

ABSTRACT
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest forms of cancer. The elevated macroautophagy/autophagy in these tumors supports growth, promotes immune evasion, and increases therapeutic resistance. Therefore, targeting autophagy is a therapeutic strategy that is being pursued to treat PDAC patients. Whereas autophagy inhibition impairs mitochondrial metabolism in PDAC, the specific metabolite(s) that becomes limiting when autophagy is inhibited has not been identified. We report that loss of autophagy specifically results in intracellular cysteine depletion under nutrient-replete conditions. Mechanistically, we show that PDAC cells utilize the autophagy machinery to regulate the activity and localization of the cystine transporter SLC7A11 at the plasma membrane. Upon inhibition of autophagy, SLC7A11 is localized to lysosomes in an MTORC2-dependent manner. Our findings reveal a novel connection between autophagy and cysteine metabolism in pancreatic cancer.
KEYWORDS: Autophagy, cysteine, lysosome, metabolism, pancreatic ductal adenocarcinoma, SLC7A11
In our recent report [1], we investigated the previously unknown metabolic contribution of autophagy that supports PDAC metabolism. Consistent with our previous work, loss of autophagy (pharmacologically using chloroquine [CQ] or RNAi to target essential autophagy genes) causes a significant decrease in proliferation and clonogenic growth which can be rescued by N-acetyl-L-cysteine (NAC). Interestingly, none of the other antioxidants tested are able to rescue the growth inhibition. We initially hypothesized that unlike other antioxidants, NAC might serve as a source of cysteine (Cys). Consistent with this, we found that pharmacological or genetic inhibition of autophagy in PDAC cells leads to a significant drop of intracellular Cys under nutrient-replete conditions, whereas other amino acids are only minimally affected. In PDAC, cystine (oxidized dimer of cysteine) transport is critical and is regulated by system xc− (cystine/glutamate antiporter) which is comprised of SLC7A11/xCT and SLC3A2. We found that there is a significant accumulation of SLC7A11 in multiple PDAC cell lines upon autophagy inhibition. This is also seen in murine PDAC tumors in vivo by immunohistochemical analysis, where autophagy is inhibited either by expressing an ATG4B dominant negative mutant, CQ treatment, or RNAi to Atg7. Given the drop in intracellular Cys, despite an accumulation of SLC7A11 after autophagy inhibition, we hypothesized that the activity of SLC7A11 may be impaired. Along these lines, we found the cystine uptake and glutamate secretion, representing the activity of SLC7A11, demonstrate a significant decline after autophagy inhibition. Importantly, we also observed that this drop in activity is accompanied by the cytoplasmic translocation of SLC7A11 from the plasma membrane to lysosomes. We determined that the trafficking of SLC7A11 requires lipidated LC3 (LC3-II) interacting with the intracellular tubulin network.
Furthermore, the previously identified MTOR (mechanistic target of rapamycin kinase) complex 2 (MTORC2) inhibitory phosphorylation site on SLC7A11 (Ser26) is enhanced after autophagy inhibition and drives the cytoplasmic translocation of SLC7A11. Consistent with this, RICTOR or MTOR knockdown prevent the colocalization of SLC7A11 with lysosomes in the setting of autophagy-inhibited PDAC cells. Our work establishes that the transport function of SLC7A11 in PDAC requires intact autophagy to ensure plasma membrane localization, whereas autophagy inhibition promotes SLC7A11 phosphorylation by MTORC2 and lysosomal translocation (Figure 1). Although the role of autophagy in PDAC amino acid metabolism appears to be selective to Cys, we cannot exclude its contribution to other metabolic processes given the intrinsic complexity of cellular metabolic systems in different contexts. Interestingly, in lung cancer systems, it has been shown that autophagy supports mitochondrial metabolism and nucleotide pools. Therefore, it is important to realize that the metabolic significance of autophagy may differ between cancer types despite having similar oncogenic drivers such as KRAS. Our study provides evidence that inhibition of autophagy in PDAC disrupts Cys metabolism and this contributes to the anti-tumor effect of autophagy inhibition. Future studies will identify potential therapeutic combinations that may capitalize on this metabolic vulnerability induced by autophagy inhibition.
