition in the S region. Although p400‐dependent deposition of the H3.3 variant has been reported, we were unable to detect any alteration of H3.3 level in the S region by depleting Phf5a or p400. However, Phf5a or p400 KD affected the deposition of two additional H2A variants, H2A.Bbd and macroH2A (Tolstorukov et al, 2012; Gaspar‐Maia et al, 2013). These observations collectively suggest that the Phf5a‐p400 axis modulates the H2A variant stoichiometry in the S region.
We revealed that Phf5a promotes stabilization of the p400 histone chaperone complex in the S region. Based on an extensive mutagenesis study, we also concluded that the zinc‐finger motifs of Phf5a are critically involved in CSR through interaction with p400 and Sf3b components (Fig 7; Appendix Fig S12). The unique trefoil knot structure of Phf5a may play a role in its association with large, multi‐protein complexes. However, the interaction of Phf5a with certain protein complexes such as the RNA methylation complex, Paf1/Ski8, or senataxin was found to be irrelevant in the context of CSR and H2A variant regulation. Although Phf5a strongly interacts with Sf3b splicing complex, KD of the individual subunits of this complex reveals that the role of Sf3b complex on CSR is distinct from that of Phf5a. For instance, KD of various Sf3bs but not of Phf5a drastically inhibited AID and p400 expression and/or cell proliferation (Appendix Fig S14).
Interestingly, however, the Sf3b subunits were co‐IPed with p400 along with Phf5a, H2A.Z, and H2A.X, suggesting that they are also part of the p400 complex, which is distinct from their association with the U2snRNP complex (Fig 7; Appendix Fig S12). Indeed, it is not unusual for Sf3b components of the U2snRNP complex to have splicing‐independent functions. For instance, Sf3b1 has been reported for its role in DNA repair and direct interaction with histones (Martinez et al, 2001; Heo et al, 2007; Kfir et al, 2015). Similarly, both Sf3b3 and Sf3b5 subunits are active components of the SAGA chromatin‐remodeling complex (Stegeman et al, 2016). Our work also raises an interesting possibility as to whether Phf5a impacts the stabilization of SAGA deubiquitination module, which has been reported to promote DNA repair and CSR (Ramachandran et al, 2016). Taken together, we favor the idea that the PHD zinc fingers of Phf5a anchor p400 complex along with the Sf3b subunits to the chromatin at the site of DSBs, especially since Phf5a has been described to bind to H3K4me3 (Zheng et al, 2018).
Although Phf5a interacts with U2SnRNP splicing complex, it did not inhibit AID/p400 expression, which suggested a non‐overlapping role of Phf5a with the major spliceosomal subunit Sf3b1. Since Phf5a is involved in the DNA repair phase of CSR, we analyzed more than 26 genes, including essential and key repair factors associated with CSR—for a possible splicing defect—by RT–PCR (Appendix Fig S15A). Both siControl‐ and siPhf5a‐treated samples showed identical PCR products sizes, confirming that the pre‐mRNA splicing of those genes was unperturbed upon Phf5a depletion (Appendix Fig S15B).
Although the switch germline transcript does not code for any protein, splicing of GLT has been implicated to play an important role in CSR (Lorenz et al, 1995; Hein et al, 1998; Marchalot et al, 2020). We therefore first examined the splicing of µGLT and H2A.Z and their associated transcripts in detail (Appendix Fig S16A–C), followed by comparing the unspliced versus spliced transcript for αGLT, AID, and p400 (Appendix Fig S16D and E). This analysis indicates that the processing of these transcripts was not affected by Phf5a depletion. On the other hand, Sf3b1 depletion or splicing inhibitor treatment affected expression of several genes important for CSR (Appendix Figs S14, S16E and S17). Interestingly, although not a single gene analyzed showed any reduction in the protein level, we noticed an increase in the protein expression of specific histones and their post‐translational modifications (Fig 6, Appendix Fig S6). We therefore hypothesize a unique yet possible transcriptional regulation by Phf5a similar to that of pluripotent cells (Strikoudis et al, 2016).
It is worth noting that Phf5a acetylation at position K29 has recently been reported to induce an alternatively spliced Kdm3a gene that deregulates gene expression in human tumor (Wang et al, 2019). We neither found Phf5a acetylated in CH12F3‐2A cells nor any aberrant splicing or transcriptional abnormalities in Kdm3a (Appendix Figs S10B and C, and S15A and B). This further suggests that the function of Phf5a in regulating specific subset of genes is cellular context‐dependent as previously observed (Hubert et al, 2013; Strikoudis et al, 2016), presumably due to its unique structure that allows dynamic interactions with distinct protein complexes in various physiological settings.
In conclusion, the functional similarity between Phf5a and p400, in the context of AID‐induced genomic instability, revealed an unprecedented function of Phf5a that links variant H2A regulation, DDR, and NHEJ (Fig 8F). Phf5a is expressed ubiquitously and is indispensable for NHEJ, a key DNA repair pathway in mammalian cells, and thus is important for global genomic integrity regulation. Phf5a can therefore be designated as a novel NHEJ co‐factor required for recombination at both IgH and non‐IgH loci. The regulation of H2A.Z deposition by Phf5a has immense importance as this particular histone variant has numerous functions in DNA repair and transcription (Bonisch & Hake, 2012; Xu et al, 2012; Hauer & Gasser, 2017). In addition, Phf5a KD also affected the deposition of H2A variants other than H2A.Z due to the perturbation of the Phf5a‐p400 pathway. Since the dynamics and interplay of locus‐specific H2A variants on chromatin are critical to maintain various activities including development, differentiation, and even malignancies (Gaspar‐Maia et al, 2013; Skene & Henikoff, 2013; Buschbeck & Hake, 2017), Phf5a‐mediated H2A variant regulation may have much broader implications than its impact on DNA repair and genomic instability.
