

ABSTRACT
Macroautophagy/autophagy is vital for neuronal homeostasis and functions. Accumulating evidence suggest that autophagy is impaired during cerebral ischemia, contributing to neuronal dysfunction and neurodegeneration. However, the outcomes after transient modification in autophagy machinery are not fully understood. This study investigated the effects of ischemic stress on autophagy and synaptic structures using a rat model of oxygen-glucose deprivation (OGD) in hippocampal neurons and a mouse model of middle cerebral artery occlusion (MCAO). Upon acute ischemia, an initial autophagy modification occurred in an upregulation manner. Following, the number of lysosomes increased, as well as lysosomal volume, indicating dysfunctional lysosomal storage. These changes were prevented by inhibiting autophagy via 3-methyladenine (3-MA) treatment or ATG7 (autophagy related 7) knockdown, or were mimicked by rapamycin (RAPA), a known activator of autophagy. This suggests that dysfunctional lysosomal storage is associated with the early burst of autophagy. Dysfunctional lysosomal storage contributed to autophagy dysfunction because the basal level of MTOR-dependent lysosomal biogenesis in the reperfusion was not sufficient to clear undegraded cargoes after transient autophagy upregulation. Further investigation revealed that impairment of synaptic ultra-structures, accompanied by dysfunctional lysosomal storage, may result from a failure in dynamic turnover of synaptic proteins. This indicates a vital role of autophagy-lysosomal machinery in the maintenance of synaptic structures. This study supports previous evidence that dysfunctional lysosomal storage may occur following the upregulation of autophagy in neurons. Appropriate autophagosome-lysosomal functioning is vital for maintenance of neuronal synaptic function and impacts more than the few known synaptic proteins.
Abbreviations: 3-MA: 3-methyladenine; ACTB: actin beta; AD: Alzheimer disease; ALR: autophagic lysosome reformation; ATG7: autophagy related 7; CTSB: cathepsin B; CTSD: cathepsin D; DAPI: 4ʹ,6-diamidino-2-phenylindole; DEGs: differentially expressed genes; DMEM: Dulbecco’s modified Eagle’s medium; DMSO: dimethyl sulfoxide; GO: Gene Ontology; HBSS: Hanks’ balanced salt solution; HPCA: hippocalcin; i.c.v: intracerebroventricular; KEGG: kyoto encyclopedia of genes and genomes; LAMP1: lysosomal-associated membrane protein 1; MAP1LC3B/LC3: microtubule-associated protein 1 light chain 3 beta; LSDs: lysosomal storage disorders; MAP2: microtubule-associated protein 2; MCAO: middle cerebral artery occlusion; mCTSB: mature CTSB; mCTSD: mature CTSD; MOI: multiplicity of infection; MTOR: mechanistic target of rapamycin kinase; OGD/R: oxygen-glucose deprivation/reoxygenation; PBS: phosphate-buffered saline; PRKAA/AMPKα: protein kinase AMP-activated catalytic subunit alpha; proCTSD: pro-cathepsin D; RAPA: rapamycin; RNA-seq: RNA sequencing; RPS6KB/p70S6K: ribosomal protein S6 kinase; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SIM: Structured Illumination Microscopy; SNAP25: synaptosomal-associated protein 25; SQSTM1/p62: sequestosome 1; SYN1: synapsin I; SYT1: synaptotagmin I; TBST: tris-buffered saline Tween-20; TEM: transmission electron microscopy; TFEB: transcription factor EB; tMCAO: transient middle cerebral artery occlusion; TTC: 2,3,5-triphenyltetrazolium chloride; TUBB3: tubulin, beta 3 class III.
KEYWORDS: Functional lysosomal storage, lysosome, middle cerebral artery occlusion (MCAO), neurons, oxygen-glucose deprivation (OGD), synaptic plasticity
Introduction
Strokes remain the second leading cause of death and primary cause of disability in adults globally [1]. With limited therapeutic strategies, ischemic stroke is still an urgent disease requiring investigation. Therapeutic strategies against ischemic stroke usually include restoring blood flow and increasing the resilience of cerebral cells to ischemic injury [2]. There are many processes involved in ischemic stress, including oxidative stress, inflammatory reactions, apoptosis, blood-brain barrier injuries, ionic imbalance, and autophagy [3]. Among these, autophagy has been known for years to contribute to cerebral ischemia and has been identified as an important therapeutic target to protect against cerebral damage during ischemia/reperfusion insults [4,5]. Therefore, autophagy dysfunction that occurs during ischemia has become a topic of focus. The proper activation of autophagy is beneficial for cellular survival to maintain energy and cellular homeostasis under ischemic stress conditions. However, excessive or prolonged autophagy activation induces neuronal necroptosis and apoptosis [6]. Upregulation or suppression of autophagy has been shown to improve ischemic injuries [7], suggesting that autophagy dysfunction is a promising stroke treatment target. Moreover, most studies emphasize the role of autophagy in ischemic stroke and attribute ischemic injury to impaired autophagy. Some studies suggest that autophagy dysfunction is transient. Therefore, concomitant changes that follow its early changes of autophagy-lysosomal machinery may be more important. However, this has not been investigated sufficiently. A better understanding of the role of autophagy in ischemia, including the transient upregulation/suppression of autophagy and the subsequent changes, will provide insights for the management and treatment of ischemic stroke, especially for patients outside of the stroke therapeutic window.
