

ABSTRACT
ABTL0812 is a first-in-class small molecule with anti-cancer activity, which is currently in clinical evaluation in a phase 2 trial in patients with advanced endometrial and squamous non-small cell lung carcinoma (NCT03366480). Previously, we showed that ABTL0812 induces TRIB3 pseudokinase expression, resulting in the inhibition of the AKT-MTORC1 axis and macroautophagy/autophagy-mediated cancer cell death. However, the precise molecular determinants involved in the cytotoxic autophagy caused by ABTL0812 remained unclear. Using a wide range of biochemical and lipidomic analyses, we demonstrated that ABTL0812 increases cellular long-chain dihydroceramides by impairing DEGS1 (delta 4-desaturase, sphingolipid 1) activity, which resulted in sustained ER stress and activated unfolded protein response (UPR) via ATF4-DDIT3-TRIB3 that ultimately promotes cytotoxic autophagy in cancer cells. Accordingly, pharmacological manipulation to increase cellular dihydroceramides or incubation with exogenous dihydroceramides resulted in ER stress, UPR and autophagy-mediated cancer cell death. Importantly, we have optimized a method to quantify mRNAs in blood samples from patients enrolled in the ongoing clinical trial, who showed significant increased DDIT3 and TRIB3 mRNAs. This is the first time that UPR markers are reported to change in human blood in response to any drug treatment, supporting their use as pharmacodynamic biomarkers for compounds that activate ER stress in humans. Finally, we found that MTORC1 inhibition and dihydroceramide accumulation synergized to induce autophagy and cytotoxicity, phenocopying the effect of ABTL0812. Given the fact that ABTL0812 is under clinical development, our findings support the hypothesis that manipulation of dihydroceramide levels might represents a new therapeutic strategy to target cancer.
Abbreviations: 4-PBA: 4-phenylbutyrate; AKT: AKT serine/threonine kinase; ATG: autophagy related; ATF4: activating transcription factor 4; Cer: ceramide; DDIT3: DNA damage inducible transcript 3; DEGS1: delta 4-desaturase, sphingolipid 1; dhCer: dihydroceramide; EIF2A: eukaryotic translation initiation factor 2 alpha; EIF2AK3: eukaryotic translation initiation factor 2 alpha kinase 3; ER: endoplasmic reticulum; HSPA5: heat shock protein family A (Hsp70) member 5; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; MEF: mouse embryonic fibroblast; MTORC1: mechanistic target of rapamycin kinase complex 1; NSCLC: non-small cell lung cancer; THC: Δ9-tetrahydrocannabinol; TRIB3: tribbles pseudokinase 3; XBP1: X-box binding protein 1; UPR: unfolded protein response.
KEYWORDS: Autophagy, cancer, clinical trial, dihydroceramide, ER stress, UPR
Introduction
ABTL0812 is an alpha-hydroxylated polyunsaturated fatty acid with anti-cancer activity, which is currently in clinical evaluation in a phase 2 trial in patients with advanced endometrial and squamous lung cancer (NCT03366480). ABTL0812 induces AKT-MTORC1 inhibition and autophagy-mediated cancer cell death by promoting TRIB3 expression, a pseudokinase that binds and prevents activation of AKT by upstream kinases PDPK1 (3-phosphoinositide dependent protein kinase 1) and mechanistic target of rapamycin kinase complex 2 (MTORC2) [1–3]. However, the precise mechanism by which ABTL0812 induces cytotoxic autophagy in cancer cells has not been fully elucidated.
Autophagy is a highly conserved cellular process characterized by the self-degradation of intracellular components that are included in double-membrane vesicles known as autophagosomes. Then, the autophagosome contents are transferred to the lysosomes for degradation [4,5]. The role of autophagy in cancer is still unclear as it can act as a tumor promoter or suppressor depending on cancer type and genetic context. Also, the final outcome of autophagy is highly dependent on the intensity and duration of the stimuli, and persistent activation can lead to cytotoxic autophagy [6]. In this regard, autophagy-mediated cell death has emerged as an alternative to kill cancer cells and, therefore, pharmacologic manipulation of autophagy has been proposed to design new anti-cancer therapies [7]. Several anti-tumor molecules activate cancer cell death by autophagy-dependent mechanisms, including salinomycin, resveratrol and tetrahydrocannabinol, by a mechanism that is coupled to the endoplasmic reticulum (ER) stress response [8–10].
Here, we have investigated the mechanism by which ABTL0812 induces autophagy-mediated cytotoxicity in cells. We found that ABTL0812 induced an increase in cellular levels of long-chain dihydroceramides, which caused ER stress and activation of the UPR that ultimately lead to cytotoxic autophagy in cancer cells. Interestingly, we have validated the expression of mRNAs of two ER stress-related genes (TRIB3 and DDIT3/CHOP) in blood as pharmacodynamic biomarkers for ABTL0812 therapy of patients enrolled in the ongoing clinical trial. Overall, our findings support the hypothesis that alteration of sphingolipid metabolism could be used as an effective anti-cancer therapeutic strategy.
Results
ABTL0812 induces ER stress-mediated autophagy
We previously showed that ABTL0812 induces autophagy-mediated death in a panel of cancer cells [1–3]. One of the hallmarks of autophagy is the conjugation of the soluble form of MAP1LC3B/LC3 with phosphatidylethanolamine and conversion to the autophagosomal membrane-associated form (MAP1LC3B-II). Treatment of human cancer cells with ABTL0812 resulted in increased MAP1LC3B-II levels (Figure 1(a)). Furthermore, pharmacological blockade of lysosomal content degradation with either cathepsin inhibitors or the vacuolar-type H+-ATPase inhibitor bafilomycin A1 enhanced MAP1LC3B-II upon ABTL0812 treatment in pancreatic adenocarcinoma MiaPaca2 and lung adenocarcinoma A549 cells (Figure 1(a,b)), indicating that this compound induces dynamic autophagy. Of note, ABTL0812 induced autophagy-mediated death in these cancer cells, since ATG5 silencing resulted in impaired ABTL0812-induced cell death (Figure 1(c)).
Figure 1.

ABTL0812 induces ER stress in cancer cell lines. (a, b) ABTL0812 induces dynamic autophagy. Cells were preincubated 3 h with vehicle or lysosomal protease inhibitors E64d (10 µmol/L) and pepstatin A (PA, 10 µg/mL) (a) or with inhibitor (50 nM) of the vacuolar-type ATPase, bafilomycin A1 (BafA) (b) before treatment with ABTL0812 for 24 h. Levels of lipidated and non-lipidated MAP1LC3B proteins were monitored by immunoblotting. (c) ABTL0812 induces autophagy-mediated cancer cell death. Effect of ABTL0812 treatment (48 h) in viability of MiaPaca2 or A459 stable cell lines transfected with control shRNA (shC) or ATG5-selective shRNA (shATG5). Right panels show the corresponding immunoblots. (d) AKT or MTOR inhibition does not result in the induction of autophagy. MiaPaca2 cells were treated with 25 µmol/L AZD5363 (AKT inhibitor), 0.1 µmol/L AZD2014 (MTOR inhibitor), 10 µmol/L everolimus (MTORC1 inhibitor) or 100 µmol/L ABTL0812 for 24 h. Expression of RPS6, p-RPS6, lipidated and non-lipidated MAP1LC3B and ACTB proteins were determined by immunoblotting. Similar results were obtained in four separate experiments. (e–h) ABTL0812 induces ER stress in cancer cells. (e) Cells were treated with 100 µmol/L ABTL0812 or 200 nmol/L Brefeldin-A (BrefA), total RNA isolated and cDNA synthesized by RT-PCR. XBP1 splicing was determined by PCR using primers that amplify both spliced (XBP1 s) and unspliced (XBP1 u) mRNA species. Similar results were obtained in four separate experiments. (f) MiaPaca2 and A549 cells were treated with 100 µmol/L ABTL0812, then lysed. The levels of p-EIF2A were monitored by immunoblotting. Similar results were obtained in three separate experiments. (g) Cells were treated with 100 µmol/L ABTL0812, then lysed. The levels of ER stress markers HSPA5, ATF4, DDIT3 and TRIB3 were monitored by immunoblotting. The marker of autophagy MAP1LC3B was also analyzed. Similar results were obtained in four separate experiments. (h) MiaPaca2 and A549 cells were treated with 100 µmol/L ABTL0812 for 24 h, pelleted, total RNA isolated, and ATF4, DDIT3 and TRIB3 mRNA levels were analyzed by RT-qPCR. Each value is the mean ± SD of three different experiments. **, P < 0.005; ***, P < 0.001, Student’s t-test
Upregulation of TRIB3 pseudokinase is one of the hallmarks of the anti-tumoral action of ABTL0812, causing the inhibition of the AKT-MTORC1 axis in cancer cells [1]. In this regard, stable TRIB3 knockdown A549 and MiaPaca2 cells showed impaired toxicity in response to ABTL0812 (Figure S1). However, pharmacological blockade of the AKT-MTORC1 axis by itself was not enough to induce a significant stimulation of autophagy in MiaPaca2 cells (Figure 1(d)). This observation led us to hypothesize that, together with the blockade of AKT-MTORC1 axis, ABTL induces autophagy-mediated cancer cell death via additional mechanisms. Since TRIB3 is an ER stress-related gene and this cellular process has been implicated in autophagy stimulation, we next investigated whether ER stress plays a role in ABTL0812-induced autophagy in MiaPaca2 and A549 human cancer cell lines.