Materials and Methods
B‐cell culture and CSR assay
For CSR‐related study, a derivative of mouse B‐cell line (CH12F3‐2A) expressing Bcl2 was used throughout the study (Nakamura et al, 1996; Stanlie et al, 2010). The cells were cultured and maintained in RPMI supplemented with glutamine, NCTC, FBS (10%), β‐mercaptoethanol, and penicillin/streptomycin. To induce IgM to IgA class switching, CH12F3‐2A cells were stimulated for 24–48 h by CIT, a cytokine cocktail consisting of anti‐CD40, IL‐4, and TGF‐β (Nakamura et al, 1996). The surface expression of IgM and IgA was examined by staining the cells with FITC‐conjugated anti‐mouse IgM and PE‐conjugated anti‐mouse IgA. Propidium iodide (PI) staining was included to exclude the dead cells. The flow cytometric analysis was performed with a BD FACSCalibur instrument, and the data were analyzed by CellQuest software (BD Biosciences).
A human BL2 cell line expressing AIDER was cultured in RPMI supplemented with Glutamine, MEM‐NEAA, Na‐Pyruvate, FBS (10%), β‐mercaptoethanol, and penicillin/streptomycin. To generate cells that stably expressed AIDER (Nagaoka et al, 2005; Doi et al, 2009), the cells were initially selected in puromycin to obtain clonal lines.
Gene Knockdown in CH12 and BL2 cells
To knock down Phf5a and other genes, gene‐specific stealth siRNAs (Invitrogen) were used. The SF Cell Line 96‐well Nucleofector Kit (Lonza) was used to introduce individual siRNAs or mixtures of 3 siRNAs per target gene into cells. The program #CM150 and #DS‐150 was used to electroporate CH12F3‐2A and BL2 cells, respectively.
Primary B‐cell culture and siRNA transfection
Splenocytes from 6‐ to 8‐week‐old C57BL/6 mice were prepared following standard procedure. After red blood cell removal, B cells were enriched by negative selection using BD IMag Kit. Gene‐specific knockdown was achieved by introducing by 20–40 pmoles of stealth siRNA (Thermo Fisher) following the instruction of 96‐well shuttle protocol for stimulated mouse B cells (Lonza). Briefly, 20–40 pmoles of siRNA was introduced to 6–7 × 105 B cells suspended in Nucleofector solution and using electroporation program # 96‐DI‐100. Primary B‐cell culture, stimulation for isotype switching (IgG1, LPS + IL4; IgG3, LPS), and surface IgG staining were described previously (Nagaoka et al, 2002; Shinkura et al, 2004; Muramatsu et al, 2007).
Constructs and mutagenesis
The Phf5a cDNA was obtained by RT–PCR of the CH12F3‐2A cell RNA and cloned into the SgfI/MluI sites of the pCMV6‐Entry mammalian expression vector (OriGene). The Myc (M) and Flag (F) epitopes were fused at the C‐terminus of Phf5a to generate Phf5a‐MF. To generate siRNA‐resistant Phf5a transcripts, the sequence representing the Phf5a siRNA#3 was modified in the cloned cDNA by introducing multiple point mutations (Appendix Table S2), without altering the encoded amino acids. This construct was designated as Phf5aR‐MF (https://benchling.com/s/seq‐Nab3VUShNRV5ooJkR46U), which was later used to generate a series of Cys to Ala mutants by point mutagenesis. The QuickChange Site‐Directed Mutagenesis Kit (Agilent Technologies) was used for these changes. All Phf5a constructs used in this study are listed in Appendix Table S2.
RT–PCR/qPCR
Total RNA was extracted from B cells using TRIzol (Gibco BRL), and the cDNA was synthesized using Superscript III and Oligo (dT) primer (Thermo Fisher). The real‐time PCR was performed using the PowerUp SYBR Green Master Mix (Applied Biosystems) and specific primer pairs. Using CH12F32A cell RNA, the qRT–PCR of µGLT, αGLT, and AID was previously described (Stanlie et al, 2010; Stanlie et al, 2012) and primers are listed in Appendix Table S3. The coding sequence (CDS/ORF) of 27 genes and the housekeeping control genes were PCR ‐amplified using KOD FX Neo DNA polymerase (TOYOBO) (Appendix Fig S15A). Total RNA (1 µg) extracted from siRNA‐transfected and CIT‐stimulated CH12F3‐2A cells was reverse‐transcribed, and 1 µl of 10× diluted RT sample was subjected 30 cycles of PCR. The qRT–PCR of these genes was normalized against tubulin and using 2−ΔΔCT method (Appendix Fig S15B). Primers for Appendix Fig S15A and B are listed in Appendix Table S4 and Appendix Table S5, respectively. The RT–PCR/qRT–PCR primers related to gene splicing and/or transcription (Appendix Figs S16 and S17) are listed in Appendix Table S6.