Autophagy is an evolutionarily conserved intercellular process for the degradation and recycling of cellular materials via the lysosomal pathway. The autophagy process is usually divided into five stages [8], including nucleation of the phagophore membrane, expansion, phagophore closure, fusion between autophagosomes and multivesicular endosomes or lysosomes, and degradation of autophagosome contents [9,10]. Disruption at any stage in this process, such as lysosomal dysfunction, causes autophagy dysfunction [11]. Although increasing evidence shows that ischemia-reperfusion injury leads to the upregulation of autophagy, the influenced exact stage of autophagy machinery in ischemic/reperfusion injury remains unknown [12]. In neurons, autophagy is upregulated during some stress conditions, such as decreased neuronal activity, sensory deprivation, and neurotrophic factor deficiency [13]. Reduced autophagy in the brains of patients with autism is associated with an accumulation of ubiquitinated proteins and a redundant increase in dendritic spine density [14]. Additionally, abnormal autophagy levels are associated with neurodegenerative diseases, such as Parkinson disease (PD) and Alzheimer disease (AD) [15,16]. However, impaired autophagy-lysosomal function has seldom been reported in chronic neurodegenerative diseases.
Lysosomes are vital for autophagy machinery, and maintenance of lysosomal functions is important for cellular hemostasis. Lysosomal storage disorders (LSDs) have been observed in up to 39 distinct genetic diseases, including neurodegenerative disorders, and there are few potential treatments available [17,18]. Each of these diseases is caused by a deficiency in a particular lysosomal protein or non-lysosomal protein associated with lysosomal biogenesis [19]. The accumulation of lysosomal cargoes and subsequent neurodegeneration are hallmarks of LSDs that influence cerebral functions [20]. To date, emphasis has been placed on the genetic sources of LSDs. However, to our knowledge, there are still no reports on functional lysosomal storage in ischemic injury. Furthermore, there have been few reports about subsequent events after initial autophagy dysfunction, such as the influence of the formation of autophagosomes on lysosome biogenesis and functioning, although these processes share common regulatory molecules [21].
Herein, we sought to study the role of autophagy and lysosomal storage in ischemic stroke. To address this aim, we used a rat model of hippocampal neuron ischemia, a mouse model of middle cerebral artery occlusion (MCAO), and ultra-structural analyses of downstream physiological processes after autophagy upregulation during ischemia. Transient upregulation of autophagy by ischemic insult caused a significant increase in lysosomal storage following reperfusion, which resulted in autophagy dysfunction by impairing the degradation. The autophagy-induced increase in lysosomal storage indicates that autophagy dysfunction sustained in the neurons features different synaptic phenotypes. One-third of stroke patients exhibit significant cognitive decline in addition to physical disability [22,23] and accompanying changes in synaptic morphological characteristics have been observed. In this study, functional lysosomal storage accompanied by synaptic dysfunction after ischemic stress occurred. These results will advance our understanding of ischemia-induced autophagy dysfunction and the genetics of LSDs based on their similar characteristics.
Results
OGD-induced autophagy upregulation precedes a long-term increase in LAMP1 protein levels
To address the changes of autophagy in different stages of ischemic stress, autophagy levels in rat hippocampal neurons were evaluated by assessing the conversion of the autophagy marker MAP1LC3-II to LC3-I (microtubule-associated protein 1 light chain 3 beta) and the protein levels of SQSTM1/p62 (sequestosome 1) by western blot at different time points during OGD and subsequent reperfusion (Figure 1A). The LC3-II:LC3-I ratio increased after 1 h of OGD (Figure 1A and B), but returned to basal levels following reperfusion, indicating that the increase of autophagy was transient. Furthermore, the SQSTM1 protein level increased significantly after 24 h of reperfusion, indicating a potential impairment of autophagic flux (Figure 1A and C). Because lysosomes are vital for autophagy, we evaluated the protein levels of lysosomal marker LAMP1 (lysosomal-associated membrane protein 1) to assess whether downstream autophagy processes are also disrupted. Notably, the LAMP1 protein level began to increase from 4 h of reperfusion to 24 h (Figure 1A and D).
Figure 1.
