

Abstract

Viral mRNA cap methyltransferases (MTases) are emerging targets for the development of broad-spectrum antiviral agents. In this work, we designed potential SARS-CoV-2 MTase Nsp14 and Nsp16 inhibitors by using bioisosteric substitution of the sulfonium and amino acid substructures of the cosubstrate S-adenosylmethionine (SAM), which serves as the methyl donor in the enzymatic reaction. The synthetically accessible target structures were prioritized using molecular docking. Testing of the inhibitory activity of the synthesized compounds showed nanomolar to submicromolar IC50 values for five compounds. To evaluate selectivity, enzymatic inhibition of the human glycine N-methyltransferase involved in cellular SAM/SAH ratio regulation was also determined, which indicated that the discovered compounds are nonselective inhibitors of the studied MTases with slight selectivity for Nsp16. No cytotoxic effects were observed; however, this is most likely a result of the poor cell permeability of all evaluated compounds.
Keywords: SARS-CoV-2, MTase inhibitors, Nsp14, Nsp16, SAM analogues, antiviral drugs
The global COVID-19 pandemic has exposed a shortage of therapeutic treatment options against coronaviruses. While repurposing studies of existing drugs have identified numerous candidates, their usefulness in treating the respiratory disease is limited due to poor efficacy.1 This has urged the scientific community to embark on extensive drug discovery research against the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
The genome of SARS-CoV-2 codes for 29 proteins,2 several of which have been explored as targets for development of new antiviral drugs. The most notorious of these are the RNA-dependent RNA polymerase (RdRp) Nsp12, the helicase Nsp13, the main (Mpro) and papain-like proteases (PLpro) Nsp5 and Nsp3d, the nucleocapsid protein N, as well as mRNA cap methyltransferases (MTases) Nsp14 and Nsp16.3 Although future clinical development of the identified specific SARS-CoV-2 inhibitors may take several years before approval, these efforts are also crucial to avoid the breakout of another coronavirus pandemic in the coming years.
Coronaviruses have evolved an mRNA capping apparatus to protect their 5′-ends with a cap moiety that is indistinguishable from eukaryotic mRNA cap structures.4 The capping is aided by the MTases Nsp14 and Nsp16, which modify the N7 of the guanosine cap and the 2′OH group of the two subsequent nucleotides of viral mRNA, respectively.5 Nsp16 is activated and stabilized by binding to Nsp10.6 The N7-methylguanosine (m7G) cap is required for efficient translation of viral proteins,7 whereas 2′-O-methylation of the first two nucleotides is important for evasion of the host immune response.8
Studies on SARS-CoV-1 have shown that mutations in the Nsp14 and Nsp16 genes lead to a significantly attenuated virus that is recognized by the innate immune system.7,9,10 Further data suggest that inhibition of Nsp16 can be used for coronavirus treatment but requires the activation of interferon-stimulated gene response for efficacy.11 Nsp14 and Nsp16 along with the RdRp Nsp12 and the helicase Nsp13 are among the most conserved proteins of coronaviruses with sequence identities above 50%. Therefore, drugs targeting the MTases of SARS-CoV-2 could have broad-spectrum antiviral activity also against other coronaviruses including SARS-CoV-1 and MERS-CoV. Furthermore, crystallographic structures of Nsp16 from SARS-CoV-212−14 and of Nsp14 from SARS-CoV-115 are available facilitating the use of virtual screening and structure-guided drug design approaches.
Most of the previous work to discover viral MTase inhibitors has focused on S-adenosylmethionine (SAM) analogues with minor (i.e., single/few atom) modifications in the amino acid and adenine substituents.16−18 Thus, a few highly potent inhibitors such as sinefungin (Figure 1A) have been identified that are effective against MTases from various species. However, all the discovered inhibitors lacked selectivity for coronavirus MTases compared to human MTases, and many showed poor cell permeability due to their zwitterionic nature. For this reason, none of the early viral MTase inhibitors have been advanced into clinical development. Aouadi et al. performed high-throughput screening against SARS-CoV-1 Nsp14 and identified other new classes of active compounds, mostly polyphenols.19 However, the most potent of these compounds were similarly nonselective, although some of the weaker inhibitors showed more selectivity toward one viral MTase. Recently, two preprints appeared reporting new Nsp14 inhibitors discovered by screening 161 SAM competitive MTase inhibitors and through structure-based design.20,21
Figure 1.

