

ABSTRACT
The antibody-dependent cellular cytotoxicity (ADCC) effector function of natural killer (NK) cells is one of the known mechanisms of action for rituximab-based anti-cancer immunotherapy. Inhibition of the ADCC function of NK cells through interactions between inhibitory killer cell immunoglobulin-like receptors (KIRs) and HLA class I ligands is associated with resistance of cancers to rituximab. In this study, we deeply investigated the impact of KIR, HLA class I, and CD16 genotypes on rituximab-dependent NK cell responses in both an in vitro cellular model from healthy blood donors and ex vivo rituximab-treated non-Hodgkin lymphoma (NHL) patients. We highlight that an HLA environment with limited KIR ligands is beneficial to promoting a higher frequency of KIR+ NK cells including both educated and uneducated NK cells, two NK cell compartments that demonstrate higher rituximab-dependent degranulation than KIR− NK cells. In contrast, a substantial KIR ligand environment favors a higher frequency of poorly effective KIR− NK cells and numerous functional KIR/HLA inhibitions of educated KIR+ NK cells. These phenomena explain why NHL patients with limited KIR ligands respond better to rituximab. In this HLA environment, CD16 polymorphism appears to have a collateral effect. Furthermore, we show the synergic effect of KIR2DS1, which strongly potentiates NK cell ADCC from C2− blood donors against C2+ target cells. Taken together, these results pave the way for stronger prediction of rituximab responses for NHL patients. HLA class I typing and peripheral blood KIR+ NK cell frequency could be simple and useful markers for predicting rituximab response.
KEYWORDS: KIR, hla, adcc, rituximab, nk cells, cd16, non hodgkin lymphoma
Introduction
Several mechanisms of action contribute to the efficacy of therapeutic antibodies, mainly including complement-dependent cytotoxicity, antibody-dependent cellular cytotoxicity (ADCC) and the induction of cell death.1,1,2 The ADCC effector function of natural killer (NK) cells and macrophages is widely used clinically as a mechanism of action for rituximab-based anti-cancer immunotherapy.3 Rituximab is a chimeric anti-CD20 monoclonal antibody used in the treatment of B cell lymphomas.3,4 The rituximab-mediated ADCC activity of NK cells is triggered through the interaction between CD16 (FcγRIIIa) activating receptor with the Fc fragments of this monoclonal antibody. On the clinical side, an association between the FCGR3A genotype and clinical and molecular responses to rituximab has been revealed.5 Thus, CD16 polymorphism may be evidence that ADCC is one of the mechanisms of action responsible for rituximab’s anti-tumor effects. It has been shown that rituximab had a strong affinity for the CD16 158 V allotype compared to its 158 F counterpart.5,6 Numerous studies have reported that the use of low-dose rituximab was associated with greater ADCC responses in patients with the CD16 158 VV or VF genotype compared to patients with the 158 FF genotype.7
Several mechanisms of cancer resistance to rituximab have been documented in the literature, and different strategies are used to increase the effector activity of NK cells to improve anti-tumor responses. The inhibition of the ADCC function of NK cells through interactions between inhibitory killer cell immunoglobulin-like receptors (KIRs) and HLA class I ligands has been described as one of the potential mechanisms of cancer resistance to rituximab8,9 . KIRs are mainly expressed by NK cells and modulate NK cell responses by recognizing HLA class I molecules in tumoral or virus-infected cells. The interactions between KIR and autologous HLA class I ligands during development contribute to the functional education of KIR+ NK cells.10 This functional competence gives educated KIR+ NK cells the ability to recognize and eliminate target cells that exhibit an absence of expression (missing-self) or a downregulation of HLA class I molecules. The main inhibitory KIRs are KIR2DL1, KIR2DL2/3 and KIR3DL1, which, respectively, recognize HLA-C molecules of group C2 (Lys80), HLA-C molecules of group C1 (Asn80) and Bw4 motifs bearing some HLA-A and HLA-B molecules. In contrast, the activating KIR2DS1 triggers NK cell alloreactivity by recognizing HLA-C molecules of group C2 on the surface of allogenic target cells only when KIR2DS1+ NK cells have evolved in a C2− environment.11–13 KIR genes are clonally expressed on the surface of NK cells.14 Thus, the KIR+ NK cell repertoire is diverse between individuals, since the NK cell repertoire harbors different NK cell populations with different functional orientations.
Previous studies have shown that uneducated NK cells dominate ADCC responses in neuroblastoma15 and lymphoma16 patients. By contrast, other groups have more recently shown a dominance of educated KIR2DL1+ and KIR3DL1+ NK cells in ADCC responses compared to uneducated counterparts in HIV models.17,18 Furthermore, some studies9,16 report disparities in regard to the impact of KIR/HLA interactions on the ADCC responses of NK cells. The experimental strategies and cellular models used in these studies do not necessarily represent the best means to study the activity of KIR+ NK cells, which may explain these discrepancies. Terszowki et al. used KIR ligand transfected 721.221 target cells as target cells to measure rituximab-mediated ADCC although 721.221 does not express CD20.9 Borgerding et al. developed a cellular model using a high arituximab concentration (10 µg/mL).8 A few studies have documented mainly patient outcomes related to KIR/HLA genotypes without mechanistic investigations.19,20 Thus, in this study, we revisited the in vitro influence of KIR, HLA and CD16 polymorphisms on rituximab-dependent NK cell degranulation in healthy blood donors. Then, we investigated their influence on the rituximab-based treatment response of a cohort of 74 non-Hodgkin lymphoma (NHL) patients. Our results provide an overview of the influence of KIR, HLA class I, and CD16 polymorphisms on rituximab-dependent NK cell cytotoxicity integrating genetic rules for the structural and functional formation of the NK cell repertoire.
Methods
Study design and patient samples
From November 2019 to February 2020, 80 consecutive adult patients treated in our institution for an NHL with a rituximab-based immunochemotherapy were included after providing informed consent. Peripheral blood mononuclear cells (PBMCs) were isolated as previously described.11 Routine information, such as disease status, previous treatments received (including anterior exposure to rituximab), and CMV serology were collected from the medical file. The best response to immunochemotherapy at the end of the follow-up period (July 2020) was determined using Cheason 2007 revised criteria21 for patients with CT-measurable disease. Patients treated for a Waldenström macroglobulinemia with no CT-measurable disease were evaluated using Owen 2012 criteria.22 In both cases, complete response (CR) was defined as the disappearance of all evidence of disease, partial response (PR) as ≥ 50% regression of measurable disease, progressive disease (PD) as the appearance of any new lesion or ≥ 50% increase measurable disease, and stable disease (SD) as failure to attain CR/PR or PD. At the time of analysis, 6 patients did not have an assessment of disease response and were excluded.
Cells (PBMC and cell lines)
PBMCs were isolated as previously described.11 All blood donors were recruited at the Blood Transfusion Center (EFS, Nantes, France) and informed consent was given by all individuals. The following CD20+ HLA class I homozygous EBV-transformed B lymphoblastoid cell lines were used as targets for functional assays: AMALA (HLA-B*15:01, -C*03:03, and Bw4−/C1C1), SP001 (HLA-B*44:02, -C*05:01, and Bw4+/C2C2) and RSH (HLA-B*42:01, -C*17:01, and Bw4−/C2C2). Cell lines were cultured in RPMI 1640 medium (Gibco, Paisley, Scotland, UK) containing glutamine (Gibco) and penicillin-streptomycin (Gibco) and supplemented with 10% fetal bovine serum (Gibco). Mycoplasma tests performed by PCR were negative for all cell lines.
