

Abstract
Mouse embryonic stem cells (mESCs) are biased toward producing embryonic rather than extraembryonic endoderm fates. Here, we identify the mechanism of this barrier and report that the histone deacetylase Hdac3 and the transcriptional corepressor Dax1 cooperatively limit the lineage repertoire of mESCs by silencing an enhancer of the extraembryonic endoderm‐specifying transcription factor Gata6. This restriction is opposed by the pluripotency transcription factors Nr5a2 and Esrrb, which promote cell type conversion. Perturbation of the barrier extends mESC potency and allows formation of 3D spheroids that mimic the spatial segregation of embryonic epiblast and extraembryonic endoderm in early embryos. Overall, this study shows that transcriptional repressors stabilize pluripotency by biasing the equilibrium between embryonic and extraembryonic lineages that is hardwired into the mESC transcriptional network.
Keywords: Dax1/Nr0b1, embryonic stem cell, extraembryonic endoderm, Hdac3, pluripotency
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Development & Differentiation; Regenerative Medicine
Extraembryonic transdifferentiation of ESCs is controlled by collaborative activity of transcriptional repressors.
Introduction
Binary cell fate decisions generate two distinct daughter cell types from one common progenitor. Once specified, cells need to prevent de‐differentiation and transdifferentiation in order to stay fate‐committed. The processes of lineage specification and maintenance can employ different mechanisms (Holmberg & Perlmann, 2012), yet the regulatory principles underlying these differences are not well understood.
A well‐studied developmental binary cell fate decision is the differentiation of the mouse embryonic day (E) 3.5 inner cell mass (ICM) into extraembryonic primitive endoderm (PrE) and pluripotent epiblast (EPI) (Hermitte & Chazaud, 2014). This is mediated by the lineage‐specifying transcription factors (TFs) Nanog and Gata6 that are co‐expressed in the ICM. Positive auto‐regulation and mutual inhibition of Nanog and Gata6, modulated by extrinsic fibroblast growth factor (FGF) signaling, drives segregation into Gata6‐expressing PrE and Nanog‐expressing EPI at E4.5 (Simon et al, 2018).
Developmental potency of the EPI is captured in vitro by mouse embryonic stem cells (mESCs) that predominantly give rise to embryonic cell types when injected into blastocysts (Beddington & Robertson, 1989). Although the lineage barrier toward extraembryonic endoderm is not as strictly maintained in mESCs in vitro (Canham et al, 2010; Niakan et al, 2010; Cho et al, 2012; Nigro et al, 2017; Rivron et al, 2018), the molecular mechanisms that impede PrE and favor embryonic differentiation are not well understood. Expression of Gata6 enables differentiation into extraembryonic endoderm stem cells (XEN cells) (Shimosato et al, 2007), raising the possibility that the Nanog‐Gata6 antagonism that segregates EPI and PrE during mouse pre‐implantation development maintains lineage separation in mESCs (Graf & Enver, 2009). Nanog is, however, largely dispensable for post‐implantation development and mESC self‐renewal (Chambers et al, 2007), suggesting operation of additional mechanisms that govern fate restriction (Holmberg & Perlmann, 2012).
Different mESC states are stabilized by specific extrinsic signaling conditions (Smith, 2017): A naïve pluripotent ground state in the presence of 2 inhibitors and leukemia inhibitory factor (LIF) (2iL), and a metastable pluripotent state by fetal calf serum (S) and LIF (SL). Although both states are interconvertible, mESCs in SL heterogeneously express differentiation and pluripotency markers (Smith, 2017), inefficiently differentiate into Epiblast‐like cells (EpiLCs) in vitro (Hayashi et al, 2011), and are thought to recapitulate a developmentally more advanced state than mESCs in naïve 2iL conditions (Schröter et al, 2015; Gonzalez et al, 2016).
Here, we exploit the transition from the naïve to the metastable mESC state (Schröter et al, 2015) to define the mechanism of lineage restriction. We identify inhibitors of PrE differentiation, define their interaction with pluripotent TFs, and describe how they enact competition between PrE and EPI fate. Our findings reveal that silencing of a Gata6 enhancer by transcriptional repressors antagonizes lineage plasticity of the mESC gene regulatory network (GRN) and secures the pluripotent lineage.
Results
Hdac3 inhibits PrE differentiation of mESCs
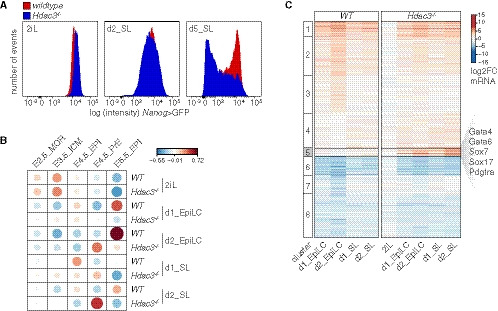
While working on a putative Hdac3 interactor, we genetically deleted Hdac3 in naïve TNG‐A mESCs that express GFP under the control of the endogenous Nanog locus (Chambers et al, 2007) (Fig EV1A). Compared with wild‐type (WT) controls, Hdac3 −/− cells expressed higher levels of the Nanog reporter in 2iL, but rapidly downregulated Nanog when converted to SL (Fig 1A) and were lost upon further passaging, indicating undue differentiation specifically in metastable conditions.
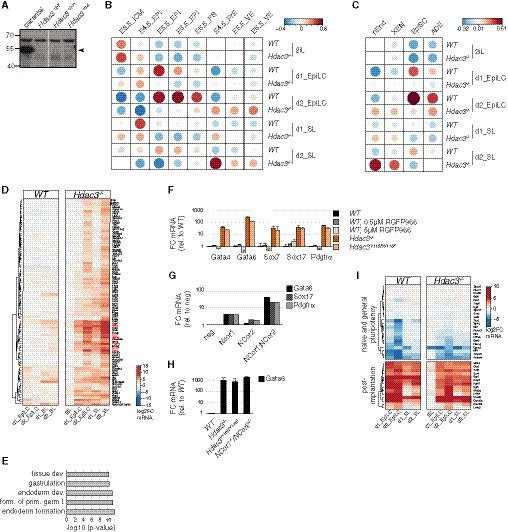
Figure EV1. Transcriptomics of Hdac3 −/− cells.
-
AAnti‐Hdac3 Western blot of naïve Hdac3 −/− TNG‐A clones. Specific Hdac3 band is indicated.
- B, C
-
DSimilar to Fig 1C with a magnified view of cluster 5. Canonical PrE marker genes are highlighted in red.
-
EGO terms enriched in cluster 5. Modified Fisher exact P‐values as determined by DAVID (Huang et al, 2008) are shown.
-
F–HIndicated mRNA levels relative to WT cells of indicated genotypes and treatments after 3 days in SLRA (F) and 2 days in SL (H). Average and standard deviation (SD) of at least two independent clones. Indicated PrE marker mRNA expression relative to negative control siRNA transfection of cells transfected with indicated siRNAs after 2 days in SL (G).
-
ISimilar to Fig 1C with a magnified view of a panel of naïve and general pluripotency, and post‐implantation markers.
Source data are available online for this figure.
Figure 1. Naïve Hdac3 −/− mESCs differentiate into PrE.
- Nanog>GFP intensity of WT and Hdac3 −/− TNG‐A cells in 2iL and at d2 and d5 of SL exposure.
- Pairwise Pearson correlations of in vitro TNG‐A and in vivo embryo (Boroviak et al, 2015) RNAseq samples.
- k‐means clustering of gene expression changes relative to naïve WT TNG‐A mESCs.
Source data are available online for this figure.
To determine the transcriptional changes underlying this phenotype, we performed RNA sequencing (RNAseq) of WT and Hdac3 −/− cells in the naïve ESC state, and after 1 and 2 days (d) in SL and epiblast‐like cell (EpiLC) (Hayashi et al, 2011) differentiation conditions. Contrasting these results with existing datasets from the early embryo (Boroviak et al, 2015; Mohammed et al, 2017) using pairwise correlation (Figs 1B and EV1B) revealed that mESCs in 2iL are most comparable to the embryonic day E3.5 ICM (Gonzalez et al, 2016). It further showed that SL and EpiLC conditions drive naïve WT mESCs into cell states that are similar to the embryonic E4.5—E6.5 pre‐ and post‐implantation EPI. In contrast, differentiating Hdac3 −/− cells, in particular in SL, transcriptionally resembled the primitive and visceral (VE), but not definitive endoderm (Anderson et al, 2017) (Fig EV1C). k‐means clustering identified a class of genes (cluster 5) that was selectively induced in differentiating mutant cells (Fig 1C). Cluster 5 is enriched for endoderm regulators by gene ontology analysis and includes the TFs Gata4, Gata6, and Sox17 that are required for PrE development in vivo (Hermitte & Chazaud, 2014) (Figs 1C and EV1D and E). Hdac3 therefore represses differentiation of mESCs into a cell state resembling the PrE upon transition from naïve to metastable conditions.
We made use of the Hdac3‐specific inhibitor RGFP966 to test the role of Hdac3 enzymatic activity. Exposure to RGFP966 did not increase transcription of the PrE markers Gata4, Gata6, Sox7, Sox17, and Pdgfrα after release from 2iL (Fig EV1F). RGFP966 may, however, only ineffectively block Hdac3 (Phelps et al, 2016) or lack specificity (Jia et al, 2016). We therefore engineered naïve mESCs to express catalytically inactive Hdac3 (Hdac3 Y118F/Y118F), or depleted Ncor1 and Ncor2 by siRNA transfection and compound knockout (Sun et al, 2013) (Table EV1). PrE markers were upregulated in these cells after SL conversion, similar to Hdac3 −/− mESCs (Fig EV1G and H). Hdac3, thus, acts as a deacetylase in complex with Ncor1/2 nuclear corepressors.
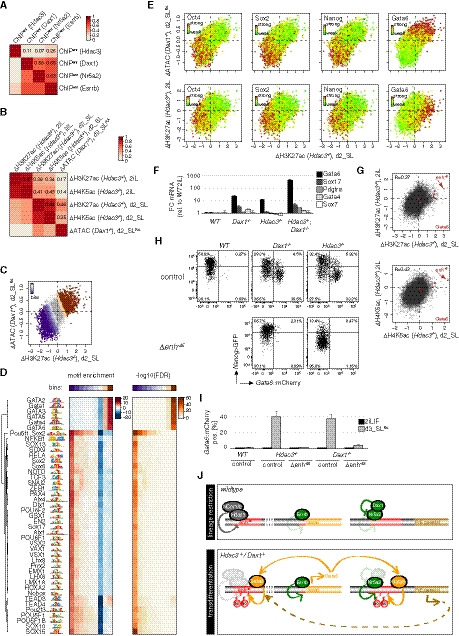
Hdac3 and Dax1 shield mESCs from PrE differentiation in response to extrinsic developmental signals
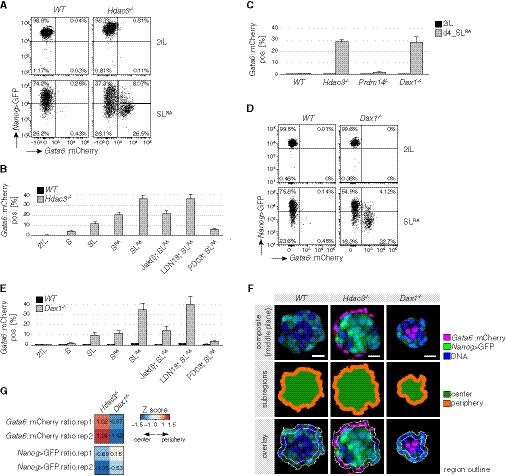
The induction of post‐implantation EPI markers in Hdac3 −/− cells was overall unperturbed (Fig EV1I), suggesting simultaneous activation of embryonic and extraembryonic gene expression. To test whether this is due to population heterogeneity, we deleted Hdac3 in a TNG‐A‐derived mESC line (G6C18) that in addition to Nanog reports endogenous transcription of Gata6 (Fig EV2A). After 3 days in the presence of SL, around 10% of mutant cells were positive for the Gata6 reporter (Fig EV2B). Addition of minimal amounts (1 nM) of retinoic acid (SLRA) increased this fraction to around 30% (Fig 2A and B). Intracellular flow cytometry analysis showed that Gata6 reporter‐positive cells homogeneously expressed the PrE markers Sox17, Dab2, and Lama1 (Hermitte & Chazaud, 2014) (Fig EV2C). Furthermore, reporter activation required LIF and FGF but not BMP signaling (Fig 2B), suggesting that PrE differentiation of Hdac3 −/− cells is driven by the same pathways that control progression of ICM into PrE in vivo (Hermitte & Chazaud, 2014; Morgani & Brickman, 2015).