(A) Molecular structures of the MTase cosubstrate SAM, known pan-MTase inhibitor sinefungin, and general structures of the compounds studied here. (B) Surface view of SAM binding pockets in the crystal structures of SARS-CoV-1 Nsp14–SAM complex (PDB ID: 5C8T), SARS-CoV-2 Nsp16–SAM complex (PDB ID: 6W4H), human glycine N-MTase–SAM complex, and human RNA guanine-N7-MTase–SAH complex (PDB ID: 3BGV). The structure of SARS-CoV-2 Nsp14 is not available; however, both strains share 100% sequence identity within 10 Å distance of the SAM binding site. Amino acid side chains within 4 Å distance of SAM/SAH are shown as lines. Hydrogen bond and π-stacking interactions are shown with yellow and cyan dashed lines. The figure was generated using PyMOL.22
The previous findings motivated us to compare the binding pockets of SAM between viral and cellular MTases. Figure 1B shows a side-by-side comparison of the SAM binding sites of SARS-CoV-1 Nsp14, SARS-CoV-2 Nsp16, and human mRNA (guanine-N7-) MTase and human glycine N-methyltransferase (GNMT). Although the binding sites of the SARS-CoV Nsp14 and Nsp16 MTases look more secluded than those of the human MTases, there are notable similarities in the shape of the binding pockets. The adenine subpocket is very narrow in all structures, whereas the amino acid binding subsite is wider and potentially able to accommodate bulkier substituents. Hence, any changes introduced in the adenine fragment have in most cases led to reduced inhibitor potency against viral MTases.16,17 In the Nsp16 structure, the amino acid binding subsite is also tightly filled; however, the shape of the methylation site would allow the introduction of bulkier or branched substituents. Based on these considerations, we decided to explore the structure–activity/selectivity relationships of the methionine fragment with the aim to achieve selectivity for the viral MTases. Bioisosteric replacements of the SAM methionine substructure were proposed, and the designed compounds were prioritized by docking against SARS-CoV-1 Nsp14 and SARS-CoV-2 Nsp16. The docking models suggested that aromatic groups replacing the aliphatic side chain can engage in interactions with the Nsp14 residues Phe401, Tyr420, Phe426, and Phe506 (Tyr47, Tyr132 in Nsp16), which make up a narrow, hydrophobic surface groove that accommodates the mRNA cap, and bioisosteres of the α-amino acid group can interact with the charged/polar amino acids Arg310, Asp331, Asn334, and Lys336 (Asn43, Lys46, Lys170, Glu203 in Nsp16). As a result, a diverse set of the top scoring compounds was prepared that could be grouped as S-aryl-5′-thioadenosine and analogues (1), S-alkylaryl(alkylhetaryl)-5̀-thioadenosines and analogues (2 and 3), and other SAM analogues (4) (Table 1).
Table 1. Enzymatic Potency against Viral and Human MTases, Cytotoxicity and Cell Permeability of SAM Analogues.

The synthesized compounds were tested for inhibition against the MTase activities of recombinant SARS-CoV-2 Nsp14, Nsp16/Nsp10,23 and human GNMT using a homogeneous time-resolved fluorescent energy transfer (HTRF) assay,19 which is based on the quantification of released S-adenosyl homocysteine (SAH) during the enzymatic reaction. GNMT was chosen as the off-target for SAM analogues because of its high abundance24 and because it competes with tRNA methyltransferases for SAM and thereby regulates the relative levels of SAM and SAH in cells.25 The pan-MTase inhibitor sinefungin was also tested for reference. Of the 11 prepared compounds, one compound (2a) showed nanomolar, four compounds (1a, 2b, 3a–b) submicromolar, three compounds (1b, 4a–b) micromolar IC50 values, and three compounds (2c, 3c, 4c) were inactive or had minimal activity (Table 1). Notably, three compounds (1a, 2a–b) were more potent than sinefungin. Although these compounds showed no selectivity for the viral MTases over human GNMT, they were more active against SARS-CoV-2 Nsp16 with selectivity factors of 2–4.
