

Abstract
Mass spectrometry has greatly improved the analysis of phosphorylation events in complex biological systems and on a large scale. Despite considerable progress, the correct identification of phosphorylated sites, their quantification, and their interpretation regarding physiological relevance remain challenging. The MS Resource Pillar of the Human Proteome Organization (HUPO) Human Proteome Project (HPP) initiated the Phosphopeptide Challenge as a resource to help the community evaluate methods, learn procedures and data analysis routines, and establish their own workflows by comparing results obtained from a standard set of 94 phosphopeptides (serine, threonine, tyrosine) and their nonphosphorylated counterparts mixed at different ratios in a neat sample and a yeast background. Participants analyzed both samples with their method(s) of choice to report the identification and site localization of these peptides, determine their relative abundances, and enrich for the phosphorylated peptides in the yeast background. We discuss the results from 22 laboratories that used a range of different methods, instruments, and analysis software. We reanalyzed submitted data with a single software pipeline and highlight the successes and challenges in correct phosphosite localization. All of the data from this collaborative endeavor are shared as a resource to encourage the development of even better methods and tools for diverse phosphoproteomic applications. All submitted data and search results were uploaded to MassIVE (https://massive.ucsd.edu/) as data set MSV000085932 with ProteomeXchange identifier PXD020801.
Keywords: phosphorylated peptides, phospho site localization, phosphopeptide enrichment, false identification rate, mass spectrometry, Human Proteome Organization (HUPO), Human Proteome Project (HPP), MS Resource Pillar, Phosphopeptide Challenge
Graphical Abstract
INTRODUCTION
Post-translational modifications (PTMs) of proteins are critical to the functional dynamics of cellular systems. PTMs are diverse in their composition and purpose, such as influencing enzymatic activity and subcellular localization. Although PTMs have been extensively studied over the past decades, resulting in a large knowledge base, we have only scratched the surface of their implications for cellular systems. Subtle changes in PTM dynamics can have drastic effects on cellular health and in some cases are indicators of disease, even where there is no obvious genetic basis for the disease.1,2 For these reasons, the importance of understanding the diversity and function of PTMs is obvious.
Protein phosphorylation generates phosphoamino acids that do not resemble any natural amino acid; they act as new chemical entities and provide a means of diversifying the chemical nature of protein surfaces. Protein function through protein–protein interaction events that depend on phosphorylated terminal hydroxyl amino acids are generally dynamic because phosphate is installed and removed by enzymes whose own activity often depends on signaling events. This observation makes the study of protein kinases and phosphoprotein phosphatases central to our understanding of signal transduction.3,4 Phosphorylation on serine, threonine, and tyrosine plays a key role in nearly every cellular process and is one of the most extensively studied PTMs. Of the many tools available, including assays utilizing 32P and a variety of antibody-based approaches, mass spectrometry (MS) has emerged as the primary method to study protein phosphorylation events on a large scale.5 High mass accuracy, high sensitivity, and high throughput (thousands of proteins within a few hours) paired with the ability to localize modifications on a specific residue have made MS the most commonly used approach for both protein-by-protein and systems approaches. Although frequently used in the field, the complete process from sample preparation through MS analysis and data interpretation can lead to a loss of phosphosite information and incorrect interpretation. As an example, the analysis of phosphotyrosine in the pathogen Mycobacterium tuberculosis (Mtb) had eluded identification for over 20 years. Its existence in this organism was first demonstrated by 4G10-antiphosphotyrosine antibody approaches but was not confirmed until optimized sample preparation and MS analysis revealed a rich landscape of phosphotyrosine residues among numerous Mtb proteins.6
Whereas ~40% of the human proteome is phosphorylated as reported to date (www.peptideatlas.org - Human Phosphoproteome 2017, PHOSIDA7) and several thousand phosphorylation sites have been reported from the analysis of different cell lines,8 phosphorylation events are generally at low stoichiometry, meaning the phospho-group is present on only a small subset of all molecules of a protein.9 As a consequence, phosphorylated peptides need enrichment after the protein digestion of lysates and complex protein mixtures, which necessitates larger quantities of starting material. For a deeper phosphoproteome analysis, sample fractionation can be performed prior to enrichment and MS data acquisition. Discovery MS, including the use of different fragmentation types, is often the method of choice to study phospho-signaling events, but targeted approaches with selected/parallel reaction monitoring (SRM/PRM) and, more recently, data-independent acquisition (DIA) have also been applied. To address the current limitations in reproducibility, robustness, and throughput, automated workflows are increasingly explored,10,11 reducing random errors from manual sample handling and reducing sample preparation times from several days to several hours. MS data need then to be analyzed by employing an appropriate database or library search strategy followed by statistical validation of the results, estimating the false discovery rate (FDR) and the accuracy of site localization of the phospho-group(s) in a peptide. The latter can be challenging on its own, especially with multiply phosphorylated residues or adjacent serine, threonine, and/or tyrosine residues.
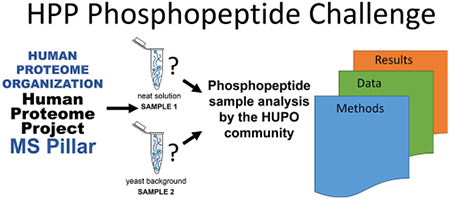
The MS Resource Pillar of the Human Proteome Organization (HUPO) Human Proteome Project (HPP) initiated the Phosphopeptide Challenge to generate a free resource to help the community evaluate methods, learn procedures and data analysis routines, and establish their own workflows by comparing results obtained from a standard set of phosphopeptides and their unphosphorylated counterparts (Figure 1). By partnering with SynPeptide (Shanghai, China, www.synpeptide.com) and ReSyn Biosciences (South Africa, www.resynbio.com), the MS Resource Pillar offered all HUPO members the unique opportunity to participate in and evaluate methods for peptide sequence analysis by mass spectrometry, phosphosite localization, phosphopeptide enrichment, and data processing. Specifically, participants received two samples and were asked to use their method(s) of choice to perform three tasks: (1) identify the number and site localization for as many phosphorylated peptides as possible in a neat sample (sample consisting of synthetic peptides and solvent to solubilize peptides, sample 1), (2) determine the relative abundance of the phosphorylated peptides by comparison with their nonphosphorylated counterparts in the neat sample, and (3) enrich for the phosphorylated peptides spiked into a yeast tryptic digest background (sample 2) and report the observed synthesized phosphorylated peptide sequences.
Figure 1.
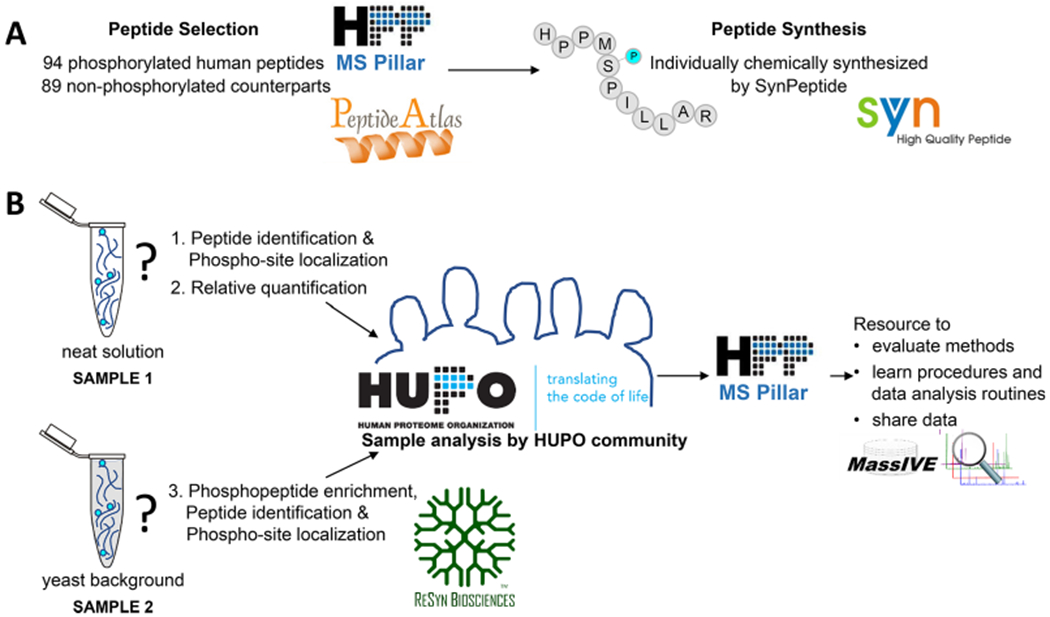
Overview of the HPP Phosphopeptide Challenge. (A) The MS Resource Pillar of the Human Proteome Project (HPP) selected 94 human phosphorylated peptides (number unknown to participants) and 89 nonphosphorylated counterparts (sequences provided to participants) considering highly observed peptides in PeptideAtlas and applied the criteria described in the Experimental Section and in Figure 2. Synthetic peptides were graciously provided by SynPeptide (www.synpeptide.com). (B) 94 phosphorylated and 89 nonphosphorylated peptides were mixed at different ratios and provided to the HUPO community as a neat solution and in a yeast background. Participants were asked to use their method(s) of choice to (1) identify the number and location of phosphorylation sites for all phosphorylated peptides in the mixture, (2) determine the relative abundance of each phosphopeptide compared with its nonphosphorylated counterpart in the neat sample, and (3) enrich for the phosphorylated peptides spiked into a yeast background and report the human phosphopeptides that were detected. ReSyn Biosciences (www.resynbio.com) graciously provided optional phosphopeptide enrichment kits to participants. The goal of the HPP MS Resource Pillar Phosphopeptide Challenge was to help the community evaluate methods, learn procedures, and improve data analysis routines for phosphopeptides. All data from this collaborative effort are shared through MassIVE and serve as resources for the development of new computational tools. SynPeptide logo reprinted with permission from SynPeptide Co., Ltd. HUPO and HPP MS Pillar logos reprinted with permission from the Human Proteome Organization. MassIVE logo reprinted with permission from Computer Science and Engineering, University of California, San Diego. ReSyn logo reprinted with permission from ReSyn Biosciences. PeptideAtlas logo reprinted with permission from the Institute for Systems Biology.