Figure EV2. Characterization of Hdac3 −/− and Dax1 −/− cells.

- Anti‐Hdac3 Western blot of naïve Hdac3 −/− G6C18 clones. Specific Hdac3 band is indicated.
- Representative Nanog>GFP and Gata6::mCherry fluorescence intensity plots in indicated genotypes after 3 days that were used for quantifications shown in Fig 2B.
- Quantification of Gata6::mCherry reporter signal, background staining, and Sox17, Dab2, and Lama1 immunofluorescence intensities in single, 3 days SLRA‐differentiated Hdac3 −/− cells binned by Gata6 reporter activity.
- Immunofluorescence and reporter expression in spheroids derived from single WT and Hdac3 −/− naïve mESCs. DNA counterstain is Hoechst. Dashed white lines indicate the embryonic Gata6::mCherry negative part. Scale bar: 10 μm.
- Similar to Fig 1D. Arrowheads indicate Dab2‐ and Gata6::mCherry‐positive PrE cells. Scale bar: 10 μm.
- Anti‐Dax1, anti‐Esrrb, and anti‐Hdac3 Western blots for the genotyping of G6C18 mutant clones. Migration behavior of targeted proteins is indicated.
- Representative Nanog>GFP and Gata6::mCherry fluorescence intensity plots in indicated genotypes and conditions that were used for quantifications shown in Fig 2C.
- Quantification of Nanog>GFP geometric mean intensities in 2iL. Average and SD of at least two independent clones.
- Representative Nanog>GFP and Gata6::mCherry fluorescence intensity plots in Dax1 ‐/‐ cells after 3 days that were used for quantifications shown in Fig 2E.
- Ratios of Nanog>GFP and Gata6::mCherry intensity (periphery/center) in spheroids of indicated genotypes. Dashed lines indicate the mean in WT cells.
Figure 2. Hdac3 and Dax1 are required for PrE lineage restriction in response to FGF, LIF, and RA.
-
A–ENanog>GFP and Gata6::mCherry fluorescence intensities of WT and Hdac3 −/− (A) and WT and Dax1 −/− (D) cells after 3 days in indicated conditions. Fraction of Gata6::mCherry‐positive cells in indicated genotypes and conditions after 3 days (B, E) and 4 days (C). Jak(i) blocks LIF, LDN19 BMP4, and PD03 FGF signaling. Average and SD of at least two independent clones.
-
F, GQuantification of spatial lineage segregation in spheroids. Representative spheroid segmentations of indicated genotypes (F). Z‐score‐normalized fluorescence distributions in mutants compared with WT spheroids (G). Positive Z‐scores indicate peripheral, outside, and negative Z‐scores central, inside, enrichment. Scale bar: 25 μm.
Source data are available online for this figure.
To explore PrE differentiation further, we turned to a 3D culture system. After 3 days in SLRA, single Hdac3 −/− but not WT cells formed spatially organized spheroids (Fig EV2D and E): Nanog reporter‐positive cells were enriched in the inside, while Gata6 reporter‐positive cells co‐expressing Sox17 and showing polarized distribution of the apical PrE marker Dab2 (Hermitte & Chazaud, 2014) were on the outside. This resembles formation of the polarized epithelial PrE cell layer on the surface of the epiblast in E4.5 embryos and indicates spatial lineage segregation in Hdac3 −/− spheroids.
To gain a more complete understanding of lineage restriction, we set out to determine the relationship of Hdac3 with two previously described inhibitors of PrE differentiation, Dax1 and Prdm14 (Khalfallah et al, 2009; Ma et al, 2010; Zhang et al, 2014). Naïve and SLRA‐exposed Prdm14 −/− cells generated in the G6C18 background were indistinguishable from WT controls. In contrast to Prdm14 −/− and similar to Hdac3 −/− cells, Dax1 mutants showed increased Nanog reporter levels in 2iL, and approximately 30% of the cells expressed the Gata6 reporter in SLRA (Figs 2C and D, and EV2F–H, Table EV1). Also, the signaling pathway dependencies for induction of the Gata6 reporter were the same for Dax1 −/− and Hdac3 −/− cells (Figs 2E and EV2I). In Dax1 mutant 3D aggregates, however, peripheral enrichment of Gata6 reporter‐expressing cells was perturbed (Figs2F and G, EV2J), suggesting different roles of Dax1 and Hdac3 in spheroid self‐organization.
We note that Dax1 knockout and knockdown mESCs in SL have been described before, reporting apart from the induction of PrE markers (Niakan et al, 2006; Zhang et al, 2014; Fujii et al, 2015), lack of viability (Yu et al, 1998), loss of pluripotency (Niakan et al, 2006; Khalfallah et al, 2009), induction of 2‐cell stage‐specific genes (Fujii et al, 2015), and multi‐lineage differentiation (Khalfallah et al, 2009). We speculate that these discrepancies arise because the absence of Dax1 in metastable, but not naïve, conditions destabilizes pluripotency and results in or exacerbates culture heterogeneity.
Conversion into a bona fide PrE state in vitro
To compare Gata6 reporter‐positive cells with embryonic PrE, we performed RNAseq of purified Nanog‐ and Gata6‐expressing mutant cells in SLRA. Principal component analysis (Fig 3A) revealed that Gata6 reporter‐positive Dax1 −/− and Hdac3 −/− cells clustered with the E4.5 PrE, while Nanog reporter‐expressing cells were more similar to the E4.5 EPI. Unsorted heterogeneous Hdac3 −/− cells (Fig 1B) resided in between. Pairwise comparison (Fig EV3A) showed that the transcriptional differences between Gata6 and Nanog reporter‐expressing cells in vitro and between the E4.5 PrE and EPI in vivo correlated as strong (Pearson correlation coefficient R = 0.42–0.50) as differences between the E4.5 PrE and EPI of two independent studies (R = 0.49) (Boroviak et al, 2015; Mohammed et al, 2017). Gene expression alterations in Hdac3 and Dax1 mutants relative to WT cells correlated in 2iL (R = 0.53), and in differentiated Nanog reporter‐ (R = 0.77) and Gata6 reporter‐ (R = 0.95) positive cells, demonstrating similar transcriptional roles of Hdac3 and Dax1 in the three cell states (Fig 3B). Notably, cluster 5 genes were deregulated in 2iL and in GFP‐positive cells, indicating PrE priming (Fig 3B). Gata6‐positive cells are therefore transcriptionally similar to embryonic E4.5 PrE.
Figure 3. Transcriptional and functional characterization of in vitro PrE cells.
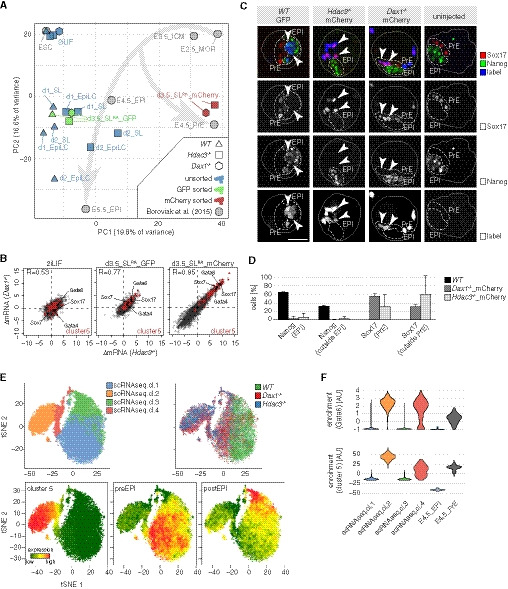
- Principal component analysis of indicated samples. GFP sorted cells express the Nanog>GFP reporter and mCherry sorted cells the Gata6::mCherry reporter. Morula (MOR).
- Scatter plots of log2 fold change (FC) gene expression changes (ΔmRNA) between naïve Hdac3 −/− or Dax1 −/− mutants and WT mESCs (left), between Nanog>GFP‐expressing Hdac3 −/− or Dax1 −/− mutants and WT cells (middle), and between Gata6::mCherry‐expressing Hdac3 −/− or Dax1 −/− mutants and Nanog>GFP‐expressing WT cells (right). Cluster 5 genes are colored and selected PrE TFs labeled.
- Representative control embryo, and embryos that were injected with labeled SLRA‐differentiated WT cells purified for Nanog>GFP expression and mutant cells purified for Gata6::mCherry expression. Embryos are outlined with continuous line. Dashed lines indicate embryonic EPI and PrE compartments. Arrowheads point to injected cells. Scale bar is 25 µm.
- Quantification of the cell fate (based on expression of Nanog and Sox17) and the localization of injected cells. Average and SD of two independent experiments with at least 5 embryos per condition and experiment.
- tSNE maps of scRNAseq of WT, Dax1, and Hdac3 mutants after 3.5 days in SLRA. Cells are color‐coded by k‐means clusters (top left), genotype (top right), and expression of indicated genesets (bottom).
- Expression distribution of Gata6 and cluster 5 genes in single cells of indicated scRNAseq clusters, and E4.5_EPI and E4.5_PrE (Mohammed et al, 2017).
Source data are available online for this figure.
Figure EV3. Characterization of mESC‐generated PrE cells.
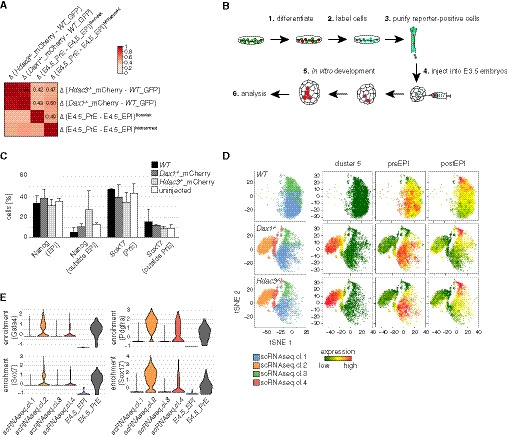
- Pairwise Pearson correlation of transcriptional differences between indicated samples. Individual correlation coefficients are indicated.
- Experimental scheme used to determine developmental competence of subpopulations.
- Quantification of the cell fate (based on expression of Nanog and Sox17) and the localization of unlabeled endogenous cells. Average and SD of two independent experiments with at least 5 embryos per condition and experiment.
- Similar to Fig 3E. Only cells of indicated genotypes are shown.
- Similar to Fig 3F, showing expression distribution of indicated transcripts.
Source data are available online for this figure.
To functionally substantiate this similarity, we decided to determine the developmental competence of mESC‐derived PrE cells. To do so, we injected labeled Nanog>GFP‐positive WT, and Gata6::mCherry‐positive Hdac3 and Dax1 mutant cells into E3.5 blastocysts (Fig EV3B). Upon in vitro development for 2 days, Nanog and Sox17 expression and localization of labeled cells in chimeric embryos were determined (Fig 3C and D). This revealed that mutant cells maintained Sox17 expression and did not contribute to the Nanog‐positive embryonic EPI, which was in contrast to WT cells that maintained Nanog expression. Notably, the spatial distribution of labeled WT and mutant cells in embryos was similar to endogenous Nanog‐ and Sox17‐expressing cells, respectively (Fig EV3C). The PrE state generated by Hdac3 and Dax1 mutant mESCs in vitro is therefore functionally similar to the embryonic PrE.