The obtained data provide some insights into the structure–activity relationships of amino acid-modified SAM analogues as SARS-CoV-2 MTase inhibitors. Replacement of the sulfonium by a thioether group leads to more potent inhibitors (1a, 2b, 3b) than by amide (1b) and amine (2c, 3c) functionalities, indicating that conformational flexibility and bulkiness rather than the positive charge of this part are required for efficient binding of the inhibitors. Installation of an aromatic or heteroaromatic group in place of the aliphatic amino acid side chain results in improved activity (compounds 1a, 2a–b, 3a–b) due to additional aromatic–aromatic interactions and reduced entropic penalty of binding (Figure 2). The observed improvements are larger for Nsp16 than for Nsp14, although the number of formed interactions is equal. A possible explanation of this is that the inhibitor’s adenine fragment binds Nsp16 in a conformation that is closer to SAM than in Nsp14 complexes. The presence of a carboxyl group 4–5 bonds apart from the S-adenosyl residue seems important for high potency, and its substitution with other hydrogen bond donors/acceptors is not beneficial (1a, 2a compared to 2b, 3a–b). In contrast, the presence of a positively charged moiety that replaces the amino group seems nonessential. This is because the aromatic interactions direct the R group toward Lys336/Arg310 and away from Asp331 (Asp130 in Nsp16, Figure 2). Finally, replacement of the transferred methyl group of SAM with larger substituents that could extend into the mRNA cap binding pocket is not well tolerated as shown by the poor activity of 4b.
Figure 2.
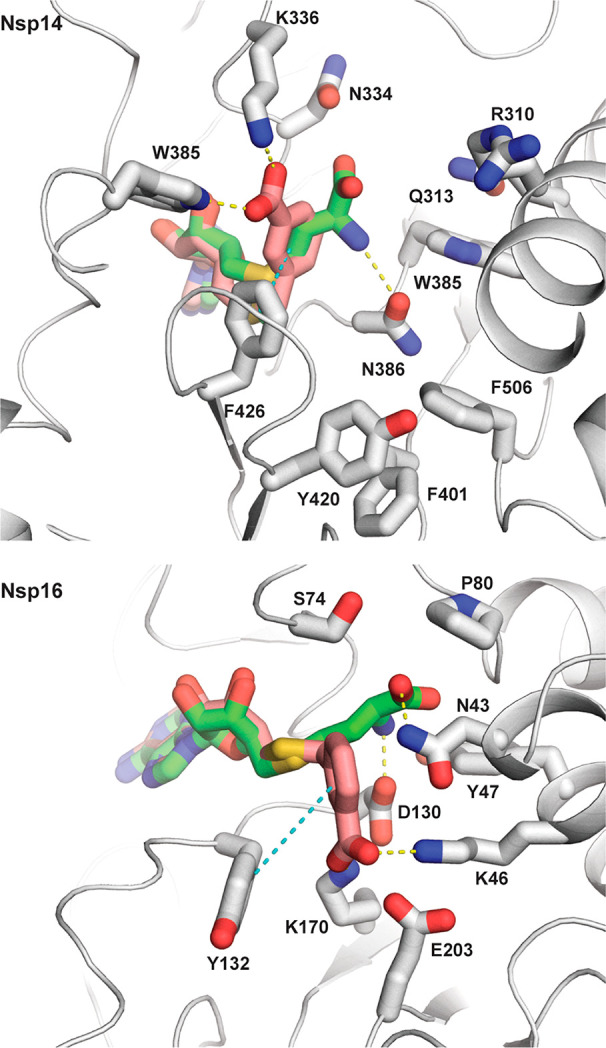
Docking models of compound 2a (colored salmon) in the X-ray crystal structures of SARS-CoV-1 Nsp14–SAM complex (PDB ID: 5C8T) and SARS-CoV-2 Nsp16–SAM complex (PDB ID: 6W4H) showing the bound SAM (green) and nearby side chains located within 5 Å from it. Hydrogen bond and π-stacking interactions are shown with yellow and cyan dashed lines. The figure was generated using PyMOL.22
Despite the high potency against human GNMT and possibly other homologous cellular MTases, the compounds exhibited virtually no cytotoxicity on mouse embryo fibroblast (NIH 3T3), human liver cancer (HepG2), and adenocarcinomic human alveolar basal epithelial (A549) cell lines. Therefore, we questioned whether the compounds are able to permeate the cell membrane. Cell permeability was tested with the A549 cell line, and compound concentrations were determined in cell lysates and culture media by mass spectrometry. The compound concentrations in the cell lysates were indeed low and often below the detection limit (Table 1). This indicates that the cell membrane crossing is a problem of this class of compounds even though several of them are not zwitterions as their parental compounds SAM and sinefungin. However, the compounds retain high polarity, which could be circumvented by utilizing derivatization or prodrug strategies to improve lipophilicity or to promote the transport of the nucleoside analogues.26
In summary, we have used SAM cosubstrate analogue design to discover S-aryl-5′-thioadenosines and S-alkylaryl(alkylhetaryl)-5̀-thioadenosines as novel classes of MTase inhibitors. These compounds show nanomolar to submicromolar potency against SARS-CoV-2 mRNA cap MTases Nsp14 and Nsp16, but they lack selectivity with respect to human GNMT and have poor cell permeability. The relatively facile synthesis and high potency of this class warranties further studies with the aim to identify novel antiviral agents with appropriate properties and selectivity toward coronavirus MTases. The availability of the crystal structures of Nsp14, Nsp16, and human GNMT should allow a structure-guided approach to design SAM analogues with increased selectivity for the coronavirus MTases.