A goal of this collaborative endeavor was to receive multiple analytical workflows, enrichment protocols, and data processing strategies so that it would be possible to identify the approaches that provide the highest level of correct phosphosite identifications in the MS Resource Pillar phosphopeptide mixture. We aimed to determine the best practices for the identification, localization, enrichment, and quantification of phosphorylated peptides, to share the collected data as a valuable resource for the assessment of new computational tools, and to ultimately contribute to improve our understanding of biological processes at the molecular level.
EXPERIMENTAL SECTION
Peptide Selection and Synthesis
Ninety-four phosphorylated human peptide sequences were selected from highly observed spectra and with a PTMProphet12 score of 1.0 in PeptideAtlas13 (Figure 2). Peptides were curated to have a roughly distributed reversed-phase chromatographic elution profile using a sequence-specific retention (SSR)14 of between 10 and 45 (a unitless measure of relative hydrophobicity), with the majority (93%) of peptides with an SSR value <40, a length of 8–20 amino acids, and an expected charge state of two (81 peptides), three (12 peptides), or four (1 peptide). Each peptide contains 1, 2, or 3 phosphorylated residues and 1–7 possible serine, threonine, or tyrosine phosphorylation sites for a total of 94 phosphorylated peptides: 1 phosphorylated site, 62 peptides; 2 sites, 29 peptides; 3 sites, 3 peptides. Six phosphorylated peptides are identical in amino acid sequence to another peptide in the set but contain an alternate placement of the phospho-groups. For the relative quantification of phosphorylation site occupancy, nonphosphorylated forms of 89 unique peptide sequences were also included in the samples.
Figure 2.
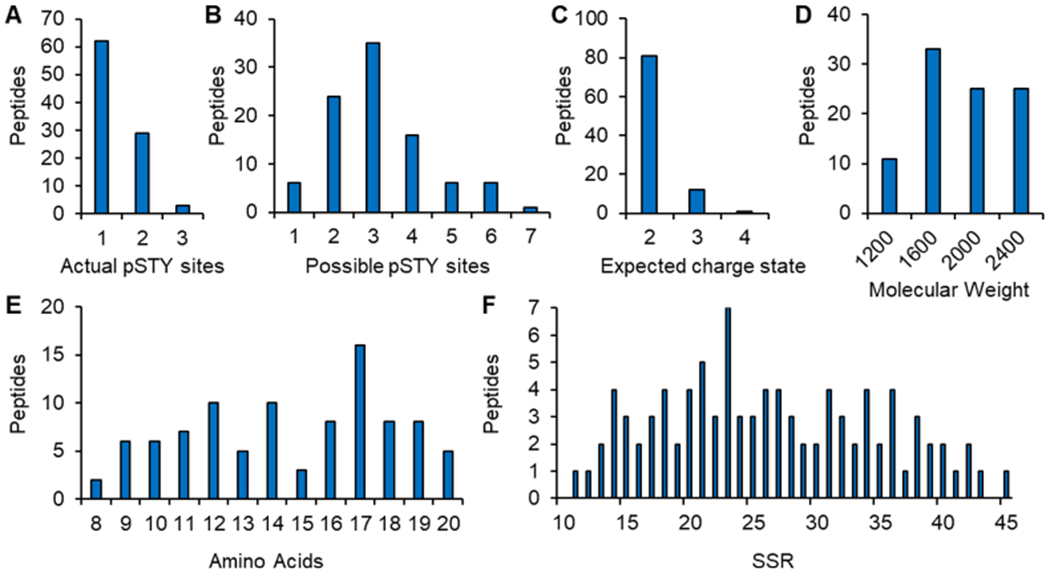
Peptide characteristics. 94 phosphorylated human peptides were selected from highly observed spectra and with a PTMProphet score of 1.0 in PeptideAtlas. Selected peptides contain (A) 1, 2, or 3 phosphorylated residues of (B) 1–7 possible serine, threonine, or tyrosine phosphorylation sites and have (C) an expected charge state of two, three, or four, (D) a molecular weight of 1000–2400 Da, (E) a length of 8–20 amino acids, and (F) a sequence-specific retention (SSR) value of 10–45 as a measure of hydrophobicity.
The synthetic peptides were graciously provided by SynPeptide. Peptides were individually chemically synthesized with free amines at the N-termini and carboxylic acids at the C-termini. All cysteine residues were carboxyamidomethylated, and specific serine, threonine, and tyrosine residues were introduced as phosphorylated building blocks. All peptides were HPLC-purified.
Phosphopeptide Neat Mixture
Each peptide was diluted to one of three different concentrations, chosen at random, before the 94 phosphorylated peptides and 89 nonphosphorylated peptides were mixed. The mixture was aliquoted and lyophilized such that when resuspended in 100 μL of solution, the peptide concentrations would be 3.3, 10, or 30 fmol/μL. A complete list of all peptide sequences, phosphorylation sites, and molar concentrations is presented in Supplementary Table S1.
Phosphopeptide Yeast Mixture
A yeast protein stock (strain EDC3, obtained from Prof. J. D. Aitchison, Seattle Children’s Research Institute, Seattle) was prepared from cells grown to mid log phase, harvested by centrifugation, and lysed by flash freezing in liquid nitrogen followed by disruption with a Retsch ball mill grinder. The protein lysate was suspended in 100 mM ammonium bicarbonate solution. The proteins were reduced with 5 mM DTT for 30 min at 60 °C and then alkylated with 10 mM iodoacetamide for 30 min at room temperature in the dark. Proteins were digested with trypsin (Promega) at a 1:250 ratio for 4 h at 37 °C; then, the digestion was stopped by heating to 95 °C for 10 min. Phosphopeptide yeast mixtures were made by adding the digest from 6 μg of yeast protein to aliquots of the phosphopeptide neat mixture. The phosphopeptide yeast mixtures were then lyophilized for storage.
Phosphopeptide Enrichment Kit
ReSyn Biosciences provided phosphopeptide enrichment kits on request to participants to be used for the enrichment component of the challenge. Kits contained 2 mL each of MagReSyn Ti-IMAC, Zr-IMAC, and HILIC polymers. Participants were given the option of using the ReSyn kit or a strategy of their choice.
Phosphopeptide Challenge Reports
Each participant was given an Excel file to complete following the sample analyses. The file contained a list of the 89 nonphosphorylated peptide sequences as well as their SSR value,14 molecular weight, and proteins of origin. Participants were asked to complete three challenges and submit the results in the Excel file.
Identify the number and site localization of as many phosphorylated peptides as possible in the neat sample.
Determine the relative abundance (as a ratio) of phosphorylated peptide and their nonphosphorylated counterpart in the neat sample.
Enrich for phosphorylated peptides from the yeast sample and report the observed synthesized phosphorylated peptide sequences.
Participants were informed that for some peptides, there is more than one phosphorylated form, that the second sample contained the same peptides in a background matrix consisting of trypsin-digested peptides from 6 μg of yeast lysate, and that the resuspension of each sample in 100 μL would result in synthetic peptide concentrations of 3–30 fmol/μL. All participants were asked to upload their reports, methods, and data files to MassIVE using an ID number that was assigned to maintain the anonymity of the submission.