To explore PrE conversion in single cells, we performed single‐cell RNAseq (scRNAseq) of WT, Hdac3, and Dax1 mutant cells in SLRA. k‐means clustering and visualization using t‐distributed stochastic neighbor embedding (tSNE) identified four scRNAseq cell clusters (Figs 3E and EV3D): Cells from all genotypes contributed to scRNAseq clusters 1 and 3. We note a shift of mutant cells along tSNE1 that likely reflects PrE priming. scRNAseq clusters 1 and 3 were enriched for the expression of genes specific to the pre‐ and post‐implantation epiblast (Boroviak et al, 2015), respectively. scRNAseq clusters 2 and 4, in contrast, were exclusively populated by mutant cells. Consistent with the uniform expression of Sox17, Dab2, and Lama1 in Gata6 reporter‐positive Hdac3 mutant cells (Fig EV2C), PrE markers, such as Gata6, Sox17, and cluster 5 genes, were homogeneously transcribed by scRNAseq cluster 2 cells, which was similar to the distribution in individual cells of the E4.5 PrE (Mohammed et al, 2017) (Figs 3F and EV3E). Moderate induction of these markers in scRNAseq cluster 4 suggests a transition state that bridges the pluripotent (scRNAseq cluster 1) and the PrE (scRNAseq cluster 2) cell states.
Taken together, our transcriptional and functional experiments demonstrate that Hdac3 and Dax1 mutant mESCs convert into a bona fide PrE state in SLRA.
Hdac3 and Dax1 independently restrict PrE fate and antagonize Nr5a2 and Esrrb
Primitive endoderm conversion upon loss of Dax1 and Hdac3 was qualitatively and quantitatively highly similar, suggesting that Dax1 and Hdac3 may act together, potentially in a protein complex. However, affinity purification coupled to label‐free quantitation by mass spectrometry (Fig 4A) revealed that Hdac3 co‐immunoprecipitated subunits of the Ncor1/Ncor2 complexes (Gps2, NCor1, NCor2, Tbl1x, Tbl1xr1), but not Dax1. Vice versa, Dax1 formed a complex with the nuclear receptors Nr5a2 and Esrrb, but not with Hdac3. To test whether Hdac3 and Dax1 mechanisms of PrE repression were truly independent, we analyzed their genetic interaction by generating compound knockout cell lines (Fig EV2F, Table EV1). After 3 days in SLRA, PrE marker levels in Hdac3 −/−; Dax1 −/− double mutants were elevated two‐ to threefold compared with single mutants (Fig 4B). Notably, this additivity was due to a doubling of the fraction of cells expressing the Gata6 reporter, reaching more than 70% (Figs 4C and EV4A). Dax1 and Hdac3 therefore act in parallel pathways that threshold the probability of single cells to exit pluripotency and activate the PrE program. Notably, Gata6 reporter‐positive cells were found at the rim and within Hdac3 −/−; Dax1 −/− spheroids (Fig EV4C and D), therefore presenting an intermediate phenotype compared with the single mutants. Although the greater number of Gata6‐expressing cells in compound versus single knockout spheroids limits direct comparison, this observation is consistent with independent roles of Dax1 and Hdac3 in spheroid self‐organization.
Figure 4. Hdac3 and Nr5a2/Dax1 are genetically and biochemically distinct pathways repressing PrE differentiation.
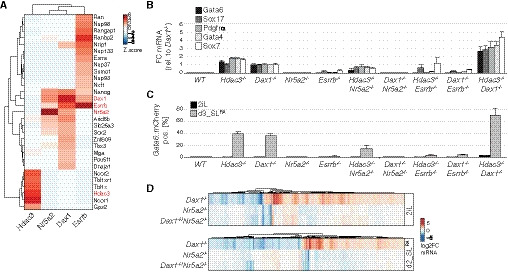
-
AZ‐scores of high‐confidence interactors coIPed by Hdac3, Dax1, Nr5a2, and Esrrb (indicated in red) in 2iL mESCs.
-
B, CExpression of PrE markers relative to Dax1 −/− cells (B), and fraction of Gata6::mCherry‐positive cells after 3 days in SLRA (C) in indicated genotypes. Average and SD of three independent clones.
-
DGene expression changes in Dax1 −/−, Nr5a2 −/−, and Dax1 −/−;Nr5a2 −/− cells compared with WT cells in 2iL (upper) and after 2 days in SLRA (lower).
Source data are available online for this figure.
Figure EV4. Characterization of compound mutant cells.
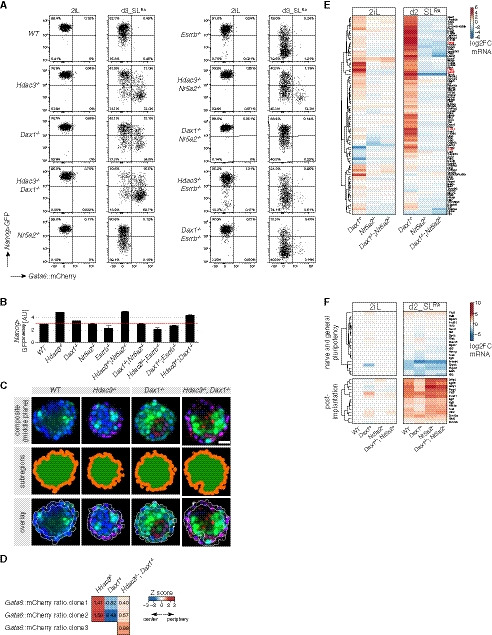
- Representative Nanog>GFP and Gata6::mCherry intensity plots of genotypes and conditions quantified in Fig 4C.
- Geometric mean intensity of Nanog>GFP reporter in 2iL. Average and SD of three independent clones.
- Similar to Fig 2F. Scale bar: 25 μm.
- Similar to Fig 2G.
- Similar to Fig 4D, but focusing on cluster 5 genes.
- Similar to Fig EV1I with a magnified view of a panel of naïve and general pluripotency, and post‐implantation markers in indicated genotypes and conditions relative to naïve WT cells in 2iL.
Source data are available online for this figure.
Since Nr5a2 and Esrrb have been implicated in modulating PrE gene expression (McDonald et al, 2014; Uranishi et al, 2016), we decided to investigate their role in more detail. Consistent with the specific binding to Dax1, deletion of Nr5a2 completely reverted upregulation of the Nanog and Gata6 reporters and of PrE marker genes in Dax1 −/− cells, but only modestly in Hdac3 −/− cells (Figs 4B and C, and EV2F, and EV4A and B, Table EV1). RNAseq in 2iL and SLRA similarly showed that the majority of transcriptional changes induced by the absence of Dax1, including those of cluster 5 genes, were reverted by co‐deletion of Nr5a2, while loss of Nr5a2 on its own caused comparatively minor defects (Figs 4D and Fig EV4E and F). Therefore, Nr5a2 is the key interactor mediating Dax1 function. In contrast to Nr5a2, deletion of Esrrb partially suppressed the upregulation of the Gata6 reporter and of PrE marker genes in both, Dax1 and Hdac3 mutants (Figs 4B and C, and EV2F, and EV4A), suggesting that Esrrb has non‐specific functions downstream of Dax1 and Hdac3. In summary, Dax1/Nr5a2 and Hdac3/Ncor1/Ncor2 form biochemically and genetically distinct complexes that antagonize PrE fate in mESCs.
Gata6 enh−45 is a mechanistic target of Hdac3 and required for PrE differentiation
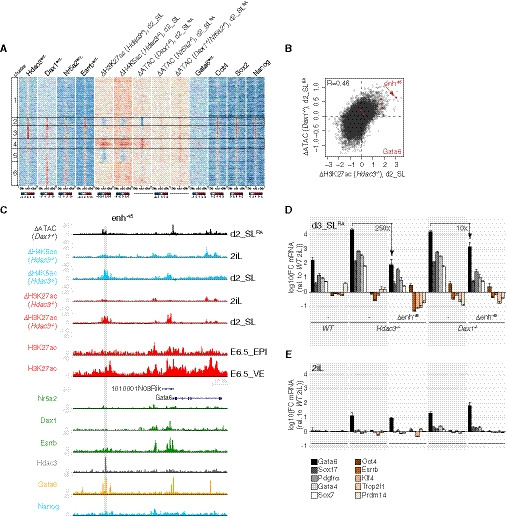
The transcriptional and phenotypic similarity of Hdac3 and Dax1 mutants indicates that they are independently acting components of the same regulatory network. To identify the mechanism of convergence, we exploited published Hdac3, Dax1, and Nr5a2 chromatin immunoprecipitation sequencing (ChIPseq) data (Beck et al, 2014; Żylicz et al, 2018; Atlasi et al, 2019), and determined cis‐regulatory elements (CRE) activity changes by performing assays for transposase‐accessible chromatin sequencing (ATACseq), and H3K27ac and H4K5ac ChIPseq in Dax1 −/− and Hdac3 −/− cells, respectively. Focusing on 29,969 regions bound by either Hdac3 or Dax1, we found that Dax1 occupancy was more similar to that of Nr5a2 (R = 0.58) than of Hdac3 (R = 0.11) (Fig EV5A), which is consistent with distinct biochemical complexes. CRE activity changes in differentiating Dax1 and Hdac3 mutants, in contrast, correlated (Fig EV5B), and clustering analysis (Fig 5A) identified two clusters of loci that were enriched for Hdac3 and Dax1 binding, and linked to CRE repression (cluster 2, decrease in accessibility, H3K27ac and H4K5ac) and activation (cluster 4, increase in accessibility, H3K27ac and H4K5ac). Notably, Nr5a2 deletion reverted CRE activity changes in Dax1 mutants (Fig 5A). Dax1/Nr5a2 and Hdac3 therefore bind to and regulate shared CREs.
Figure EV5. Epigenomic analysis and enh−45 characterization.
-
APairwise Pearson correlation of Hdac3, Dax1, Nr5a2, Esrrb occupancy at regions bound by Hdac3 and/or Dax1. Individual correlation coefficients are indicated.
-
BPairwise Pearson correlation of changes in H3K27ac and H4K5ac or accessibility in mutant relative to WT cells and in indicated conditions at regions bound by Hdac3 and/or Dax1. Individual correlation coefficients are indicated.
-
CSimilar to Fig 5B. CRE bins according to activation and repression in Dax1 and Hdac3 mutants during differentiation are colored.
-
DHeat map of high‐confidence TF motifs, enrichments, and false discovery rates (FDRs) at these bins.
-
ESimilar to Fig 5B with individual regions colored by ChIPseq signal for indicated TFs (upper panels). The same scatterplots with log2 fold H3K27ac changes in naïve Hdac3 mutants instead of accessibility changes in differentiating Dax1 −/− cells plotted on the y‐axis (lower panels).
-
FExpression changes of PrE markers in naïve mESCs of indicated genotypes relative to WT cells. Average and SD of three independent clones.
-
GScatterplots of log2 fold changes in H4K5ac and H3K27ac in Hdac3 mutants at regions bound by Dax1 and/or Hdac3 in naïve and differentiation conditions. Regions colored in red are associated with Gata6.
-
H, IRepresentative Nanog>GFP and Gata6::mCherry intensity plots (H) and quantification of Gata6::mCherry‐positive cells (I) in indicated genotypes after 3 days in SLRA. Average and SD of at least three independent clones.
-
JModel. In WT cells, Hdac3 and Dax1 form a regulatory network that inhibits lineage conversion. Hdac3/NCor1/NCor2 bind and silence enh−45, while Dax1 interacts with and antagonizes Nr5a2, and indirectly influences enh−45. Derepression of Gata6 in Dax1 and Hdac3 mutants causes Gata6 to associate with Hdac3‐ and Dax1/Nr5a2‐bound CREs, together with Esrrb forming a positive feedforward loop for PrE conversion.
Source data are available online for this figure.
Figure 5. Hdac3 and Dax1 co‐repress Gata6‐bound CREs, including the essential Gata6 enh−45 .