Detection of excised circle DNA
The excised circular DNA (αCD) produced after Sµ‐Sα recombination was detected by PCR amplification of the various Sµ‐Sα junctions present in the excised circle (Kinoshita et al, 2001). CH12F3‐2A cells were transfected with siControl or siPhf5a and stimulated by CIT for 24 h. The genomic DNA was then isolated from each transfectant, and 50 ng of the DNA was subjected to LaTaq (Takara) PCR using the appropriate forward and reverse primers designed from Sα and Sµ, respectively. After gel electrophoresis, the PCR products were visualized by EtBr staining.
SHM analysis in BL2 and CH12F3‐2A cells
A human B‐cell line BL2 stably expressing ER‐fused C‐terminally truncated AID (JP8Bdel‐ER) was used for SHM analysis of the rearranged V(D)J region (Nagaoka et al, 2005). The cells were transfected with control or Phf5a siRNA and then 24 h later were stimulated with 1 mM OHT for another 24 h. The cells were then transferred to fresh medium and cultured for an additional 48 h without OHT. The total RNA was isolated from the cells and subjected to RT–PCR using PrimeSTAR HS DNA polymerase (TaKaRa). A 426‐bp fragment representing the variable region (VH4–39DJH5) was cloned as described (Denepoux et al, 1997).
In order to analyze SHM in the S region, genomic DNA was isolated from CH12F3‐2A cells treated with siControl or siPhf5a and stimulated with CIT for 24 h. A 564 bp region, 5′ of the core Sµ, was PCR‐amplified, cloned, and sequenced as described previously (Nagaoka et al, 2002). Several independent clones were sequenced for V‐SHM and Sµ‐SHM analyses. Clones with unique mutations were counted, and the mutation frequency was calculated from the number of mutations per total bases analyzed.
Detection of IgH/c‐myc translocation
CH12F3‐2A cells transfected with control or gene‐specific siRNA were stimulated with CIT 48 h prior to genomic DNA isolation. PCR amplification of the IgH/c‐Myc translocation junctions (der15) was performed as described previously (Ramiro et al, 2004; Boboila et al, 2012b). In brief, using the appropriate set of primer pairs and the Expand Long Taq PCR system (Roche) two rounds of PCR were performed. Each of the PCRs used 750 ng of gDNA isolated from individual samples, and the PCR condition and cycles were same as described previously (Boboila et al, 2012b). The samples were separated by gel electrophoresis, and then, amplified PCR products were subjected to Southern hybridization using a cMyc probe.
Chromatin immunoprecipitation assay
ChIP analysis of histones, Phf5a, p400, and various DNA repair proteins was performed using the ChIP‐IT Express Kit (Active Motif) and as described previously (Stanlie et al, 2010; Stanlie et al, 2014). Real‐time PCR of the input DNA and the ChIPed DNA was performed using the PowerUp SYBR Green Master Mix (Applied Biosystems) in duplicate, and each ChIP was performed twice or thrice. The enrichment of H2A family histones and other proteins at the IgH loci was normalized to the input DNA. The background signals obtained by IgG ChIP in the respective genomic locations were subtracted. The antibodies for ChIP are listed in Appendix Table S2, and the primers used are described previously (Stanlie et al, 2010; Stanlie et al, 2014; Husain et al, 2016).
NHEJ and HR assays
The NHEJ and HR assays were performed as described previously (Stanlie et al, 2014). In brief, The I‐SceI meganuclease‐expressing plasmid (pCBASce) and the siRNA of interest were co‐transfected into NHEJ or HR reporter‐expressing cell lines, using Lipofectamine 2000 (Invitrogen). The two cell lines H1299dA3‐1 (Ogiwara & Kohno, 2011) and GM7166VA (Sakamoto et al, 2007) were a kind gift from Dr. T. Kohno at the National Cancer Center Research Institute, Tokyo. Transfected cells were harvested 48 h later for FACS analysis and genomic DNA isolation. To examine the NHEJ junctions, duplicate PCRs were performed using 100 ng of genomic DNA and Pyrobest DNA polymerase (Takara). The PCR conditions were 95°C for 5 min; and 98°C for 10 s and 68°C for 2 min for 20 cycles. Genomic DNA isolated from the cells that underwent successful NHEJ produced two major products representing the intact (~1.4 kb) and the recombined (~400 bp) target regions. The 400‐bp fragment was cloned, sequenced, and analyzed in reference to the inserted I‐SceI sites in the reporter gene (Ogiwara & Kohno, 2011; Ogiwara et al, 2011).
Immunoblotting and immunoprecipitation
For the routine immunodetection of various proteins in CH12F3‐2A cells, whole‐cell extract (WCE) was prepared by lysing 1 × 106 cells in 500 µl of WCE buffer (30 mM Tris–HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 10% glycerol, 1% Triton X‐100, and 0.05% Na‐deoxycholate) supplemented with EDTA‐free protease inhibitor cocktail (Roche). The cells were mixed well in the lysis buffer, incubated on ice for 30 min, and clarified by centrifuging (13,000 x g) for 15 min at 4°C. The cleared lysate (40 µl) was mixed with SDS sample buffer and heated at 85°C for 8 min. The denatured samples were separated by electrophoresis in 4–20% SDS–PAGE (Bio‐Rad) and subjected to standard Western blot analysis.