Mass Spectrometry Reanalysis with PTM Localization
Raw data from four participants were converted to mzML format using MSConvert from ProteoWizard (version 3.0.9974)15 and searched with Comet (version 2018.01 rev. 4).16 The four data sets were chosen for their characteristic features (e.g., precision and sensitivity) that represented a broad range of the results. Spectra were searched against the UniProt yeast reviewed and unreviewed proteome (downloaded on December 4, 2015, 6627 sequences), the full complement of synthesized phosphopeptide sequences, and shuffled sequences with repeat sequences preserved.17 Comet parameters included a fixed modification of +57.021464 Da on cysteine, a variable modification of +15.994915 Da on methionine, and a variable modification of +79.966331 Da on serine, threonine, or tyrosine. The precursor mass tolerance was set to 20 ppm; a fragment bin tolerance of 0.02 and a fragment bin offset of 0 were used. Complete enzymatic cleavage was set, allowing for up to two missed cleavages. Peptide-spectrum matches (PSMs) were analyzed using the Trans-Proteomic Pipeline (TPP, version 5.2.0 Flammagenitus)18 to assign peptide probabilities using PeptideProphet19 and iProphet.20 PTM localization probabilities were assigned using PTMProphet (SVN revision 7986 of the TPP code repository).12 PTMProphet was executed with the options “MINPROB = 0.2 MAXTHREADS = 16 STY:79.9663,MW:15.994”, which limited PTMProphet to the localization of phosphorylation modifications of serine, threonine, and tyrosine and of oxidation modifications of methionine and tryptophan, on PSMs with probabilities of 20% and higher, utilizing 16 computational threads.
Recalibration of MS1 Data
All submitted MS1 data were recalibrated using a previously published algorithm for Fourier transform ion cyclotron resonance (FTICR) recalibration21 extended to time-of-flight (TOF) and Orbitrap data; confidently identified (FDR 1%) nonphosphopeptides from the yeast background were used as internal calibrants. The recalibration component, mzRecal, was inserted between two consecutive executions of the Comet-based reanalysis workflow listed above, differing only in mass measurement and amino acid mass modifications considered. The first pass used 10 ppm mass tolerance and static cysteine carbamidomethylation and variable methionine oxidation modifications. The second pass used 2 ppm mass tolerance and the same amino acid modifications plus the phosphorylation of serine, threonine, and tyrosine. mzRecal is available on GitHub (https://github.com/524D/mzrecal).
RESULTS AND DISCUSSION
Each participant of the Phosphopeptide Challenge received two sample tubes—one containing a neat mixture of peptides (phosphorylated and unphosphorylated counterparts) and the other with the same peptides in a yeast background, as described in the Experimental Section. Participants analyzed the samples using their preferred methods and data analysis pipelines. Twenty-two laboratories submitted their results using the Excel file that had been provided to them. Two participants submitted more than one report, documenting their results for the use of multiple instruments and workflows. One participant submitted only their data but no report. In total, 23 reports were received for which peptides and phosphorylation sites were provided. Of these, 22 also completed the quantitation challenge, and 21 completed the enrichment challenge.
Participants made use of a diverse range of instrumentation (Table 1), with most opting to collect high-resolution MS/MS spectra (even when they had the ability to use either high or low resolution). Extended details of the instrumentation parameters and data analysis platforms and parameters used by participants are provided in Supplementary Tables S2 and S3, respectively. The most common fragmentation method was higher-energy collisional dissociation (HCD), followed by collision-induced dissociation (CID) and electron-transfer dissociation (ETD). Liquid chromatography (LC) gradients ranged from 30 to 300 min using different C18 columns with 15–50 cm length. A diverse set of computational tools was used for data processing. Some participants used MaxQuant22 for identification, FDR estimation, phosphorylation site localization, and peptide quantification. Other data analysis methods were built from combinations of freely available and commercial software. Users of Proteome Discoverer built their pipelines from various nodes. The algorithm used for the task at each node is listed in Table 1, if reported. Thresholds for FDR estimation and phosphorylation site localization are also included in Table 1, if reported. Participants used a variety of different search databases, such as combined human and yeast proteomes, a yeast proteome with only the human proteins that could identify the synthesized peptides, or a yeast proteome plus the synthesized peptide sequences. Various protocols and materials were used for phosphopeptide enrichment, many utilizing titanium, such as TiO2, Ti4+-IMAC, and MagReSyn Ti-IMAC. Because of the large number of variables in the instrument and analytical workflows, our analysis of the submitted results focused on methods for phosphopeptide analysis that apply generally to any analytical platform.
Table 1:
Summary of Instrumentation and Methods Used By Participants
| Report | Instrument | MS/MS Fragmentation | MS/MS Search Tool | FDR Estimation Tool | Phosphosite Localization Software | Quantitation Software | Enrichment Material |
|---|---|---|---|---|---|---|---|
| 01195 | Thermo Fusion | HCD | MaxQuant | MaxQuant <1% | MaxQuant | MaxQuant | unknown |
| 04345 | Thermo QE-HF | HCD | Proteome Discoverer (SEQUEST) | Percolator | ptmRS | Spectral Counting | TiO2 (GL Sciences) |
| 04704 | Thermo LTQ-OrbiXL | CID | MS-GF+ | n/a | LuciPHOr | MS peak intensity (in-house method) | TiO2 (GL Sciences) |
| 10104 | Thermo QE | HCD | PeptideShaker (X!Tandem) | PeptideShaker | PeptideShaker | Skyline | TiO2 (GL Sciences) |
| 11235 | Bruker Maxis HD UHR-TOF | CID | PEAKS | PEAKS <0.1% | PEAKS (AScore) | n/a | TiO2 |
| 12914 | Thermo QE-HF | HCD | MaxQuant | MaxQuant | MaxQuant | MaxQuant | unknown |
| 13273 | Thermo Lumos | HCD | MaxQuant | MaxQuant <1% | MaxQuant | MaxQuant | TiO2 (GL Sciences) |
| 13579 | Thermo QE-HFX | HCD | Proteome Discoverer (SEQUEST) | Percolator <1% | phosphorRS > 95% | Skyline | TiO2 (GL Sciences) |
| 23721 | Thermo QE-HFX | HCD | Proteome Discoverer | - | - | - | Fe(III)-NTA IMAC (AssayMap) |
| 23854-P238 | Thermo Lumos | HCD | Proteome Discoverer | - | - | - | unknown |
| 23854-P77 | Thermo Lumos | HCD | Proteome Discoverer | - | - | - | unknown |
| 26402 | Sciex 5600 | CID | Mascot | Mascot | Mascot | Skyline | TiO2 (GL Sciences) |
| 27573-Fusion | Thermo Fusion | CID | Proteome Discoverer (Mascot) | Percolator | ptmRS >75% | Minora | TiO2 (GL Sciences) |
| 27573-QEHF | Thermo QE-HFX | HCD | Proteome Discoverer (Mascot) | Percolator | ptmRS >75% | Minora | TiO2 (GL Sciences) |
| 30417 | Bruker amaZon/Impact | CID + ETD | Mascot | Mascot <5% | Mascot | Bruker Data Analysis | n/a |
| 31245 | Thermo QE-HFX | HCD | Comet | PeptideProphet | - | Skyline | n/a |
| 38060 | Thermo Fusion | CID, ETD, HCD | Mascot | Mascot <5% | Mascot | - | TiO2+ZrO2 (Glygen) |
| 66666 | Thermo Lumos | HCD | Comet | PeptideProphet | PTMProphet | TPP/Quantic | MagResyn Ti-IMAC, ResynBio |
| 81990 | Thermo QE-HF | HCD | Proteome Discoverer (Mascot) | Mascot | PhosphoRS (<1% FDR) | n/a | ERLIC |
| 84931 | Thermo QE | HCD | MaxQuant | MaxQuant | MaxQuant | MaxQuant | Ti4 (J&K Scientific) |
| 86749 | Thermo QE+ | HCD | PEAKS | PEAKS | PEAKS | - | TiO2 (Thermo) |
| 95051 | Agilent 6550 | CID + ECD | Byonic | Byonic <5% | - | - | Fe(III)-NTA IMAC (AssayMap) |
| 97867 | Thermo QE | HCD | MaxQuant | MaxQuant | MaxQuant | MaxQuant | Ti4 (MagResyn Ti-IMAC, ResynBio) |
| 99588 | Thermo Lumos | HCD | Mascot | Mascot <5% | Mascot | - | n/a |
Challenge #1
All participants in the Phosphopeptide Challenge completed the first task of identifying the number and sites of phosphorylation on peptides in the neat mixture (Figure 3). Of the 89 nonphosphorylated peptide sequences in the neat mixture, 88 were present in a phosphorylated form, 6 of which were also phosphorylated in a second configuration, for a total of 94 uniquely phosphorylated peptides. Each identification of one of these forms counted as a true-positive identification, while missing identifications counted as false-negatives. Any identification of a sequence with the incorrect localization of a phospho-group was counted as a false-positive. Participants were asked to report “yes” or “no” for the observation of phosphorylation at a potential site; they set their own criteria for resolving ambiguity among the sites. Because the nonphosphorylated peptide sequences were given to all participants a priori, the two primary indicators of success in the identification challenge are (1) a high number of correctly localized phosphorylation sites and (2) a low number of false localizations. The submitted results were assessed using the following
Figure 3.