-
Ak‐means clustered heatmap showing enrichment of TFs, and changes of chromatin marks or accessibility in indicated mutants relative to WT at Hdac3‐ and/or Dax1‐bound regions. Repressed and induced clusters 2 and 4 are marked.
-
BScatterplot of log2 fold accessibility changes in d2_SLRA Dax1 −/− cells and of log2 fold H3K27ac ChIP signal changes in d2_SL Hdac3 −/− cells relative to WT controls at Hdac3‐ and/or Dax1‐bound regions. Regions colored in red are associated with Gata6.
-
CGenome‐browser view of the Gata6 locus showing accessibility and chromatin mark changes in indicated mutants, and of ChIPseq signals of indicated TFs and H3K27ac in E6.5 VE and EPI. The gray area indicates enh−45.
-
D, EFC of PrE and naïve pluripotency markers relative to WT cells in 2iL of indicated genotypes after 3 days in SLRA (D) and in 2iL (E). Average and SD of at least three independent clones.
Source data are available online for this figure.
To determine the molecular features that are associated with co‐regulated CREs, we scanned underlying TF motifs (Fig EV5C and D). This revealed enrichment of the GATA and OCT4‐SOX2 consensus motifs at CREs that were activated and inactivated in mutant cells, respectively. Comparison with ChIPseq confirmed binding of Oct4, Sox2 and Nanog (Marson et al, 2008) to CREs that are ectopically silenced in naïve and differentiating Hdac3 and Dax1 mutants, and binding of Gata6 (Wamaitha et al, 2015) to CREs that are ectopically activated in differentiating but not naïve mutant cells data (Figs 5A and EV5E). Hdac3 and Dax1 therefore converge on regulating CREs co‐bound by core pluripotency TFs, and on repressing PrE‐specific CREs that are occupied by Gata6 and activated during conversion.
As Gata6 overexpression is sufficient to convert mESCs into XEN cells in vitro (Shimosato et al, 2007), we hypothesized a causative role for Gata6 in PrE differentiation of Dax1 and Hdac3 mutants. This possibility is supported by specific upregulation of Gata6 in naïve single and compound mutants (Fig EV5F) and by the fact that a region 45 kb upstream of the Gata6 gene (enh−45) was among the most strongly activated CREs in mutant cells both during differentiation and in 2iL (Figs 5B and C, and EV5G), and decorated with H3K27ac in the E6.5 VE but not EPI (Xiang et al, 2019) (Fig 5C). Enh−45 is bound by Gata6 and Hdac3, raising the possibility that it is a relevant direct Hdac3 target. To test this, we deleted the 500bp region that is occupied by Hdac3 (Δenh−45) in Hdac3‐mutant cells (Δenh−45; Hdac3 −/− cells) (Table EV1), which fully suppressed activation of the Gata6 reporter upon SLRA transition (Fig EV5H and I) and reduced PrE marker expression to levels observed in WT cells (Fig 5D). Enh−45 therefore mediates PrE conversion in the absence of Hdac3. Gata6 transcription was, however, unaffected in naïve mESCs (Fig 5E), suggesting that PrE priming is independent of enh−45 activation. PrE differentiation was also suppressed in Δenh−45; Dax1 −/− cells, although the reversion of Gata6 transcription was more efficient in Hdac3 than Dax1 compound Δenh−45 mutants (Fig 5D and EV5H and I). This was surprising, since Dax1 does not interact with enh−45 (Fig 5C). Furthermore, removal of enh−45 triggered downregulation of the Nanog reporter and of the naïve pluripotency markers Klf4, Esrrb, Tfcp2l1, and Prdm14 in Hdac3 but not Dax1 mutants in SL (Fig 5D). This indicates specific roles of Hdac3 in the stability of the pluripotent GRN that are masked by enh−45 regulation and not shared with Dax1. We conclude that direct and indirect suppression of enh−45 by Hdac3 and Dax1, respectively, is crucial for repressing Gata6 transcription and, consequentially, PrE conversion upon SL transition.
Discussion
Here, we show that the transcriptional repressors Hdac3 and Dax1 set the lineage barrier that prevents mESCs from differentiating into PrE upon exposure to developmental signals. In naïve conditions, Hdac3‐ and Dax1‐mutant mESCs are stable, which allowed us to exploit the transition from 2iL to SL conditions as paradigm for PrE conversion (Schröter et al, 2015). Even in WT cells, exposure to SL induced transcription of PrE marker genes (Fig 5D) and of Gata6 reporter‐positive cells, although at low frequency (Fig 2A). This is reminiscent of PrE‐primed subpopulations expressing Sox17, Hhex, and Pdgfrα (Canham et al, 2010; Cho et al, 2012; Nigro et al, 2017) in SL cultures. Induction of Pdgfra and Sox17 but not of Hhex in differentiating Hdac3 and Dax1 mutants required enh−45 (Figs 3B and 5D), suggesting that Pdgfrα and Sox17 expression in established SL cultures (Cho et al, 2012; Nigro et al, 2017) is inhibited by Hdac3 and Dax1, while Hhex‐linked PrE priming (Canham et al, 2010) is independent.
Adaption of the SL differentiation regime to 3D results in formation of Hdac3‐mutant spheroids in which pluripotent and PrE lineages are spatially segregated. In Dax1 −/− and, less so, in compound Dax1−/−; Hdac3−/− spheroids, segregation is perturbed. This setup may therefore be amenable to dissecting the molecular requirement underlying PrE differentiation and sorting. However, sorting of mESC‐derived Dax1‐ and Hdac3‐mutant PrE cells in blastocysts is indistinguishable. We speculate that this is because segregation defects that are exposed in mutant spheroids are non‐cell autonomously rescued in chimeric embryos.
Hdac3 and Dax1 cooperatively inhibit PrE differentiation. Notably, conversion of naïve Hdac3 −/− and Dax1 −/− mESCs occurs without overexpression of PrE‐specifying TFs, such as Sox17 (McDonald et al, 2014), Gata4 or Gata6 (Fujikura et al, 2002), or use of selective culture conditions that confer expanded potential (Yang et al, 2017; Sozen et al, 2019) or select the survival and/or proliferation of PrE cell types (Anderson et al, 2017), but upon transition to SL, which supports both, EPI and PrE fates. The pluripotent gene regulatory network is therefore permissive to lineage switching in the absence of ectopic reprogramming TFs or developmental reversion.
Dax1 and Hdac3 act in independent protein complexes that converge on repressing Gata6. We show that lineage conversion in the absence of Dax1 and Hdac3 requires activation of a single Gata6 enhancer, enh−45, that is directly targeted by Hdac3. Recruitment of epigenetic regulators, such as Sin3a (Mall et al, 2017) or Groucho (Muhr et al, 2001; Kutejova et al, 2016) is associated with the repression of alternate lineages, but this study links the physical binding and enzymatic activity of a transcriptional repressor, Hdac3, at a single CRE with suppression of lineage switching. Enh−45 is occupied by Gata6 during PrE conversion, indicating a positive feedforward loop that is characteristic of cell type‐specifying TFs (Crews & Pearson, 2009) and can stabilize cell identity (Leyva‐Díaz & Hobert, 2019). Our findings therefore suggest that the continuous silencing of autoregulated enhancers of competing master TFs is an endogenous mechanism for maintaining lineage restriction.
We find that Hdac3 and Dax1 repress PrE differentiation by modulating the pluripotent GRN network, specifically Nr5a2 and Esrrb, which are TFs that regulate mESC self‐renewal (preprint: Festuccia et al, 2020) and iPSC reprogramming (Feng et al, 2009; Guo & Smith, 2010). Nr5a2 is a well‐established interactor of Dax1 (Sablin et al, 2008). Co‐deletion of Nr5a2 rescues PrE conversion and most transcriptional and epigenetic alterations in Dax1 but not Hdac3 mutants. Therefore, Nr5a2 and Dax1 operate as a functional unit where Nr5a2 is the transcriptional activator and Dax1 the repressor. The direct targets of Nr5a2 driving PrE differentiation remain unclear, since Nr5a2 and Dax1 do not bind to enh−45. Lack of Esrrb, in contrast, partially blocks PrE conversion of Dax1 and Hdac3 mutants. Although Esrrb interacts and shares genomic binding with Dax1 and Nr5a2 (Figs 4A and 5A, and EV5A), incomplete phenotypic rescue and lack of specificity for Dax1 suggests a role of Esrrb in PrE conversion beyond the Nr5a2/Dax1 axis, e.g., by binding (Fig 5C) and activating the Gata6 promoter (Uranishi et al, 2016) or, indirectly, by stabilizing pluripotency (Martello et al, 2012). Taken together, we propose that Nr5a2 and Esrrb support both the EPI and the PrE fate within a coherent TF network that integrates extrinsic signals with transcriptional repressors to maintain lineage choice.
Gata6 overexpression also induces PrE gene transcription in human ESCs (Wamaitha et al, 2015). However, naïve human ESCs are less lineage‐restricted than mouse naïve cells and efficiently differentiate into both, extraembryonic ectoderm (Guo et al, 2021) and extraembryonic endoderm (Linneberg‐Agerholm et al, 2019). Furthermore, DAX1 is not significantly expressed in human EPI (Boroviak et al, 2018) and ESCs (Guo et al, 2016). Distinct mechanisms may therefore regulate lineage segregation in mouse and human pluripotent stem cells. The role of HDAC3 therein remains to be determined.
In summary, we uncover how the pluripotent lineage is safeguarded in mESCs. We find that both, Gata6 and Nanog, are induced in naïve Hdac3 −/− and Dax1 −/− mESCs, and that enh−45 is not occupied by Nanog (Fig 5C). This is inconsistent with mutual inhibition of Gata6 by Nanog and indicates that maintenance of the EPI‐PrE segregation is independent of direct TF antagonism. We, instead, suggest operation of a GRN that (i) epigenetically silences Gata6, the master TF of the competing PrE fate, and (ii) balances the activities of lineage‐divergent TFs, such as Nr5a2 and Esrrb (Fig EV5J). We speculate that this regulatory network is established during developmental progression of pluripotency to stabilize the initially labile lineage segregation set by antagonism between Nanog and Gata6.
Materials and Methods
Mouse ESCs
Male TNG‐A mESCs where a GFP‐IRES‐Puromycin‐N‐acetyltransferase is knocked into one of the Nanog alleles (Chambers et al, 2007) are a gift from Austin Smith (Wellcome—MRC Cambridge Stem Cell Institute and Living Systems Institute, University of Exeter).
Cell culture
Mouse embryonic stem cells were cultured on gelatin‐coated plates in N2B27 medium (DMEM/F12 medium (Life Technologies) and Neurobasal medium (Gibco) 1:1, N2 supplement 1/200 (homemade), B‐27 Serum‐Free Supplement 1/100 (Gibco), 2 mM L‐glutamine (Gibco), 0.1 mM 2‐mercaptoethanol (Sigma)), 1 µM PD0325901, 3 µM CHIR99021 (Steward lab, Dresden), and mLIF (Smith lab, Cambridge; Chao lab, FMI). For medium switch and differentiation experiments, cells were washed with PBS, detached in Accutase (Sigma), centrifuged at 300 g for 3 min in DMEM/F12 with 0.1% BSA (Gibco), resuspended in the appropriate medium, counted using Fast Read 102 counting chambers (Kova), diluted, and plated. Cells were plated in Serum LIF (SL) medium (GMEM (Sigma), 10% fetal bovine serum (Sigma), 1 mM sodium pyruvate (Gibco), 2 mM L‐glutamine (Gibco), 0.1 mM non‐essential amino acids (Gibco), 0.1 mM 2‐mercaptoethanol (Sigma), and mLIF), plus Retinoic acid (1 nM, Sigma), JAK Inhibitor I (1 µM, Calbiochem) and LDN193189 (100 nM, Sigma), PD0325901, where specified, at 10,000 cells/cm2 on gelatin‐coated plates, or in EpiLC medium (N2B27 medium, 20 ng/ml activin A and 12 ng/ml bFGF (Smith lab, Cambridge), and 1% KSR (Life Technologies)) at 25,000 cells/cm2 on fibronectin‐coated plates. For pharmacological inhibition of Hdac3, WT mESCs were grown in 2iL in the presence of 5 or 0.5 µM RGFP966 (Abcam, ab144819) for 48 h and then switched to SLRA with 5 or 0.5 µM RGFP966 for 72 h, as described above. To generate spheroids, 1,200 cells were plated as described above in 1.5 ml SL plus 1 nM Retinoic acid in a well of an AggreWell 400 plate (Stemcell Technologies), prepared with anti‐adherence rinsing solution (Stemcell Technologies) according to manufacturer’s instructions, and incubated for 3 days.