HLA, KIR, and FcγRIIIa (CD16) genotyping
HLA class I allele assignment was performed for all blood donor volunteers and lymphoma patients as previously described.23 KIR generic typing was performed on all blood donors using a KIR multiplex PCR-SSP method as previously described24 using primers provided by Ketevan Gendzekhadze. The presence or absence of KIR2DL1, 2DL2, 2DL3, 2DL5, 3DL1, 2DS1, 2DS2, 2DS3, 2DS4/1D, 2DS5 and 3DS1 genes was assigned. Donor KIR genotypes were then determined based on the presence or absence of activating KIR. Thus, a KIR AA genotype was defined by the presence of only KIR2DS4 as activating KIR gene and a KIR B+ genotype by the presence of several activating KIR genes.25 KIR ligands A3/A11, Bw4 (HLA-A and/or HLA-B), C1, and C2 were defined for all blood donors and lymphoma patients based on allelic HLA-A, -B, and -C typing. FcγRIIIa V158F polymorphism was typed for 14 healthy blood donors and 80 lymphoma patients by PCR-SSP as previously described.26
Cell phenotypic analysis by flow cytometry
The NK cell phenotype from healthy blood donors was determined by eight-color multiparameter flow cytometry (MFC) on PBMC using the following mouse anti-human mAbs: anti-CD3-APC-Cy7 (SK7, Sony Biotechnology, San Jose, CA, USA), anti-CD56-BV510 (HCD56, Sony), anti-CD16-PerCP-Cy5.5 (3G8, Sony), anti-KIR2DL1-FITC (143211, R&D Systems, Minneapolis, MN, USA) anti-KIR2DL1/S1 (EB6, Beckman Coulter, Immunotech, Marseilles, France), anti-KIR2DL2/3/S2-PE-Cy7 (GL183, Beckman Coulter), anti-KIR3DL1/S1-PE (Z27, Beckman Coulter), and anti-NKG2C-APC (REA205, Miltenyi Biotec, Paris, France). The NK cell surface phenotype from patients was determined by seven-color multiparameter flow cytometry (MFC) on PBMCs using the following mouse anti-human mAbs: anti-CD3-APC-Cy7 (SK7, Sony), anti-CD56-BV510 (HCD56, Sony), anti-KIR2DL1/S1 (EB6, Beckman Coulter), anti-KIR2DL2/3/S2-PE-Cy7 (GL183), anti-KIR3DL1/S1-PE (Z27), anti-NKG2A-PC7 (Z199, Beckman Coulter) and anti-NKG2C-APC (REA205, Miltenyi Biotec, Paris, France).
CD20 and HLA class I cell surface expressions were confirmed on EBV-transformed B lymphoblastoid cell lines by flow cytometry using the following mouse anti-human mAbs: anti-CD20-FITC and anti-HLA class I-FITC (F41-IE3H1D2, EFS Nantes).
CD107a mobilization assay by flow cytometry
EBV-B cell lines were pre-incubated with the anti-CD20 mAb (Roche, Basel, Switzerland) to a final concentration of 0.1, 1, or 10 µg/mL. PBMCs from healthy blood donors were mixed with EBV-B target cells in the presence of anti-CD107a-BV421 (H4A3, Sony) mAb, and NK-cell (CD3− CD56+) degranulation was assessed after incubation for 5 h alone (negative control) or with different target cells (E:T ratio = 1:1) with brefeldin A (Sigma) at 10 μg/mL for the last 4 h. Then, the NK cell phenotype was obtained as previously indicated. All flow cytometry data were collected using an FACSCanto II (BD Biosciences) and analyzed using FlowJoTM 10.6 software (FlowJo LLC, Ashland, OR, USA).
Statistical analysis
Comparison of data in two series was performed using the Student’s t-test. Statistical differences between different groups were analyzed using one-way ANOVA. All statistical analyses were performed using GraphPad Prism v6.0 software (San Diego, CA, USA). P values <.05 were considered statistically significant.
Results
Inhibitory KIR2DL/HLA-C interaction brings rituximab-dependent educated KIR2DL+ NK cell degranulation down to the degranulation baseline of uneducated counterpart
We evaluated the modulation of rituximab-dependent NK cell cytotoxicity in a cohort of healthy blood donors using the main inhibitory KIR2DL1, KIR2DL2/3 and KIR3DL1 and activating KIR2DS1. The KIR genotypes and HLA class I types were determined as well as CD16 V158F polymorphism, in order to include only donors with CD16 158 VV or VF (supplemental table S1). Seven C1C1, three C1C2 and four C2C2 were studied from 14 blood donors. Notably, a majority of donors (11/14) were HLA-A and/or HLA-B Bw4+ (supplemental table S1). We used three homozygous HLA class I EBV-B cell lines that did or did not express Bw4, C1, or C2 KIR ligands. All EBV-B cell lines expressed CD20 and a similar level of HLA class I molecules (figure S1).
From blood donor PBMCs, NK cells were identified as CD3− CD56+ cells. Most of them were CD56dim. A KIR-specific mAb combination led us to target three NK cell subsets expressing either KIR2DL1+ others− (KIR2DS1−/2DL2/3−/3DL1−), KIR2DL2/3+ others− (KIR2DL1/S1−/3DL1−), or KIR3DL1+ others− (KIR2DL1/2/3−2DS1−), and one NK cell subset expressing only KIR2DS1+ (KIR2DL1−/2DL2/3−/3DL1−) as illustrated in Figure 1a with a representative donor expressing KIR2DL1, KIR2DS1, KIR2DL3, and KIR3DL1.
Figure 1.
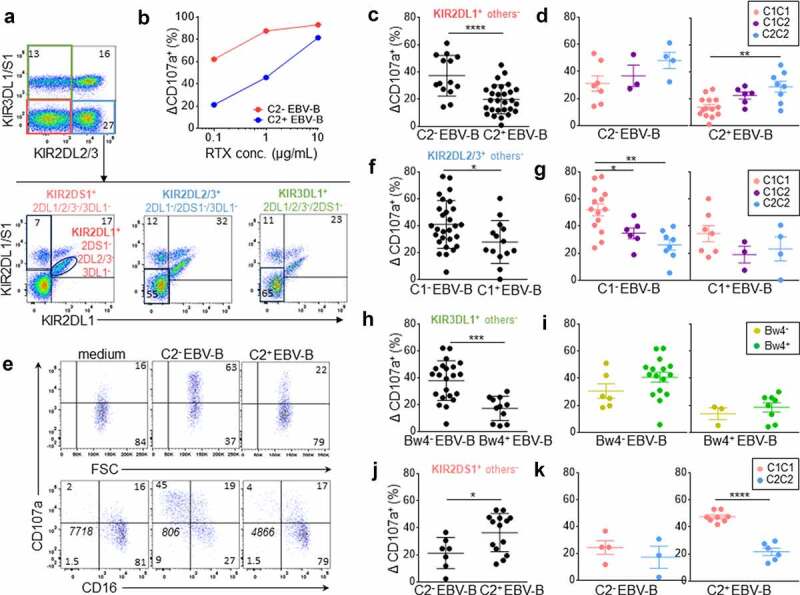
Inhibitory KIR-HLA interaction brings rituximab-dependent educated KIR+ NK cell degranulation down to the degranulation baseline of uneducated counterpart. (a) Representative density plots illustrating the strategy to target three NK cell subsets expressing either KIR2DL1+ others− (KIR2DS1−/2DL2/3−/3DL1−), KIR2DL2/3+ others− (KIR2DL1/S1−/3DL1+) or KIR3DL1+ others− (KIR2DL1/2/3−2DS1−) and one NK cell subset expressing only KIR2DS1+ (KIR2DL1−/2DL2/3−/3DL1−) from PBMC as illustrated for a representative donor expressing KIR2DL1, KIR2DS1, KIR2DL3 and KIR3DL1. A first mAb combination led to exclude KIR3DL1/S1+ and KIR2DL2/3+ NK cells (red gate) and a second mAb combination led to select either KIR2DL1+ 2DS1− NK cells or KIR2DS1+ 2DL1+ NK cells. Similarly, a first mAb combination led to select KIR3DL1+ NK cells and to exclude KIR2DL2/3+ NK cells (green gate) and a second mAb combination to exclude KIR2DL1+ and KIR2DS1+ NK cells. To target KIR2DL2/3+ others−, the first combination led to target KIR2DL2/3+ and exclude KIR3DL1/S1+ NK cells and the second one led to exclude KIR2DL1+ and KIR2DS1+ NK cells. (b) Determination of the optimal rituximab (RTX) concentration to observe inhibiting KIR2DL1-C2 interaction on NK cell ADCC. RTX-dependent KIR2DL1+ others− NK cell degranulation (Δ CD107a+) frequency against C2− and C2+ EBV-B cell lines for a representative blood donor determined by flow cytometry at different RTX concentrations (0.1, 1, and 10 µg/mL) using 1:1 PBMC:EBV-B cell line ratio. (c) RTX dependent KIR2DL1+ others− NK cell degranulation (Δ CD107a+) frequency against C2− (n = 14) and C2+ (Bw4−, n = 14 and Bw4+, n = 14) EBV-B cell lines for all blood donors determined by flow cytometry. Specific CD107a expression is reported as ΔCD107a calculating the difference of CD107a+ NK cell frequency of the test (EBV-B + RTX) and