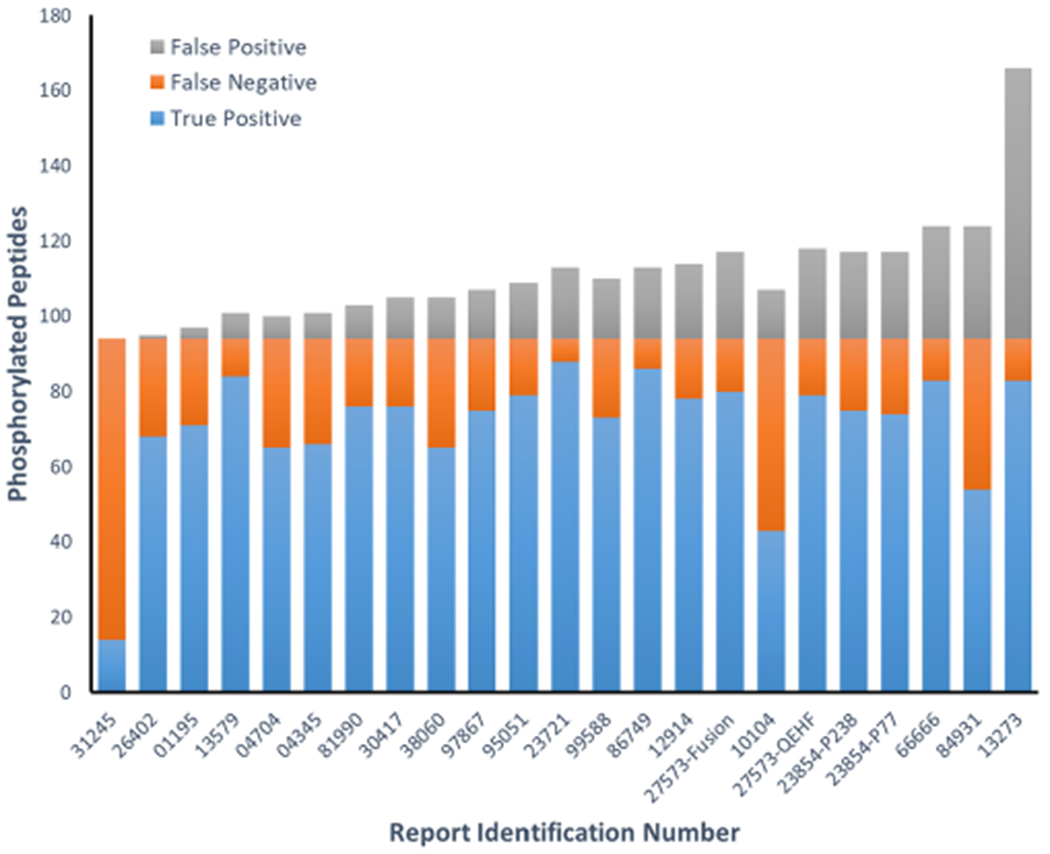
Summary of the 23 reports submitted for the phosphopeptide challenge. Correct identifications of the 94 different synthesized phosphorylated peptides are marked in blue, while those not identified are shown in orange. False identifications are marked in gray and indicate the incorrect placement of the phospho groups.
precision = true-positives/(true-positives + false-positives)
sensitivity = true-positives/(true-positives + false-negatives)
The results are shown in Figure 4. In one report, there was perfect precision, but only 15% of the sequences were identified. Conversely, in another report, almost all sequences were identified (88%), but only 54% of the localizations were correct. These two reports represent different extremes. The ideal results can be found in the upper rightmost corner of the plot in Figure 4, where all but four of the reports were clustered. However, it is worth noting that within this cluster, the ranges of precision (73–99%) and sensitivity (69–94%) are somewhat wide.
Figure 4.
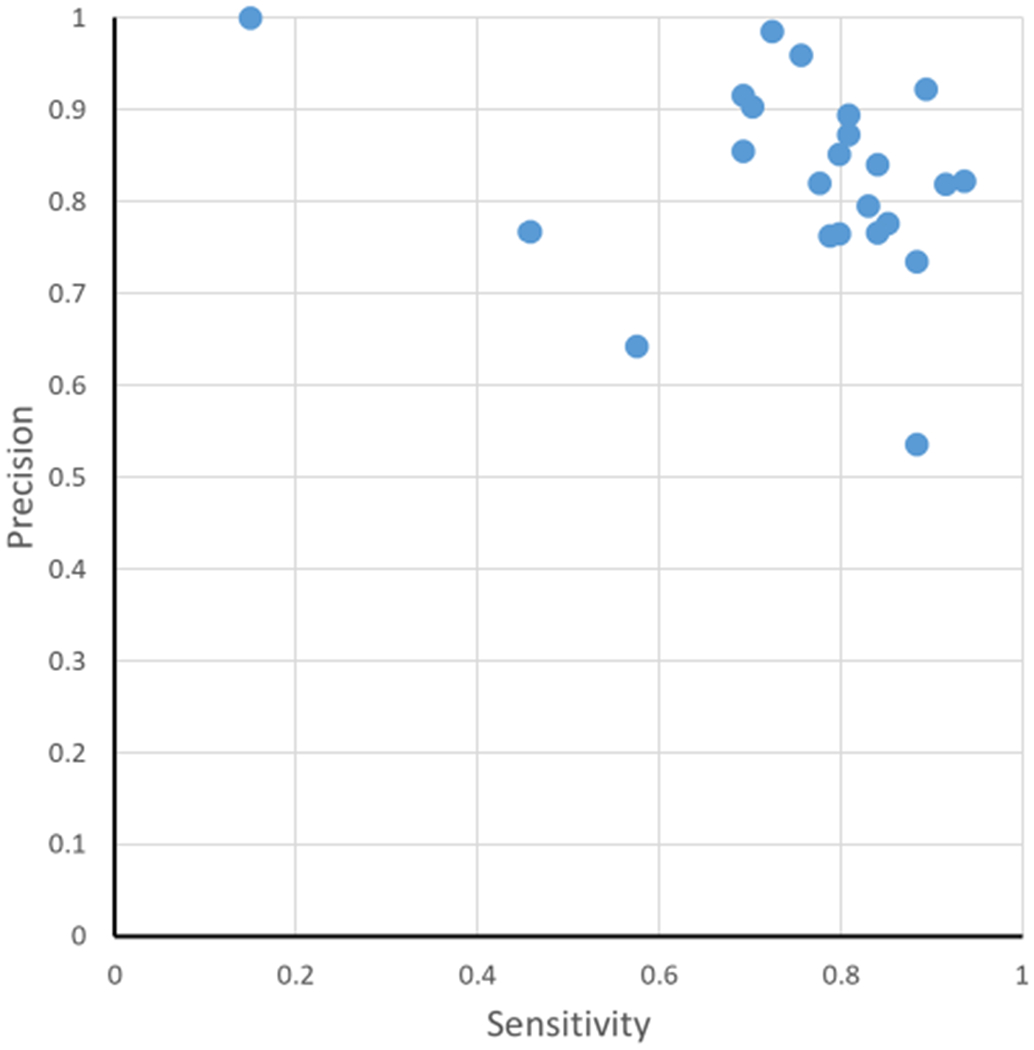
Precision–sensitivity graph for each report. The percentage of correct results (precision) is on the y axis, and the percentage of possible correct results (sensitivity) is on the x axis.
Although the selected phosphopeptides are highly observed in PeptideAtlas, the detection of some of them seem to be more challenging than that of others based on the results obtained. The majority of the phosphorylated peptides (82 of 94) were correctly identified in more than half of the submitted reports. Thirty-five peptides were correctly reported by >90% of participating laboratories, and eight phosphopeptides were correctly reported by everyone. The eight most successfully identified peptides have only one phosphorylated amino acid of one to three possible serine, threonine, or tyrosine residues. In six of these eight peptides with more than one possible phosphorylation site, the sites were at least four amino acids apart, and all but one peptide was present at the medium (1 fmol/μL) or high (3 fmol/μL) concentration in the mix. As an example, peptide GDVTAEEAAGApSPAK is phosphorylated on serine, has a second possible phosphorylation site on threonine located seven amino acids apart from serine, and was identified by all participating laboratories. Whereas peptides with phosphorylated serine, threonine, or tyrosine residues in close proximity were successfully identified (e.g., SVAAEGALLPQpTPPpSPR was successfully identified by 91% of participating laboratories), the 12 peptides with the lowest identification rate (<50% of participants) expose and confirm the expected challenges to correctly localize the phospho-group(s). All but 2 of these 12 peptides have 3 to 5 possible phosphorylation sites (serine, threonine, or tyrosine), which for most of these peptides are either adjacent (3), one amino acid (3), or two amino acids (3) apart. These 12 peptides include the 3 peptides with 3 phosphorylated residues. Only 2 of these 12 sequences were present in the mix at the lowest concentration, so abundance probably does not contribute to their low identification rate. Fifteen peptides with an equal number of phosphorylated residues and possible phosphorylation sites highlight that other factors such as the concentration or fragmentation behavior also likely contribute to their overall lower identification rates. For example, peptide HPPVLpTPPDQEVIR is singly phosphorylated, and threonine is the only possibility, yet it was identified by only 48% of the participants. The three most hydrophilic peptides (TAQVPp-SPPR, SSR 10.8; AQpTPPGPSLSGSK, SSR 11.1; VGGpSDEEASGIPSR, SSR 12.5) were identified by >91% of the participants, whereas the 7 most hydrophobic peptides (with an SSR ≥ 40) were identified in 17–70% of the reports, with SLSQpSFENLLDEPAYGLIQK being the most hydrophobic peptide (SSR 45.0) and one of the least identified peptides (17% of participating laboratories). Overall, correct localization is clearly more difficult in peptides with several possible phosphorylation sites, especially when these are adjacent or in close proximity, that is, less than three amino acids apart.