Different situations, including the accumulation of misfolded proteins, the emptying of ER Ca2+ stores or the increased accumulation of certain lipids, can affect the normal functioning of the ER leading to ER stress. The UPR is activated to restore ER and cellular homeostasis. It relies on a specific signaling network that is controlled by three transmembrane ER stress protein sensors, namely ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1), EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3) and ATF6 (activating transcription factor 6) [11]. Therefore, to investigate whether ABTL induced ER stress in cancer cells, we asked whether this compound modified the activity of these ER stress sensors. In response to ER stress, ERN1 excises a 26-nucleotide intron of the of XBP1 (X-box binding protein 1) RNA, resulting in an unconventional mRNA spliced form [12]. ABTL0812 induced the splicing of XBP1 after 2 h (A549 cells) or 4 h (MiaPaca2 cells) treatment (Figure 1(e)). We also observed unconventional XBP1 splicing after 24–36 h treatment, indicating that ABTL0812 induced a sustained ER stress in these cells (Figure S2). Another hallmark of ER stress is the phosphorylation of EIF2A initiation factor at Ser51, which results in attenuation of general protein synthesis while enhancing ATF4 mRNA translation and activation of DDIT3 and TRIB3 expression [11]. ABTL0812 treatment resulted in increased phosphorylation of EIF2A (Figure 1(f)), as well as on the expression of HSPA5/GRP78/BiP, ATF4, DDIT3 and TRIB3 (Figure 1(g)). Interestingly, 1 h treatment of ABTL0812 induced expression of ATF4 and DDIT3 UPR markers without activating autophagy, indicating that ER stress preceded autophagy (Figure 1(g)). Also, RT-qPCR analysis showed an increase in ATF4, DDIT3 and TRIB3 mRNA levels in response to ABTL0812 (Figure 1(h)). ABTL0812-treated cells also presented dilated ER [10], as shown by electron microscopy (Figure S3(a)) and immunostaining of the ER luminal marker protein disulfide isomerase (Figure S3(b)). Moreover, ABTL0812 induced hallmarks of ER stress in vivo, as tumors from MiaPaca2 and A549 xenograft mice treated with ABTL0812 showed increased ATF4 and HSPA5 expression, respectively (Figure 2(a)). Likewise, immunohistochemical analysis of these tumors also showed augmented DDIT3 expression (Figure 2(b)). Overall, these results show that ABTL0812 induces ER stress in cancer cells.
Figure 2.

ABTL0812 induces ER stress in human lung and pancreatic xenografts. Athymic nude mice injected with A549 or MiaPaCa2 cells were treated for 33 d with vehicle or 120 mg/kg ABTL0812 by oral gavage. (a) Tumor xenograft samples were collected and expression of ATF4 or HSPA5 ER stress markers were analyzed by immunoblotting. Left graphs show growth curves of tumors referred to initial values (fold-change ± SEM). Right histograms show the corresponding quantifications referred to ACTB. (b) Expression of DDIT3 protein from MiaPaca2 and A549 tumors was also evaluated by immunohistochemical analysis. Scale bars, 100 µm. Right histogram shows the corresponding quantification. *, P < 0.05; **, P < 0.005; ***, P < 0.001 from vehicle-treated cells, Student’s t-test
To determine whether ABTL0812 induces autophagy via ER stress-mediated EIF2A-ATF4 axis, we used mouse embryonic fibroblast (MEF) EIF2AS51A knock-in cells. This EIF2A mutant cannot be phosphorylated, and therefore ATF4 expression is blocked in these cells. ABTL0812 did not induce ATF4 or TRIB3 expression in EIF2AS51A knock-in MEF cells, compared with wild type cells (Figure 3(a)). EIF2A could be mainly phosphorylated at Ser51 by EIF2AK3 in response to ER stress, or by the EIF2AK4/GCN2 (eukaryotic translation initiation factor 2 alpha kinase 4) kinase in response to amino acid deficiency [11]. Silencing of EIF2AK3 with two different siRNAs, but not silencing of EIF2AK4, resulted in a significant decrease in the ATF4 levels induced by ABTL0812 treatment in MiaPaca2 and A549 cells (Figure 3(b,c)). Interestingly, silencing (siRNA) of ATF4 resulted in a dramatically decreased ABTL0812-induced expression of TRIB3 and MAP1LC3B-II (Figure 3(d)). Overall, these results indicated that activation of EIF2AK3-EIF2A-ATF4 axis mediates TRIB3 upregulation and the subsequent induction of autophagy triggered by ABTL0812.
Figure 3.
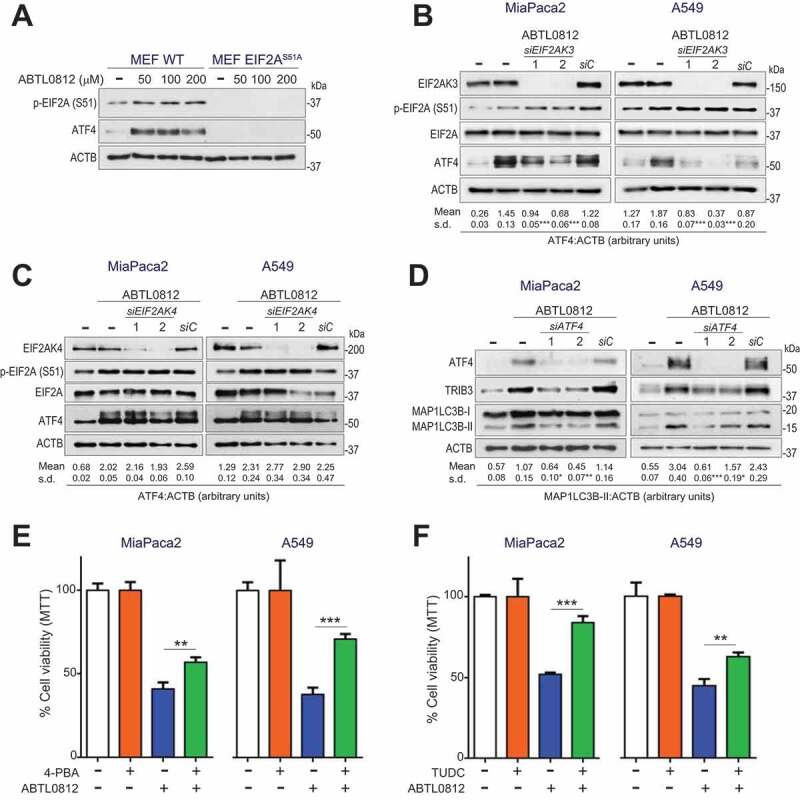
ER stress mediates ABTL0812-induced autophagy and cytotoxicity in cancer cells. (a–d) EIF2A-ATF4 axis mediates in ABTL0812-induced autophagy. (a) ABTL0812 does not induce ATF4 in EIF2AS51A knock-in cells. Levels of phosphorylated EIF2A and ATF4 were monitored by immunoblotting. Similar results were obtained in three separate experiments. (b,c) EIF2AK3 silencing, but not EIF2AK4 silencing, prevents ABTL0812-induced ATF4 expression. Cells transfected with control siRNA (siC) or EIF2AK3- or EIF2AK4-selective siRNAs (siEIF2AK3 in (b) or siEIF2AK4 in (c)) were treated with 100 µmol/L ABTL0812 for 4 h, lysed and the indicated proteins were monitored by immunoblotting. The corresponding quantification of MAP1LC3B-II levels referred to ACTB are indicated under the panels. Results representative of three separate experiments. (d) ATF4 silencing prevents ABTL0812-induced autophagy. Cells were transfected with scramble siRNA (siC) or two different ATF4-selective siRNAs, and then treated with 100 µmol/L ABTL0812 for 18 h. Levels of ATF4, TRIB3, MAP1LC3B and ACTB protein expression were analyzed by immunoblotting. The corresponding quantification of MAP1LC3B-II levels referred to ACTB are indicated under the panels. Results representative of four separate experiments. (e,f) Pharmacological inhibition of ER response with molecular chaperones impairs ABTL0812-induced cytotoxicity. Cells were preincubated 3 h with 1 mmol/L 4-BPA (e) or 150 µmol/L sodium tauroursodeoxycholate (TUDC) (f) before treatment with 30 µmol/L ABTL0812 for 48 h. Cell viability was determined by MTT assay. Each value is the mean ± SD of three different experiments. *, P < 0.05; **, P < 0.005; ***, P < 0.001 from ABTL0812-treated cells, one-way ANOVA Bonferroni
To set the role of ER stress on the cytotoxicity induced by ABTL0812, we next treated cells with two different chemical chaperones, 4-phenylbutyrate (4-PBA) and tauroursodeoxycholate (TUDC), which facilitate protein folding and therefore attenuate ER stress [13]. 4-PBA or TUDC significantly mitigated the cytotoxicity induced by ABTL0812 in A549 and MiaPaca2 cells (Figure 3(e,f)). Furthermore, persistent ER stress activation by the inhibitor of ER-Golgi protein transport Brefeldin-A resulted in dynamic autophagy and cytotoxicity, showing that sustained ER stress could jeopardize the viability of these cells (Figure S4).
Blood DDIT3 and TRIB3 mRNAs are pharmacodynamic markers for ABTL0812 in NSCLC and endometrial cancer patients
ABTL0812 is currently in phase 2 clinical trials in patients with advanced endometrial and squamous non-small cell lung (NSCLC) cancers, in combination with paclitaxel and carboplatin chemotherapy (NTC03366480). Given the robust activation of ER stress observed in cells treated with ABTL0812, we decided to monitor levels of the UPR markers DDIT3 and TRIB3 in blood from patients enrolled in the clinical trials to test them as potential pharmacodynamic biomarkers. We measured TRIB3 and DDIT3 mRNA levels by RT-qPCR assay. Firstly, we set up experimental conditions using peripheral blood mononuclear cells (Figure S5). However, we could not obtain enough amount of mRNA to perform RT-PCR analysis for all the cases. This prompted us to perform RT-qPCR analysis directly from whole blood RNA samples.
Figure 4(a) shows the overall results for 14 patients enrolled in the clinical trial: TRIB3 and DDIT3 mRNA levels were significantly increased after 8 h of ABTL0812 intake and remained augmented at day 7 and 28. Interestingly, 13 out of 14 patients showed a significant increase in either DDIT3 or TRIB3 (or both) mRNA levels. Specifically, 8 patients showed increased levels of both DDIT3 and TRIB3 mRNAs, whereas five patients showed an increase in either DDIT3 or TRIB3 mRNA (Figure 4(b)). These results show that ABTL0812 activated ER stress and UPR in human patients, and strongly support the use of blood DDIT3 and TRIB3 expression as surrogated pharmacodynamic markers.
Figure 4.