Glossary
Abbreviations
- COVID-19
coronavirus disease 2019
- SARS-CoV
severe acute respiratory syndrome coronavirus
- MERS-CoV
Middle East respiratory syndrome coronavirus
- Nsp
nonstructural protein
- MTase
methyltransferases
- GNMT
human glycine N-methyltransferase
- SAM
S-adenosylmethionine
- SAH
S-adenosylhomocysteine
- HTRF
homogeneous time-resolved fluorescent energy transfer
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00140.
Description of synthesis of compounds 1–4, their NMR spectra, description of molecular modeling, protein expression and purification, description of enzymatic, cytotoxicity, and cell permeability assays (PDF)
This work was supported by the Latvian Council of Science (grant No. VPP-COVID-2020/1–0014). R.B. acknowledges European Regional Development Fund project No. 1.1.1.2/VIAA/2/18/379 for financial support. O.B. acknowledges European Regional Development Fund project No. 1.1.1.2/VIAA/4/20/747 for financial support.
The authors declare no competing financial interest.
This article is made available via the ACS COVID-19 subset for unrestricted RESEARCH re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for the duration of the World Health Organization (WHO) declaration of COVID-19 as a global pandemic.
Supplementary Material
References
- Singh T. U.; Parida S.; Lingaraju M. C.; Kesavan M.; Kumar D.; Singh R. K. Drug Repurposing Approach to Fight COVID-19. Pharmacol. Rep. 2020, 72 (6), 1479–1508. 10.1007/s43440-020-00155-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; Lee J.-Y.; Yang J.-S.; Kim J. W.; Kim V. N.; Chang H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181 (4), 914–921.e10. 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajarshi K.; Khan R.; Singh M. K.; Ranjan T.; Ray S.; Ray S. Essential Functional Molecules Associated with SARS-CoV-2 Infection: Potential Therapeutic Targets for COVID-19. Gene 2021, 768, 145313. 10.1016/j.gene.2020.145313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decroly E.; Ferron F.; Lescar J.; Canard B. Conventional and Unconventional Mechanisms for Capping Viral MRNA. Nat. Rev. Microbiol. 2012, 10 (1), 51–65. 10.1038/nrmicro2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet M.; Debarnot C.; Imbert I.; Selisko B.; Snijder E. J.; Canard B.; Decroly E. In Vitro Reconstitution of SARS-Coronavirus MRNA Cap Methylation. PLoS Pathog. 2010, 6 (4), e1000863 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decroly E.; Debarnot C.; Ferron F.; Bouvet M.; Coutard B.; Imbert I.; Gluais L.; Papageorgiou N.; Sharff A.; Bricogne G.; Ortiz-Lombardia M.; Lescar J.; Canard B. Crystal Structure and Functional Analysis of the SARS-Coronavirus RNA Cap 2’-O-Methyltransferase Nsp10/Nsp16 Complex. PLoS Pathog. 2011, 7 (5), e1002059 10.1371/journal.ppat.1002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Cai H.; Pan J.; Xiang N.; Tien P.; Ahola T.; Guo D. Functional Screen Reveals SARS Coronavirus Nonstructural Protein Nsp14 as a Novel Cap N7Methyltransferase. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (9), 3484–3489. 10.1073/pnas.0808790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S.; Szretter K. J.; Schriewer J.; Li J.; Youn S.; Errett J.; Lin T.-Y.; Schneller S.; Zust R.; Dong H.; Thiel V.; Sen G. C.; Fensterl V.; Klimstra W. B.; Pierson T. C.; Buller R. M.; Gale M.; Shi P.