TNG‐A mESCs stably transfected with pPB‐LR51‐EF1a‐bsdr2ACas9 (modified from (Koike‐Yusa et al, 2013)) were reverse transfected with U6>sgRNA plasmids (George Church, Addgene plasmid #41824) as follows: 3 µl of Lipofectamin2000 (Life Technologies) were mixed with 250 µl of OPTIMEM (Gibco) and incubated for 5 min at RT, 350 ng of sgRNA plasmid diluted in 250 µl of OPTIMEM was added to the Lipofectamin2000 mix and incubated for 30 min at room temperature (RT), the transfection mix was added to freshly resuspended 200,000 cells in 2 ml of medium in a well of a 6‐well plate. The next day medium was changed. 3 days after transfection, single cells were sorted in 96‐well plates for clonal isolation. Parental cell line, sgRNA sequences, genotyping strategy, and sequencing results/Western blots for every knockout clone used in this study are detailed in Table EV1 and supplementary figure panels. The Hdac3Y118F knock‐in cell lines were generated by reverse transfection as above of 400 ng Hdac3_gRNA7 plasmid and 10 pmol of oligo Hdac3YF_ki_f (for clone 1034‐15) or oligo Hdac3YF_ki_r (for clone 1035‐6). The Gata6::mCherry knock‐in cell line G6C18 was generated by reverse transfection as above of 300 ng Gata6‐C gRNA#1 plasmid and 800 ng of Gata6_3xGS_mCherry homologous recombination template. sgRNAs, oligonucleotide sequences, genotyping strategies, and sequencing results for knock‐in clones are detailed in Table EV1.
siRNA transfections
Cells were reverse transfected in 2iL as follows: 2.4 µl of Lipofectamine RNAiMAX (Life Technologies) were mixed with 200 µl of OPTIMEM (Gibco) and incubated for 5 min at RT, 2.4 µl of 20 µM Ncor1 siRNA mix and Ncor2 siRNA mix or AllStars Neg. Control siRNA (FlexiTube GeneSolution, Qiagen) diluted in 200 µl of OPTIMEM were added to the Lipofectamine RNAiMAX mix and incubated for 30 min at RT, the transfection mix was added to freshly resuspended 100,000 cells in 1.6 ml of medium in a well of a 12‐well plate. The next day the cells were detached and transferred to fresh 2iL or Serum LIF. siRNA sequences are displayed in Table EV1.
Western blotting
Cells were lysed in 1× RIPA buffer (50 mM Tris pH7.5, 150 mM NaCl, 1% IGEPAL, 0.5% Na Deoxycholate, 0.1% SDS, 2 mM EDTA), protein concentration was determined by the BCA protein assay (Pierce), and 10 µg of protein per sample were loaded on 10% polyacrylamide gels. Primary antibodies used: anti‐HDAC3 antibody, ab7030, by Abcam, 1/5,000; DAX1/NR0B1 antibody (mAb), Clone: 1DA‐2F4, Active Motif, Catalog No: 39983, 1/10,000; Human ERR beta/NR3B2 Antibody, PP‐H6705‐0, by R&D Systems, 1/10,000.
Quantitative PCR (qPCR)
RNA was purified with the RNeasy Mini Kit with on‐column DNase digest (Qiagen). 1 µg of RNA was reverse transcribed using SuperScript III Reverse Transcriptase (Life Technologies), and qPCR was performed with the TaqMan Fast Universal PCR Master Mix (Thermo Fisher) and the following TaqMan probes: GAPDH (4352339E), Gata6 (Mm00802636_m1), Gata4 (Mm00484689_m1), Pdgfra (Mm00440701_m1), Sox7 (Mm00776876_m1), Sox17 (Mm00488363_m1), Oct4 (Mm03053917_g1), Esrrb (Mm00442411_m1), Klf4 (Mm00516104_m1), Tfcp2l1 (Mm00470119_m1), and Prdm14 (Mm01237814_m1).
Co‐immunoprecipitations (IPs)
Endogeneous Hdac3 was IP’ed from nuclear extract of G6C18 cells in 2iL using anti‐HDAC3 antibody (ab7030, by Abcam) and rabbit normal IgGs (Santa Cruz) as control. N‐3xFLAG‐3GS‐Dax1, Nr5a2‐3GS‐3xFLAG‐C, and Esrrb‐3GS‐3xFLAG‐C cloned in pPB‐CAG‐DEST‐pgk‐hph (Betschinger et al, 2013) were expressed in G6C18 cells in 2iL and IP’ed from nuclear extracts using anti‐FLAG M2 antibody (Sigma) using un‐transfected cells as control. Briefly, cells from a confluent T‐75 flask were harvested and nuclei isolated by hypotonic buffer (10 mM Tris–HCl pH7.5, 10 mM KCl, 1 mM dithiothreitol (DTT), 0.5% IGEPAL) for 20 min on ice. Nuclei were lysed by rotation for 90 min at 4°C in 1 ml of lysis buffer (20 mM Tris–HCl pH7.5, 100 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 10% glycerol, 1× Protease Inhibitor Tablet (Roche), 2× Phosphatase Inhibitor Tablets (Roche), 0.5% Triton X‐100, 250 units/ml Benzonase (Sigma)). Clarified lysates were incubated with 1.2 µg of antibody pre‐bound to 5 µl protein Dynabeads (Life Technologies) for 6 h at 4°C. Beads were washed 4 times with wash buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5% IGEPAL) and 3 times with wash buffer without detergent, and digested with 0.2 µg Lys‐C (WAKO) and 0.2 µg modified porcine trypsin (Promega) before subjecting to mass spectrometry as described before (Villegas et al, 2019).
Flow cytometry
Live cells were washed with PBS, detached in Accutase, centrifuged at 300 g for 3 min in DMEM/F12 with 0.1% BSA, resuspended in DMEM/F12 with 0.1% BSA, run on an LSRII SORP Analyzer (Becton Dickinson), and analyzed using FlowJo (FlowJo, LLC). Quantification of Gata6::mCherry‐positive cells (Figs 2B and E, 4C, and EV5I) was performed on experimental triplicates, with gates shown in fluorescence intensity plots. Nanog>GFP intensities (Figs EV2H and EV4B) are the average of geometric GFP mean intensities of experimental triplicates.
For purification of Gata6::mCherry and Nanog>GFP‐positive cells for RNAseq, WT, Hdac3 −/−, and Dax1 −/− naïve mESCs were differentiated in SLRA as described above for 86 h, detached as described above, and sorted on a Becton Dickinson FACSAria III cell sorter.
For indirect flow cytometry, cells were detached as described above, fixed for 20 min in 4%PFA (Electron Microscopy Science, Cat# 15713) in PBS, washed twice with PBS with 0.1% Tween 20, and permeabilized with 0.1% Triton X‐100 in PBS for 15 min at RT. After blocking for 3 h with blocking solution (PBS with 1% BSA (Sigma, A2153) and 0.1% Tween 20), cells were incubated overnight at 4°C in blocking solution with the primary antibodies: anti‐Dab2 monoclonal antibody (D709T), #12906S Cell Signaling Technology, 1/200; Human SOX17 Antibody, #AF1924 R&D Systems, 1/100; anti‐Lama1 rabbit polyclonal antibody, L9393 Sigma‐Aldrich, 1/1,000. Cells were washed 3 times with blocking solution, incubated for 1 h at RT with secondary antibody in blocking solution (goat anti‐rabbit Alexa Fluor 647 and donkey anti‐goat Alexa Fluor 647, 1/500, Thermo Fisher). After 3 washes in PBS‐0.1% Tween 20, cells were analyzed on a LSRII SORP Analyzer (Becton Dickinson). Primary data were analyzed using the Bioconductor package flowCore (Hahne et al, 2009): Gata6::reporter‐positive and Gata6::reporter‐negative populations were identified, and the distribution of antibody staining intensities in the two populations was plotted.
Blastocyst injection and maturation
Seven‐week‐old CD‐1 females (Janvier laboratory) were superovulated by injection of pregnant mare serum gonadotropin (PMSG, 5U, MSD Cat#A207A01) and of human chorionic gonadotropin (hCG, 5U, MSD Cat#A201A01) and mated to CD‐1 males. 2‐cell stage embryos were recovered from oviducts by flushing and cultured in KSOM medium (Millipore Cat#MR‐106‐D) covered with mineral oil (Sigma) at 37°C with 5% CO2 and 5% O2 air.
WT, Hdac3 −/−, and Dax1 −/− naïve mESCs were cultured in SLRA as described above for 72 h, detached as described above, and stained with 5 μM Tag‐it Violet Proliferation and Cell Tracking Dye (BioLegend, 425101) in PBS for 20 min at RT. The staining solution was quenched with 1 volume of SLRA, and the cells were washed once in SLRA prior to sorting Hdac3 −/− and Dax1 −/− Gata6::mCherry‐positive cells and WT Nanog>GFP‐positive cells on a Becton Dickinson FACSAria III cell sorter. 10 to 15 FACS‐sorted cells were injected into a blastocyst (E3.5) by Piezo‐assisted micromanipulation, followed by 48 h in vitro culture until E5.5. All experiments were approved by the veterinary authority of the canton Basel‐Stadt, Switzerland.
Antibody staining of blastocysts and image analysis
Embryos were fixed in 4% paraformaldehyde (PFA, Electron Microscopy Science, Cat# 15713) in PBS for 20 min and permeabilized with 0.5% Triton X‐100 in PBS for 15 min at RT. After two washes of 10 min in PBS with 0.1% Tween 20, embryos were blocked for 2 h in PBS containing 0.1% Tween 20 and 2% horse serum at RT. Embryos were incubated with primary antibodies in PBS containing 0.1% Tween 20 overnight at 4°C (rabbit anti‐NANOG, Cosmo Bio, Cat# REC‐RCAB004P‐F, 1:700 dilution; goat anti‐SOX17, R&D, Cat# AF1924, 1:300 dilution). After three washes of 10 min in PBS with 0.1% Tween 20, embryos were incubated with secondary antibodies diluted 1:500 in PBS with 0.1% Tween 20 for 1 h at RT (Alexa Fluor donkey anti‐rabbit IgG 488, Thermo Fisher Cat# A‐21206; Alexa Fluor donkey anti‐goat IgG 647, Thermo Fisher Cat# A‐21447). After three washes of 10 min in PBS with 0.1% Tween 20, embryos were transferred on a glass slide (VWR, Cat# 631‐0453), mounted with Vectashield medium (Vector Laboratories, Cat# H‐1000), and covered by coverslips. Embryos were imaged using a Zeiss Axio Imager M2 microscope equipped with a Yokogawa CSU W1 Dual camera T2 spinning disk confocal scanning unit using a 63× objective. Z‐stacks were acquired with a step size of 0.5 μm.
Endogenous and injected cells within each embryo were scored for Nanog and Sox17 staining and for being part of EPI or PrE (Figs 3D and EV3C): Nanogpositive and Sox17positive cells were counted on the basis of immunofluorescence signal. Assignment to either EPI or PrE was done by visual inspection, firstly identifying the location of the inner cell mass (ICM) inside each embryo, also with respect to the position of the blastocoel. Subsequently, cells that were clustered together in the inner part of the ICM were considered as belonging to the EPI, and cells that were positioned between the EPI and the blastocoel were defined as belonging to the PrE. All other Nanog+ and Sox17+ cells that did not fulfill these spatial requirements were scored as being outside. Injected cells were scored as “dead” and removed from the analysis whenever they were just a bright fluorescent dot without expression of Nanog or Sox17.