of the NK alone (medium). (d) Frequencies of RTX-dependent KIR2DL1+ others− NK cell degranulation (Δ CD107a+) against one C2− EBV-B cell line for C1C1 (n = 7), C1C2 (n = 3) and C2C2 (n = 4) blood donors and two C2+ (Bw4− and Bw4+) EBV-B cell lines for C1C1 (n = 14), C1C2 (n = 6) and C2C2 (n = 8) blood donors. (e) Representative density plots of RTX-dependent KIR2DL1+ others− NK cell degranulation (CD107a) against C2− and C2+ EBV-B cell lines. CD16 expression is evaluated on NK cells. The frequencies are indicated on the density plot and the mean fluorescent intensity of CD16 on NK cells is indicated in italic. (f) RTX-dependent KIR2DL2/3+ others− NK cell degranulation (Δ CD107a+) frequency against C1− (Bw4−, n = 14 and Bw4+, n = 14) and C1+ (n = 14) EBV-B cell lines for all blood donors. (g) RTX-dependent KIR2DL2/3+ others− NK cell degranulation against C1− EBV-B cell lines for C1C1 (n = 14), C1C2 (n = 6) and C2C2 (n = 8) blood donors and against C1+ EBV-B cell lines for C1C1 (n = 7), C1C2 (n = 3) and C2C2 (n = 4) blood donors. (h) RTX-dependent KIR3DL1+ others− NK cell degranulation against Bw4− (Bw4−, n = 11 and Bw4+, n = 11) and Bw4+ (n = 11) EBV-B cell lines for all blood donors. (i) RTX-dependent KIR3DL1+ others− NK cell degranulation against Bw4− EBV-B cell lines for Bw4− (n = 6) and Bw4+ (n = 16) blood donors and against Bw4+ EBV-B cell lines for Bw4− (n = 3) and Bw4+ (n = 8) blood donors. (j) RTX-dependent KIR2DS1+ others− NK cell degranulation (% CD107a+) against C2− (n = 7) and C2+ (Bw4−, n = 7 and Bw4+, n = 7) EBV-B cell lines for all blood donors. (k) RTX-dependent KIR2DS1+ others− NK cell degranulation against 2 C2+ (Bw4− and Bw4+) EBV-B cell lines for C1C1 (n = 8) and C2C2 (n = 6) blood donors and against one C2− EBV-B cell line for C1C1 (n = 4) and C2C2 (n = 3) blood donors. *p < .05, **p < .01, ***p < .001, ****p < .0001
Different rituximab concentrations were evaluated to determine the best one to assess the modulator effect of inhibitory KIR/HLA interactions in our in vitro cellular model. PBMCs were incubated with the different EBV-B cell lines, NK cells were identified as CD3− CD56+, and the degranulation (CD107a) of each NK cell subset was determined by flow cytometry. At 10 µg/mL of antibody, the CD16-mediated activation masked the inhibitory effect of KIR2DL1 on NK cell cytotoxicity against the C2+ EBV-B cell line (Figure 1b). However, at 1 and 0.1 µg/mL, the inhibitory KIR2DL1 signal was measurable and thus the concentration of rituximab was 0.1 µg/mL in all subsequent experiments in this study. At this concentration, we observed lower rituximab-dependent degranulation of all NK cells from CD16-158 FF blood donors compared to CD16-158 VV and VF blood donors and no difference between the last two groups (data not shown).
Rituximab-dependent KIR2DL1+ others− NK cell degranulation was inhibited against both C2+ EBV-B cell lines compared to the C2− EBV-B cell line (p < .0001) (Figure 1c). The degranulation of C2C2 KIR2DL1+ others− NK cells was significantly higher than that of their C1C1 counterparts against the C2+ EBV-B cell line (p = .001) due to their educated status (Figure 1d). Indeed, the inhibition ratio against C2+ EBV-B cell lines compared to the C2− EBV-B cell line was similar for C1C1, C1C2 and C2C2 donors (data not shown) and better spontaneous degranulation of educated KIR2DL1+ NK cells against the C2− EBV-B cell line was observed (Figure 1d). Interestingly, although the KIR2DL1/C2 interaction inhibited the rituximab-dependent educated KIR2DL1+ others− NK cell degranulation (C2+ target condition), the latter was brought down to the degranulation level of uneducated KIR2DL1+ others− NK cells without KIR2DL1/C2 inhibition (C2− target condition). The rituximab-dependent NK cell degranulation was associated with decreased expression of CD16 on the cell surface for all studied NK cell subsets, as illustrated for KIR2DL1+ others− NK cells (Figure 1e) and as previously demonstrated.27,28
The rituximab-dependent KIR2DL2/3+ others− NK cell degranulation was reduced with the C1+ EBV-B cell line compared with both C1− EBV-B cell lines (p = .02) (figure 1f). The lower degranulation potential of uneducated C2C2 KIR2DL2/3+ others− NK cells was clearly observed against C1− EBV-B cell lines (p = .0007) and to a lesser extent with only one C1 ligand present to educate KIR2DL2/3+ others− NK cells (p = .02) (Figure 1g). Although the KIR2DL/HLA-C interaction inhibited the rituximab-dependent educated KIR2DL+ others− NK cell degranulation, the latter was brought down to a higher level compared to uneducated KIR2DL+ others− NK cells. Together, these results show a significant inhibitory effect of KIR2DL on NK ADCC and the hyporesponsiveness of uneducated KIR2DL+ NK cells.
Inhibitory KIR3DL1-Bw4 interaction brings rituximab-dependent NK cell degranulation down to the degranulation baseline of uneducated KIR3DL1+ NK cells
Rituximab-dependent KIR3DL1+ others− NK cell degranulation was significantly inhibited against the Bw4+ EBV-B cell line compared with the Bw4− EBV-B cell line after rituximab NK cell activation (p = .0002) (Figure 1h). Educated KIR3DL1+ NK cells displayed better degranulation than uneducated ones against Bw4− and Bw4+ target cells (Figure 1i). However, the differences in degranulation were not significant probably due to the broad polymorphism of KIR3DL1, which is known to drastically impact its membrane expression and inhibitory effect. As previously observed for KIR2DL, although the KIR3DL1/HLA-Bw4 interaction inhibited the rituximab-dependent educated KIR3DL1+ others− NK cell degranulation, the latter was brought down to a higher level compared to uneducated KIR3DL1+ others− NK cells underlying the hyporesponsiveness of these uneducated NK cells.
Activating KIR2DS1 signal enhances rituximab-dependent NK cell degranulation from C2− blood donors
The activating KIR2DS1 recognizes HLA-C C2 ligands as its inhibitory homolog KIR2DL1.11,12,29,30 Using the same KIR-specific mAb combination, we further investigated the activating potential of KIR2DS1 on rituximab-dependent NK cell degranulation. The degranulation of KIRDS1+ others− (KIR2DL1/2/3−/3DL1−) NK cell subset was higher after rituximab NK cell activation against both C2+ EBV-B cell lines compared to the C2− EBV-B cell line (p = .0246) (Figure 1j). We observed two groups of responders against the C2+ EBV-B cell line. Indeed, this observation was particularly obvious for C1C1 compared to C2C2 blood donors (p < .0001) (Figure 1k) in accordance with the fact that only KIR2DS1+ NK cells from C2− blood donors are C2 alloreactive.