Closer examination of the results revealed that in 20 of the 23 reports, one or more of the false-positives was due to the lack of finding an expected site on a multiply phosphorylated peptide. Missed detection was minimal in some data sets (1 of 9 incorrect results in report no. 81990) and extensive in others (40 of 72 incorrect results in report no. 13273). Reports nos. 84931 and #23721 showed fractions of incorrect results due to the lack of detection of a phosphosite in an excess of 80%. Because these peptide identifications were derived from precursor m/z values that did not correspond to any of the synthesized peptides, it is possible that the sequence identification and phosphorylation site are indeed correct but cannot be explained at this time given the diversity of methods used in MS data acquisition and the disparate frequencies of detection of these peptides across all results. Supplementary Figure 1 summarizes the submitted results (after the removal of peptide identifications that the lacked detection of an expected phosphosite). When considering only the correct number of phosphorylation sites on each peptide, there was a marked improvement in precision, with all but 2 reports in excess of 80%, 15 of which are >90%. The remaining incorrect peptides in the reports are due specifically to false localization, which was at most 28%.
To illustrate the difficulty of phosphorylation site localization, the peptides were divided into groups based on the number of phosphorylation sites (Figure 5A–C). Peptides containing only a single phosphorylated residue (and one to seven possible pSTY sites) had the highest identification rate, with a median of 91%. Peptides with two or three phosphorylated residues were more problematic, with median identification rates of 65 and 26%, respectively. Similarly, the false-positive rate of localization increased for multiply phosphorylated peptides, from a median of 4% (single) to a median of 9% (double). No incorrect localizations of triply phosphorylated peptides were reported; however, the sample contained only three such peptides, only one of which contained a single alternate site. The observed FDR increased with the number of potential phosphorylation sites per peptide (Figure 5C). This highlights the difficulty in correctly localizing the PTM when challenged with multiple sites and emphasizes the need for stringent validation of the results of the spectral analysis.
Figure 5.
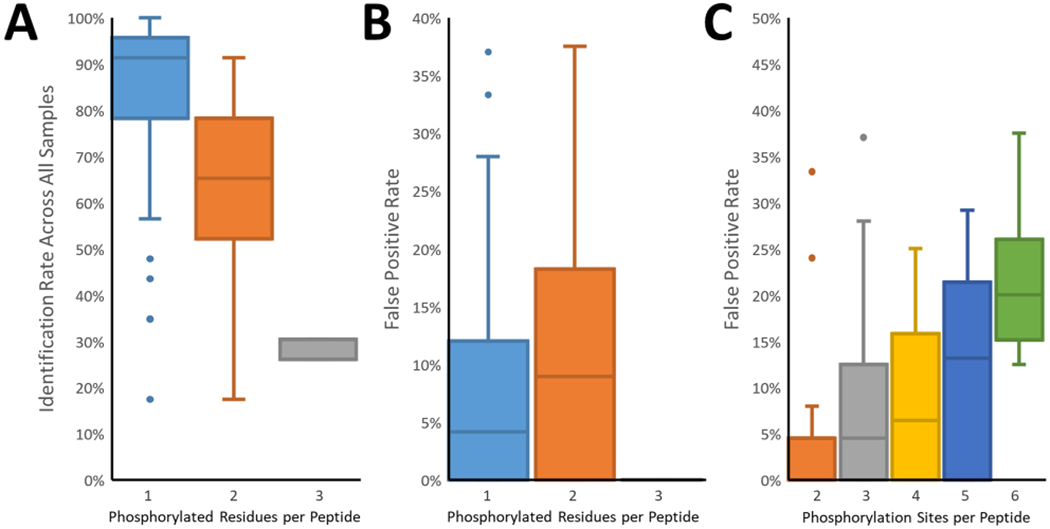
Analysis of the correct phosphorylation site identification. (A) The frequency of correct identification across all reports decreased as the number of phosphorylated residues per peptide increased. Here the identification required both sequence and localization to be correct. (B) The false localization rate was higher among multiply phosphorylated peptides. The exception was the triply phosphorylated peptides, where only a single alternate site existed on only one of the three peptides. (C) False localization rates increased in parallel with the number of potential sites (serine, threonine, tyrosine) per peptide.
The observance of high false localization rates (FLRs) is likely attributable to the interpretation of the results rather than a failure of any particular informatics tool, and underscores the importance of PTM localization regardless of the analytical platform. For example, report nos. 01195 and 13273 had some of the lowest and highest FLRs (1 and 28%, respectively, Supplementary Figure 1) despite using the same software (MaxQuant) with similar parameters (e.g., 0.01 Site FDR) and differing only in the use of “Match Between Runs” and the sequence database for the searches. Report no. 27573-QEHF helpfully provided detailed methods of the analysis using Proteome Discoverer and indicated that all phosphorylation localizations with a “ptmRS Phospho Site Probability” of 75% and above were reported, resulting in a moderately high 17% FLR. Because this is the FLR on the level of individual spectra, false unique phosphorylation sites will quickly accumulate when considering hundreds to thousands of PSMs. Report no. 13579 used Proteome Discoverer with a similar analysis pipeline as report no. 27573 but chose a phosphoRS threshold of 95%, resulting in a much lower 2% FLR. One analysis (report no. 30417) manually validated phosphorylation sites, resulting in an 8% FLR, similar to the results from methods that relied on computational approaches (Figure 3). Despite the fact that the majority of reports indicated that PTM site localization was performed, few participants provided explicit details about the parameters or thresholds used in their analysis, which may explain the diversity in the FLR among participants using the same analysis platform.
To further illustrate the potential issues associated with phosphorylation site localization, the PSM identification results of the neat mixtures of four reports were analyzed using PTMProphet23 to assign probabilities to each potential phosphorylation site, compute the information content of site localizations, and test different thresholds for the information content. PTMProphet computes normalized per-modification information content for each type of PTM analyzed in each PSM. This statistic ranges between 0 and 1 and estimates the portion of modifications (phosphorylation on serine, threonine, and tyrosine for these analyses) that can be confidently positioned within the peptide given the parameters of the PSM. It can be used to directly compare PTM localizations in PSMs having different numbers of sites and phospho-groups. Figure 6 summarizes the analysis of the submitted data when considering only the peptides that were identified with the correct number of phosphorylated residues. Report no. 13273 contained 83 correct localizations but also 32 incorrect localizations. Using a PTMProphet information content threshold of 50%, incorrect localizations dropped to 24, and at a 90% threshold, incorrect localizations were reduced to 6. The increase in specificity comes at the expense of sensitivity, and at the 90% threshold, the number of correct localizations decreased to 73. Although fewer correct localizations are reported, the FLR dropped from 28 to 8%. This trend was also observed in report no. 84931, where incorrect localizations were reduced from 5 to just 1 while maintaining 49 of 54 (91%) correct phosphosite localizations. Report no. 31245 was noteworthy for having no incorrect localizations, but it also reported the fewest that were correct; there was an increase in correct phosphorylated peptide identifications for this data set following PTMProphet analysis. This is likely due to the differences applied in the scoring thresholds between PTMProphet and the tool used in that analysis (not provided). Report no. 23721 is interesting in that false localizations using PTMProphet increased at 50% information content but then rapidly decreased at higher information content thresholds. This behavior is likely because the 50% threshold was less selective than that used in the report. Also, report no. 23721 listed phosphorylated peptides not observed among the PSMs submitted for the neat mixture. This is most likely because the report contained a combination of results observed in both the neat and yeast mixtures, whereas the PTMProphet analysis was limited to just the PSMs from the results of the neat analysis. Therefore, an in-depth reanalysis of the neat mixtures starting from a database search was performed (Supplementary Figure 2). This analysis also showed the trend of reduced incorrect localizations at higher information content thresholds. These results show the effect of setting various ambiguity tolerances. Where the precise localization of sites is prioritized, stringent thresholds should be applied when performing PTM site localization, with rigorous FLR estimation to minimize errors in the results. Whereas this study focused on the identification and correct site localization of phosphorylated peptides, the presence of other PTMs that are possibly expected in a sample needs to be likewise considered, and additional PTMs need to be correctly localized simultaneously.
Figure 6.