Antibody staining of spheroids
Fixed spheroids were incubated in blocking solution (3% donkey serum (Sigma), 1% BSA in PBST) for 1 h at RT, stained overnight at 4°C with antibodies (anti‐Dab2 monoclonal antibody (D709T), #12906S Cell Signaling Technology, 1/200; Human SOX17 Antibody, #AF1924 R&D Systems, 1/200) in blocking solution, washed three times with PBST, stained with secondary antibodies (goat anti‐rabbit Alexa Fluor 647 and donkey anti‐goat Alexa Fluor 647, 1/500, Thermo Fisher) in PBST for 1 h at RT, washed twice with PBST and once with PBST‐Hoechst 33342, transferred in PBS to a µCLEAR 96‐well plate (Greiner bio‐one), and imaged on a Zeiss LSM710.
High‐throughput imaging of spheroids
To fix the spheroids, 1mL of medium was removed from each well and 500 µl of 8% PFA (Alfa Aesar) in PBS were added to the remaining 500 µl of medium and incubated for 20 min at 37°C. The spheroids were then harvested with a 1,000 µl pipette, washed twice with PBST (PBS‐0.1% Tween 20 (Sigma)) and once with PBST‐Hoechst 33342 (1/5,000, Life Technologies), transferred in PBS to a µCLEAR 96‐well plate (Greiner Bio‐One), and imaged on a Yokogawa CV7000s high throughput confocal microscope at 40× magnification. Images were acquired in confocal mode as z‐stack multiplane images over a z distance of 100 µm with 5 µm step width. Subsequently, images were stitched to generate a single image per z‐plane, channel, and well, which was used for object segmentation and feature extraction.
Native histone ChIPseq
Nuclear lysates were prepared from 10 million cells as for coIPs above. Clarified lysates were incubated with 4 µg of primary antibody for 2 h and then SDS concentration was brought to 0.1% and EDTA to 5 mM to inactivate benzonase. Lysates were clarified again and 40 µl of protein G Dynabeads were added and rotated for 6 h at 4°C. Beads were washed twice with RIPA buffer, four times with RIPA buffer with 500 mM NaCl, and once with 10 mM Tris–HCl pH8.0 with 1 mM EDTA. DNA was eluted by proteinase K (MACHEREY‐NAGEL) treatment in 0.5% SDS for 3 h at 55°C and purified with Qiagen MinElute PCR Purification Kit. Primary antibodies used: H4K5ac (Merck‐Millipore, 07‐327) and H3K27ac (Active Motif, 39135). Sequencing libraries for biological duplicates of each assayed histone modification and input per condition were prepared using the ChIP‐Seq NEB Ultra (Dual Indexes) Kit (New England Biolabs) and sequenced on an Illumina HiSeq2500 (50 bp single‐end reads). In total, ~ 1.05 billion reads (accounting for ca 45 million reads per replicate) were generated. After demultiplexing, reads were aligned against the mouse genome (GRCb38/mm10) using the QuasR Bioconductor library (Gaidatzis et al, 2014) with default parameters. The overall alignment rate was ~95%.
ATACseq
ATACseq was performed as described before (Buenrostro et al, 2015). Briefly, freshly isolated nuclei from 50,000 cells were subjected to transposition using the Nextera Tn5 Transposase for 30’ at 37°C. DNA was purified with the Qiagen MinElute PCR Purification Kit and amplified for a total of 12 cycles with the Q5 PCR mix (NEB). Libraries of experimental triplicates were purified with AMPure‐XP beads (Beckman Coulter) and sequenced on an Illumina NextSeq500 machine (75 bp paired‐end reads). In total, ~ 0.96 billion reads (accounting to ca 80 million reads or 40 million read‐pairs per replicate) were generated. After demultiplexing, reads were aligned against the mouse genome (GRCb38/mm10) using the QuasR Bioconductor library (Gaidatzis et al, 2014) with default parameters. The overall alignment rate was ~ 94%.
RNAseq
RNA of biological triplicates was purified with the RNeasy Mini Kit with on‐column DNase digest (Qiagen), RNAse treatment etc. Libraries were prepared using TruSeq mRNA Library preparation kit (Illumina) and sequenced on an Illumina HiSeq2500 machine (50 bp single‐end reads). In total, ~ 1.6 billion reads (accounting for ca 30 million reads per replicate) were generated. After demultiplexing, reads were aligned against the mouse genome and guided by transcriptome annotation (GRCb38/mm10, GENCODE release M4) using STAR (Dobin et al, 2012) version 2.5.0 with command line parameters:
‐‐outSJfilterReads Unique ‐‐outFilterType BySJout ‐‐outFilterMultimapNmax 10 ‐‐alignSJoverhangMin 6 ‐‐alignSJDBoverhangMin 3 ‐‐outFilterMismatchNoverLmax 0.1 ‐‐alignIntronMin 20 ‐‐alignIntronMax 1000000 ‐‐outFilterIntronMotifs RemoveNoncanonicalUnannotated ‐‐seedSearchStartLmax 50 ‐‐twopassMode Basic
The overall alignment rate was consistently ~ 90%.
scRNAseq
Wild‐type, Hdac3 −/−, and Dax1 −/− naïve mESCs were differentiated in SLRA as described above for 86 h, detached as described above, and sorted on a Becton Dickinson FACSAria III cell sorter. Sorted single‐cell suspensions were processed onto the 10× Genomics Chromium platform for GEM and cDNA generation, followed by sequencing library preparation according to the manufacturer protocol, using the 10× Chromium Single Cell 3ʹ GEM, Library & Gel Bead Kit v3 reagents. Libraries were sequenced on the NextSeq500 platform using 75 cycles v2.5 reagents (R1: 28 bp, R2 56 bp).
Integration of external embryonic RNAseq datasets, and pairwise correlation and principal component analysis
GRCm38 alignments from (Boroviak et al, 2015) were downloaded (E‐MTAB‐2958), and exonic gene counts were obtained on the GENCODE M4 transcriptome annotation using the Genomic Features Bioconductor package (Lawrence et al, 2013). Single‐cell RNAseq filtered count matrices from (Mohammed et al, 2017) were downloaded (GSE100597) and gene annotation was lifted from ENSEMBL to GENCODE M4 discarding ambiguous mappings. Assignments of individual cells to specific lineages were obtained through personal communication with the study authors and bulk gene counts for each stage and lineage were calculated by aggregating the corresponding single‐cell data. Gene counts from all datasets were library normalized and converted to CPMs. Microarray data from (Anderson et al, 2017) were downloaded (GSE77783) and analyzed using the limma Bioconductor package (Ritchie et al, 2015). Pairwise correlation values (Figs 1B and EV1B and C) between individual samples of two datasets were calculated after gene‐wise mean normalization of the log2 gene expression values.
For the principal component analysis using the prcomp function (parameters: center = TRUE; scale = TRUE) of datasets from different labs (Fig 3A), we converted gene expression values of a sample i in dataset d to log2 fold gene expression changes (S′) relative to a matching reference sample R (WT 2iL and mESC (Boroviak et al, 2015)):
We used the intersection of the approximately top 5,000 most variable genes, according to a within‐dataset mean‐variance trend fit for RNAseq experiments and a log2FC‐based selection for microarray experiments (provided in source data), for pairwise correlation and principal component analysis.
Differential gene expression analysis and gene clustering
Differential gene expression (Figs 3B and 4D, and EV4E) was calculated using the Bioconductor edgeR package (Robinson et al, 2009; McCarthy et al, 2012) (provided in source data). Briefly, for each experimental design a common negative binomial dispersion parameter was estimated using the estimateGLMCommonDisp function and a negative binomial generalized log‐linear model was fit per gene using the glmFit function with a prior count of 1 in order to shrink log‐fold change (LFC) effect sizes of lowly expressed genes toward zero. Reported P‐values are the Benjamini–Hochberg‐adjusted estimates for multiple comparisons.
Clusters of genes with consistent expression profiles (Fig 1C) were identified with k‐means clustering using as input features the gene expression log2FC of WT and Hdac3 −/− TNG‐A‐derived clones in d2_SL and d2_EpiLC relative to WT TNG‐A 2iL cells, focusing on the approximately top 5,000 most variable genes as described above, and applying a pseudocount of 1 to shrink effect sizes of lowly expressed genes. Only genes with a maximum absolute log2FC > 1.5 in any of the four contrasts were considered (provided in source data). The calculation was carried out using the base R k‐means function implementation with centers = 8 and 200 random starts. Analyses of enriched gene sets (Fig EV1E) were performed using DAVID (Huang et al, 2008) for GO terms of biological processes.
scRNAseq analysis
The single‐cell RNAseq data were quantified using CellRanger (Zheng et al, 2017) (version 4.0.0), and a reference transcriptome generated from the mouse mm10 genome and the GENCODE genome annotation. All subsequent processing operations and analyses were performed in R (version 4.0.3). Quantifications were imported using the DropletUtils package (Lun et al, 2019) (version 1.4.3), and quality control was performed using scater version 1.18.3 (McCarthy et al, 2017), and normalization and log‐transformation of UMI counts with scran version 1.18.1 (Lun et al, 2016). After inspection of the distribution of cells across an array of quality indices, we excluded from further analyses cells with a total feature (gene) count below 400 or above 6,000, a mitochondrial gene count fraction above 10% as well as cells outside the 1st percentile of assigned 2 days density estimated from the total UMI count per cell and the percentage of ribosomal protein gene counts. The final total numbers of retained cells for the WT, Hdac3 KO, and Dax1 KO conditions were 6,030, 3,951, and 4,707, respectively. The corresponding median library sizes were 7,840, 11,238, and 9,318. The minimum and maximum library sizes across all three conditions were 4,012 and 23,918, respectively. Batch library normalization across the three conditions was performed with batchelor version 1.6.2 (function multiBatchNorm).
Prior to dimensionality reduction, we excluded ribosomal protein and mitochondrial genes, genes detected in < 10% cells in all three libraries as well as the bottom 75% of the remaining genes in terms of their modeled combined variance (scran functions modelGeneVar, combineVar, and getTopHVGs). We finally excluded the scran collection of mouse cell‐cycle markers in order to reduce the cell‐cycle originating variance. Multi‐sample PCA and mutual‐nearest‐neighbor batch‐effect correction (Haghverdi et al, 2018) across the three libraries was performed with batchelor. The final corrected low‐dimensional coordinates were passed to Rtsne to obtain a harmonized projection for the three conditions. Cell clusters were defined using k‐means clustering (function k‐means, centers = 4) on the common, batch‐corrected low‐dimensional space (Table EV1). Pre‐EPI is the combination of the EPI and ICM+EPI markers, and post‐EPI the PE geneset (Boroviak et al, 2015). For calculating enrichment scores (Figs 3F and EV3E), library normalized single‐cell gene counts were log2‐transformed using a pseudocount of 1 and subsequently mean‐centered across all single cells of a particular dataset. Enrichment scores are the sum of these transformed counts for the indicated genesets or genes per cell.