For the immunoprecipitation of Phf5a and proteomics analysis, CH12F3‐2A cells transfected with Phf5a‐MycFlag were used. Transfected cells were stimulated with CIT for 24 h prior to harvest. Nuclear and chromatin‐bound nuclear extracts were prepared using the Subcellular Protein Fractionation Kit (Thermo Fisher) according to the manufacturer’s instructions. To pull down the Phf5a‐associated proteins from nuclear and chromatin fractions, anti‐FLAG IP was performed. The immunoprecipitated protein samples were separated by electrophoresis on SDS–PAGE (4–20%), and the gel was silver‐stained using the Silver Quest Staining Kit (Invitrogen). Individual gel lanes were sliced out and cut into several pieces, destained, and subjected to in‐gel digestion with trypsin. The obtained peptides were subjected to LC‐MS/MS analyses as described previously (Husain et al, 2016).
For routine co‐IP or reciprocal IP, the nuclear extract was prepared as described above from transfected CH12F3‐2A cells. Anti‐Flag and anti‐p400 antibodies were used to pull down Phf5a‐MycFlag and p400, respectively. In the case of 293T cells, WCE was prepared as described above from mock‐ and plasmid‐transfected cells. Cleared WCE was used for subsequent co‐IP and immune blot analyses.
Detection of deletional and inversional joining by DC‐PCR
Both deletional and inversional joining signatures in the IgH locus can be detected by the Digestion Circularization PCR assay (Chu et al, 1992). Due to the presence of the rearranged Sµ‐Sα, the detection of Sµ‐Sα recombination in the productive IgH allele by DC‐PCR used to be difficult. Here, we used a derivative line of CH12F3 cells in which the rearranged Sµ‐Sα was deleted (non‐productive allele ΔSµ‐Sα) from the inactive IgH allele (Dong et al, 2015). Genomic DNA was isolated from CH12F3 cells stimulated by CIT for IgA switching for 24 h. Approximately 2 µg of DNA, from siControl‐ and siPhf5a‐treated cells, was digested overnight with EcoRI and heat inactivated. To promote circularized ligation, the digested DNA samples were diluted (2 ng/µl), and multiple ligation reactions were set up to cover up to 1 µg of DNA. The ligation reactions were combined and purified through a Wizard DNA purification column (Promega). The DNA was eluted in 100 µl of water, and 0.5–1 µl of the elute was subjected to rTaq PCR in a 50‐µl volume. The primers for the nAchRe (nicotinic acetylcholine receptor epsilon‐subunit) control, Sµ‐Sα‐direct, and Sµ‐Sα‐inversional joining PCR are described earlier. The gel‐purified PCR products were cloned and sequenced, and the direct and inversional joining was confirmed by the DC‐PCR method.
DNA break assays: Biotin‐dUTP end labeling and LM‐PCR
CH12F3‐2A cells were transfected with control or gene‐specific siRNAs for 24 h and then stimulated with CIT for another 24 h. The cells were harvested and passed through a Percoll gradient to remove the dead cells. The live cells were then mildly fixed, permeabilized, and incubated with DNA end‐labeling mix containing Biotin‐16‐dUTP and T4 DNA polymerase (Takara) as described previously (Doi et al, 2009). After the DNA labeling in situ, the genomic DNA was isolated by phenol/chloroform extraction and digested with HindIII. The DNA break ends with incorporated biotin‐dUTP were captured from the digested DNA pool by streptavidin magnetic beads. The magnetic beads were then washed three times and were subjected to real‐time PCR for the target genomic region in the IgH locus.
CH12F3‐2A cells were stimulated for CSR as described above, and dead cells were removed by standard Percoll gradient. The live cells were mixed with low‐melting agarose to prepare agarose plugs containing 1 × 106 cells, which were processed as described previously (Rush et al, 2004; Guikema et al, 2007). The washed plugs containing high molecular weight intact DNA were stored in TE (50 µl/plug) or melted for enzymatic treatments and linker ligation. Heat inactivation of the T4 polymerase (Takara), exonuclease 1 (NEB), or RecJF (NEB) was performed according to the manufacturers’ instructions and as described previously (Yamane et al, 2013). Linker ligation was performed overnight at 16°C, and the reaction was terminated by heating the samples at 70°C for 10 min. The samples were subjected to PCR using KOD FX Neo DNA polymerase (TOYOBO). Semi‐quantitative PCR was performed for the target loci using an Sµ‐specific primer (forward) and a linker‐specific primer (reverse). To confirm the amplification of Sµ‐specific DSBs, Southern blot analysis of the PCR products was performed using a DIG‐labeled Sµ probe. PCR of the GAPDH locus from the same sample served as a normalization control.
DNA end‐resection assay
The DNA end‐resection assay was adopted for Sµ (close to and 5′ of the core Sµ) from previously described methods (Zhou et al, 2014; Gao et al, 2020; Nath & Nagaraju, 2020). Genomic DNA isolated from CIT‐stimulated CH12F3‐2A cells, with and without Phf5a knockdown, was subjected to single (EcoRI) or double (EcoRI and AluI) restriction endonuclease digestion. The target region contains AluI but not EcoRI site and is prone to AID‐induced DNA breaks. The EcoRI digestion was employed as an alternative of sonication to reduce the genomic DNA complexity. Digested DNA samples were subjected to phenol:chloroform extraction and ethanol precipitation. For each sample, 50 ng of purified digested DNA was used for PCR (30 cycles) using KOD FX Neo high fidelity DNA polymerase (TOYOBO). PCR products (1/3rd) were resolved on a 2% agarose gel and visualized by ethidium bromide staining. The sequences of the primers used to amplify the target Sµ region are shown in Appendix Table S3.