Lower modulator effect of KIR2DL2/3 than KIR2DL1, KIR3DL1 and KIR2DS1 on rituximab-dependent NK cell degranulation
After considering each KIR+ NK cell subset, we further compared them against each EBV-B target cell. The KIR− NK cell subset was included in our analysis (supplemental Figure S2A) as a control cell subset, since its rituximab-dependent degranulation was similar against all EBV-B cell lines whatever their HLA class I genotype in terms of KIR ligands (supplemental figure S2B).
Inhibitory KIR+ NK cells displayed similar rituximab-dependent degranulation against the C1C1 Bw4− EBV-B cell line despite potential inhibition mediated by the KIR2DL2/3/C1 interaction (Figure 2a). Of note, KIR2DS1+ others− and KIR− NK cell subsets showed lower rituximab-dependent degranulation (p = .02 between KIR2DS1+ others− and KIR2DL1+ others− NK cell subsets, p = .005 between KIR− and KIR2DL1+ others− NK cell subsets). In contrast, KIR2DL1/C2 inhibition and KIR2DS1/C2 activation limited rituximab dependent NK cell degranulation against the C2C2 Bw4− EBV-B cell line (Figure 2b) (p < .0001 between KIR2DL1+others− and KIR3DL1+ others− NK cell subsets). Finally, KIR2DL1/C2 and KIR3DL1/Bw4 inhibition and KIR2DS1/C2 activation also reduced rituximab-dependent NK cell degranulation against the C2C2 Bw4+ EBV-B cell line (Figure 2c). Once more, KIR− NK cell subsets showed low rituximab-dependent NK cell degranulation against these last EBV-B cell lines (Figure 2b-c). Taken together, these results highlight a lower modulator effect of KIR2DL2/3 with its C1 ligands on NK cell rituximab-dependent NK cell degranulation compared to inhibitory KIR2DL1 and KIR3DL1 and activating KIR2DS1.
Figure 2.
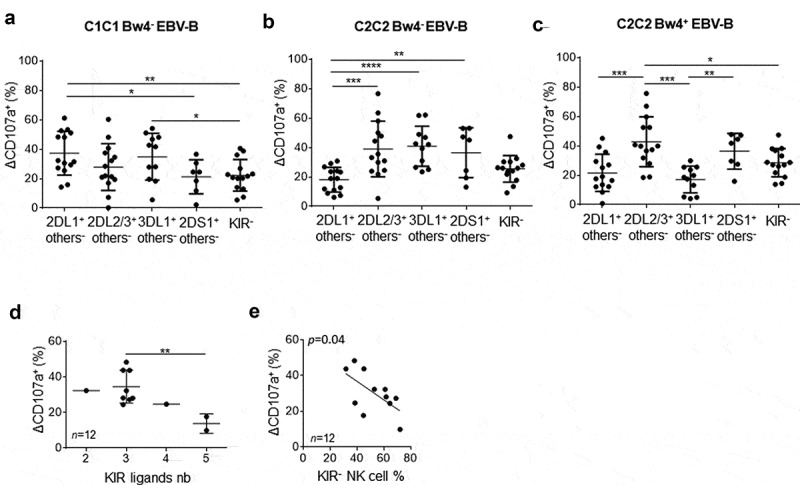
Lower modulator effect of KIR2DL2/3 than KIR2DL1, KIR3DL1 and KIR2DS1 on rituximab-dependent NK cell degranulation. RTX-dependent NK cell degranulation (Δ CD107a+) frequency for each targeted NK cell subset: 2DL1− others−, 2DL2/3 others−, 3DL1+ others−, 2DS1+ others− and KIR- NK cells against (a) C1C1 Bw4− EBV-B cell line, (b) C2C2 Bw4− EBV-B cell line, and (c) C2C2 Bw4+ EBV-B cell line. ANOVA test. (d) RTX-dependent NK cell degranulation (Δ CD107a+) frequency following KIR ligands number. The Student’s t-test was performed between the series with three and five KIR ligands. (e) Correlation between percentage of RTX-dependent NK cell degranulation (Δ CD107a+) frequency and the KIR− NK cell frequency. *p < .05, **p < .01, ***p < .005, ****p < .0001
In order to obtain an overview of how KIR and HLA class I molecules affect rituximab-dependent NK cell degranulation, we analyzed the NK cell degranulation of all NK cells taking into account the number of KIR ligands and the frequency of peripheral blood KIR− and KIR+ NK cells in studied blood donors. For this, we chose NK cell degranulation against the EBV-B cell line, which does not express any KIR ligand susceptible to inhibiting NK cell functions. Thus, both C1C1 Bw4− and C2C2 Bw4− EBV-B cell lines were identified as target cells with only one KIR ligand (C1 or C2). In parallel, we excluded all C1C2 blood donors as they possess educated KIR2DL1 and KIR2DL2/3 NK cells. We kept only the results obtained for C1C1 or C2C2 blood donors in this analysis. For instance, NK cell degranulation from C1C1 and C2C2 Bw4− or Bw4+ blood donors was retained against C2C2 Bw4− and C1C1Bw4− EBV-B cell lines, respectively. Interestingly, the higher the cell degranulation, the more the number of KIR ligands limits the frequency of rituximab-dependent CD107a+ NK cells (p = .01) (Figure 2d). Of note, the frequency of KIR− NK cells correlates inversely with rituximab-dependent CD107a+ NK cells (p = .04) (Figure 2e).
Lower KIR ligand numbers and peripheral blood KIR− NK cell frequency are good prognostic markers of rituximab responses in lymphoma patients
The influence of CD16, the number of KIR ligands, and the frequency of peripheral blood KIR− and KIR+ NK cells were evaluated in a cohort of 74 NHL patients treated with rituximab as part of immunochemotherapy (Table 1).
Table 1.
Characteristics of patients
| Characteristics | CR (N = 34) |
PR (N = 29) |
PD/SD (N = 11) |
Total (N = 74) |
p Value |
|---|---|---|---|---|---|
| Gender | |||||
| Female Male |
15 (44) 19 (47.5) |
15 (44) 14 (35) |
4 (12) 7 (17.5) |
34 (46) 40 (54) |
|
| Age (years) | |||||
| <65 >65 |
13 (56) 21 (41) |
8 (35) 21 (41) |
2 (9) 9 (18) |
23 (31) 51 (69) |
|
| Disease | 0.0085 | ||||
| High grade NHL Other low grade NHL |
20 (67) 14 (32) |
6 (20) 23 (52) |
4 (13) 7 (16) |
30 (41) 44 (59) |
|
| CMV status | |||||
| Positive Negative ND |
13 (52) 13 (45) 8 (40) |
9 (36) 13 (45) 7 (35) |
3 (12) 3 (10) 5 (25) |
25 (34) 29 (39) 20 (27) |
|
| CD16 genotype | 0.4656 | ||||
| VV VF FF |
3 (37.5) 14 (41) 17 (53) |
3 (37.5) 13 (38) 13 (41) |
2 (25) 7 (21) 2 (6) |
8 (11) 34 (46) 32 (43) |
|
| HLA-C ligands | 0.569 | ||||
| C1C1 C1C2 C2C2 |
10 (39) 19 (50) 5 (50) |
11 (42) 13 (34) 5 (50) |
5 (19) 6 (16) 0 (0) |
26 (35) 38 (51) 10 (14) |
|
| HLA-A/B Bw4 ligands | 0.5216 | ||||
| Bw4+ Bw4- |
22 (43) 12 (52) |
21 (41) 8 (35) |
8 (16) 3 (13) |
51 (69) 23 (31) |
|
| HLA-A3/A11 ligands | 0.8716 | ||||
| A3/A11+ A3/A11- |
10 (42) 24 (48) |
10 (42) 19 (38) |
4 (16) 7 (14) |
24 (32) 50 (68) |
Data are presented as n (%).
Abbreviations: CR: Complete response; PR: partial response; SD: stable disease; PD: progressive disease; CMV: cytomegalovirus; HLA: Human Lymphocytic Antigen.
The distribution of CD16 genotypes in the cohort (10% VV, 47% VF, and 43% FF) was consistent with known frequencies.5,26 Of note, no association between CD16-158V/F polymorphism and the rituximab response was observed (Figure 3a).
Figure 3.