ABTL0812 treatment induces TRIB3 and DDIT3 mRNA levels in blood from patients enrolled in phase 2 clinical trial (NCT03366480). (a) Blood TRIB3 and DDIT3 mRNA levels from patients enrolled in phase 2 clinical trial (N = 14 for TRIB3; N = 12 for DDIT3). Values represented in the scatter plot correspond to the mean ± SEM of 2−△△Ct values. Values show fold-changes of mRNA levels, referred to as “0” value. (b) Whole blood TRIB3 and DDIT3 mRNA levels of nine patients before (0) or after 1,300 mg t.i.d. ABTL0812 oral daily treatment as monotherapy (8 h and 7 d) or in combination with chemotherapy (28 d). mRNA levels were evaluated by RT-qPCR. Each value is the mean ± SD of three technical replicates. Patients 6 and 7 showed increased TRIB3 mRNA levels only, whereas patients 8 and 9 showed increased DDIT3 mRNA levels only. Values show fold-changes of mRNA levels, referred to as “0” value. Statistical analyses were performed using the △△Ct values. A one-way ANOVA Tukey test was applied. *, P < 0.05; **, P < 0.005; ***, P < 0.001 compared to day 0 sample
ABTL0812 induces the accumulation of cellular long-chain dihydroceramides in cancer cells and tumor xenografts
Sphingolipids are bioactive molecules synthesized in the ER from non-sphingolipid precursors. Sphingolipids are implicated in the regulation of ER stress [10,14], autophagy [15] and cell death [16,17], and it has been reported that several compounds induce autophagy-mediated cancer cell death by altering sphingolipid metabolism, such as △9-tetrahydrocannabinol [18,19] or resveratrol [9,15]. To explore if this was also the case for ABTL0812, we next quantified changes on sphingolipids in response to ABTL0812. Pancreatic cancer Miapaca2 cells were treated at different times, and sphingolipid content was determined by UPLC-MS analysis. ABTL0812 induced an increase in cellular dihydroceramides after 6 h (2.9-fold increase), without affecting ceramide levels (Figure 5(a,b)). Longer (24 h) incubation resulted in changes in both dihydroceramides and ceramides levels, but the increase in dihydroceramides (5.3-fold) was larger than that observed for ceramides (1.6-fold). ABTL0812 did not induce changes in levels of other sphingolipids, such as sphingomyelins, glucosylceramides or lactosylceramides (Figure S6). Of note, since ABTL0812 induced ER stress and autophagy at 6 h (Figure 1(e)), these results suggest an implication of dihydroceramides, but not ceramides, in triggering those cellular processes. Detailed analysis revealed that ABTL0812 (6 h) specifically induced an increase of long-chain dihydroceramides C16 (3.3-fold), C22 (2.3-fold), C24 (2.4-fold) and C24:1 (3.2-fold) (Figure 5(c)). Higher increases on these dihydroceramides were observed after 24 h treatment (5.9-fold, 3.4-fold, 5.7-fold and 5.6-fold for C16, C22, C24 and C24:1, respectively). Importantly, we also observed similar changes in long-chain dihydroceramides in endometrial cancer Ishikawa cells treated with ABTL0812 (Figure 5(d)). Finally, to investigate the relevance of these observations in vivo, we performed sphingolipidomic analysis of Miapaca2 tumor xenografts. ABTL012 induced a significant increase in tumor dihydroceramide levels, which correlated with reduced tumor growth (Figure 5(e)). Specifically, ABTL0812 induced a significant accumulation of C24 and C24:1 species (Figure 5(f)). Detailed amounts for each sphingolipid determination are given in Supplementary Tables.
Figure 5.

ABTL0812 induces accumulation of long-chain dihydroceramides in cancer cells and tumors. (a) Scheme representing the pathway of de novo sphingolipid biosynthesis. Serine palmitoyltransferase (SPT) catalyzes the condensation of palmitoyl-CoA and serine to produce 3-ketosdihydrosphingosine. KDSR (3-ketodihydrosphingosine reductase) reduces 3-ketodihydrosphingosine to dihydrosphingosine. Then, ceramide synthases (CERSs) convert dihydrosphingosine into the different molecular species of dihydroceramides, which are transformed to ceramides by the insertion of a 4,5-trans double bond catalyzed by the enzyme DEGS1. (b) ABTL0812 effect on levels of total cellular ceramides and dihydroceramides. MiaPaca2 cells were treated with 100 µmol/L ABTL0812 for the times indicated, pelleted and lipid content extracted and analyzed by UPLC-TOF-Ms. Data are expressed in pmol of sphingolipid per 106 cells. Each value is the mean ± SD of three different determinations. (c) Levels of molecular species of dihydroceramides at 6 h (left histogram) and 24 h of ABTL0812 treatment (right histogram) in MiaPaca2 cells. Each value is the mean ± SD of three different determinations. (d) Levels of dihydroceramides in endometrial cancer Ishikawa cells after 6 h of ABTL0812 treatment. Total dihydroceramides and molecular species are shown in the left and right panels, respectively. Each value is the mean ± SD of three different determinations. (e,f) ABTL0812 effect on levels of dihydroceramides in tumors. *, P < 0.05; **, P < 0.005; ***, P < 0.001 from vehicle-treated cells
Increased levels of cellular dihydroceramides induce ER stress, autophagy and cytotoxicity
Accumulation of dihydroceramides activates ER stress and autophagy, as well as cytotoxicity, in glioblastoma and gastric cancer cells [15,20]. Given our results, we next investigated whether high levels of dihydroceramides could mimic the cellular response to ABTL0812. To do so, we used a dideuterated dihydroceramide analog, d2C8dhCer, which is desaturated to ceramide at a much slower rate than natural dihydroceramides due to the primary isotope effect. Therefore, incubation of cells with d2C8dhCer results in the accumulation of dideuterated dihydroceramides that are eventually transacylated to long-chain species [21]. Treatment of MiaPaca2 and A549 cells with d2C8dhCer resulted in enhanced expression of ATF4 and TRIB3 ER stress markers, as well as in the accumulation of the lipidated MAP1LC3B-II form (Figure 6(a)) and cytotoxicity (Figure 6(b)). These results suggest that cellular dihydroceramides might mediate in the ER stress, autophagy and cytotoxicity observed in response to ABTL0812.
Figure 6.
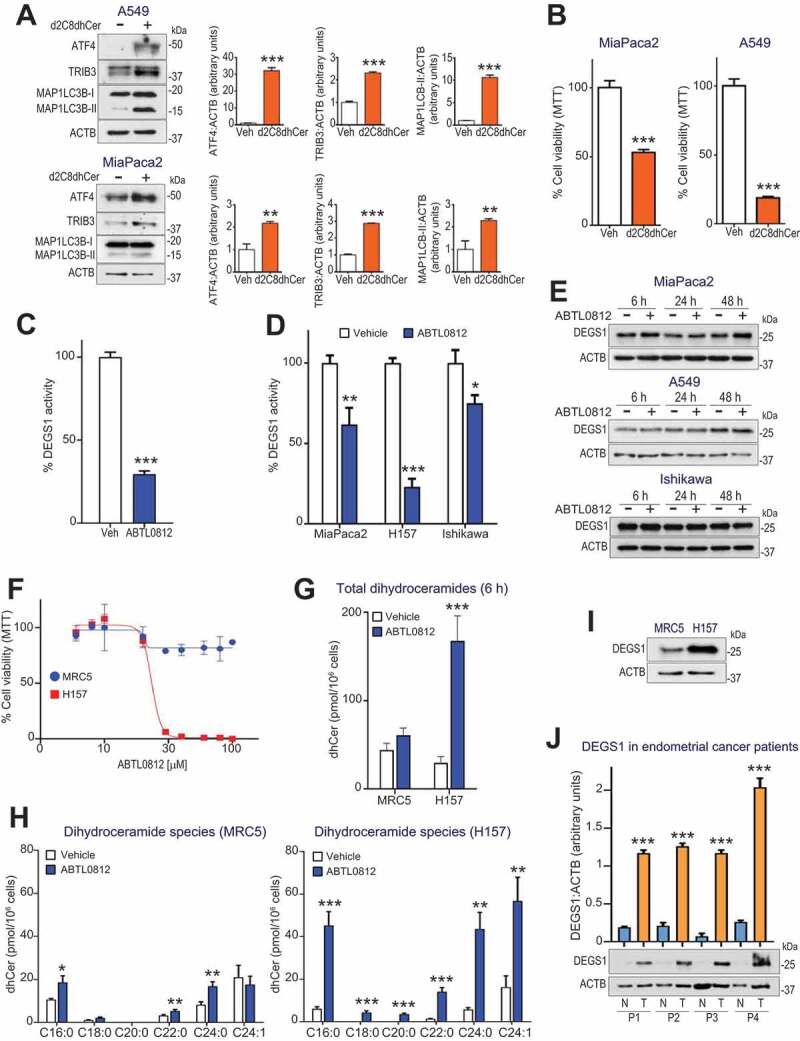
Dihydroceramide accumulation by ABTL0812 treatment induces ER stress, autophagy and selective cancer cell death. (a) Cells were treated with 25 µmol/L of the dideuterated dihydroceramide analog d2C8dhCer for 48 h, then lysed. The levels of ER stress markers ATF4 and TRIB3, as well as MAP1LC3B lipidation, were monitored by immunoblotting. Histograms show quantification of ATF4, TRIB3 and MAP1LC3B-II levels normalized to ACTB levels and represent the mean fold-change relative to vehicle-treated cells. (b) MiaPaca2 and A549 cells were treated 48 h with 25 µmol/L of d2c8dhCer, and cell viability was determined by MTT assay. Each value is the mean ± SD of two different experiments performed in triplicates. (c,d) ABTL0812 treatment induces DEGS1 inhibition. (c) A549 cell lysates were treated with vehicle or 100 µmol/L ABTL0812 for 4 h. (d) Cultured MiaPaca2 (pancreatic), H157 (SNSCLC) and Ishikawa (endometrial) cancer cells were treated 6 h with vehicle or 100 µmol/L ABTL0812. DEGS1 activity was determined as described in the Materials and Methods Section using DHCerC6NBD as substrate. Data expressed in percentage of the CerC6NBD pick area. Each value is the mean ± SD of three different determinations. (e) DEGS1 levels visualized by immunoblotting. (f) Selectivity of ABTL0812 cytotoxic effect for cancer cells. Human lung fibroblast MRC5 and squamous NSCLC H157 cells were treated with ABTL0812 for 48 h, and cell viability determined by MTT assay. Results representative of three separate experiments. (g,h) ABTL0812 does not induce accumulation of dihydroceramides in a non-tumoral cell line. (g) Levels of total dihydroceramides in MRC5 fibroblasts and H157 cancer cells, after 6 h of ABTL0812 treatment. (h) Levels of molecular dihydroceramide species. Each value is the mean ± SD of three different determinations. (i) DEGS1 protein levels (immunoblot) in MRC5 and H157 cells. (j) Human endometrial tumors express higher levels of DEGS1 protein than adjacent non-tumoral tissue. Immunoblot analysis of samples (tumor area [T] and non-tumor area [N]) from 4 different patients with endometrial cancer. *, P < 0.05; **, P < 0.005; ***, P < 0.001 from vehicle-treated cells
ABTL0812 increases cellular levels of dihydroceramides by impairing desaturase-1 activity
Last step of de novo synthesis of ceramides is catalyzed by DEGS1/dihydroceramide desaturase (delta [4]-desaturase, sphingolipid 1), which introduces a 4,5-trans-double bond in the sphingolipid backbone of dihydroceramides to generate ceramides. Therefore, we next asked whether the increase in the levels of dihydroceramides observed was due to an impairment of DEGS1 activity. Interestingly, we also observed enhanced levels of cellular dihydrosphingomyelins in response to ABTL0812 (Figure S6), a feature that is always observed after DEGS1 inhibition [22]. We first performed an in vitro assay treating cell lysates with ABTL0812 and monitoring DEGS1 activity by HPLC using DHCerC6NBD as substrate. DEGS1 activity from cell lysates was impaired by ABTL0812 (Figure 6(c)). We also performed parallel experiments in which cells were treated with ABTL0812, and DEGS1 activity was measured. ABTL0812 treatment resulted in reduced DEGS1 activity in MiaPaca2 (pancreatic), Ishikawa (endometrial) and H157 (squamous lung NSCLC) cancer cells (Figure 6(d)) without affecting DEGS1 protein expression levels (Figure 6(e)).