Figure 1.
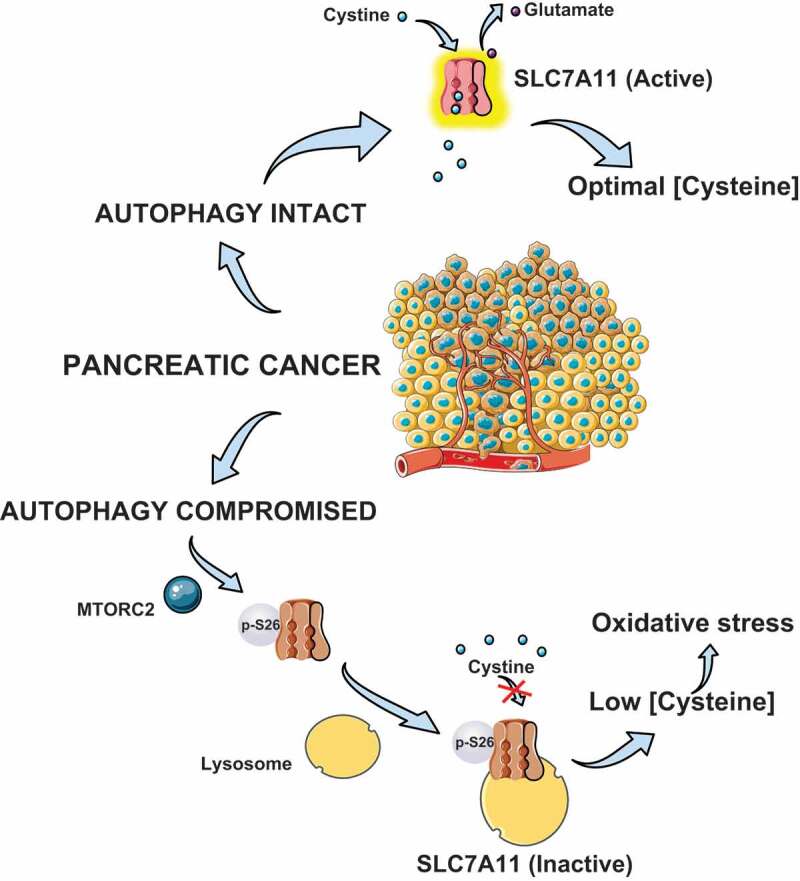
PDAC utilize autophagy for maintaining cysteine homeostasis by regulating SLC7A11. Under autophagy-compromised conditions, SLC7A11 is phosphorylated at Ser26 by MTORC2, which promotes the translocation from the plasma membrane to the lysosome leading to its inactivation. Disruption of autophagy leads to pancreatic tumor shrinkage thereby highlighting the therapeutic significance. This figure is adapted from Fig. 5 in ref [1]
Acknowledgments
Figure 1 was created with the help of Servier Medical Art, which is licensed under a Creative Commons Attribution 3.0 Unported License.
Funding Statement
This work was supported by National Cancer Institute Grants [R01CA157490, R01CA188048, P01CA117969, and R35CA232124]; NIH Grant [R01GM095567]; and the Lustgarten Foundation, and SU2C (Stand-Up-To-Cancer) (to A.C.K.).
Disclosure statement
A.C.K. is a founder and has financial interests in Vescor Therapeutics, LLC. A.C.K. is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, targeting GOT1 as a therapeutic approach and the autophagic control of iron metabolism. A.C.K is on the SAB of Rafael/Cornerstone Pharmaceuticals. A.C.K. is a consultant for Deciphera and Abbvie. All other authors declare no competing financial interests.
Reference
- [1].Mukhopadhyay S, Biancur DE, Parker SJ, et al. Autophagy is required for proper cysteine homeostasis in pancreatic cancer through regulation of SLC7A11. Proc Natl Acad Sci U S A. 2021. February 9;118(6):e2021475118. . PMID: 33531365. [DOI] [PMC free article] [PubMed] [Google Scholar]