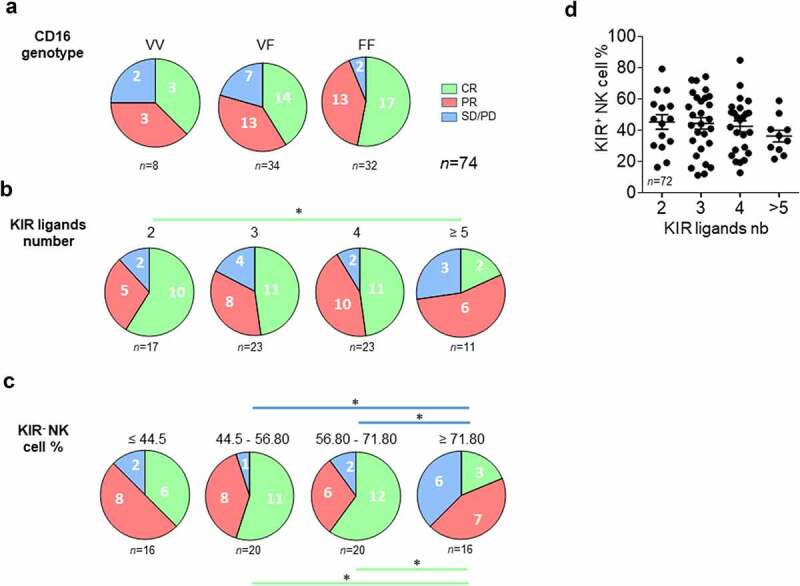
Lower KIR ligand numbers and peripheral blood KIR− NK cell frequency are good prognostic markers of rituximab responses in lymphoma patients. (a) Pie charts representing the distribution of lymphoma patients (n = 74) following clinical outcome (CR, PR, and SD/PD) and CD16 polymorphism (VV, VF, and FF). (b) Pie charts representing the distribution of lymphoma patients following clinical outcome (CR, PR, and SD/PD) and the number of KIR ligands (2, 3, 4 or ≥5). (c) Pie charts representing the distribution of lymphoma patients following clinical outcome (CR, PR, and SD/PD) and the KIR− NK cell frequency (≤ 44.5, 44.5–56.80, 56.80–71.80 and ≥ 71.80). (d) KIR+ NK cell frequencies following the number of KIR ligands for all lymphoma patients. The green and blue lines determine the significant difference between 2 groups for CR and SD/PD outcomes *p < .05
The clinical response to rituximab was analyzed according to the number of KIR ligands and the frequency of peripheral blood KIR− NK cells. A phenotype of NK cells was obtained from patient PBMCs to determine whether KIR2DL1, KIR2DL2/3, and KIR3DL1 were expressed (supplemental Figure S3). A KIR-specific mAb combination led us to determine KIR− and KIR+ NK cell frequencies. NKG2A and NKG2C were also evaluated. KIR2DL1 and KIR2DL2/3 were always expressed on NK cells from all studied patients and only six Bw4+ patients did not express a membrane KIR3DL1, as illustrated for a representative patient in supplementary figure S3C-D. In parallel, from their HLA typing, we deduced the number of KIR ligands (A3/A11, Bw4, C1, and C2). We determined the number of KIR ligands for which the corresponding KIR was expressed in NK cells. C1, C2, and A3/A11 were always identified as KIR ligands as the cognate receptor was expressed in NK cells of all patients. In contrast, HLA-A and HLA-B molecules with a Bw4 motif were identified as KIR ligands only for patients expressing membrane KIR3DL1. Thus, four groups of patients were determined with two, three, four or more than 5 KIR ligands (Figure 3b). Complete responses were significantly higher in the group of patients displaying only 2 KIR ligands compared to those with more than five KIR ligands (p = .03) (Figure 3b).
To evaluate the impact of KIR− NK cell frequency on the clinical outcome, we determined 4 four groups of patients including around 16 and 20 patients with lower to higher frequencies (Figure 3c). PD is more frequent in patients displaying a high frequency of KIR− NK cells (≥ 71.80%) compared to low frequency (44.5–56.80%, p = .01; 56.80–71.80%, p = .04). In contrast, CR is less frequent in patients displaying a high frequency of KIR− NK cells (≥ 71.80%) compared to low frequency (44.5–56.80%, p = .02; 56.80–71.80%, p = .01) (Figure 3c). Even though it was nonsignificant, we observed that the frequency of KIR+ NK cells decreased with an increased number of KIR ligands (Figure 3d).
Low KIR ligand numbers and peripheral blood KIR− NK cell frequency are good prognostic markers of rituximab responses in low-grade lymphoma patients
Details on the treatment and pathologies of NHL patients are presented in supplemental table S2. Forty-four patients were treated for low grade (LG) NHL (FL, n = 24; WM, n = 16; other, n = 4) and 30 were treated for high-grade (HG) (DLBCL, n = 16; MCL, n = 12; other, n = 2). As expected, given the intensity of treatments received, a higher rate of CR was observed in patients treated for a HG NHL than LG NHL (p = .003) (Figure 4a). The distribution of CD16 genotype, number of KIR ligands, and frequency of peripheral blood KIR− NK cells were comparable between patients treated for HG and LG NHL (Figure 4b-d).
Figure 4.
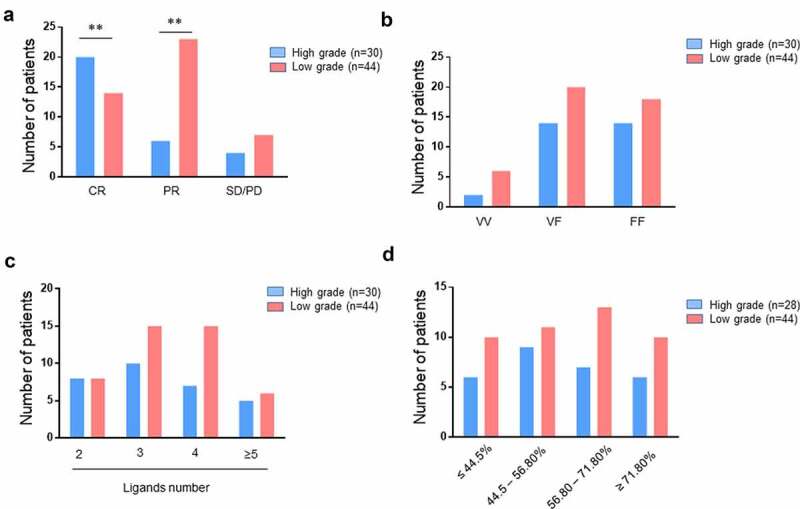
Distribution of lymphoma patients following clinical grade level. histograms representing the distribution of high-grade and low-grade lymphoma patients (n = 74) following (a) clinical outcome (CR, PR, and SD/PD), (b) CD16 polymorphism (VV, VF, and FF), (c) the number of KIR ligands (2, 3, 4 or ≥5) and (d) the KIR− NK cell frequency (≤ 44.5, 44.5–56.80, 56.80–71.80 and ≥ 71.80). *p < .05
The same analysis in the LG subgroup showed analogous results, with a 75% CR rate in patients displaying 2 KIR ligands vs. 0% in patients displaying ≥5 KIR ligands (p = .03) (Figure 5a). HG subgroup analysis showed an unfavorable trend for patients with ≥5 KIR ligands, with no significant difference, probably due to the small sample size (Figure 5b). The same result was observed when analyzing the LG subgroup; patients exhibiting higher frequency of KIR− NK cells (≥ 71.80%) had lower CR (p = .01) and higher PD (p = .01) (Figure 5c). By contrast, the frequency of KIR− NK cells in the HG subgroup was not an important factor for explaining the classification of patients based on their clinical outcome (Figure 5d).
Figure 5.