-Y.; Diamond M. S. 2’-O Methylation of the Viral MRNA Cap Evades Host Restriction by IFIT Family Members. Nature 2010, 468 (7322), 452–456. 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Tao J.; Sun Y.; Wu A.; Su C.; Gao G.; Cai H.; Qiu S.; Wu Y.; Ahola T.; Guo D. Structure-Function Analysis of Severe Acute Respiratory Syndrome Coronavirus RNA Cap Guanine-N7-Methyltransferase. J. Virol. 2013, 87 (11), 6296–6305. 10.1128/JVI.00061-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V. D.; Yount B. L.; Josset L.; Gralinski L. E.; Scobey T.; Agnihothram S.; Katze M. G.; Baric R. S. Attenuation and Restoration of Severe Acute Respiratory Syndrome Coronavirus Mutant Lacking 2’-o-Methyltransferase Activity. J. Virol. 2014, 88 (8), 4251–4264. 10.1128/JVI.03571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V. D.; Debbink K.; Baric R. S. Coronavirus Non-Structural Protein 16: Evasion, Attenuation, and Possible Treatments. Virus Res. 2014, 194, 191–199. 10.1016/j.virusres.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Lemus M.; Minasov G.; Shuvalova L.; Inniss N. L.; Kiryukhina O.; Brunzelle J.; Satchell K. J. F.. High-Resolution Structures of the SARS-CoV-2 2’-O-Methyltransferase Reveal Strategies for Structure-Based Inhibitor Design. Sci. Signal. 2020, 13 ( (651), ), 10.1126/scisignal.abe1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan T.; Arya S.; Chan S.-H.; Qi S.; Dai N.; Misra A.; Park J.-G.; Oladunni F.; Kovalskyy D.; Hromas R. A.; Martinez-Sobrido L.; Gupta Y. K. Structural Basis of RNA Cap Modification by SARS-CoV-2. Nat. Commun. 2020, 11 (1), 3718. 10.1038/s41467-020-17496-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafcikova P.; Silhan J.; Nencka R.; Boura E. Structural Analysis of the SARS-CoV-2 Methyltransferase Complex Involved in RNA Cap Creation Bound to Sinefungin. Nat. Commun. 2020, 11 (1), 3717. 10.1038/s41467-020-17495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; Wu L.; Shaw N.; Gao Y.; Wang J.; Sun Y.; Lou Z.; Yan L.; Zhang R.; Rao Z. Structural Basis and Functional Analysis of the SARS Coronavirus Nsp14-Nsp10 Complex. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (30), 9436–9441. 10.1073/pnas.1508686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh C. S.; Borchardt R. T.; Stone H. O. Inhibition of Newcastle Disease Virion Messenger RNA (Guanine-7-)-Methyltransferase by Analogues of S-Adenosylhomocysteine. Biochemistry 1977, 16 (17), 3928–3932. 10.1021/bi00636a032. [DOI] [PubMed] [Google Scholar]
- Pugh C. S.; Borchardt R. T. Effects of S-Adenosylhomocysteine Analogues on Vaccinia Viral Messenger Ribonucleic Acid Synthesis and Methylation. Biochemistry 1982, 21 (7), 1535–1541. 10.1021/bi00536a011. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Wang Z.; Tao J.; Wang Y.; Wu A.; Yang Z.; Wang K.; Shi L.; Chen Y.; Guo D. Yeast-Based Assays for the High-Throughput Screening of Inhibitors of Coronavirus RNA Cap Guanine-N7-Methyltransferase. Antiviral Res. 2014, 104, 156–164. 10.1016/j.antiviral.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouadi W.; Eydoux C.; Coutard B.; Martin B.; Debart F.; Vasseur J. J.; Contreras J. M.; Morice C.; Quérat G.; Jung M.-L.; Canard B.; Guillemot J.-C.; Decroly E. Toward the Identification of Viral Cap-Methyltransferase Inhibitors by Fluorescence Screening Assay. Antiviral Res. 2017, 144, 330–339. 10.1016/j.antiviral.2017.