Peak calling
| Accession | ChIPseq description | Reference |
|---|---|---|
| SRR8058260 | Hdac3 | Żylicz et al (2018) |
| SRR8058264 | Hdac3, control | |
| SRR2141930 | Dax1, rep1 | Beck et al (2014) |
| SRR2141931 | Dax1, rep2 | |
| SRR2141933 | Dax1, control | |
| SRR5110917 | Nr5a2 | Atlasi et al (2019) |
| SRR5110908 | Nr5a2, input | |
| SRR5077736 | Esrrb | Chronis et al (2017) |
| SRR5077675 | Esrrb, input | |
| SRR2043315 | Gata6, rep1 | Wamaitha et al (2015) |
| SRR2043317 | Gata6, rep2 | |
| SRR2043316 | Gata6, control1 | |
| SRR2043318 | Gata6, control2 | |
| SRR713340 | Oct4 | Marson et al (2008) |
| SRR713341 | Sox2 | |
| SRR713342 | Nanog |
Chromatin immunoprecipitation sequencing data were realigned to GRCb38/mm10 using the QuasR Bioconductor library (Gaidatzis et al, 2014) with default parameters. Dax1 and Hdac3 peaks of significantly high read density, taken as a proxy of protein binding, were called using the csaw software (Lun & Smyth, 2015a) and according to the workflow outlined in (Lun & Smyth, 2015b). Briefly, the average fragment length was computed (function correlateReads) and reads were counted in windows of width w = 78 bp both for the precipitates and the input (where available). Windows of significant enrichment were filtered against a local background in regions of 5 kbp centered around the 78 bp windows (threefold enrichment, function filterWindowsLocal), and against the matching control sample (threefold enrichment, function filterWindowsControl). Finally, nearby identified peaks, closer than 750nt apart, were merged (function mergeWindows). Blacklisted regions that often yield artifactual high signal in ChIPseq experiments were obtained from the ENCODE project (https://www.encodeproject.org/files/ENCFF547MET/) and excluded from all read‐counting operations (read parameter discard). Peaks were assigned to their proximal‐most genes and distances to transcription start‐sites (TSSs) were calculated according to the GENCODE release M4 annotation. A peak was considered as a “promoter peak” if it was within 1 kbp distance of an annotated TSS.
Differential ChIPseq/ATACseq enrichment
Chromatin immunoprecipitation sequencing and ATACseq signal was quantified in 1 kb regions at the 29,969 peaks that are the union of Hdac3 (24,249) and Dax1 (10,671) peaks (provided in source data). In all cases, differential ChIPseq or ATACseq enrichments (Figs 5A and B, and EV5A–C, E and G) between conditions are stated, the values refer to changes in library normalized read counts, averaged, when available, over biological replicates. enr: denotes enrichment over control or input.
Clusters of peaks (Fig 5A) were identified with k‐means clustering using as input features the enrichment of Hdac3 and Dax1 ChIPseq over controls, respectively, and differential H3K27ac ChIPseq and ATACseq signals in Hdac3 and Dax1 mutants, respectively, compared with WT cells after 2 days of differentiation in SL. The calculation was carried out using the base R k‐means function implementation with centers = 6 and 200 random starts. The Bioconductor ComplexHeatmap package (Gu et al, 2016) was used to plot enrichments in 10 kb, peak‐centered windows.
Figure 5C was assembled using the UCSC genome browser interface (https://genome.ucsc.edu).
Motif enrichment analysis
Motif enrichment (Fig EV5D) at Hdac3 and Dax1 peaks as a function of CRE activity was performed as described before (Barisic et al, 2019). Briefly, H3K27ac (log2(Hdac3 −/−)—log2(WT), d2_SL) and ATAC (log2(Dax1 −/−)—log2(WT), d2_SLRA) changes between mutant and wild‐type cells were calculated at each of the 29,969 peaks of the Hdac3/Dax1 peak union set. log2FCs of both, H3K27ac and ATAC signals, were used to group peaks into nine discrete bins containing 1,319 peaks each. Peaks with absolute log2FCs less than 0.5 were grouped into a single bin (unchanged CRE activity). The nine color‐coded bins are shown in Fig EV5C. Positional weight matrices (PWMs) for 519 vertebrate transcription factors available from the JASPAR database through the Bioconductor JASPAR2016 package (Mathelier et al, 2015) were used to determine motif enrichment in each bin with the findMontifsGenome.pl script of the HOMER package (Heinz et al, 2010) using all other bins as background and parameters ‐nomotif ‐size 500. A cutoff for the expression of TFs in mESCs and during differentiation, and for the enrichment significance of their motifs was employed. Reported enrichment significance values are FDRs corrected for multiple testing.
Mass spectrometry analysis
Relative protein quantifications of three independent affinity purifications (1: experimental duplicates of anti‐Dax1, anti‐Esrrb, and anti‐Nr5a2 coIPs, 2: experimental triplicates of anti‐Dax1, anti‐Esrrb, anti‐Nr5a2, and anti‐Hdac3 coIPs, 3: experimental triplicates of anti‐Hdac3 coIPs) were merged and missing values filled in with a 1.8‐fold downshift and a 0.3‐fold distribution width of the actual distribution of detected proteins, as described (Tyanova et al, 2016). Only proteins detected in all experimental replicates of any coIP were considered. Log2FC enrichments were calculated over the respective experimental negative controls, and Z‐scores per each coIP and experiment determined (provided in source data). Only proteins with Z‐scores > 2 in both independent affinity purifications are considered (Fig 4A).
Segmentation and quantification
Object segmentation was performed using image processing libraries available for Python 3. To identify the middle plane of spheroids, segmentation masks were first created for the maximum intensity projections (MIPs) of the DNA channel by automated thresholding with 2‐class Otsu algorithm. Entire z‐stacks were then cropped to segmentation outlines obtained from the MIP segmentation. Subsequently, sum intensity of DNA staining in each plane was measured and used to identify the z‐plane with maximum sum intensity of DNA signal, corresponding to the middle plane of the object. Segmentation masks were then adjusted in‐plane by automated thresholding and used for subregion analysis. Each segmentation mask of the object middle plane was subdivided into 2 regions: A 50 pixel‐wide concentric region along the object periphery and a central region, ranging from the object center to the border of the peripheral region. To exclude debris, objects where the central subregion was less than 30% of the object area were eliminated from the analysis. Areas and mean intensities of the Nanog>GFP and Gata6::mCherry channels were then separately calculated for the two regions, and used for quantitative analysis. Distribution of intensity between the peripheral and central regions was calculated as the ratio of mean intensity in the peripheral and central region (provided in source data). Extracted features describing object area and intensity were normalized to the WT controls within corresponding biological replicates using z‐score transformation and unified into a cross‐comparable dataset (Figs 2F and EV4C, provided in source data).
Quantification and statistical analysis
Details for quantification and statistical analysis are specified in the figure legends, including the number of biological replicates. Data are presented as the average and standard deviation.
Author contributions
Study conception and experiments: DO; Bioinformatical analysis: PP and JB; RT–qPCRs: MR; Mass spectrometry: DH; Spheroid image analysis: IL; Embryo injection: YKK; Embryo staining and quantification: EC; Supervision of sequencing library generation and sequencing: SAS; Bioinformatical analysis: MBS; Embryo work: AHFMP; Manuscript writing: DO and JB.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank H. Kohler (FMI) for cell sorting, Sirisha Aluri (FMI) for 10X Genomics scRNAseq, Karolina Guja for technical support, V. Iesmantavicius (FMI) for help with computational analysis of mass spectrometry data, Min Jia and Jeff Chao (FMI) for providing LIF, and Austin Smith (Wellcome‐MRC Cambridge Stem Cell Institute) for providing TNG‐A cells. We are grateful to F. Mohn, D. Schuebeler (FMI), and S. Stricker (University Munich) for comments on the manuscript. D.O. was supported by EMBO (ALTF 1632‐2014) and Marie Curie Actions (LTFCOFUND2013, GA‐2013‐609409). Research in the laboratory of A.H.F.M.P. is supported by the Novartis Research Foundation and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program grant agreement no. 695288—Totipotency. Research in the laboratory of J.B. is supported by the Novartis Research Foundation.
The EMBO Journal (2021) 40: e106818.
See also: A Janiszewski et al (June 2021)
Contributor Information
Daniel Olivieri, Email: daniel.olivieri@fmi.ch.
Joerg Betschinger, Email: joerg.betschinger@fmi.ch.
Data availability
All sequencing data are available at ArrayExpress (E‐MTAB‐9446, E‐MTAB‐9447, E‐MTAB‐0450, E‐MTAB‐9453, E‐MTAB‐10147 and E‐MTAB‐10150; http://www.ebi.ac.uk/arrayexpress/experiments/).
References
- Anderson KGV, Hamilton WB, Roske FV, Azad A, Knudsen TE, Canham MA, Forrester LM, Brickman JM (2017) Insulin fine‐tunes self‐renewal pathways governing naive pluripotency and extra‐embryonic endoderm. Nat Cell Biol 19: 1164–1177 [DOI] [PubMed] [Google Scholar]
- Atlasi Y, Megchelenbrink W, Peng T, Habibi E, Joshi O, Wang S‐Y, Wang C, Logie C, Poser I, Marks H et al (2019) Epigenetic modulation of a hardwired 3D chromatin landscape in two naive states of pluripotency. Nat Cell Biol 21: 568–578 [DOI] [PubMed] [Google Scholar]
- Barisic D, Stadler MB, Iurlaro M, Schübeler D (2019) Mammalian ISWI and SWI/SNF selectively mediate binding of distinct transcription factors. Nature 569: 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S, Lee B‐K, Rhee C, Song J, Woo AJ, Kim J (2014) CpG island‐mediated global gene regulatory modes in mouse embryonic stem cells. Nat Commun 5: 5490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beddington RS, Robertson EJ (1989) An assessment of the developmental potential of embryonic stem cells in the midgestation mouse embryo. Dev Camb Engl 105: 733–737 [DOI] [PubMed] [Google Scholar]
- Betschinger J, Nichols J, Dietmann S, Corrin PD, Paddison PJ, Smith A (2013) Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 153: 335–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Loos R, Lombard P, Okahara J, Behr R, Sasaki E, Nichols J, Smith A, Bertone P (2015) Lineage‐specific profiling delineates the emergence and progression of naive pluripotency in mammalian embryogenesis. Dev Cell 35: 366–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroviak T, Stirparo GG, Dietmann S, Hernando‐Herraez I, Mohammed H, Reik W, Smith A, Sasaki E, Nichols J, Bertone P (2018) Single cell transcriptome analysis of human, marmoset and mouse embryos reveals common and divergent features of preimplantation development. Development 145: dev167833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ (2015) ATAC‐seq: a method for assaying chromatin accessibility genome‐wide. Curr Protoc Mol Biol 109: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canham MA, Sharov AA, Ko MSH, Brickman JM (2010) Functional heterogeneity of embryonic stem cells revealed through translational amplification of an early endodermal transcript. Plos Biol 8: e1000379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, Vrana J, Jones K, Grotewold L, Smith A (2007) Nanog safeguards pluripotency and mediates germline development. Nature 450: 1230–1234 [DOI] [PubMed] [Google Scholar]
- Cho LTY, Wamaitha SE, Tsai IJ, Artus J, Sherwood RI, Pedersen RA, Hadjantonakis A‐K, Niakan KK (2012) Conversion from mouse embryonic to extra‐embryonic endoderm stem cells reveals distinct differentiation capacities of pluripotent stem cell states. Development 139: 2866–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, Plath K (2017) Cooperative binding of transcription factors orchestrates reprogramming. Cell 168: 442–459.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews ST, Pearson JC (2009) Transcriptional autoregulation in development. Curr Biol 19: R241–R246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2012) STAR: ultrafast universal RNA‐seq aligner. Bioinform Oxf Engl 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng BO, Jiang J, Kraus P, Ng J‐H, Heng J‐C, Chan Y‐S, Yaw L‐P, Zhang W, Loh Y‐H, Han J et al (2009) Reprogramming of fibroblasts into induced pluripotent stem cells with orphan nuclear receptor Esrrb. Nat Cell Biol 11: 197–203 [DOI] [PubMed] [Google Scholar]