Analysis of the CSR junctions in CH12F3‐2A cells
CH12F3‐2A cells transfected with control or gene‐specific siRNA were stimulated with CIT for 24 h. Since CSR was severely reduced in Phf5a KD cells, IgA (+) cells were combined from duplicate sets of transfectants. The PE‐stained IgA (+) cells were isolated from siControl‐ and siPhf5a‐transfected cells using anti‐PE Magnetic Particle‐DM (BD Biosciences). Genomic DNA was isolated from the IgA (+) cells and subjected to PCR using forward and reverse primers located in 5′Sμ and 3′Sα, respectively (Stanlie et al, 2014). Two rounds of PCR were conducted using PrimeSTAR HS DNA polymerase (Takara). Four PCRs were set up using 200 ng of genomic DNA per reaction for each genomic DNA sample, and the PCR conditions were 98°C for 2 min; and 98°C for 10 s, 55°C for 10 s, and 72°C for 2 min for 20 cycles. The first PCR products were purified using the Wizard PCR clean up system (Promega) and subjected to second‐round PCR. The PCR products were separated by gel electrophoresis, and the products ranging from 1.5 kb to 300 bp were gel‐purified and cloned. Several clones were sequenced from each reaction set and analyzed by Sequencher (Genecodes) software to eliminate vector sequences and common clones. The sequence of each unique clone was subjected to pair‐wise nBLAST at NCBI against Sµ‐and Sα‐harboring IgH locus clones. Any region of conflict was verified with the genomic DNA sequence derived from non‐stimulated CH12F3‐2A cells.
Author contributions
NAB, FH, AS, AH, MN, and SM designed and performed experiments; NAB and NM provided methodology. TT and HT performed the MS analysis; NAB and TH conceptualized and supervised the project and wrote the manuscript with comments from the authors. AS, AH, and FH organized and reviewed the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 1B,D,F
Source Data for Figure 2B
Source Data for Figure 3C,E
Source Data for Figure 4C
Source Data for Figure 6C
Source Data for Figure 7C,D
Source Data for Figure 8B
Acknowledgements
This work was supported by funding from the Japan Society for the Promotion of Science; Grant‐in‐Aid for Scientific Research (S) 15H05784 to T.H. and Grant‐in‐Aid for Scientific Research (C) 16K07214 and 21K06015 to N.A.B. We also would like to thank Dr. Jianliang Xu for providing the template for CtIP and Mre11 constructs. The graphical abstract image was created with BioRender.com.
The EMBO Journal (2021) 40: e106393.
Data availability
This study includes no data deposited in external repositories.
References
- Adkins NL, Niu H, Sung P, Peterson CL (2013) Nucleosome dynamics regulates DNA processing. Nat Struct Mol Biol 20: 836–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum NA, Honjo T (2012) Evolutionary comparison of the mechanism of DNA cleavage with respect to immune diversity and genomic instability. Biochemistry 51: 5243–5256 [DOI] [PubMed] [Google Scholar]
- Begum NA, Stanlie A, Nakata M, Akiyama H, Honjo T (2012) The histone chaperone Spt6 is required for activation‐induced cytidine deaminase target determination through H3K4me3 regulation. J Biol Chem 287: 32415–32429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettridge J, Na CH, Pandey A, Desiderio S (2017) H3K4me3 induces allosteric conformational changes in the DNA‐binding and catalytic regions of the V(D)J recombinase. Proc Natl Acad Sci USA 114: 1904–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz M (2006) The PHD finger, a nuclear protein‐interaction domain. Trends Biochem Sci 31: 35–40 [DOI] [PubMed] [Google Scholar]
- Boboila C, Alt FW, Schwer B (2012a) Classical and alternative end‐joining pathways for repair of lymphocyte‐specific and general DNA double‐strand breaks. Adv Immunol 116: 1–49 [DOI] [PubMed] [Google Scholar]
- Boboila C, Oksenych V, Gostissa M, Wang JH, Zha S, Zhang Y, Chai H, Lee C‐S, Jankovic M, Saez L‐MA et al (2012b) Robust chromosomal DNA repair via alternative end‐joining in the absence of X‐ray repair cross‐complementing protein 1 (XRCC1). Proc Natl Acad Sci USA 109: 2473–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonisch C, Hake SB (2012) Histone H2A variants in nucleosomes and chromatin: more or less stable? Nucleic Acids Res 40: 10719–10741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschbeck M, Hake SB (2017) Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol 18: 299–314 [DOI] [PubMed] [Google Scholar]
- Ceppi I, Howard SM, Kasaciunaite K, Pinto C, Anand R, Seidel R, Cejka P (2020) CtIP promotes the motor activity of DNA2 to accelerate long‐range DNA end resection. Proc Natl Acad Sci USA 117: 8859–8869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR (2017) Non‐homologous DNA end joining and alternative pathways to double‐strand break repair. Nat Rev Mol Cell Biol 18: 495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CC, Paul WE, Max EE (1992) Quantitation of immunoglobulin mu‐gamma 1 heavy chain switch region recombination by a digestion‐circularization polymerase chain reaction method. Proc Natl Acad Sci USA 89: 6978–6982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, Jimenez‐Sainz J, Wang W, Miller AS, Xue X, Nguyen KA, Jensen RB, Sung P (2017) Enhancement of BLM‐DNA2‐mediated long‐range DNA end resection by CtIP. Cell Rep 21: 324–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, Filsuf D, Chen H‐T, Gazumyan A, Yamane A et al (2010) PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science 329: 917–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denepoux S, Razanajaona D, Blanchard D, Meffre G, Capra JD, Banchereau J, Lebecque S (1997) Induction of somatic mutation in a human B cell line in vitro. Immunity 6: 35–46 [DOI] [PubMed] [Google Scholar]
- Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO (2009) Multiple functions of MRN in end‐joining pathways during isotype class switching. Nat Struct Mol Biol 16: 808–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T, Kato L, Ito S, Shinkura R, Wei M, Nagaoka H, Wang J, Honjo T (2009) The C‐terminal region of activation‐induced cytidine deaminase is responsible for a recombination function other than DNA cleavage in class switch recombination. Proc Natl Acad Sci USA 106: 2758‐2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Panchakshari RA, Zhang T, Zhang Yu, Hu J, Volpi SA, Meyers RM, Ho Y‐J, Du Z, Robbiani DF et al (2015) Orientation‐specific joining of AID‐initiated DNA breaks promotes antibody class switching. Nature 525: 134–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccleston J, Yan C, Yuan K, Alt FW, Selsing E (2011) Mismatch repair proteins MSH2, MLH1, and EXO1 are important for class‐switch recombination events occurring in B cells that lack nonhomologous end joining. J Immunol 186: 2336–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster ER, Downs JA (2005) Histone H2A phosphorylation in DNA double‐strand break repair. FEBS J 272: 3231–3240 [DOI] [PubMed] [Google Scholar]
- Gao M, Guo G, Huang J, Kloeber JA, Zhao F, Deng M, Tu X, Kim W, Zhou Q, Zhang C et al (2020) USP52 regulates DNA end resection and chemosensitivity through removing inhibitory ubiquitination from CtIP. Nat Commun 11: 5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar‐Maia A, Qadeer ZA, Hasson D, Ratnakumar K, Adrian Leu N, Leroy G, Liu S, Costanzi C, Valle‐Garcia D, Schaniel C et al (2013) MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat Commun 4: 1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevry N, Hardy S, Jacques PE, Laflamme L, Svotelis A, Robert F, Gaudreau L (2009) Histone H2A.Z is essential for estrogen receptor signaling. Genes Dev 23: 1522–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guikema JE, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, Schrader CE (2007) APE1‐ and APE2‐dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med 204: 3017–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer MH, Gasser SM (2017) Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev 31: 2204–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein K, Lorenz MG, Siebenkotten G, Petry K, Christine R, Radbruch A (1998) Processing of switch transcripts is required for targeting of antibody class switch recombination. J Exp Med 188: 2369–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo K, Kim B, Kim K, Choi J, Kim H, Zhan Y, Ranish JA, An W (2007) Isolation and characterization of proteins associated with histone H3 tails in vivo. J Biol Chem 282: 15476–15483 [DOI] [PubMed] [Google Scholar]
- Hubert CG, Bradley RK, Ding Y, Toledo CM, Herman J, Skutt‐Kakaria K, Girard EJ, Davison J, Berndt J, Corrin P et al (2013) Genome‐wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes Dev 27: 1032–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain A, Begum NA, Taniguchi T, Taniguchi H, Kobayashi M, Honjo T (2016) Chromatin remodeller SMARCA4 recruits topoisomerase 1 and suppresses transcription‐associated genomic instability. Nat Commun 7: 10549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliakis G, Murmann T, Soni A (2015) Alternative end‐joining repair pathways are the ultimate backup for abrogated classical non‐homologous end‐joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat Res Genet Toxicol Environ Mutagen 793: 166–175 [DOI] [PubMed] [Google Scholar]
- Jiang Y, Qian Xu, Shen J, Wang Y, Li X, Liu R, Xia Y, Chen Q, Peng G, Lin S‐Y et al (2015) Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat Cell Biol 17: 1158–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kfir N, Lev‐Maor G, Glaich O, Alajem A, Datta A, Sze SK, Meshorer E, Ast G (2015) SF3B1 association with chromatin determines splicing outcomes. Cell Rep 11: 618–629 [DOI] [PubMed] [Google Scholar]
- Kinoshita K, Harigai M, Fagarasan S, Muramatsu M, Honjo T (2001) A hallmark of active class switch recombination: transcripts directed by I promoters on looped‐out circular DNAs. Proc Natl Acad Sci USA 98: 12620–12623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee‐Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J (2011) CtIP promotes microhomology‐mediated alternative end joining during class‐switch recombination. Nat Struct Mol Biol 18: 75–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, White CA, Lam T, Pone EJ, Tran DC, Hayama KL, Zan H, Xu Z, Casali P (2013) Combinatorial H3K9acS10ph histone modification in IgH locus S regions targets 14‐3‐3 adaptors and AID to specify antibody class‐switch DNA recombination. Cell Rep 5: 702–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M, Jung S, Radbruch A (1995) Switch transcripts in immunoglobulin class switching. Science 267: 1825–1828 [DOI] [PubMed] [Google Scholar]