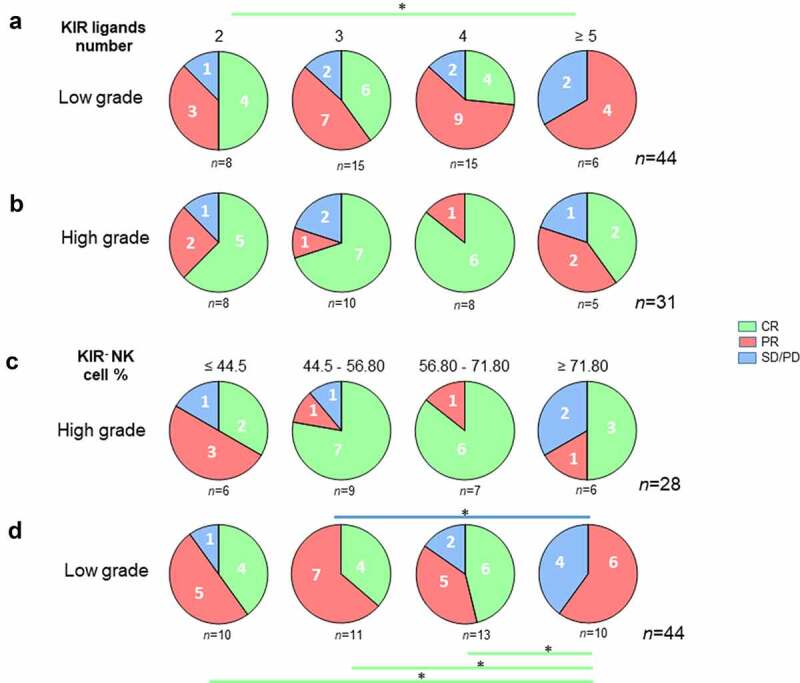
Lower KIR ligand numbers and peripheral blood KIR− NK cell frequency are good prognostic markers of rituximab responses in low-grade lymphoma patients. Pie charts representing the distribution of (a) low-grade and (b) high-grade lymphoma patients following clinical outcome (CR, PR, and SD/PD) and the number of KIR ligands (2, 3, 4 or ≥5). The green line determines the significant difference between 2 groups for CR outcome. *p < .05. Pie charts representing the distribution of (c) low-grade and (d) high-grade lymphoma patients following clinical outcome (CR, PR, and SD/PD) and the KIR− NK cell frequency (≤ 44.5, 44.5–56.80, 56.80–71.80 and ≥ 71.80). The green and blue lines determine the significant difference between 2 groups for CR and SD/PD outcomes. *p < .05
Discussion
In our in vitro cellular model, we determined the optimal rituximab concentration (0.1 µg/mL), in accordance with previous studies,16 leading to an assessment of KIR/HLA inhibition of rituximab-dependent NK cell degranulation from blood donors with CD16-158VV or VF genotype to rule out the influence of CD16-158 FF polymorphism. All inhibitory KIR investigated in our cellular model inhibited rituximab-dependent NK cell degranulation, as previously observed by other groups with different cellular models.9,16,17 Nonetheless, we observed a differential contribution of inhibitory KIR, as KIR2DL1 had a greater effect than KIR2DL2/3 or KIR3DL1. Educated KIR NK cells had higher responsiveness compared to their uneducated counterparts. However, the inhibitory effect was similar for both NK cell subsets. Thus, despite the inhibitory signal mediated by KIR/HLA interactions, educated NK cells constitute the best subset to mediate high ADCC rituximab-dependent NK cell degranulation. Our observations are along the same lines as those previously reported in HIV models.17,18 However, they disagree with previous studies showing a dominance of uneducated NK cells in anti-GD215 and in rituximab-dependent NK ADCC responses.16 Finally, we observed a negative correlation between rituximab-dependent NK cell degranulation and the number of KIR ligands and between degranulation and KIR+ NK cell frequency.
To elaborate further on the clinical side, we observed an increased frequency of KIR− NK cells and inversely decreased frequency of KIR+ NK cells associated with disease progression in lymphoma patients. These results are in accordance with the NK cell dysfunction observed in chronic lymphocytic leukemia (CLL) associated with a striking reduction in the frequency of NK cells expressing KIR2DL1 and/or KIR3DL1, which progresses over time in most patients.31 Our observation can be explained by the poor contribution of KIR− NK cells to rituximab-dependent ADCC against all EBV-B cell lines in our cellular model in contrast to educated and uneducated KIR+ NK cells. It is known that the induction of ADCC resistance can be explained in part by the formation of non-optimal immune synapse.32 Genetic diversity in KIRs affects membrane-proximal signaling and the formation of activating immune synapses.33 KIR− NK cells may present a non-optimal immune synapses due to the absence of education via KIR/HLA interactions. Moreover, our study emphasizes the influence of the number of KIR ligands on the frequency of KIR+ NK cells, showing that the frequency of cells decreases with the number of ligands, and inversely for KIR− NK cells. Thus, KIR+ NK cells appear essential to drive efficient ADCC rituximab-dependent degranulation, whatever their education status, as even uneducated, KIR+ NK cells demonstrate higher responsiveness than KIR− NK cells in rituximab-dependent degranulation (Figure 6).
Figure 6.
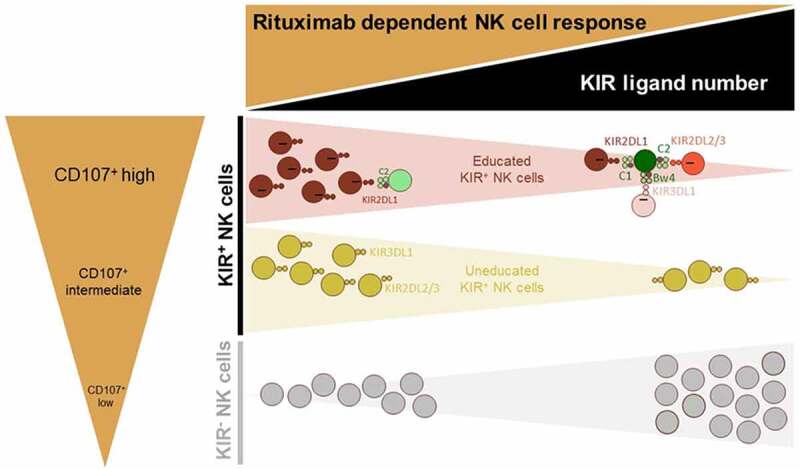
Graphic overview of the functional articulation between the number of KIR ligands and main NK cell subsets in modulating rituximab-dependent NK ADCC responses. A sizable KIR ligand environment favors a higher frequency of inefficient KIR− NK cells and numerous functional KIR/HLA inhibitions of educated KIR+ NK cells. By contrast, a low number of KIR ligands favor higher frequency of uneducated and educated NK cells that harbor a better rituximab-dependent ADCC than KIR− NK cells
Furthermore, we describe for the first time the triggering effect of the KIR2DS1-C2 interaction on rituximab-dependent NK cell degranulation. Although the triggering effect of KIR2DS1 on the rituximab-dependent total NK cell response is limited due to the low frequency of KIR2DS1+ NK cells, it can be an advantage to select KIR2DS1+ C2− NK cells with the overarching goal of obtaining an efficient CD16+ cell line for ADCC, as previously described.34
The deleterious influence of the CD16-158F/F genotype has been described with regard to the clinical outcomes of NHL patients after rituximab treatment.5 Against this HLA background, CD16 polymorphism appears to have a collateral effect, as no association between CD16-158V/F polymorphism and rituximab responses was observed in 74 NHL patients. This discrepancy may be due to the lower number of NHL patients (n = 49) in the original study and the focus only on the CD16-158V/F polymorphism, as the authors themselves suggested other potential genetic polymorphisms to explain their observations. Moreover, it has been shown that this functional difference of NK cells between VV and FF genotyped patients is hidden when high rituximab concentrations are used.7 Delgado et al. showed an association between relapsed/refractory neuroblastoma (NB) patients with KIR/KIR-ligand mismatches and better response/improvement to humanized anti-GD2 monoclonal antibody linked to human IL2 in a phase II trial.35 Interestingly, in their study, autologous KIR/KIR-ligand mismatches were more meaningful than the CD16 polymorphism in this study. Due to the limited number of patients and the low frequency of the CD16 158-V/V variant, no significant association between this genotype and response was determined.