In agreement with the clinical data showing low toxicity in patients treated with ABTL0812, this compound shows selective cytotoxic effect for tumor cells. We previously reported low cytotoxic effect in primary astrocyte cultures, compared with glioblastoma cell lines [1,2]. We confirmed these observations using a model of lung cancer: compared with squamous NSCLC H157 cells, human lung fibroblast cell line MRC-5 were resistant to ABTL0812 treatment (Figure 6(f)). Moreover, ABTL0812 treatment induced an increase in dihydroceramide levels in tumoral H157 cells but not in MRC-5 fibroblasts (Figure 6(g,h)), which correlated with a higher expression of the DEGS1 protein in tumor cells (Figure 6(i)). Moreover, a preliminary immunoblot analysis of samples from patients with endometrial cancer showed enhanced DEGS1 expression in tumor tissue compared with non-tumoral surrounding tissue (Figure 6(j)).
Thus far, our results suggested that ABTL0812 treatment impaired DEGS1 activity, resulting in the accumulation of dihydroceramides that ultimately leads to activation of ER stress and autophagy preceding cancer cell death. If this hypothesis is correct, DEGS1 might play an important role in the mechanism of the anti-tumoral action of ABTL0812. Therefore, we undertook experiments to test whether pharmacological modulation of DEGS1 activity reproduced the cellular signature of ABTL0812: i) accumulation of long-chain dihydroceramides; ii) ER stress induction; iii) activation of dynamic autophagy; iv) autophagy-mediated cell death; and, v) selectivity for cancer cells. Treatment of cancer cells with the specific DEGS1 inhibitor GT11 [23] induced similar changes on the sphingolipid profile than those produced by ABTL0812 (Figure 7(a)), and resulted in enhanced expression of ATF4 and TRIB3 proteins, as well as lipidated MAP1LC3B-II form, at similar levels than treatment with ABTL0812 (Figure 7(b)). Importantly, DEGS1 inhibition induced dynamic autophagy (Figure 7(c,d)) and cytotoxicity (Figure S7) in A549 and MiaPaca2 cells, similar to that reported for other tumoral cells such as U87 human glioblastoma cells [20]. Furthermore, ATF4 silencing dramatically decreased GT11-induced autophagy (Figure 7(e)), as it happened for ABTL0812 treatment (Figure 3(d)). We also providedgenetic evidence showing that DEGS1 inhibitor GT11 induced autophagy-mediated cancer cell death. Autophagy-deficient atg5−/- transformed MEF cells were more resistant to GT11-induced cell death, compared to wild type MEF cells (Figure S8). Finally, A549 cancer cells were more sensitive to DEGS1 inhibition, compared to lung fibroblast MRC-5 cells (Figure 7(f)). Together, these results suggest an important role of DEGS1 activity in the anti-tumor action of ABTL0812.
Figure 7.
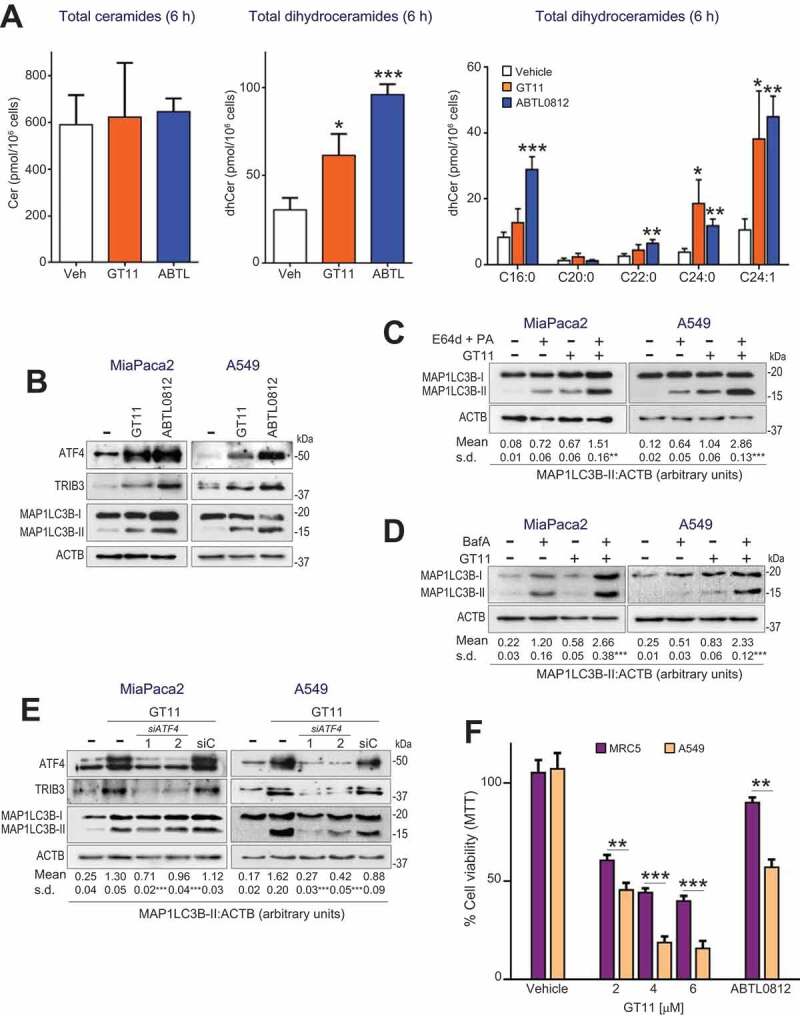
DEGS1 specific inhibitor GT11 induces accumulation of dihydroceramides, ER stress, dynamic autophagy and selective cancer cell death. (a) MiaPaca2 cells were treated for 6 h with vehicle, 6 µmol/L GT11, or 100 µmol/L ABTL0812, and total ceramides, dihydroceramides and molecular species of dihydroceramides were quantified. Each value is the mean ± SD of three different experiments. (b–e) DEGS1 inhibition induces ER stress-mediated dynamic autophagy. (b) cCells were treated 48 h with 5 µmol/L GT11 and levels of ER stress markers ATF4 and TRIB3 and MAP1LC3B lipidation analyzed by immunoblotting. Similar results were obtained in three separate experiments. (c,d) Cells were preincubated 3 h with vehicle or lysosomal protease inhibitors E64d (10 µmol/L) and pepstatin-A (PA, 10 µg/mL) (c), or 50 nm bafilomycin-A (BafA) (d) before treatment with 6 µmol/L GT11 for 24 h. Cells were lysed and MAP1LC3B lipidation was visualized by immunoblotting. Similar results were obtained in three different experiments. (e) ATF4 silencing prevents GT11-induced autophagy. Cells were transfected with scramble siRNA (siC) or two different ATF4-selective siRNAs, and then treated with 100 µmol/L ABTL0812 for 18 h. Levels of ATF4, TRIB3, MAP1LC3B and ACTB protein expression were analyzed by immunoblotting. The corresponding quantification of MAP1LC3B-II levels referred to ACTB are indicated under the panels. Results representative of two separate experiments. (f) Non-tumoral cells show lower sensitivity to DEGS1 inhibitor GT11. Human lung fibroblast MRC5 and squamous NSCLC H157 cells were treated with the 30 µM ABTL0812 or with the indicated concentration of GT11 for 48 h, and cell viability was determined by MTT assay. Results representative of three separate experiments. *, P < 0.05; **, P < 0.005; ***, P < 0.001 from vehicle-treated cells
Dihydroceramide accumulation collaborates with MTORC1 inhibition to promote autophagy and cancer cell death
ABTL0812 induces inhibition of the AKT/MTORC1 axis in cancer cells [1]. Given the results presented here showing that ABTL0812 induces ER stress by uprising cellular long-chain dihydroceramides species, we next addressed the question of whether dihydroceramides collaborate with MTORC1 inhibition to activate autophagy and cytotoxicity. MTORC1 inhibitor everolimus enhanced the effect of GT11 on the expression of lipidated MAP1LC3B-II, thus resulting in higher autophagy (Figure 8(a)). More importantly, DEGS1 and MTORC1 inhibition synergized to promote death in A549 and MiaPaca2 cells (Figure 8(b), see combination index values lower than 1).
Figure 8.