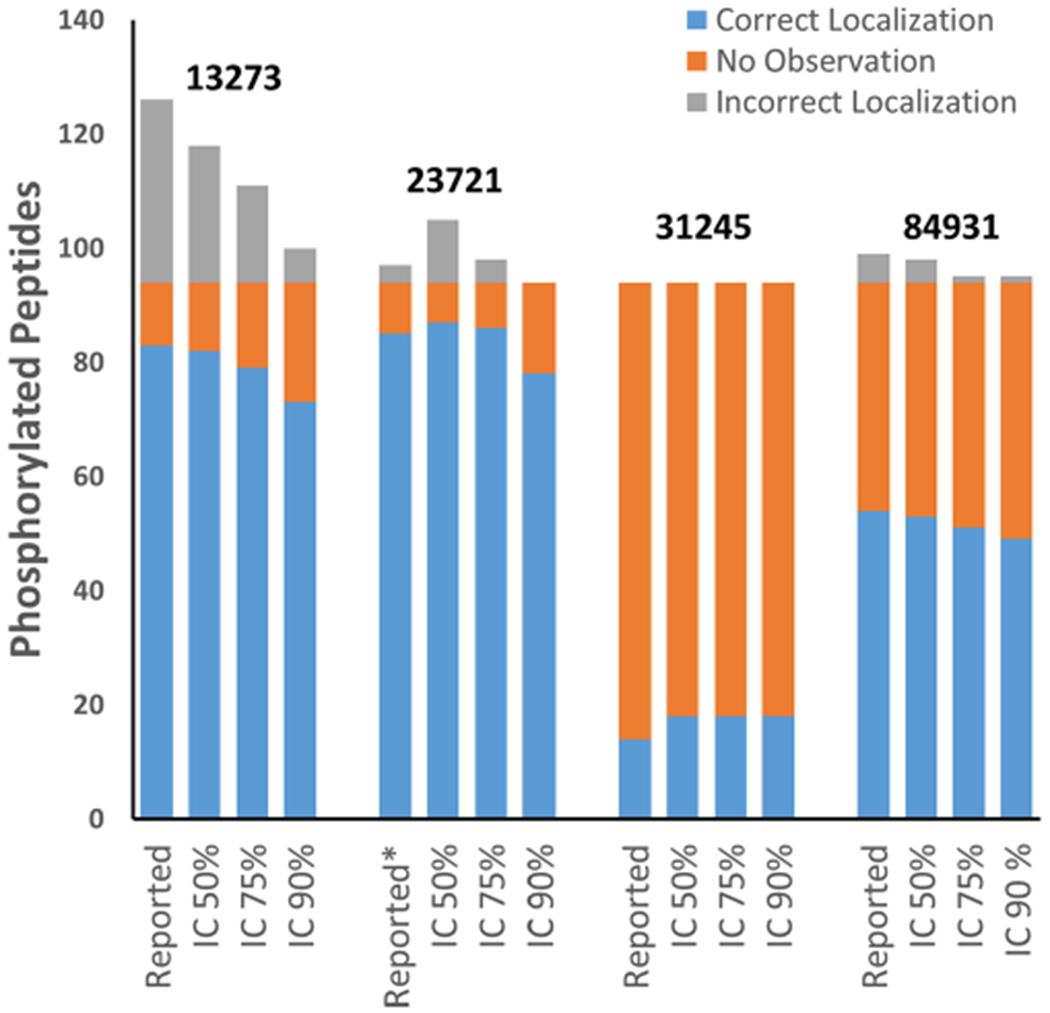
Effect of PTMProphet normalized information content thresholds on the numbers of correct and incorrect phosphosite localizations. The neat sample search results from four reports were analyzed using PTMProphet to compute the localization statistics of the potential phosphorylation sites of each PSM. Increasing the normalized information content threshold from 50 to 75% decreased the incorrect localizations reported.
Challenge #2
The second part of the Phosphopeptide Challenge evaluated the methodology for the relative quantification of phosphopeptides, which has biological implications given that protein activity and localization are often altered through phosphorylation and dephosphorylation events at specific sites. For each phosphorylated peptide in the neat sample, its nonphosphorylated form of the peptide was also included. The ratios between the phosphorylated and nonphosphorylated forms of each peptide were 9:1, 3:1, 1:1, 1:3, or 1:9. Participants were asked to report the ratio between an observed phosphorylated peptide and its nonphosphorylated counterpart they found by their method of choice (e.g., peak area, spectral counts).
Twenty-two reports included phosphorylated peptide relative quantification, summarized in Figure 7. To assess the accuracy of each peptide quantity estimate, the log2 of the ratio between the observed and expected ratios was computed, and a distribution was plotted for each report. Ideally, the spread would center around zero with minimal variance, indicating that the observed ratios closely matched the expected ratios. Most reports had median accuracy values below zero and variances of three times to six times different from those expected. In addition, frequently there were fewer quantified phosphopeptides than identifications, likely due to the difficulty of observing the nonphosphorylated form when present at low concentration. Few reports specified the software that was used for quantification. Therefore, unless otherwise indicated, whenever MaxQuant or Proteome Discoverer was used for identification, it was assumed that they were also used for quantification. The node used for quantification in Proteome Discoverer was usually not provided. See Table 1 for our best assumptions of the pipelines used for quantification.
Figure 7.
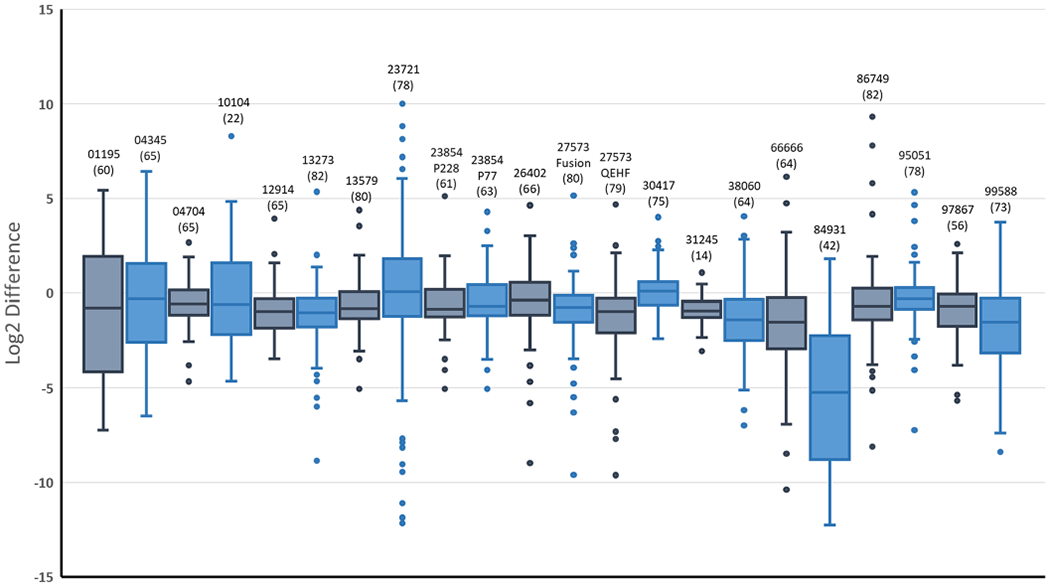
Box-and-whisker plots summarizing the relative quantification part of the challenge. Participants were asked to compute the ratio between the phosphorylated and nonphosphorylated forms of each peptide sequence (present in the sample from 1:9 to 9:1). Shown are the log2 fold changes between the expected and observed ratios. Each report is identified by its number above the whiskers, and the analytical method used is referenced in Supplementary Table S3. In parentheses below each report identifier is the number of phosphorylated peptides they were able to quantify.
Some generalizations about quantification could be deduced from the reports. As with identification, the use of any particular algorithm did not ensure accurate results. For example, report nos. 84931 and 12914 both used MaxQuant but had variances of 13.0 and 1.9, respectively. It was difficult to ascertain from the provided information whether the primary factor is the algorithm/workflow version, the user-selected parameters, or the data quality, in particular, because all precursor spectra in these reports were acquired using similar Orbitrap instrumentation with high resolving power. Similar disparate results were observed in workflows utilizing Skyline24 (e.g., report no. 10104 vs 95051), but it is worth noting that there was a high degree of diversity in the instrumentation used for the submissions reporting Skyline results. Of note, an in-house, precursor ion-based approach (report no. 04704) performed as well as the publicly available software tools. Report no. 04345 was the only submission in which spectral counting was used for relative quantification; the results exhibited lower quantitative accuracy. A possible explanation could be the low number of spectra acquired for each peptide due to the use of dynamic exclusion (which is typical of shotgun proteomic analysis). In protein-level quantification using spectral counting, such limitations can be mitigated by summing the accumulated spectra from many peptides to estimate the protein quantity.25 In many protein quantification workflows, quantitative values from multiple peptides are assembled to generate the protein-level value. Here quantification necessarily was based on a single pair of peptide signals for each sequence, which would be expected to give greater variability. The use of replicate injections can yield improved quantity estimates, as done in report no. 95051.