In this study, we focused our investigations on KIR-HLA interactions; however, CD94/NKG2A-HLA-E interaction contributes to inhibiting NK cell function and educating NK cells. It has been shown that NKG2A+ NK cell frequency is inversely correlated with KIR+ NK cell frequency and KIR number.36 Our results suggest that rituximab-dependent NK ADCC is improved when KIR+ NK cell frequency is high and indirect when CD94/NKG2A+ NK cell frequency is low. Deep investigations into the effect of CD94/NKG2A on rituximab-dependent NK ADCC should be conducted in order to evaluate the potential interest in monalizumab to improve lymphoma treatment.37 Indeed, additive approaches may improve the poor clinical prognosis of NHL patients on rituximab treatment.38 For instance, it has been demonstrated that the combination of rituximab and inhibitory KIR blocking mAbs yields improved NK cell-mediated lysis over rituximab alone.39,40,41
KIR and HLA gene families constitute the most polymorphic and polygenic receptor/ligand pair in humans, and their functional interactions drive a large diversity of NK cells with a limited number of germline-encoded genes.41 Our results illustrate in part the major impact of KIR and HLA polymorphism on the structural and functional formation of the NK cell repertoire, as was recently shown (Makanga et al. on Cancer). Numerous approaches to NK cell immunotherapy are developed using mainly autologous or allogeneic NK cell bulk without taking into account all genetic and allelic KIR and HLA polymorphisms. More investigations are required to better define the most efficient NK cell subsets and the NK cell donors from genetic markers to improve promising NK cell immunotherapies.
Taken together, our results permit an overview on the influence of KIR, KIR ligands and CD16 polymorphisms on rituximab-dependent NK cell cytotoxicity, integrating genetic rules of the structural and functional formation of the NK cell repertoire. These results pave the way for strengthening the prediction of rituximab responses in NHL patients. Indeed, two digit HLA class I types (HLA-A, -B, and -C) determined by molecular biology and/or KIR+ NK cell frequency determined by flow cytometry could be simple and useful markers for predicting rituximab responses.
Supplementary Material
Acknowledgments
We thank all blood donor volunteers and lymphoma patients who accepted to participate in the study. We also thank Dr Catherine Pellat-Deceunynck and Dr Fabienne Haspot for their advice.
Funding Statement
This work was financially supported by the Etablissement Français du Sang (EFS)/Centre Pays de la Loire and by grants from la Ligue contre le Cancer (comité de Loire-Atlantique et comité de Charente Maritime), Leucémie Espoir Atlantique Famille (LEAF) and l’Agence de Biomédecine (ABM). DRM is a PhD student supported by Industrial Agreement for Training Through Research Grants 2017/0850. LD is a PhD student supported by la Région Pays de la Loire/EFS Centre Pays de la Loire (N°2018-09766);Région Centre Pays de la Loire/EFS [N°2018-09766];ANRT [2017/0850];
Author’s contributions
Study design: BC and CR; Acquisition and analysis of flow cytometry data: DRM, GD and CW; HLA typing: AW; KIR genotyping: NL, LD and KG; FcγRIIIa (CD16) genotyping: DRM, NL and GD; Patient’s sample collection and clinical documentation: MJ, CT, TG, BT, SLG, BM and PC; Data interpretation: DRM, MJ and CR; Figure and table design: DRM, MJ and CR. Manuscript writing: DRM, MJ and CR; All authors read and approved the final manuscript.
Abbreviations
HLA, human leucocyte antigen; KIR, killer cell immunoglobulin-like receptor; NK, natural killer; PBMC, peripheral blood mononuclear cell; ADCC, antibody dependent cellular cytotoxicity
Ethics approval and consent to participate
The study complies with the Declaration of Helsinki. In addition, declaration of the preparation and conservation of a bio-collection (DC-2014-2340) was submitted to the French Ministry of Research and had received agreement from the IRB (2015- DC-1) to study NK cell reconstitution after transplant. This study was approved by the Ethics Review Board of the Nantes University Hospital and all included patients and blood donors provided informed consent.
Consent for publication
Not required
Availability of data and material
The data that support the findings of this study are included in the article or uploaded as supplementary information. Data are available from the corresponding author on reasonable request.
Competing interests
The authors declare that they have no competing interests.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Cartron G, Watier H, Golay J, Solal-Celigny P.. From the bench to the bedside: ways to improve rituximab efficacy. Blood. 2004;104(9):2635–12. doi: 10.1182/blood-2004-03-1110. [DOI] [PubMed] [Google Scholar]
- 2.Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. 2007;44(16):3823–3837. doi: 10.1016/j.molimm.2007.06.151. [DOI] [PubMed] [Google Scholar]
- 3.Alderson KL, Sondel PM. Clinical Cancer Therapy by NK Cells via Antibody-Dependent Cell-Mediated Cytotoxicity. J Biomed Biotechnol. 2011;2011:1–7. doi: 10.1155/2011/379123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker PSA, Suck G, Nowakowska P, Ullrich E, Seifried E, Bader P, Tonn T, Seidl C. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol Immunother. 2016;65(4):477–484. doi: 10.1007/s00262-016-1792-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–758. doi: 10.1182/blood.V99.3.754. [DOI] [PubMed] [Google Scholar]
- 6.Veeramani S, Wang S-Y, Dahle C, Blackwell S, Jacobus L, Knutson T, Button A, Link BK, Weiner GJ. Rituximab infusion induces NK activation in lymphoma patients with the high-affinity CD16 polymorphism. Blood. 2011;118(12):3347–3349. doi: 10.1182/blood-2011-05-351411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dall’Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, Watier H, Thibault G. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004;64(13):4664–4669. doi: 10.1158/0008-5472.CAN-03-2862. [DOI] [PubMed] [Google Scholar]
- 8.Borgerding A, Hasenkamp J, Engelke M, Burkhart N, Trümper L, Wienands J, Glass B. B-lymphoma cells escape rituximab-triggered elimination by NK cells through increased HLA class I expression. Exp Hematol. 2010;38(3):213–221. doi: 10.1016/j.exphem.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 9.Terszowski G, Klein C, Stern M. KIR/HLA interactions negatively affect rituximab- but not GA101 (obinutuzumab)-induced antibody-dependent cellular cytotoxicity. J Immunol Baltim Md. 2014;192(12):5618–5624. 1950 doi: 10.4049/jimmunol.1400288. [DOI] [PubMed] [Google Scholar]
- 10.Anfossi N, André P, Guia S, Falk CS, Roetynck S, Stewart CA, Breso V, Frassati C, Reviron D, Middleton D, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25(2):331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 11.Morvan M, David G, Sébille V, Perrin A, Gagne K, Willem C, Kerdudou N, Denis L, Clémenceau B, Folléa G, et al. Autologous and allogeneic HLA KIR ligand environments and activating KIR control KIR NK-cell functions. Eur J Immunol. 2008;38(12):3474–3486. doi: 10.1002/eji.200838407. [DOI] [PubMed] [Google Scholar]
- 12.Chewning JH, Gudme CN, Hsu KC, Selvakumar A, Dupont B. KIR2DS1-positive NK cells mediate alloresponse against the C2 HLA-KIR ligand group in vitro. J Immunol Baltim Md. 2007;179(2):854–868. 1950 [DOI] [PubMed] [Google Scholar]
- 13.Pittari G, Liu X-R, Selvakumar A, Zhao Z, Merino E, Huse M, Chewning JH, Hsu KC, Dupont B. NK cell tolerance of self-specific activating receptor KIR2DS1 in individuals with cognate HLA-C2 ligand. J Immunol Baltim Md. 2013;190(9):4650–4660. 1950 doi: 10.4049/jimmunol.1202120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, Tyan D, Lanier LL, Parham P. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7(6):753–763. doi: 10.1016/S1074-7613(00)80394-5. [DOI] [PubMed] [Google Scholar]