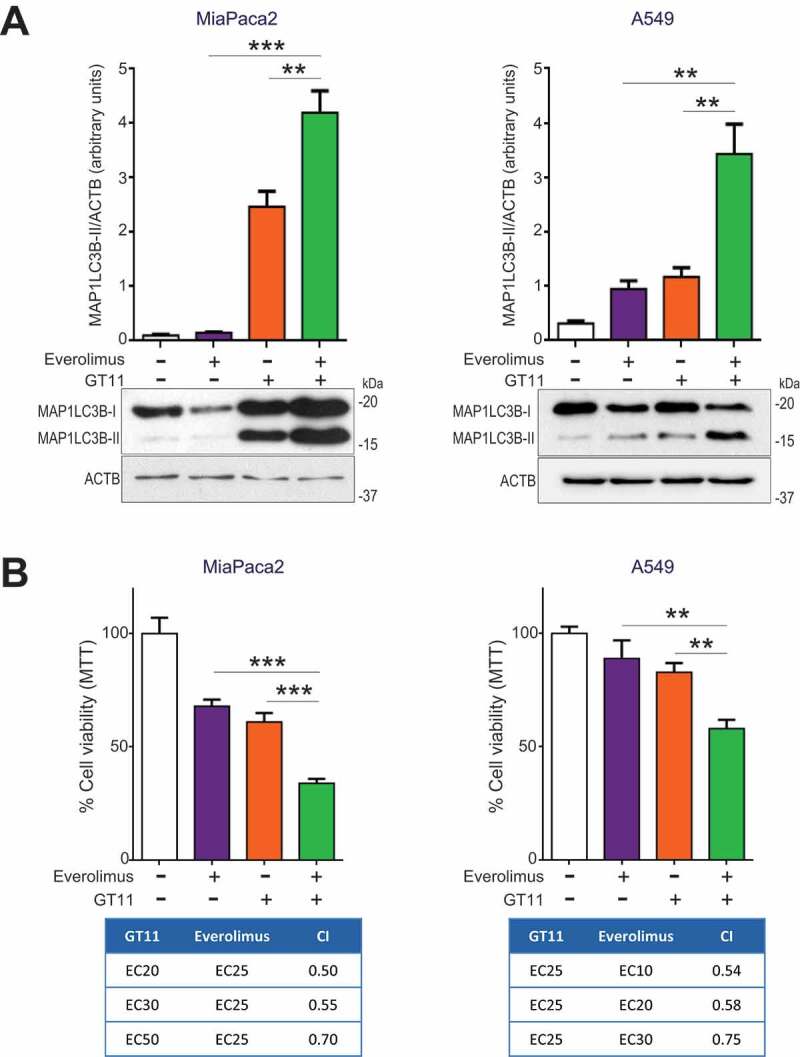
DEGS1 inhibition collaborates with MTORC1 inhibition to promote autophagy and cancer cell death. (a) MiaPaca2 (left histogram) and A549 (right histogram) cells were treated with a combination of the DEGS1 inhibitor GT11 and the MTORC1 inhibitor everolimus for 24 h. Cell viability was determined by MTT assay. Each value is the mean ± SD of three different experiments. Lower tables show the combination index (CI) analysis for different concentrations of GT11 and everolimus. CIs were obtained using the indicated doses of inhibitors and the Compusyn software (see Methods Section) (CI > 1, antagonism; CI = 1, summary effect; CI < 1, synergism). Similar results were obtained in three separate determinations. (b) MiaPaca2 (left) and A549 (right) cells were treated with a combination of the DEGS1 inhibitor GT11 and the MTORC1 inhibitor everolimus for 24 h, then lysed. MAP1LC3B lipidation was visualized by immunoblotting. MAP1LC3B-II levels were normalized to ACTB levels and estimated by densitometric units. Similar results were obtained in three separate experiments. **, P < 0.005; ***, P < 0.001 from everolimus and GT11-treated cells, one-way ANOVA
Collectively, the results presented here suggest that ABTL0812 exert its anti-tumoral action by activating ER stress through dihydroceramide accumulation and inhibiting MTORC1, two signaling pathways that might cooperate to promote autophagy-mediated cancer cell death.
Discussion
Here, we report that ABTL0812 induces robust and sustained ER stress in cancer cells, leading to the activation of autophagy and cell death. Of note, ABTL0812 induced ER stress in a panel of different human cancer cell lines, including pancreatic, endometrial, cholangiocarcinoma, glioblastoma, and adenocarcinoma and squamous NSCLC cancers (Figure S9). Importantly, we found that ER stress precedes autophagy upon ABTL0812 treatment (Figure 1(g)) and provided evidence showing that silencing the UPR genes EIF2AK3, EIF2A or ATF4 results in impaired autophagy (Figure 3). The ER stress response is activated to protect cells from ER stress-induced damage. However, if cells do not recover from the stress, this response can lead to cell death. Thus, the balance between intensity (and duration) of the ER stress and the UPR dictates cell fate [24]. In this sense, persistent UPR activity correlates with sustained increased levels of the DDIT3 transcription factor, a critical executor of the pro-death arm of the UPR [24,25]. This notion seems to be the case for ABTL0812, since it induced XBP1 splicing and DDIT3 overexpression after 36 h of treatment, and it also induced apoptosis [2] or necrosis [1], depending on the cancer cell model.
We previously showed that tumor cells are more sensitive to ABTL0812 than non-tumoral cells. For instance, concentrations of ABTL0812 that exert a cytotoxic effect in glioblastoma LN-18, cholangiocarcinoma EGi-1 or endometrial Hec-1A cells do not affect the viability of the corresponding non-tumor cells (primary astrocytes, cholangiocytes or endometrial epithelial cells) [1,2]. Here, we show that ABTL0812 induced cytotoxicity in H157 squamous NSCLC cells, but not in MRC5 human lung fibroblasts (Figure 6(f)). Activation of the ER stress pathway might contribute to the selectivity for tumor cells showed by ABTL0812. Cancer cells have evolved to use the UPR to survive the ER stress induced by the hostile conditions of the tumor microenvironment (hypoxia, low glucose, intracellular acidification, etc.) To do so, cancer cells express elevated levels of certain genes of the UPR, such as the chaperone HSPA5 and the ATF6 transcription factor [26–28], which correlate with increased tolerance to ER stress and with poor tumor prognosis [29]. The use of drugs that exacerbate basal ER stress of cancer cells can result in an overpass of the cytoprotective effect of the UPR, leading to activation of the pro-apoptotic arm (DDIT3) and cell death. This is the case for some drugs such as salinomycin [8], eeyarestatin 1 [30] or Δ9-tetrahydrocannabinol (THC) [18], which exert their anti-tumoral action by inducing ER stress-mediated apoptotic cell death. On the contrary, non-tumoral cells show negligible levels of ER stress and, therefore, possess a broader margin to resist stress-induced cytotoxicity [31]. In agreement with this, ABTL0812 induced ER stress in both tumoral and primary non-tumoral cells at the concentrations tested, but only exerted cytotoxicity in tumor cells (Figure 7(f)). Furthermore, we detected increased DDIT3 and TRIB3 mRNA levels in blood cells from patients treated with ABTL0812 (Figure 4). Interestingly, these blood cells were healthy and viable regardless of having high expression levels of UPR genes. In agreement with this observation, patients treated with ABTL0812 did not show a decrease in peripheral blood cell counts during previous clinical phase 1 [32] or current clinical phase 2 trials (NTC03366480). Further research is needed to clarify the exact role of the intensity of UPR in the selectivity of ABTL0812 for tumor cells.
Elevated levels of UPR-related genes have also been implicated in chemoresistance of tumor cells [33], and drugs that exacerbate the UPR can exhibit contradictory effects by either sensitizing cancer cells to chemotherapy or increasing their resistance to chemotherapy. For instance, the ER stressor tunicamycin sensitizes ovarian cancer cells to cisplatin and carboplatin [34,35], but it reduces the chemotherapy-induced apoptosis in hepatocellular carcinoma cells [35,36]. Interestingly, ABTL0812 synergizes with carboplatin and paclitaxel to reduce tumor growth of patient-derived xenografts of endometrial cancer [2], and of human squamous lung cancer xenograft models [3]. Therefore, it is likely that the aggravation of UPR observed in cancer cells treated with ABTL0812 could lead to potentiate the anti-tumoral effect of chemotherapy in those in vivo tumor models.
Notably, we propose the expression of TRIB3 and DDIT3 mRNAs in the blood as pharmacodynamic biomarkers for ABTL0812 therapy of patients enrolled in the ongoing phase 2 clinical trial (Figure 4 and S5). Future studies will determine if the expression of these mRNAs also correlates with the efficacy of the ABTL0812 treatment. To our knowledge, this is the first time that ER stress markers TRIB3 and DDIT3 are reported to change in human blood in response to any drug treatment. Our results suggest that the quantification of the levels of UPR gene products in the blood could be an option to monitor the biological activity of compounds that activate ER stress in humans.
In this work, we show for the first time that ABTL0812 treatment of human pancreatic, endometrial, adenocarcinoma and squamous NSCLC cancer cells resulted in impaired DEGS1 activity (Figure 6(c,d)). Consequently, ABTL0812 induced an increase on the levels of cellular long-chain dihydroceramides in those cells (Figures 5(b–d) and 6(g,h)) without affecting ceramide levels at short treatment times, similar to that observed for short-time treatment with the DEGS1 specific inhibitor GT11 (Figure 7(a)) or for the cancer cells where DEGS1 was silenced [37,38]. Importantly, DEGS1 inhibitor GT11 recapitulated ABTL0812 mechanism of action, since it induced ER stress-dependent autophagy and selective cytotoxicity (non-tumor cells are less sensitive than tumor cells) (Figure 7). According to this, ablation of DEGS1 results in cancer cell cycle arrest at G0/G1 [39] and apoptosis [37], and DEGS1 inhibition has been associated with the anti-tumoral effect of compounds, such as the natural molecules resveratrol and THC [20]. In this regard, we found that tumor cell lines express higher DEGS1 protein than non-tumor ones (human lung MRC5 fibroblasts compared to NSCLC H157 cells [Figure 6(i)] or human NHC3 cholangiocytes compared to EGi-1, TFK-1 and Witt cholangiocarcinoma cells [Fig. S10]). This notion would be in agreement with the observation that MRC5 fibroblasts do not accumulate dihydroceramides in response to ABTL0812 (Figure 6(g)). More importantly, our preliminary data showed that tumors from patients with endometrial cancer expressed higher DEGS1 levels than the surrounding non-tumoral tissues (Figure 6(j)). Further work is required to expand this observation to other human cancers, as well as to address the exact role of DEGS1 in tumor biology. Nevertheless, it is likely that elevated levels of long-chain dihydroceramides observed in cells treated with ABTL0812 should play a fundamental role in mediating cancer cell death. In this regard, human xenograft tumors from mice treated with ABTL0812 showed elevated levels of long-chain dihydroceramides (Figure 5(e,f)), as it has been described for glioma tumors in mice treated with THC [20]. Recent improvement of quantitative mass spectrometry analysis of plasma sphingolipids has allowed the proposal of plasma long-chain dihydroceramides as a biomarker for diabetes susceptibility [40]. In this regard, quantification of dihydroceramide levels in plasma from patients enrolled in clinical trials could be of interest to develop new pharmacodynamic biomarkers for ER stress-activating drugs, such as ABTL0812.