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota K.; Schapira M.; Perveen S.; Yazdi A. K.; Li F.; Chau I.; Ghiabi P.; Hajian T.; Loppnau P.; Bolotokova A.; Satchell K. J. F.; Wang K.; Li D.; Liu J.; Smil D.; Luo M.; Jin J.; Fish P. V.; Brown P. J.; Vedadi M.. Probing the SAM Binding Site of SARS-CoV-2 Nsp14 in Vitro Using SAM Competitive Inhibitors Guides Developing Selective Bi-Substrate Inhibitors; preprint; bioRxiv, 2021, 10.1101/2021.02.19.424337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otava T.; Šála M.; Li F.; Fanfrlík J.; Devkota K.; Pakarian P.; Hobza P.; Vedadi M.; Boura E.; Nencka R.. The Structure-Based Design of SARS-CoV-2 Nsp14 Methyltransferase Ligands Yields Nanomolar Inhibitors; preprint; 2021, 10.26434/chemrxiv.14075408.v1. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System; Schrödinger, LLC. [Google Scholar]
- Altincekic N.; Korn S. M.; Qureshi N. S.; Dujardin M.; Ninot-Pedrosa M.; Abele R.; Abi Saad M. J.; Alfano C.; Almeida F. C. L.; Alshamleh I.; de Amorim G. C.; Anderson T. K.; Anobom C. D.; Anorma C.; Bains J. K.; Bax A.; Blackledge M.; Blechar J.; Böckmann A.; Brigandat L.; Bula A.; Bütikofer M.; Camacho-Zarco A. R.; Carlomagno T.; Caruso I. P.; Ceylan B.; Chaikuad A.; Chu F.; Cole L.; Crosby M. G.; de Jesus V.; Dhamotharan K.; Felli I. C.; Ferner J.; Fleischmann Y.; Fogeron M.-L.; Fourkiotis N. K.; Fuks C.; Fürtig B.; Gallo A.; Gande S. L.; Gerez J. A.; Ghosh D.; Gomes-Neto F.; Gorbatyuk O.; Guseva S.; Hacker C.; Häfner S.; Hao B.; Hargittay B.; Henzler-Wildman K.; Hoch J. C.; Hohmann K. F.; Hutchison M. T.; Jaudzems K.; Jović K.; Kaderli J.; Kalniņš G.; Kaņepe I.; Kirchdoerfer R. N.; Kirkpatrick J.; Knapp S.; Krishnathas R.; Kutz F.; Zur Lage S.; Lambertz R.; Lang A.; Laurents D.; Lecoq L.; Linhard V.; Löhr F.; Malki A.; Bessa L. M.; Martin R. W.; Matzel T.; Maurin D.; McNutt S. W.; Mebus-Antunes N. C.; Meier B. H.; Meiser N.; Mompeán M.; Monaca E.; Montserret R.; Mariño Perez L.; Moser C.; Muhle-Goll C.; Neves-Martins T. C.; Ni X.; Norton-Baker B.; Pierattelli R.; Pontoriero L.; Pustovalova Y.; Ohlenschläger O.; Orts J.; Da Poian A. T.; Pyper D. J.; Richter C.; Riek R.; Rienstra C. M.; Robertson A.; Pinheiro A. S.; Sabbatella R.; Salvi N.; Saxena K.; Schulte L.; Schiavina M.; Schwalbe H.; Silber M.; Almeida M. da S.; Sprague-Piercy M. A.; Spyroulias G. A.; Sreeramulu S.; Tants J.-N.; Ta̅rs K.; Torres F.; Töws S.; Treviño M. Á.; Trucks S.; Tsika A. C.; Varga K.; Wang Y.; Weber M. E.; Weigand J. E.; Wiedemann C.; Wirmer-Bartoschek J.; Wirtz Martin M. A.; Zehnder J.; Hengesbach M.; Schlundt A. Large-Scale Recombinant Production of the SARS-CoV-2 Proteome for High-Throughput and Structural Biology Applications. Front. Mol. Biosci. 2021, 8, 653148. 10.3389/fmolb.2021.653148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo E. J.; Wagner C. Tissue Distribution of Glycine N-Methyltransferase, a Major Folate-Binding Protein of Liver. Proc. Natl. Acad. Sci. U. S. A. 1994, 91 (1), 210–214. 10.1073/pnas.91.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luka Z.; Mudd S. H.; Wagner C. Glycine N-Methyltransferase and Regulation of S-Adenosylmethionine Levels. J. Biol. Chem. 2009, 284 (34), 22507–22511. 10.1074/jbc.R109.019273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Maag H.; Alfredson T. Prodrugs of Nucleoside Analogues for Improved Oral Absorption and Tissue Targeting. J. Pharm. Sci. 2008, 97 (3), 1109–1134. 10.1002/jps.21047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.