- 15.Tarek N, Le Luduec J-B, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, Modak S, Heller G, Dupont B, Cheung N-KV, et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Invest. 2012;122(9):3260–3270. doi: 10.1172/JCI62749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du J, Lopez-Verges S, Pitcher BN, Johnson J, Jung S-H, Zhou L, Hsu K, Czuczman MS, Cheson B, Kaplan L, et al. CALGB 150905 (Alliance): rituximab broadens the antilymphoma response by activating unlicensed NK cells. Cancer Immunol Res. 2014;2(9):878–889. doi: 10.1158/2326-6066.CIR-13-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lisovsky I, Kant S, Tremblay-mclean A, Isitman G, Kiani Z, Dupuy FP, Gilbert L, Bruneau J, Shoukry NH, Lebouché B, et al. Differential contribution of education through KIR2DL1, KIR2DL3, and KIR3DL1 to antibody-dependent (AD) NK cell activation and ADCC. J Leukoc Biol. 2019;105(3):551–563. doi: 10.1002/JLB.4A0617-242RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kristensen AB, Kent SJ, Parsons MS, Silvestri G. Contribution of NK Cell Education to both Direct and Anti-HIV-1 Antibody-Dependent NK Cell Functions. J Virol. 2018;92(11):11. doi: 10.1128/JVI.02146-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forlenza CJ, Boudreau JE, Zheng J, Le Luduec J-B, Chamberlain E, Heller G, Cheung N-KV, Hsu KC. KIR3DL1 Allelic Polymorphism and HLA-B Epitopes Modulate Response to Anti-GD2 Monoclonal Antibody in Patients With Neuroblastoma. J Clin Oncol Off J Am Soc Clin Oncol. 2016;34(21):2443–2451. doi: 10.1200/JCO.2015.64.9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erbe AK, Wang W, Carmichael L, Hoefges A, Grzywacz B, Reville PK, Ranheim EA, Hank JA, Kim K, Seo S, et al. Follicular lymphoma patients with KIR2DL2 and KIR3DL1 and their ligands (HLA-C1 and HLA-Bw4) show improved outcome when receiving rituximab. J Immunother Cancer. 2019;7(1):70. doi: 10.1186/s40425-019-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, Coiffier B, Fisher RI, Hagenbeek A, Zucca E, et al. Revised response criteria for malignant lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. 2007;25(5):579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 22.Owen RG, Kyle RA, Stone MJ, Rawstron AC, Leblond V, Merlini G, Garcia-Sanz R, Ocio EM, Morra E, Morel P, et al. Response assessment in Waldenström macroglobulinaemia: update from the VIth International Workshop. Br J Haematol. 2013;160(2):171–176. doi: 10.1111/bjh.12102. [DOI] [PubMed] [Google Scholar]
- 23.Willem C, Makanga DR, Guillaume T, Maniangou B, Legrand N, Gagne K, Peterlin P, Garnier A, Béné MC, Cesbron A, et al. Impact of KIR/HLA Incompatibilities on NK Cell Reconstitution and Clinical Outcome after T Cell–Replete Haploidentical Hematopoietic Stem Cell Transplantation with Posttransplant Cyclophosphamide. J Immunol. 2019;202(7):2141–2152. doi: 10.4049/jimmunol.1801489. [DOI] [PubMed] [Google Scholar]
- 24.Sun JY, Gaidulis L, Miller MM, Goto RM, Rodriguez R, Forman SJ, Senitzer D. Development of a multiplex PCR-SSP method for Killer-cell immunoglobulin-like receptor genotyping. Tissue Antigens. 2004;64(4):462–468. doi: 10.1111/j.1399-0039.2004.00303.x. [DOI] [PubMed] [Google Scholar]
- 25.Hsu KC, Chida S, Geraghty DE, Dupont B. The killer cell immunoglobulin-like receptor (KIR) genomic region: gene-order, haplotypes and allelic polymorphism. Immunol Rev. 2002;190(1):40–52. doi: 10.1034/j.1600-065X.2002.19004.x. [DOI] [PubMed] [Google Scholar]
- 26.Leppers-van de Straat FG, Van Der Pol WL, Jansen MD, Sugita N, Yoshie H, Kobayashi T, van de Winkel JG. A novel PCR-based method for direct Fc gamma receptor IIIa (CD16) allotyping. J Immunol Methods. 2000;242(1–2):127–132. doi: 10.1016/S0022-1759(00)00240-4. [DOI] [PubMed] [Google Scholar]
- 27.Bowles JA, Weiner GJ. CD16 polymorphisms and NK activation induced by monoclonal antibody-coated target cells. J Immunol Methods. 2005;304(1–2):88–99. doi: 10.1016/j.jim.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 28.Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, Luo X, Cooley S, Verneris M, Walcheck B, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood. 2013;121(18):3599–3608. doi: 10.1182/blood-2012-04-425397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fauriat C, Ivarsson MA, Ljunggren H-G, K-j M, Michaëlsson J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood. 2010;115(6):1166–1174. doi: 10.1182/blood-2009-09-245746. [DOI] [PubMed] [Google Scholar]
- 30.Foley B, De Santis D, Lathbury L, Christiansen F, Witt C. KIR2DS1-mediated activation overrides NKG2A-mediated inhibition in HLA-C C2-negative individuals. Int Immunol. 2008;20(4):555–563. doi: 10.1093/intimm/dxn013. [DOI] [PubMed] [Google Scholar]
- 31.MacFarlane AW, Jillab M, Smith MR, Alpaugh RK, Cole ME, Litwin S, Millenson MM, Al-Saleem T, Cohen AD, Campbell KS. NK cell dysfunction in chronic lymphocytic leukemia is associated with loss of the mature cells expressing inhibitory killer cell Ig-like receptors. Oncoimmunology. 2017;6(7):e1330235. doi: 10.1080/2162402X.2017.1330235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aldeghaither DS, Zahavi DJ, Murray JC, Fertig EJ, Graham GT, Zhang Y-W, O’Connell A, Ma J, Jablonski SA, Weiner LM. A Mechanism of Resistance to Antibody-Targeted Immune Attack. Cancer Immunol Res. 2019;7(2):230–243. doi: 10.1158/2326-6066.CIR-18-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennedy PR, Barthen C, Williamson DJ, Pitkeathly WTE, Hazime KS, Cumming J, Stacey KB, Hilton HG, Carrington M, Parham P, et al. Genetic diversity affects the nanoscale membrane organization and signaling of natural killer cell receptors. Sci Signal. 2019;12(612):612. doi: 10.1126/scisignal.aaw9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clémenceau B, Vivien R, Pellat C, Foss M, Thibault G, Vié H. The human natural killer cytotoxic cell line NK-92, once armed with a murine CD16 receptor, represents a convenient cellular tool for the screening of mouse mAbs according to their ADCC potential. mAbs. 2013;5(4):587–594. doi: 10.4161/mabs.25077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delgado DC, Hank JA, Kolesar J, Lorentzen D, Gan J, Seo S, Kim K, Shusterman S, Gillies SD, Reisfeld RA, et al. Genotypes of NK cell KIR receptors, their ligands, and Fcγ receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res. 2010;70(23):9554–9561. doi: 10.1158/0008-5472.CAN-10-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fauriat C, Andersson S, Björklund AT, Carlsten M, Schaffer M, Björkström NK, Baumann BC, Michaëlsson J, Ljunggren H-G, Malmberg K-J. Estimation of the Size of the Alloreactive NK Cell Repertoire: studies in Individuals Homozygous for the Group A KIR Haplotype. J Immunol. 2008;181(9):6010–6019. doi: 10.4049/jimmunol.181.9.6010. [DOI] [PubMed] [Google Scholar]
- 37.André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Bléry M, Bonnafous C, Gauthier L, Morel A, et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell. 2018;175(7):1731–1743.e13. doi: 10.1016/j.cell.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM, Cell-Mediated Antibody-Dependent NK. Cellular Cytotoxicity in Cancer Immunotherapy. Front Immunol. 2015;6:368. doi: 10.3389/fimmu.2015.00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, Weiner LM. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J Immunol Baltim Md. 2008;180(9):6392–6401. 1950 doi: 10.4049/jimmunol.180.9.6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, Fuseri N, Bonnafous C, Czerwinski D, Rajapaksa A, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. 2014;123(5):678–686. doi: 10.1182/blood-2013-08-519199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parham P. Co-evolution of lymphocyte receptors with MHC class I. Immunol Rev. 2015;267(1):1–5. doi: 10.1111/imr.12338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are included in the article or uploaded as supplementary information. Data are available from the corresponding author on reasonable request.