- Festuccia N, Owens N, Chervova A, Dubois A, Navarro P (2020) The combined action of Esrrb and Nr5a2 is essential for naïve pluripotency. bioRxiv 10.1101/2020.06.05.134999 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Fujii S, Nishikawa‐Torikai S, Futatsugi Y, Toyooka Y, Yamane M, Ohtsuka S, Niwa H (2015) Nr0b1 is a negative regulator of Zscan4c in mouse embryonic stem cells. Sci Rep 5: 9146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikura J, Yamato E, Yonemura S, Hosoda K, Masui S, Nakao K, Miyazaki J, Niwa H (2002) Differentiation of embryonic stem cells is induced by GATA factors. Gene Dev 16: 784–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D, Lerch A, Hahne F, Stadler MB (2014) QuasR: quantification and annotation of short reads in R. Bioinformatics 31: 1130–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JM, Morgani SM, Bone RA, Bonderup K, Abelchian S, Brakebusch C, Brickman JM (2016) Embryonic stem cell culture conditions support distinct states associated with different developmental stages and potency. Stem Cell Rep 7: 177–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf T, Enver T (2009) Forcing cells to change lineages. Nature 462: 587–594 [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, Schlesner M (2016) Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32: 2847–2849 [DOI] [PubMed] [Google Scholar]
- Guo G, von Meyenn F, Santos F, Chen Y, Reik W, Bertone P, Smith A, Nichols J (2016) Naive pluripotent stem cells derived directly from isolated cells of the human inner cell mass. Stem Cell Rep 6: 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Smith A (2010) A genome‐wide screen in EpiSCs identifies Nr5a nuclear receptors as potent inducers of ground state pluripotency. Development 137: 3185–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G, Stirparo GG, Strawbridge S, Spindlow D, Yang J, Clarke J, Dattani A, Yanagida A, Li MA, Myers S et al (2021) Human naïve epiblast cells possess unrestricted lineage potential. Cell Stem Cell 10.1016/j.stem.2021.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Lun ATL, Morgan MD, Marioni JC (2018) Batch effects in single‐cell RNA‐sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36: 421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, LeMeur N, Brinkman RR, Ellis B, Haaland P, Sarkar D, Spidlen J, Strain E, Gentleman R (2009) flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinform 10: 106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Ohta H, Kurimoto K, Aramaki S, Saitou M (2011) Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146: 519–532 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermitte S, Chazaud C (2014) Primitive endoderm differentiation: from specification to epithelium formation. Philos Trans R Soc Lond B Biol Sci 369: 20130537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg J, Perlmann T (2012) Maintaining differentiated cellular identity. Nat Rev Genet 13: 429–439 [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2008) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Wang Y, Morris CD, Jacques V, Gottesfeld JM, Rusche JR, Thomas EA (2016) The Effects of Pharmacological Inhibition of Histone Deacetylase 3 (HDAC3) in Huntington’s disease mice. PLoS One 11: e0152498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalfallah O, Rouleau M, Barbry P, Bardoni B, Lalli E (2009) Dax‐1 knockdown in mouse embryonic stem cells induces loss of pluripotency and multilineage differentiation. Stem Cells 27: 1529–1537 [DOI] [PubMed] [Google Scholar]
- Koike‐Yusa H, Li Y, Tan E‐P, Velasco‐Herrera MDC, Yusa K (2013) Genome‐wide recessive genetic screening in mammalian cells with a lentiviral CRISPR‐guide RNA library. Nat Biotechnol 32: 267–273 [DOI] [PubMed] [Google Scholar]
- Kutejova E, Sasai N, Shah A, Gouti M, Briscoe J (2016) Neural progenitors adopt specific identities by directly repressing all alternative progenitor transcriptional programs. Dev Cell 36: 639–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ (2013) Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyva‐Díaz E, Hobert O (2019) Transcription factor autoregulation is required for acquisition and maintenance of neuronal identity. Development 146: dev177378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linneberg‐Agerholm M, Wong YF, Herrera JAR, Monteiro RS, Anderson KGV, Brickman JM (2019) Naïve human pluripotent stem cells respond to Wnt, Nodal, and LIF signalling to produce expandable naïve extra‐embryonic endoderm. Development 146: dev180620 [DOI] [PubMed] [Google Scholar]
- Lo Nigro A, de Jaime‐Soguero A, Khoueiry R, Cho DS, Ferlazzo GM, Perini I, Abon Escalona V, Aranguren XL, Chuva de Sousa Lopes SM, Koh KP et al (2017) PDGFRα+ cells in embryonic stem cell cultures represent the in vitro equivalent of the pre‐implantation primitive endoderm precursors. Stem Cell Rep 8: 318–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Bach K, Marioni JC (2016) Pooling across cells to normalize single‐cell RNA sequencing data with many zero counts. Genome Biol 17: 75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, Jamboree Participants in the 1st HCA , Marioni JC (2019) EmptyDrops: distinguishing cells from empty droplets in droplet‐based single‐cell RNA sequencing data. Genome Biol 20: 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Smyth GK (2015a) csaw: a Bioconductor package for differential binding analysis of ChIP‐seq data using sliding windows. Nucleic Acids Res 44: e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Smyth GK (2015b) From reads to regions: a Bioconductor workflow to detect differential binding in ChIP‐seq data. F1000Res 4: 1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Swigut T, Valouev A, Rada‐Iglesias A, Wysocka J (2010) Sequence‐specific regulator Prdm14 safeguards mouse ESCs from entering extraembryonic endoderm fates. Nat Struct Mol Biol 18: 120–127 [DOI] [PubMed] [Google Scholar]
- Mall M, Kareta MS, Chanda S, Ahlenius H, Perotti N, Zhou Bo, Grieder SD, Ge X, Drake S, Euong Ang C et al (2017) Myt1l safeguards neuronal identity by actively repressing many non‐neuronal fates. Nature 544: 245–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J et al (2008) Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134: 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello G, Sugimoto T, Diamanti E, Joshi A, Hannah R, Ohtsuka S, Göttgens B, Niwa H, Smith A (2012) Esrrb is a pivotal target of the Gsk3/Tcf3 axis regulating embryonic stem cell self‐renewal. Cell Stem Cell 11: 491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Fornes O, Arenillas DJ, Chen C‐Y, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley‐Hunt R et al (2015) JASPAR 2016: a major expansion and update of the open‐access database of transcription factor binding profiles. Nucleic Acids Res 44: D110–D115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Campbell KR, Lun ATL, Wills QF (2017) Scater: pre‐processing, quality control, normalization and visualization of single‐cell RNA‐seq data in R. Bioinformatics 33; 1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res 40: 4288–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ACH, Biechele S, Rossant J, Stanford WL (2014) Sox17‐mediated XEN cell conversion identifies dynamic networks controlling cell‐fate decisions in embryo‐derived stem cells. Cell Rep 9: 780–793 [DOI] [PubMed] [Google Scholar]
- Mohammed H, Hernando‐Herraez I, Savino A, Scialdone A, Macaulay I, Mulas C, Chandra T, Voet T, Dean W, Nichols J et al (2017) Single‐cell landscape of transcriptional heterogeneity and cell fate decisions during mouse early gastrulation. Cell Rep 20: 1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgani SM, Brickman JM (2015) LIF supports primitive endoderm expansion during pre‐implantation development. Development 142: 3488–3499 [DOI] [PubMed] [Google Scholar]
- Muhr J, Andersson E, Persson M, Jessell TM, Ericson J (2001) Groucho‐mediated transcriptional repression establishes progenitor cell pattern and neuronal fate in the ventral neural tube. Cell 104: 861–873 [DOI] [PubMed] [Google Scholar]
- Niakan KK, Davis EC, Clipsham RC, Jiang M, Dehart DB, Sulik KK, McCabe ERB (2006) Novel role for the orphan nuclear receptor Dax1 in embryogenesis, different from steroidogenesis. Mol Genet Metab 88: 261–271 [DOI] [PubMed] [Google Scholar]
- Niakan KK, Ji H, Maehr R, Vokes SA, Rodolfa KT, Sherwood RI, Yamaki M, Dimos JT, Chen AE, Melton DA et al (2010) Sox17 promotes differentiation in mouse embryonic stem cells by directly regulating extraembryonic gene expression and indirectly antagonizing self‐renewal. Gene Dev 24: 312–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps MP, Bailey JN, Vleeshouwer‐Neumann T, Chen EY (2016) CRISPR screen identifies the NCOR/HDAC3 complex as a major suppressor of differentiation in rhabdomyosarcoma. Proc National Acad Sci USA 113: 15090–15095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivron NC, Frias‐Aldeguer J, Vrij EJ, Boisset J‐C, Korving J, Vivié J, Truckenmüller RK, van Oudenaarden A, van Blitterswijk CA, Geijsen N (2018) Blastocyst‐like structures generated solely from stem cells. Nature 557: 106–111 [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2009) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablin EP, Woods A, Krylova IN, Hwang P, Ingraham HA, Fletterick RJ (2008) The structure of corepressor Dax‐1 bound to its target nuclear receptor LRH‐1. Proc National Acad Sci USA 105: 18390–18395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröter C, Rué P, Mackenzie JP, Arias AM (2015) FGF/MAPK signaling sets the switching threshold of a bistable circuit controlling cell fate decisions in embryonic stem cells. Development 142: 4205–4216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimosato D, Shiki M, Niwa H (2007) Extra‐embryonic endoderm cells derived from ES cells induced by GATA Factors acquire the character of XEN cells. BMC Dev Biol 7: 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon CS, Hadjantonakis A, Schröter C (2018) Making lineage decisions with biological noise: lessons from the early mouse embryo. Wiley Interdiscip Rev Dev Biol 7: e319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A (2017) Formative pluripotency: the executive phase in a developmental continuum. Development 144: 365–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozen B, Cox AL, Jonghe JD, Bao M, Hollfelder F, Glover DM, Zernicka‐Goetz M (2019) Self‐organization of mouse stem cells into an extended potential blastoid. Dev Cell 51: 698–712.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Feng D, Fang B, Mullican S, You S‐H, Lim H‐W, Everett L, Nabel C, Li Y, Selvakumaran V et al (2013) Deacetylase‐independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell 52: 769–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Uranishi K, Akagi T, Koide H, Yokota T (2016) Esrrb directly binds to Gata6 promoter and regulates its expression with Dax1 and Ncoa3. Biochem Biophys Res Commun 478: 1720–1725 [DOI] [PubMed] [Google Scholar]
- Villegas F, Lehalle D, Mayer D, Rittirsch M, Stadler MB, Zinner M, Olivieri D, Vabres P, Duplomb‐Jego L, De Bont ESJM et al (2019) Lysosomal signaling licenses embryonic stem cell differentiation via inactivation of Tfe3. Cell Stem Cell 24: 257–270.e8 [DOI] [PubMed] [Google Scholar]
- Wamaitha SE, del Valle I, Cho LTY, Wei Y, Fogarty NME, Blakeley P, Sherwood RI, Ji H, Niakan KK (2015) Gata6 potently initiates reprograming of pluripotent and differentiated cells to extraembryonic endoderm stem cells. Gene Dev 29: 1239–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Zhang Yu, Xu Q, Zhou C, Liu B, Du Z, Zhang Ke, Zhang B, Wang X, Gayen S et al (2019) Epigenomic analysis of gastrulation identifies a unique chromatin state for primed pluripotency. Nat Genet 52: 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ryan DJ, Wang W, Tsang J‐H, Lan G, Masaki H, Gao X, Antunes L, Yu Y, Zhu Z et al (2017) Establishment of mouse expanded potential stem cells. Nature 550: 393–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RN, Ito M, Saunders TL, Camper SA, Jameson JL (1998) Role of Ahch in gonadal development and gametogenesis. Nat Genet 20: 353–357 [DOI] [PubMed] [Google Scholar]
- Zhang J, Liu G, Ruan Y, Wang J, Zhao Ke, Wan Y, Liu B, Zheng H, Peng T, Wu W et al (2014) Dax1 and Nanog act in parallel to stabilize mouse embryonic stem cells and induced pluripotency. Nat Commun 5: 5042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J et al (2017) Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 14049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Żylicz JJ, Bousard A, Žumer K, Dossin F, Mohammad E, da Rocha ST, Schwalb B, Syx L, Dingli F, Loew D et al (2018) The implication of early chromatin changes in X chromosome inactivation. Cell 176: 182–197.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
All sequencing data are available at ArrayExpress (E‐MTAB‐9446, E‐MTAB‐9447, E‐MTAB‐0450, E‐MTAB‐9453, E‐MTAB‐10147 and E‐MTAB‐10150; http://www.ebi.ac.uk/arrayexpress/experiments/).