For many years, dihydroceramides were considered ineffective sphingolipids, but recently have been implicated in active signaling. Of interest, elevated levels of cellular dihydroceramides can induce ER stress and UPR-mediated autophagy. Thus, treatment of human gastric carcinoma cancer HGC-27 cells with the DEGS1 inhibitor XM462 resulted in dihydroceramide accumulation-mediated ER stress and autophagy [41]. Similarly, THC induced dihydroceramide-mediated UPR activation and cytotoxic autophagy in human glioblastoma cancer cell and tumor models [20]. Here, we show that manipulation of levels of cellular dihydroceramides mimicked the effect of ABTL0812 in activating UPR and autophagy, as well as cytotoxicity in both pancreatic and lung adenocarcinoma cells (Figure 6). This observation supports the hypothesis that dihydroceramides could mediate (at least in part) the anti-tumoral effect of ABTL0812. However, the mechanism by which dihydroceramides induce ER stress and UPR remains to be elucidated. We speculate that accumulation of long-chain dihydroceramides could result in increased rigidity of the ER membrane and in reduced transbilayer lipid movement, which ultimately would compromise the folding capacity of the ER machinery. It will be important to establish whether ABTL0812 also induces the accumulation of dihydroceramides in ER membranes of cancer cells.
How ABTL0812 induces cytotoxic autophagy remains to be elucidated, but the changes in dihydroceramide cellular content reported in this work might be relevant. In this regard, it has been reported that THC induces an increase in dihydroceramide:ceramide ratio in the ER membrane of human glioma cells, resulting in enhanced rigidity of this organelle membranes. This alteration is transmitted to the nascent autophagosomal membranes, resulting in membrane destabilization that ultimately results in lysosomal membrane permeabilization (LMP), cathepsin release and cell death [20]. Interestingly, ABTL0812 also induced a sustained increase in dihydroceramide:ceramide ratio in pancreatic adenocarcinoma MiaPaca2, squamous NSCLC H157 and endometrial cancer Ishikawa cells, as well in xenograft tumors, but not in non-tumoral MRC5 cells (Figure S11). Also, ABTL0812 induced cytosolic release of cathepsin-B in cancer cells (data not shown). Since cathepsin inhibitors E64d and pepstatin-A prevent ABTL0812 cytotoxicity [1], these observations point to LMP as a major player in ABTL00812-induced cancer cell death.
We previously showed that ABTL0812 inhibits the AKT-MTORC1 axis in cancer cells and tumor models by upregulating TRIB3 [1–3]. Unlike AKT or MTOR inhibitors, ABTL0812 also activates UPR-mediated cytotoxic autophagy by uprising levels of cellular dihydroceramides. Importantly, MTORC1 inhibition and dihydroceramides accumulation synergize to induce autophagy and cytotoxicity (Figure 8), reproducing the effect of ABTL0812 in cancer cells. Further work is necessary to establish the molecular mechanisms implicated in this synergistic effect, but the activation of UPR by MTORC1 inhibition could account for this. In this respect, it has been recently reported that MTORC1 inhibitors rapamycin and temsirolimus induce the EIF2A-ATF4-DDIT3 arm of the UPR in human sarcoma cells [42], as well as EIF2AK3-EIF2A axis in cells derived from neuroendocrine intestinal tumors [43]. However, and unlike ABTL0812, MTORC1 inhibitors do not induce cytotoxic UPR in cancer cells [43]. In this scenario, it is likely that the non-cytotoxic ER stress induced by MTORC1 inhibition could converge with that one induced by accumulation of dihydroceramides to surpass the capacity of the UPR to recover cell homeostasis and, therefore, to activate UPR-mediated cytotoxic autophagy. If so, it is tempting to speculate that pharmacological manipulation of cellular dihydroceramides might be useful to improve the efficacy of MTOR inhibitors. This is the case for ABTL0812, a new drug that is under clinical evaluation in patients with advanced cancer.
Materials and Methods
Reagents
ABTL0812 (Ability Pharma SL, in-house synthesized; stock concentration 100 mM) was prepared weekly, diluted in ethanol and kept at −20°C. The dihydroceramide N-octanoyl-D-erythro-[4,4–2 H2]sphinganine (d2C8dhCer) was in-house synthesized. Brefeldin A and bafilomycin A1 (Sigma-Aldrich, B7651 and B1793), the DEGS1 inhibitor GT11 (Avanti Polar Lipids, 857395P), and the cathepsin inhibitors E64d (Sigma-Aldrich, E8640) and pepstatin A (Sigma-Aldrich, P5318) were diluted in ethanol. 4-phenylbutyrate (4-PBA; Sigma-Aldrich, SML0309) was diluted in phosphate-buffered saline (PBS; Sigma-Aldrich, 806,552), whereas sodium tauroursodeoxycholate (TUDC; Sigma-Aldrich, T0622) was diluted in water. Everolimus (Sigma-Aldrich, SML2282), AZD5363 (Medchem-Express, S8019) and AZD2014 (Medchem-Express, S2783) were diluted in DMSO.
Cell culture and cell viability assay
Cell lines were cultured at 37°C under a humidified atmosphere (5% CO2). Human cancer cell lines A549 (lung adenocarcinoma), MiaPaca2 (pancreas adenocarcinoma) and MRC5 (human lung fibroblasts) were purchased from American Type Culture Collection (ATCC®, CRM-CCL-185, CRM-CRL-1420 and CCL-171, respectively; ATCC-authentication by isoenzymes analysis). Ishikawa (endometrial adenocarcinoma) and H157 (squamous NSCLC) cells were from European Collection of Authenticated Cell Cultures (ECACC®, 99,040,201 and 07030901, respectively). Cells were grown by under standard conditions in Dulbecco’s modified Eagle’s medium with 1% L-glutamine, 4.5 g/l glucose (DMEM; Thermo Fisher Scientific, 41,965,039) supplemented with 10% fetal bovine serum (Sigma-Aldrich, F0926). T-large antigen-transformed MEFs atg5+/+ and atg5−/- were generously provided by Dr. Mizushima (Tokyo Medical University, Tokyo, Japan). EIF2A/eIF2α WT and EIF2A/eIF2αS51A T-large antigen MEFs were provided by Dr. Kaufman and Dr. de Haro and Dr. Berlanga (Centro Biología Molecular Severo Ochoa, Autonoma University, Madrid, Spain), respectively. MEF fibroblasts were maintained in DMEM supplemented with 10% FBS.
Cell viability was determined by MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium Blue; Sigma-Aldrich, M2128) reduction assay as described previously [44].
Immunoblotting
Cell lines were lysed in ice-cold radio-immunoprecipitation assay (RIPA) buffer containing 25 mM Tris-HCl pH 7.9, 150 mM NaCl, 1 mM EGTA (Sigma-Aldrich, E3889), 1% NP-40 (Sigma-Aldrich, 74,385), 0.1% SDS (Sigma-Aldrich, L4509), 0.5% deoxycholic acid (Sigma-Aldrich, D2510), 5 mM sodium pyrophosphate (Sigma-Aldrich, S6422), 50 mM sodium fluoride (Sigma-Aldrich, 1,614,002), 1 mM sodium orthovanadate (Sigma-Aldrich, 450,243). Lysed cells were sonicated and stored at −20°C until use. Protein amount was quantified using the Pierce BCA Protein Assay Kit (Thermo-Fischer Scientific, 1,856,209), following the manufacturer’s protocol. Immunoblot analysis was performed following standard procedures as describe previously [45]. Briefly, proteins were resolved by SDS-PAGE gels and transferred to nitrocellulose membrane (Merck Millipore, HATF00010). Membranes were blocked with TBS-T (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.2% Tween-20 [Sigma-Aldrich, P9416] containing 5% skimmed milk [Sigma-Aldrich, 70,166]), and incubated with the indicated primary antibody. Detection was performed using a horseradish peroxidase-conjugated secondary antibody (Thermo Fisher Scientific, 31,460 [anti-rabbit] and 31,430 [anti-mouse]) and enhanced chemiluminescence reagent (Bio-Rad, 170–5061). The following rabbit polyclonal antibodies were from Cell Signaling Technology: anti-ATF4 (11,815), anti-DDIT3 (2895), anti-ACTB (4970), anti-RPS6/S6 (2217), anti-phospho-RPS6/S6 (4858), anti-EIF2A (2103), anti-EIF2AK3 (3192), anti-ATG5 (12,994), anti-EIF2AK4/GCN2 (3302) and anti-phospho-EIF2A (3597). Rabbit polyclonal anti-TRIB3 (ab50516), anti-DEGS1 (ab167169) and anti-MAP1LC3B (ab48394) were from Abcam. Mouse polyclonal anti-HSPA5 was from BD Transduction Laboratories (610,978).
Transfection of siRNAs
Control siRNA (5ʹ-GUAAGACACGACUUAUCGC-3ʹ) and ATF4 siRNAs (ATF4-1: 5ʹ-GCCUAGGUCUCUUAGAUGA-3ʹ and ATF4-2: 5ʹ-CCAGAUCAUUCCUUUAGUUUA-3ʹ), EIF2AK3 siRNAs (EIF2AK3-1: 5ʹ-CCAAUGGGAUAGUGACGAA-3-3ʹ LaEIF2AK3-2: 5K3 GAUAGUGACGAA-3-3ʹ LaboratoriEIF2AK4 siRNAs (GCN2-1: 5 s CACCGTCAAGATTACGGACTA-3ʹ and EIF2AK4-2: 5ʹ- GGGAAAUGUAUUGGCAGUGUU-3GA were from Sigma-Aldrich. siRNAs were transfected into cells using Lipofectamine 2000TM (Thermo Fisher Scientific, 11,668,027). Cells were incubated with siRNAs and after 4 h the medium was changed, and cells were incubated further 18 h prior to treatment with ABTL0812.
Generation of stable ATG5 and TRIB3 knockdown cell lines with shRNAs
MiaPaca2 and A549 cells were transfected with control or vectors encoding shRNA for ATG5 (Santa Cruz Biotechnology, sc-41,445-V) or TRIB3 (Sigma-Aldrich, MISSION shRNA TRCN0000196756). Clones stably expressing the shRNAs were selected with 2 µg/mL puromycin (Sigma-Aldrich, P8833). Stable silencing was confirmed by immunoblot analysis.