- Makharashvili N, Tubbs A, Yang S‐H, Wang H, Barton O, Zhou Yi, Deshpande R, Lee J‐H, Lobrich M, Sleckman B et al (2014) Catalytic and noncatalytic roles of the CtIP endonuclease in double‐strand break end resection. Mol Cell 54: 1022–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchalot A, Ashi MO, Lambert JM, Carrion C, Lecardeur S, Srour N, Delpy L, Le Pennec S (2020) Uncoupling splicing from transcription using antisense oligonucleotides reveals a dual role for I exon donor splice sites in antibody class switching. Front Immunol 11: 780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E, Palhan VB, Tjernberg A, Lymar ES, Gamper AM, Kundu TK, Chait BT, Roeder RG (2001) Human STAGA complex is a chromatin‐acetylating transcription coactivator that interacts with pre‐mRNA splicing and DNA damage‐binding factors in vivo. Mol Cell Biol 21: 6782–6795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews AGW, Kuo AJ, Ramón‐Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN et al (2007) RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450: 1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C (2004) ATP‐driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303: 343–348 [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T (2000) Class switch recombination and hypermutation require activation‐induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563 [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Nagaoka H, Shinkura R, Begum NA, Honjo T (2007) Discovery of activation‐induced cytidine deaminase, the engraver of antibody memory. Adv Immunol 94: 1–36 [DOI] [PubMed] [Google Scholar]
- Nagaoka H, Ito S, Muramatsu M, Nakata M, Honjo T (2005) DNA cleavage in immunoglobulin somatic hypermutation depends on de novo protein synthesis but not on uracil DNA glycosylase. Proc Natl Acad Sci USA 102: 2022–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka H, Muramatsu M, Yamamura N, Kinoshita K, Honjo T (2002) Activation‐induced deaminase (AID)‐directed hypermutation in the immunoglobulin Smu region: implication of AID involvement in a common step of class switch recombination and somatic hypermutation. J Exp Med 195: 529–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Kondo S, Sugai M, Nazarea M, Imamura S, Honjo T (1996) High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int Immunol 8: 193–201 [DOI] [PubMed] [Google Scholar]
- Nath S, Nagaraju G (2020) FANCJ helicase promotes DNA end resection by facilitating CtIP recruitment to DNA double‐strand breaks. PLoS Genet 16: e1008701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig A, Nussenzweig MC (2010) Origin of chromosomal translocations in lymphoid cancer. Cell 141: 27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obri A, Ouararhni K, Papin C, Diebold M‐L, Padmanabhan K, Marek M, Stoll I, Roy L, Reilly PT, Mak TW et al (2014) ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 505: 648–653 [DOI] [PubMed] [Google Scholar]
- Ogiwara H, Kohno T (2011) Essential factors for incompatible DNA end joining at chromosomal DNA double strand breaks in vivo. PLoS One 6: e28756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, Yasui A, Yokota J, Kohno T (2011) Histone acetylation by CBP and p300 at double‐strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non‐homologous end joining factors. Oncogene 30: 2135–2146 [DOI] [PubMed] [Google Scholar]
- Onozawa M, Zhang Z, Kim YJ, Goldberg L, Varga T, Bergsagel PL, Kuehl WM, Aplan PD (2014) Repair of DNA double‐strand breaks by templated nucleotide sequence insertions derived from distant regions of the genome. Proc Natl Acad Sci USA 111: 7729–7734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah‐Sobrinho C, Resch W, Yamane A, San‐Martin BR, Barreto V et al (2010) Activation‐induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143: 122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Haddad D, Li C, Le MX, Ling AK, So CC, Nepal RM, Gommerman JL, Yu K, Ketela T et al (2016) The SAGA deubiquitination module promotes DNA repair and class switch recombination through ATM and DNAPK‐mediated gammaH2AX formation. Cell Rep 15: 1554–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen‐Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC (2004) AID is required for c‐myc/IgH chromosome translocations in vivo. Cell 118: 431–438 [DOI] [PubMed] [Google Scholar]
- Reina‐San‐Martin B, Chen J, Nussenzweig A, Nussenzweig MC (2007) Enhanced intra‐switch region recombination during immunoglobulin class switch recombination in 53BP1‐/‐ B cells. Eur J Immunol 37: 235–239 [DOI] [PubMed] [Google Scholar]
- Reina‐San‐Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC (2003) H2AX is required for recombination between immunoglobulin switch regions but not for intra‐switch region recombination or somatic hypermutation. J Exp Med 197: 1767–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM (1999) Megabase chromatin domains involved in DNA double‐strand breaks in vivo. J Cell Biol 146: 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Roon AM, Loening NM, Obayashi E, Yang JC, Newman AJ, Hernandez H, Nagai K, Neuhaus D (2008) Solution structure of the U2 snRNP protein Rds3p reveals a knotted zinc‐finger motif. Proc Natl Acad Sci USA 105: 9621–9626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush JS, Fugmann SD, Schatz DG (2004) Staggered AID‐dependent DNA double strand breaks are the predominant DNA lesions targeted to S mu in Ig class switch recombination. Int Immunol 16: 549–557 [DOI] [PubMed] [Google Scholar]
- Rzymski T, Grzmil P, Meinhardt A, Wolf S, Burfeind P (2008) PHF5A represents a bridge protein between splicing proteins and ATP‐dependent helicases and is differentially expressed during mouse spermatogenesis. Cytogenet Genome Res 121: 232–244 [DOI] [PubMed] [Google Scholar]