Challenge #3
The third part of the Phosphopeptide Challenge explored phosphopeptide enrichment from a complex sample. Phosphopeptide enrichment is a key component of many phosphoproteomic studies, and numerous different procedures and materials exist. In hopes of obtaining information about a variety of the available enrichment methods, a second sample (Phosphopeptide-Yeast) was provided to all participants in which the peptides in the “Phosphopeptide-Neat” samples were mixed with 6 μg of trypsin-digested yeast protein. The yeast background matrix masks the detection of the synthetic phosphopeptides. Participants were asked to use their preferred enrichment procedure with the Phosphopeptide-Yeast sample and report if they were able to identify the sequences they detected in the Phosphopeptide-Neat sample (Challenge #1). Twenty-one reports included results for this challenge (Supplementary Table S4). Ten of the reports listed methods that used TiO2, with alternatives including Fe(III)-NTA, Ti-IMAC, electrostatic repulsion–hydrophilic interaction chromatography (ERLIC), and combination materials (e.g., TiO2 + ZrO2). Four reports did not disclose the materials used for enrichment.
Overall, the enrichment procedures resulted in 60–100% recovery of phosphopeptides observed in the neat samples. Eighteen of the 20 reports had at least 80% recovery, and 14 had more than 90% recovery. TiO2 was used by 10 participants and showed the highest variability in recovery (used for submissions with both the highest and lowest recovery). The alternative materials showed 85–100% recovery; however, the small number of participants using each individual material makes it difficult to assess if the results are representative. Interestingly, four of the seven reports that showed lower recovery (<85%) also had high FLRs (>20%) in the first part of the study, although FLRs for these reports were all lower (<20%) following enrichment.
Role of Mass Measurement Accuracy
In 2007, Gygi and coworkers investigated the importance of a number of parameters in phosphopeptide identification, including the accuracy of peptide (MS1) mass measurement.26 At the time, low-resolution ion traps were still the most commonly used instruments in proteomics. Using a hybrid ion trap–FTICR instrument, Gygi and coworkers found a 2.4-fold increase in the phosphopeptide identification rate when going from an uncertainty of 2.1 Da (ion trap data) to 10 ppm (FTICR data) for the precursor mass measurement for the same ion trap tandem mass spectra. Many of the submissions in the challenge also used a 10 ppm mass measurement tolerance in their analyses, and for some, this was necessary given the residual mass measurement errors. However, this does not reflect the accuracy and precision achievable in the instrumentation used, which has improved considerably since 2007. To revisit the role of mass measurement accuracy in phosphopeptide identification, we recalibrated all submitted MS1 data using a previously published algorithm for FTICR recalibration21 extended to TOF and Orbitrap data and confidently identified (FDR 1%) nonphosphopeptides from the yeast background as internal calibrants. The recalibration component, mzRecal, was inserted between two consecutive executions of our Comet-based reanalysis workflow, differing only in mass measurement tolerance (10 ppm in the first pass and 2 ppm in the second) and modifications considered (only fixed cysteine carbamidomethylation and methionine oxidation in the first pass, phosphorylation of serine, threonine, and tyrosine added in the second). The recalibration eliminated the mass measurement bias and improved the precision in most data sets, which allowed them to be searched within 2 ppm without losing precursors, which would not have been possible in the original data. However, unlike the 2.4-fold improvement in the identification rate going from 2.1 Da to 10 ppm observed by the Gygi group,26 we observed only minor improvement, in the number of identified phosphopeptides, from 10 to 2 ppm using our reanalysis workflow. Even in open searches, the identification rates were virtually the same. However, the 2 ppm searches were considerably faster, and we do not rule out the notion that recalibration can improve phosphopeptide identification when considering noncanonical phosphorylation27 or additional PTMs as well as their chromatographic alignment across data sets (“Match Between Runs”) and label-free quantitation.
Data Exploration in MassIVE
All submitted data and search results were uploaded to MassIVE (https://massive.ucsd.edu/) as data set MSV000085932 with ProteomeXchange identifier PXD020801. Where possible, results provided in or convertible to standardized formats were included in the data set and can thus be interactively explored and visualized through a web browser. Individual sets of results from each participant can be visualized by clicking “Browse Results” (Supplementary Figure 3) from the MassIVE data set page to show a table of search results that can be filtered by participant identifier. Clicking on the number for PSMs, Peptides, or Proteins will show a table with the corresponding set of search results. On both PSMs and Peptide results views, it is possible to visualize the original spectra annotated with the assigned identification by clicking on the spectrum icon on the leftmost column of the table. This expands the corresponding row to show a Lorikeet spectrum viewing panel, where it is possible to zoom in/out of the annotated spectrum, select ion types to annotate the spectrum, including annotating losses of phosphate from modified residues, and compare experimental ion masses (in the spectrum) with theoretical ion masses (in the ion table to the right) (Supplementary Figure 4). Different possibilities of site localization can also be explored by manually editing the peptidoform string in the text box right above the spectrum panel (i.e., cut/paste the modification from one site to an alternative site) and clicking “Update” to refresh the spectrum panel to show the spectrum annotated with the alternative site. This is illustrated with a spectrum submitted by participant 84931 for peptide ANS+79.966331FVGTAQYVSPELLTEK by MassIVE resolution of the Universal Spectrum Identifier (USI) mzspec:PXD020801:84931_ppep_yeast_enriched.mz-ML:scan:18909:ANS+79.966331FVGTAQYVSPELLTEK to show a strong y-ion series for the reported site (a strong series of b-ions is also shown if clicking the option for “H3PO4 (p)” neutral losses on the left-side ion panel) (Supplementary Figure 4), which is substantially reduced by switching the modification site to the right-most threonine on the peptide (ANSFVGTAQYVSPELLT+79.966331EK). Finally, it is possible to see all study spectra submitted by all participants for each peptide by using the MassIVE Search interface at https://massive.ucsd.edu/ProteoSAFe/massive_search.jsp and filtering by the peptide of interest and by data set MSV000085932, then clicking the “Search PSMs” button to see all data set PSMs for the peptide. For example, this link shows all spectra submitted for the same example peptide as above (Supplementary Figure 5).
CONCLUSIONS
The results presented here provide a first look at this extensive, community-driven phosphopeptide analysis and data set. The results indicate the challenges faced in the correct identification and localization of phosphorylation on peptides, in particular, when there are multiple potential phosphorylation sites. Although TiO2 was by far the most popular material for phosphopeptide enrichment, successful capture and analysis of the phosphorylated peptides was observed after the use of diverse enrichment materials. Regardless of the tools and methodologies, care must be taken in the interpretation of results to ensure site localization accuracy, as was shown by the benefits of applying stricter thresholds that result in reduced FLRs. Furthermore, the broad range of quantitative results may be indicative of challenges faced in providing the quantitative analysis of phosphorylation stoichiometry. The data herein offer further opportunities to explore, compare, and test different hypotheses and analytical methodologies to better characterize phosphorylation with mass spectrometry. All report submissions and their data are available on ProteomeXchange28 (PXD020801), and, where possible, the data files can be directly examined in MassIVE. For researchers interested to contribute to this compendium of methods and results, the HPP Phosphopeptide mixtures can be requested by email to the HUPO Office (office@hupo.org). The exemplar nature of this community challenge for peptide phospho-site determination also provides the opportunity for other community-directed challenges, and we invite interested researchers to get in touch with members of the Human Proteome Project of HUPO and discuss their ideas and take part in developing a new proteome challenge for community assessment.
Supplementary Material
Supplementary Figure 1. Summary of submitted results after the removal of peptide identifications showing the loss of a phosphoryl group. Supplementary Figure 2. Reanalysis of neat mixtures starting from the database search. Supplementary Figure 3. Data set page in MassIVE to browse results. Supplementary Figure 4. Universal Spectrum Identifier (USI) resolution page in MassIVE illustrating Lorikeet interactions with the annotated spectrum. Supplementary Figure 5. MassIVE search interface (PDF)
Supplementary Table S1. Phosphorylated peptides with their characteristics and identification success (XLSX)
Supplementary Table S2. Detailed instrument platforms and parameters (XLSX)
Supplementary Table S3. Detailed spectral analysis platforms and parameters (XLSX)
Supplementary Table S4. Details of phosphopeptide enrichment (XLSX)
ACKNOWLEDGMENTS
We thank SynPeptide for generously providing the synthetic peptides and Justin Jordaan and Isak Gerber from ReSyn Biosciences for providing the MagReSyn phosphopeptide enrichment kits. We thank John Aitchison (Seattle Children’s Research Institute) for providing the yeast protein stock, Sarah Li (Moritz Lab, ISB) for help with sample formulation, and Jeremy Carver (UCSD) for supporting the data implementation in MassIVE. This work was funded in part by the National Institutes of Health, National Institute of General Medical Sciences grant R01GM087221, the Office of the Director S10OD026936, the National Institute of Allergy and Infectious Diseases grant R21AI133335, and the National Institute on Aging grant U19AG023122 (R.L.M.); by NSF award 1920268 and ABI 1759980 and NIH awards NIH-NLM 1R01LM013115, P41GM103484, and R24GM127667 (N.B.); and by NIH P30ES017885 and U24CA210967 (G.S.O.). Finally, we thank all the HPP MS Pillar Phosphopeptide Challenge participants that contributed expertise and reported their methods and results for the community to evaluate and learn from this process.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00648.