Real-time, reverse-transcription polymerase chain reaction (RT-qPCR)
Total RNA was isolated from cells by using GeneJet RNA purification kit (Thermo Fisher Scientific, K0761). cDNA was obtained using RevertAid cDNA Synthesis Kit (Thermo Fisher Scientific, K1621). Real-time quantitative PCR assays were performed using TaqMan Gene Expression Master Mix (Thermo Fisher Scientific, 4,369,016) and the following probes (Thermo Fisher Scientific): human ATF4 (Hs00909569_g1), human DDIT3 (Hs99999172_m1), human GAPDH (Hs03929097_g1) and human TRIB3 (Hs01083394_m1). Amplifications were run in a Bio-Rad CFX96 real-time PCR system (Bio-Rad) using the following protocol: 50°C for 2 min, 95°C for 10 min, 39 cycles of 95°C for 15 s, 55°C for 1 min, plate read. Each value was normalized to GAPDH levels. Relative expression levels were determined using the 2−ΔΔCt method. Real-time PCR was controlled by the Bio-Rad CFX Manager v 3.1 software.
Human blood samples from patients enrolled in the clinical trial were used for the pharmacodynamic biomarker studies [32]. Briefly, total RNA was isolated from whole blood using the RiboPure RNA purification kit for whole blood (Thermo Fisher Scientific, AM1928). cDNA synthesis and real-time quantitative PCR assays for TRIB3 and DDIT3 mRNA levels were performed as described above.
XBP1 splicing assay
Total RNA isolation and cDNA synthesis was performed as described above. XBP1 cDNA was amplified by PCR with TaqMan polymerase (Thermo Fisher Scientific, EP0401) using the primers 5ʹ-TTACGAGAGAAAACTCATGGCC-3ʹ and 5ʹ-GGGTCCAAGTTGTCCAGAATGC-3ʹ. These primers amplify spliced and unspliced isoforms of XBP1. Amplifications were run using the following protocol: 95°C for 5 min, 34 cycles of 95°C for 1 min, 55°C for 1 min, 72°C for 1 min, and 5 min 72°C. Amplified DNA fragments were separated by electrophoresis on an 8% acrylamide gel and visualized by ethidium bromide (Sigma-Aldrich, E1510) staining.
Xenograft models
For MiaPaca2 and A549 xenograft models, athymic female nude mice (n = 8 per group) were injected subcutaneously with 5 × 106 cells into one flank. When tumors reached 80–100 mm3, mice were randomly distributed into treatment groups and administered the corresponding treatments. ABTL0812 was administered 5 times per week by oral gavage at 120 mg/kg in water 5% glycerol. Mice body weight was measured three times a week. Tumors volumes were measured as (length x width2)/2, thrice a week. When tumors reached more than 1000 mm3 or bodyweight decreased 20% of the initial measure, mice were euthanized by CO2 inhalation. All procedures involving animals were performed with approval of the UAB Animal Experimentation Committee, according to Spanish official regulations.
Immunohistochemistry of xenograft samples
Samples from tumor xenografts were dissected out, formalin-fixed and paraffin-embedded, as described previously [1]. Sections (4-µm thickness) were stained with hematoxylin and eosin and analyzed by immunohistochemistry using a standard protocol. Briefly, deparaffinized samples were subjected to heat-induced antigen retrieval in citrate buffer. After blocking endogenous peroxidase with 3% H202, and reducing unspecific binding by incubation with and 5% goat serum (Sigma-Aldrich, G9023) and 1% Triton X-100 (Sigma-Aldrich, 1.08643), samples were then incubated with anti-DDIT3 antibody (Sigma-Aldrich, HPA068416) and further processed with secondary antibody (Dako, k0609).
Lipid extraction and sphingolipidomic analysis (UPLC-TOF-Ms)
1.6x106 cells were seeded on 60 mm dishes and were allowed to adhere for 24 h. After the indicated treatment, medium was removed, cells washed with PBS and harvested by trypsinization. Tumors were dissected, washed and mechanically lysed in PBS. To extract lipids, cells or tumors homogenates were mixed with 0.5 ml methanol and 0.25 ml chloroform (Scharlab, Spain), and internal standards were added (0.2 nmol N-dodecanoylsphingosine, N-dodecanoylglucosylsphingosine and N-dodecanoylsphingosyl phosphorylcholine; Avanti Polar Lipids, Alabaster, AL). Samples were incubated at 48°C overnight. The following day, 75 µl 1 M KOH in methanol was added and incubated for 2 h at 37°C. Lipid extracts were then neutralized with 75 µl 1 M acetic acid, dried under nitrogen stream and solubilized in 150 µl methanol. Lipids were analyzed using a liquid chromatography-mass spectrometer consisted of a Waters Acquity UPLC system connected to a Waters LCT Premier Orthogonal Accelerated Time-of-Flight Mass Spectrometer (Waters, Millford, MA), operated in positive and negative electrospray ionization mode. Full scan spectra from 50 to 1500 Da were obtained. Mass accuracy and reproducibility were maintained by using an independent reference spray via Lock Spray, using a 100 mm x 2.1 mm id, 1.7 µm C8 Acquity UPLC BEH (Waters) analytical column. The mobile phases were 1 mM ammonium formate in methanol (phase A) and 2 mM ammonium formate in H2O (phase B), both phases with 0.05 mM formic acid. The gradient was run at 30°C (0 min 80% A; 3 min 90% A, 6 min 90% A; 15 min 99% A; 18 min 99% A; 20 min 80% A). Quantification was carried out using the ion chromatogram obtained for each compound using 50 mDa windows. Linear dynamic range was determined by injection of standard mixtures. Positive identification of compounds was based on the accurate mass measurement with an error <5 ppm and its LC retention time, compared to that of a standard (± 2).
DEGS1 enzyme assay
DEGS1 assays were performed in intact cells as well as in cell lysates. 6-(N-[7-nitro-2,1,3-benzoxadiazol-4-yl]amino) hexanoylsphinganine (DHCerC6NBD; in-house synthesized) was used as DEGS1 substrate. This compound is desaturated in cells to the ceramide analog 6-(N-[7-nitro-2,1,3-benzoxadiazol-4-yl]amino) hexanoylsphingosine (CerC6NBD), which can be determined by HPLC analysis [46]. To determine DEGS1 activity in cell lysates, 106 cells were pelleted, washed with PBS, resuspended in 0.2 M phosphate buffer (pH 7.4) and sonicated. ABTL0812 or ethanol (vehicle) and the substrate mix (10 µM DHCerC6NBD, 2 mg/ml NADH) were added to cell lysates. The reaction took place at 37°C for 4 h, and was stopped by adding methanol. Samples were mixed by vortex and kept at 4°C overnight. After centrifugation, clear supernatants were transferred to HPLC vials and 25 µl were injected. To determine the effect of ABTL0812 on DEGS1 activity in intact cells, 1.5 × 106 cells were plated and, after treatment, media was removed and 10 µM DHCerC6NBD was added in fresh medium. After 4 h incubation at 37°C, media were collected and cells harvested by trypsinization, resuspended in water and sonicated. Reaction was stopped by adding methanol and kept at 4°C overnight. Either 25 µl (media) or 0.1 ml (cells) were injected into the HPLC. Instrumental analysis was carried out by HPLC with a fluorimetric detector as reported before [46].
Clinical trial and sample collection from patients
ABTL0812 is currently in clinical evaluation in a phase 2 trial in patients with advanced endometrial and squamous lung cancer. This study was approved by the local ethics committees of the participating sites and by the Spanish Agency of Medicines and Medical Devices, and it is registered at www.clinicaltrials.gov (identifier: NCT03366480). The study was conducted in compliance with the protocol, regulatory requirements, good clinical practice and the ethical principles of the Declaration of Helsinki as adopted by the World Medical Association. All subjects received written and verbal information regarding the study and signed an informed consent document.
ABTL0812 was orally administered (1,300 mg tid) as the first line of treatment in combination with chemotherapy (carboplatin and paclitaxel). ABTL0812 administration started a week before the first chemotherapy cycle and lasted until the patient left the study. Blood samples for the biomarker study were obtained by venipuncture with a 21 G needle and using tubes containing citrate. Peripheral blood mononuclear cells were isolated by Ficoll-Paque PLUS (Sigma-Aldrich, GE17-1440-03) density centrifugation, pelleted and stored at −80°C until RT-qPCR analysis.
Endometrial patient tumor collection
Patients with no previous therapy (50–80 years) underwent surgery for endometrial carcinoma at Hospital Vall d’Hebron (Barcelona). The protocol was approved by the Institutional Review Boards, and informed consent was obtained from the patients involved. After surgery, normal tissue and tumor samples were collected from each patient and stored frozen (−80°C) until analysis. Samples were lysed in 7 M urea (Sigma-Aldrich, U5378), 2 M thiourea (Sigma-Aldrich, T7875), 4% CHAPS (Sigma-Aldrich, C5070), 30 mM Tris, pH 8.5., sonicated and centrifuged (12,000 g x 3 min).
Statistical analysis
All in vitro data were assessed using one-way ANOVA followed by Bonferroni multiple comparison test or two-tailed Student’s t-test. Tumor volumes of mice were compared using ANOVA followed by t-test. Statistical significance between the groups was assessed with the log-rank test (GraphPad software). Data in the figures are presented as mean ± SD, unless otherwise indicated, with the result of the statistical test, with *p < 0.05, **p < 0.01, ***p < 0.001. Statistical significance was set at p < 0.05. Statistical analyses of RT-qPCR data from clinical samples were analyzed by the one-way ANOVA Tukey test. Synergism analysis was performed using the Compusyn software [47].
Supplementary Material
Acknowledgments
We are grateful to Cristina Gutierrez, Neus Ontiveros and Enrique Claro for tissue culture and electron microscopy, respectively. We also thank Franziska Pueschel for technical assistance, Eva Colás for providing human tissue samples, and the following Services of UAB: Servei de Cultius Cel·lulars INc, Laboratori de Luminiscència Espectroscopia de Biomolècules, Servei de Microscopia and Servei Genòmica Informàtica.