- Sakamoto S, Iijima K, Mochizuki D, Nakamura K, Teshigawara K, Kobayashi J, Matsuura S, Tauchi H, Komatsu K (2007) Homologous recombination repair is regulated by domains at the N‐ and C‐terminus of NBS1 and is dissociated with ATM functions. Oncogene 26: 6002–6009 [DOI] [PubMed] [Google Scholar]
- Sanchez R, Zhou MM (2011) The PHD finger: a versatile epigenome reader. Trends Biochem Sci 36: 364–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450: 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J (2005) Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J Exp Med 202: 561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard EC, Morrish RB, Dillon MJ, Leyland R, Chahwan R (2018) Epigenomic modifications mediating antibody maturation. Front Immunol 9: 355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkura R, Ito S, Begum NA, Nagaoka H, Muramatsu M, Kinoshita K, Sakakibara Y, Hijikata H, Honjo T (2004) Separate domains of AID are required for somatic hypermutation and class‐switch recombination. Nat Immunol 5: 707–712 [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S (2013) Histone variants in pluripotency and disease. Development 140: 2513–2524 [DOI] [PubMed] [Google Scholar]
- Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA (2010) Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc Natl Acad Sci USA 107: 22190–22195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanlie A, Begum NA, Akiyama H, Honjo T (2012) The DSIF subunits Spt4 and Spt5 have distinct roles at various phases of immunoglobulin class switch recombination. PLoS Genet 8: e1002675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanlie A, Yousif AS, Akiyama H, Honjo T, Begum NA (2014) Chromatin reader Brd4 functions in Ig class switching as a repair complex adaptor of nonhomologous end‐joining. Mol Cell 55: 97–110 [DOI] [PubMed] [Google Scholar]
- Stavnezer J, Bjorkman A, Du L, Cagigi A, Pan‐Hammarstrom Q (2010) Mapping of switch recombination junctions, a tool for studying DNA repair pathways during immunoglobulin class switching. Adv Immunol 108: 45–109 [DOI] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JE, Schrader CE (2008) Mechanism and regulation of class switch recombination. Annu Rev Immunol 26: 261–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegeman R, Spreacker PJ, Swanson SK, Stephenson R, Florens L, Washburn MP, Weake VM (2016) The spliceosomal protein SF3B5 is a novel component of Drosophila SAGA that functions in gene expression independent of splicing. J Mol Biol 428: 3632–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strikoudis A, Lazaris C, Trimarchi T, Galvao Neto AL, Yang Y, Ntziachristos P, Rothbart S, Buckley S, Dolgalev I, Stadtfeld M et al (2016) Regulation of transcriptional elongation in pluripotency and cell differentiation by the PHD‐finger protein Phf5a. Nat Cell Biol 18: 1127–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng T, Tsai JHC, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B et al (2017) Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A‐SF3b complex. Nat Commun 8: 15522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstorukov MY, Goldman JA, Gilbert C, Ogryzko V, Kingston RE, Park PJ (2012) Histone variant H2A.Bbd is associated with active transcription and mRNA processing in human cells. Mol Cell 47: 596–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trappe R, Ahmed M, Glaser B, Vogel C, Tascou S, Burfeind P, Engel W (2002) Identification and characterization of a novel murine multigene family containing a PHD‐finger‐like motif. Biochem Biophys Res Commun 293: 816–826 [DOI] [PubMed] [Google Scholar]
- Wang Z, Yang X, Liu C, Li X, Zhang B, Wang Bo, Zhang Yu, Song C, Zhang T, Liu M et al (2019) Acetylation of PHF5A modulates stress responses and colorectal carcinogenesis through alternative splicing‐mediated upregulation of KDM3A. Mol Cell 74: 1250–1263.e6 [DOI] [PubMed] [Google Scholar]
- Xie Y, Zheng M, Chu X, Chen Y, Xu H, Wang J, Zhou H, Long J (2018) Paf1 and Ctr9 subcomplex formation is essential for Paf1 complex assembly and functional regulation. Nat Commun 9: 3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ayrapetov MK, Xu C, Gursoy‐Yuzugullu O, Hu Y, Price BD (2012) Histone H2A.Z controls a critical chromatin remodeling step required for DNA double‐strand break repair. Mol Cell 48: 723–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane A, Robbiani D, Resch W, Bothmer A, Nakahashi H, Oliveira T, Rommel P, Brown E, Nussenzweig A, Nussenzweig M et al (2013) RPA accumulation during class switch recombination represents 5′‐3′ DNA‐end resection during the S‐G2/M phase of the cell cycle. Cell Rep 3: 138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y‐Z, Xue M‐Z, Shen H‐J, Li X‐G, Ma D, Gong Y, Liu Y‐R, Qiao F, Xie H‐Y, Lian Bi et al (2018) PHF5A epigenetically inhibits apoptosis to promote breast cancer progression. Cancer Res 78: 3190–3206 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Caron P, Legube G, Paull TT (2014) Quantitation of DNA double‐strand break resection intermediates in human cells. Nucleic Acids Res 42: e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 1B,D,F
Source Data for Figure 2B
Source Data for Figure 3C,E
Source Data for Figure 4C
Source Data for Figure 6C
Source Data for Figure 7C,D
Source Data for Figure 8B
Data Availability Statement
This study includes no data deposited in external repositories.