The authors declare the following competing financial interest(s): L.H. and B.X. are employees of SynPeptide Co., S.H.S is an employee of ReSyn Biosciences.
All submitted data and search results were uploaded to MassIVE (https://massive.ucsd.edu/) as data set MSV000085932 with ProteomeXchange identifier PXD020801.
Contributor Information
Michael R. Hoopmann, Institute for Systems Biology, Seattle, Washington 98109, United States.
Ulrike Kusebauch, Institute for Systems Biology, Seattle, Washington 98109, United States.
Magnus Palmblad, Center for Proteomics and Metabolomics, Leiden University Medical Center, 2300 RC Leiden, The Netherlands.
Nuno Bandeira, Department of Computer Science and Engineering University of California, San Diego, La Jolla, California 92093, United States.
David D. Shteynberg, Institute for Systems Biology, Seattle, Washington 98109, United States
Lingjie He, Synpeptide Co., Ltd., Shanghai 201204, China.
Bin Xia, Synpeptide Co., Ltd., Shanghai 201204, China.
Stoyan H. Stoychev, ReSyn Biosciences, Pretoria 0184, South Africa
Gilbert S. Omenn, Institute for Systems Biology, Seattle, Washington 98109, United States; Departments of Computational Medicine and Bioinformatics, Internal Medicine, and Human Genetics and School of Public Health, University of Michigan, Ann Arbor, Michigan 48109, United States.
Susan T. Weintraub, Department of Biochemistry and Structural Biology, The University of Texas Health Science Center at San Antonio, San Antonio, Texas 78229, United States.
Robert L. Moritz, Institute for Systems Biology, Seattle, Washington 98109, United States.
REFERENCES
- (1).Noberini R; Sigismondo G; Bonaldi T The contribution of mass spectrometry-based proteomics to understanding epigenetics. Epigenomics 2016, 8 (3), 429–45. [DOI] [PubMed] [Google Scholar]
- (2).Simithy J; Sidoli S; Garcia BA Integrating Proteomics and Targeted Metabolomics to Understand Global Changes in Histone Modifications. Proteomics 2018, 18 (18), No. 1700309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hunter T Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc, B 2012, 367 (1602), 2513–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Thorner J; Hunter T; Cantley LC; Sever R Signal transduction: From the atomic age to the post-genomic era. Cold Spring Harbor Perspect. Biol 2014, 6 (12), No. a022913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Junger MA; Aebersold R Mass spectrometry-driven phosphoproteomics: patterning the systems biology mosaic. Wiley Interdiscip Rev. Dev Biol. 2014, 3 (1), 83–112. [DOI] [PubMed] [Google Scholar]
- (6).Kusebauch U; Ortega C; Ollodart A; Rogers RS; Sherman DR; Moritz RL; Grundner C Mycobacterium tuberculosis supports protein tyrosine phosphorylation. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (25), 9265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gnad F; Gunawardena J; Mann M PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 2011, 39 (suppl_1), D253–D260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).von Stechow L; Francavilla C; Olsen JV Recent findings and technological advances in phosphoproteomics for cells and tissues. Expert Rev. Proteomics 2015, 12 (5), 469–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Olsen JV; Mann M Status of Large-scale Analysis of Post-translational Modifications by Mass Spectrometry. Mol. Cell. Proteomics 2013, 12 (12), 3444–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tape CJ; Worboys JD; Sinclair J; Gourlay R; Vogt J; McMahon KM; Trost M; Lauffenburger DA; Lamont DJ; Jørgensen C Reproducible Automated Phosphopeptide Enrichment Using Magnetic TiO2 and Ti-IMAC. Anal. Chem. 2014, 86 (20), 10296–10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Leutert M; Rodriguez-Mias RA; Fukuda NK; Villen J R2-P2 rapid-robotic phosphoproteomics enables multidimensional cell signaling studies. Mol. Syst. Biol. 2019, 15 (12), No. e9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shteynberg DD; Deutsch EW; Campbell DS; Hoopmann MR; Kusebauch U; Lee D; Mendoza L; Midha MK; Sun Z; Whetton AD; Moritz RL PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. J. Proteome Res. 2019, 18 (12), 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Desiere F; Deutsch EW; King NL; Nesvizhskii AI; Mallick P; Eng J; Chen S; Eddes J; Loevenich SN; Aebersold R The Peptide Atlas project. Nucleic Acids Res. 2006, 34 (1), D655–D658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Krokhin OV Sequence-Specific Retention Calculator. Algorithm for Peptide Retention Prediction in Ion-Pair RP-HPLC: Application to 300- and 100-Å Pore Size C18 Sorbents. Anal. Chem. 2006, 78 (22), 7785–7795. [DOI] [PubMed] [Google Scholar]
- (15).Adusumilli R; Mallick P Data Conversion with ProteoWizard msConvert. Methods Mol. Biol 2017, 1550, 339–368. [DOI] [PubMed] [Google Scholar]
- (16).Eng JK; Jahan TA; Hoopmann MR Comet: an open-source MS/MS sequence database search tool. Proteomics 2013, 13 (1), 22–4. [DOI] [PubMed] [Google Scholar]
- (17).Moosa JM; Guan S; Moran MF; Ma B Repeat-Preserving Decoy Database for False Discovery Rate Estimation in Peptide Identification. J. Proteome Res 2020, 19 (3), 1029–1036. [DOI] [PubMed] [Google Scholar]
- (18).Deutsch EW; Mendoza L; Shteynberg D; Slagel J; Sun Z; Moritz RL Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics: Clin. Appl 2015, 9 (7–8), 745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Keller A; Nesvizhskii AI; Kolker E; Aebersold R Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem 2002, 74 (20), 5383–92. [DOI] [PubMed] [Google Scholar]
- (20).Shteynberg D; Deutsch EW; Lam H; Eng JK; Sun Z; Tasman N; Mendoza L; Moritz RL; Aebersold R; Nesvizhskii AI iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 2011, 10 (12), M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Palmblad M; Bindschedler LV; Gibson TM; Cramer R Automatic internal calibration in liquid chromatography/Fourier transform ion cyclotron resonance mass spectrometry of protein digests. Rapid Commun. Mass Spectrom 2006, 20 (20), 3076–80. [DOI] [PubMed] [Google Scholar]
- (22).Cox J; Mann M Max Quant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26 (12), 1367–72. [DOI] [PubMed] [Google Scholar]
- (23).Shteynberg DD; Deutsch EW; Campbell DS; Hoopmann MR; Kusebauch U; Lee D; Mendoza L; Midha MK; Sun Z; Whetton AD; Moritz RL PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. Journal of Proteome Research 2019, 18 (12), 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26 (7), 966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nahnsen S; Bielow C; Reinert K; Kohlbacher O Tools for label-free peptide quantification. Mol. Cell. Proteomics 2013, 12 (3), 549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bakalarski CE; Haas W; Dephoure NE; Gygi SP The effects of mass accuracy, data acquisition speed, and search algorithm choice on peptide identification rates in phosphoproteomics. Anal. Bioanal. Chem 2007, 389 (5), 1409–19. [DOI] [PubMed] [Google Scholar]
- (27).Hardman G; Perkins S; Brownridge PJ; Clarke CJ; Byrne DP; Campbell AE; Kalyuzhnyy A; Myall A; Eyers PA; Jones AR; Eyers CE Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO J. 2019, 38 (21), No. e100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Deutsch EW; Csordas A; Sun Z; Jarnuczak A; Perez-Riverol Y; Ternent T; Campbell DS; Bernal-Llinares M; Okuda S; Kawano S; Moritz RL; Carver JJ; Wang M; Ishihama Y; Bandeira N; Hermjakob H; Vizcaino JA The Proteome Xchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45 (D1), D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Summary of submitted results after the removal of peptide identifications showing the loss of a phosphoryl group. Supplementary Figure 2. Reanalysis of neat mixtures starting from the database search. Supplementary Figure 3. Data set page in MassIVE to browse results. Supplementary Figure 4. Universal Spectrum Identifier (USI) resolution page in MassIVE illustrating Lorikeet interactions with the annotated spectrum. Supplementary Figure 5. MassIVE search interface (PDF)
Supplementary Table S1. Phosphorylated peptides with their characteristics and identification success (XLSX)
Supplementary Table S2. Detailed instrument platforms and parameters (XLSX)
Supplementary Table S3. Detailed spectral analysis platforms and parameters (XLSX)
Supplementary Table S4. Details of phosphopeptide enrichment (XLSX)