This work was supported by the Spanish Ministry of Economy and Competitiveness (MINECO) grants INNPACTO/IPT-2012-0614-010000, RETOS RTC-2017-6261-1, SAF2015-64237-R, and cofunded by the European Regional Development Fund (ERDF). GV’s laboratory was supported by PI18/00442 and PI15/00339 grants by Instituto de Salud Carlos III (ISCIII), ERDF, “Fundació La Marató de TV3” (20134031) and Marie Skłodowska-Curie Innovative Training Network (TRAIN GA721532). GF’s and CM-P’s laboratories were supported by grants from Spanish Ministry of Science, Innovation and Universities (CTQ2017-85378-R), and MINECO BFU2016-78154-R, respectively. Ability Pharmaceuticals was supported by grants from MINECO RTC-2014-1532-1/RTC-2015-3821-1/RTC-2017-6261-1/EMPLEA/EMP-TU-2015-4576, and from CDTI (INNOGLOBAL/20171061).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was supported by the Centre for Industrial Technological Development [CDTI,INNOGLOBAL/20171061]; European Regional Development Fund [PI18/00442 and PI15/00339]; European Regional Development Fund [INNPACTO/IPT-2012-0614-010000, RETOS RTC-2017-6261-1, SAF2015-64237-R]; Fundació la Marató de TV3 [20134031]; H2020 Marie Skłodowska-Curie Actions [TRAIN GA721532]; Instituto de Salud Carlos III [PI15/00339]; Instituto de Salud Carlos III [PI18/00442]; Ministerio de Ciencia, Innovación y Universidades [CTQ2017- 85378-R]; Ministerio de Economía y Competitividad [RTC-2015-3821-1]; Ministerio de Economía y Competitividad [RTC-2017-6261-1]; Ministerio de Economía y Competitividad [EMP-TU-2015-4576]; Ministerio de Economía y Competitividad [RETOS RTC-2017-6261-1]; Ministerio de Economía y Competitividad [BFU2016-78154-R]; Ministerio de Economía y Competitividad [INNPACTO/IPT-2012-0614-010000]; Ministerio de Economía y Competitividad [SAF2015-64237-R]; Ministerio de Economía y Competitividad [RTC-2014-1532-1].
Disclosure statement
PM-G, SS, EM-R; HP-M, MY-V, CD and JA are Ability Pharmaceuticals employees; CD holds shares of the company; JML is advisory member of Ability Pharmaceuticals.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Erazo T, Lorente M, Lopez-Plana A, et al. The new antitumor drug ABTL0812 inhibits the Akt/mTORC1 axis by upregulating tribbles-3 pseudokinase. Clin Cancer Res. 2016;22:2508–2519. [DOI] [PubMed] [Google Scholar]
- [2].Felip I, Moiola CP, Megino-Luque C, et al. Therapeutic potential of the new TRIB3-mediated cell autophagy anticancer drug ABTL0812 in endometrial cancer. Gynecol Oncol. 2019;153:425–435. [DOI] [PubMed] [Google Scholar]
- [3].Lopez-Plana A, Fernandez-Nogueira P, Munoz-Guardiola P, et al. The novel pro-autophagy anticancer drug ABTL0812 potentiates chemotherapy in adenocarcinoma and squamous non-small cell lung cancer. Int J Cancer. 2020. DOI: 10.1002/ijc.32865. [DOI] [PubMed] [Google Scholar]
- [4].Boya P, Reggiori F, Codogno P.. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Marino G, Niso-Santano M, Baehrecke EH, et al. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang X, Gocek E, Novik V, et al. Inhibition of Cot1/Tlp2 oncogene in AML cells reduces ERK5 activation and up-regulates p27Kip1 concomitant with enhancement of differentiation and cell cycle arrest induced by silibinin and 1,25-dihydroxyvitamin D(3). Cell Cycle. 2010;9:4542–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li T, Su L, Zhong N, et al. Salinomycin induces cell death with autophagy through activation of endoplasmic reticulum stress in human cancer cells. Autophagy. 2013;9:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Puissant A, Robert G, Fenouille N, et al. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010;70:1042–1052. [DOI] [PubMed] [Google Scholar]
- [10].Salazar M, Carracedo A, Salanueva IJ, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119:1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. [DOI] [PubMed] [Google Scholar]
- [12].Yoshida H, Matsui T, Yamamoto A, et al. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. [DOI] [PubMed] [Google Scholar]
- [13].Cao SS, Zimmermann EM, Chuang BM, et al. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology. 2013;144:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer. 2018;18:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Signorelli P, Munoz-Olaya JM, Gagliostro V, et al. Dihydroceramide intracellular increase in response to resveratrol treatment mediates autophagy in gastric cancer cells. Cancer Lett. 2009;282:238–243. [DOI] [PubMed] [Google Scholar]
- [16].Chipuk JE, McStay GP, Bharti A, et al. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 2012;148:988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Morales A, Lee H, Goni FM, et al. Sphingolipids and cell death. Apoptosis. 2007;12:923–939. [DOI] [PubMed] [Google Scholar]
- [18].Carracedo A, Gironella M, Lorente M, et al. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006;66:6748–6755. [DOI] [PubMed] [Google Scholar]
- [19].Salazar M, Carracedo A, Salanueva IJ, et al. TRB3 links ER stress to autophagy in cannabinoid anti-tumoral action. Autophagy. 2009;5:1048–1049. [DOI] [PubMed] [Google Scholar]
- [20].Hernandez-Tiedra S, Fabrias G, Davila D, et al. Dihydroceramide accumulation mediates cytotoxic autophagy of cancer cells via autolysosome destabilization. Autophagy. 2016;12:2213–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Savile CK, Fabrias G, Buist PH. Dihydroceramide delta(4) desaturase initiates substrate oxidation at C-4. J Am Chem Soc. 2001;123:4382–4385. [DOI] [PubMed] [Google Scholar]
- [22].Fabrias G, Munoz-Olaya J, Cingolani F, et al. Dihydroceramide desaturase and dihydrosphingolipids: debutant players in the sphingolipid arena. Prog Lipid Res. 2012;51:82–94. [DOI] [PubMed] [Google Scholar]
- [23].Triola G, Fabrias G, Casas J, et al. Synthesis of cyclopropene analogues of ceramide and their effect on dihydroceramide desaturase. J Org Chem. 2003;68:9924–9932. [DOI] [PubMed] [Google Scholar]
- [24].Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69:169–181. [DOI] [PubMed] [Google Scholar]
- [25].Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. Febs J. 2016;283:2640–2652. [DOI] [PubMed] [Google Scholar]
- [26].Shuda M, Kondoh N, Imazeki N, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38:605–614. [DOI] [PubMed] [Google Scholar]
- [27].Wu X, Xin Z, Zhang W, et al. A missense polymorphism in ATF6 gene is associated with susceptibility to hepatocellular carcinoma probably by altering ATF6 level. Int J Cancer. 2014;135:61–68. [DOI] [PubMed] [Google Scholar]
- [28].Cerezo M, Lehraiki A, Millet A, et al. Compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell. 2016;30:183. [DOI] [PubMed] [Google Scholar]
- [29].Niu Z, Wang M, Zhou L, et al. Elevated GRP78 expression is associated with poor prognosis in patients with pancreatic cancer. Sci Rep. 2015;5:16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang Q, Mora-Jensen H, Weniger MA, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci USA. 2009;106:2200–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schonthal AH. Endoplasmic reticulum stress and autophagy as targets for cancer therapy. Cancer Lett. 2009;275:163–169. [DOI] [PubMed] [Google Scholar]
- [32].Alfon J, Vidal L, Gaba L, et al. Determination of recommended phase II dose of ABTL0812, a novel regulator of Akt/mTOR axis, by pharmacokinetic-pharmacodynamic modelling. Ann Oncol. 2016;27(Suppl 6):378P. [Google Scholar]
- [33].Avril T, Vauleon E, Chevet E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis. 2017;6:e373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yamada M, Tomida A, Yun J, et al. Cellular sensitization to cisplatin and carboplatin with decreased removal of platinum-DNA adduct by glucose-regulated stress. Cancer Chemother Pharmacol. 1999;44:59–64. [DOI] [PubMed] [Google Scholar]
- [35].Nagelkerke A, Bussink J, Sweep FC, et al. The unfolded protein response as a target for cancer therapy. Biochim Biophys Acta. 2014;1846:277–284. [DOI] [PubMed] [Google Scholar]
- [36].Hsu JL, Chiang PC, Guh JH. Tunicamycin induces resistance to camptothecin and etoposide in human hepatocellular carcinoma cells: role of cell-cycle arrest and GRP78. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:373–382. . [DOI] [PubMed] [Google Scholar]
- [37].Breen P, Joseph N, Thompson K, et al. Dihydroceramide desaturase knockdown impacts sphingolipids and apoptosis after photodamage in human head and neck squamous carcinoma cells. Anticancer Res. 2013;33:77–84. [PMC free article] [PubMed] [Google Scholar]
- [38].Siddique MM, Bikman BT, Wang L, et al. Ablation of dihydroceramide desaturase confers resistance to etoposide-induced apoptosis in vitro. PLoS One. 2012;7:e44042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kraveka JM, Li L, Szulc ZM, et al. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J Biol Chem. 2007;282:16718–16728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wigger L, Cruciani-Guglielmacci C, Nicolas A, et al. Plasma dihydroceramides are diabetes susceptibility biomarker candidates in mice and humans. Cell Rep. 2017;18:2269–2279. [DOI] [PubMed] [Google Scholar]
- [41].Gagliostro V, Casas J, Caretti A, et al. Dihydroceramide delays cell cycle G1/S transition via activation of ER stress and induction of autophagy. Int J Biochem Cell Biol. 2012;44:2135–2143. [DOI] [PubMed] [Google Scholar]
- [42].Briggs JW, Ren L, Chakrabarti KR, et al. Activation of the unfolded protein response in sarcoma cells treated with rapamycin or temsirolimus. PLoS One. 2017;12:e0185089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Freis P, Bollard J, Lebeau J, et al. mTOR inhibitors activate PERK signaling and favor viability of gastrointestinal neuroendocrine cell lines. Oncotarget. 2017;8:20974–20987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Erazo T, Moreno A, Ruiz-Babot G, et al. Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex. Mol Cell Biol. 2013;33:1671–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rodriguez-Asiain A, Ruiz-Babot G, Romero W, et al. Brain specific kinase-1 BRSK1/SAD-B associates with lipid rafts: modulation of kinase activity by lipid environment. Biochim Biophys Acta. 2011;1811:1124–1135. [DOI] [PubMed] [Google Scholar]
- [46].Munoz-Olaya JM, Matabosch X, Bedia C, et al. Synthesis and biological activity of a novel inhibitor of dihydroceramide desaturase. ChemMedChem. 2008;3:946–953. [DOI] [PubMed] [Google Scholar]
- [47].Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.