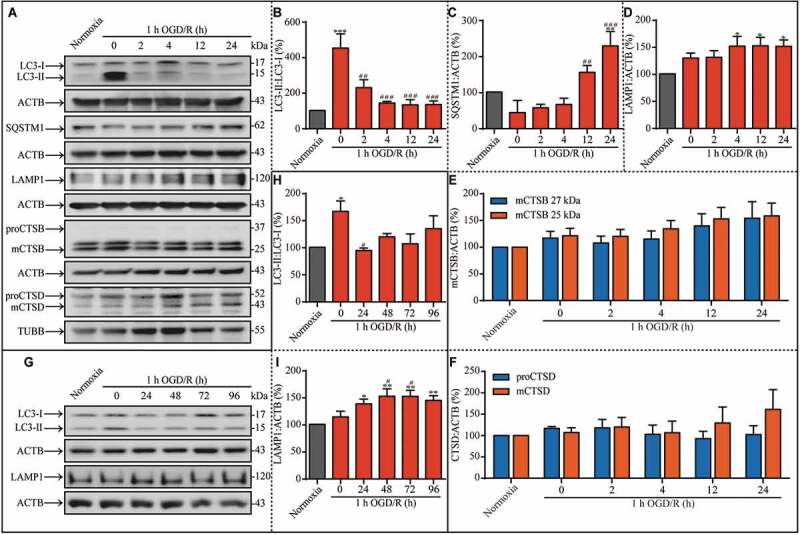
Autophagic dysfunction is transiently induced by OGD insult, and a prolonged increase in LAMP1 expression occurs during the subsequent reperfusion. (A) Representative western blots showing the LC3-II:LC3-I ratio and the expression of SQSTM1, LAMP1, mCTSB, proCTSD, and mCTSD under the indicated treatment conditions. (B-F) The analysis results of LC3-II:LC3-I ratio, SQSTM1, LAMP1, mCTSB, and proCTSD and mCTSD in hippocampal neurons cultured under the indicated conditions. (G) Representative western blots showing the LC3-II:LC3-I ratio and the expression of LAMP1 under different experimental conditions. The neuronal (H) LC3-II:LC3-I ratio and (I) LAMP1 level in the indicated groups. ACTB was used as the loading control in these experiments. (B, n = 6; C, n = 4; D, n = 6; E, n = 6; F, n = 6; H, n = 3; I, n = 5; *p < 0.05, **p < 0.01 or ***p < 0.001 vs. the normoxia group; #p < 0.05, ##p < 0.01, or ###p < 0.001 vs. the 1-h OGD group). Statistical comparisons were carried out with one-way ANOVA. Data are shown as the mean ± SEM
Because autophagic flux was disrupted in ischemia-reperfusion injury producing a concomitant increase in lysosomal marker protein accumulation, we next sought to determine if the lysosomal degradation process is disrupted. Therefore, we analyzed the levels of lysosomal proteases during ischemia and reperfusion. The degradation of cargoes within autolysosomes depends on a family of lysosomal proteases named cathepsins, including the cysteine proteinases CTSB (cathepsin B), CTSF, and CTSL, and the aspartyl protease CTSD. As previously reported, CTSD deficiency or CTSB and cathepsin L double-deficiency leads to a strong blockage of autophagic flux [24]. First, we examined the protein levels of mCTSB (mature cathepsin B) (Figure 1A and E), proCTSD (pro-cathepsin D) (Figure 1A and F), and mCTSD (mature cathepsin D) (Figure 1A and F). When ACTB and TUBB were used as the loading control based on the molecular weight of target proteins, there were no distinct differences among the experimental groups. Furthermore, when the reperfusion duration was prolonged to 96 h (Figure 1G), LC3-II levels remained stable (Figure 1G and H). Additionally, the protein levels of LAMP1 remained increased throughout this prolonged reperfusion duration (Figure 1G and I). In summary, the prolonged increase in LAMP1 protein levels indicates that there may be a significant lysosome-associated dysfunction during reperfusion.
Lysosomal enzyme dysfunction induces impairment of lysosomal function
Because lysosomes are vital executors of cellular component degradation, we next investigated the underlying mechanism of the long-lasting lysosome-associated dysfunction following a transient upregulation in autophagy during reperfusion. Based on the increase in LAMP1 protein expression, we further explored whether this phenomenon is related to morphological changes of lysosomes. We created three groups representing three time points of interest: the normoxia group, the 1-h OGD group, and the 1-h OGD/24-h R group, and we acquired LAMP1 and MAP2 (microtubule-associated protein 2) images by structured illumination microscopy (SIM) to address the lysosomes in the cytoplasm of neurons (Figure 2A). MAP2-positive puncta were used to assess cell size, and the quantity of LAMP1-positive puncta per unit area in the neuronal cytoplasm was analyzed. There were significantly more LAMP1-positive puncta in neurons of the 1-h OGD/24-h R group than the other two groups (Figure 2B), which again suggested an accumulation of lysosomes. Our above results (Figure 1A, E, and F) showed that the expression levels of mCTSB, proCTSD, and mCTSD did not change in coordination with the LAMP1 levels, even though their expression should align under physiological conditions. Therefore, the protein levels of mCTSB, proCTSD, and mCTSD were normalized to the expression of LAMP1 as the loading control and compared among the three groups (Figure 2C and Fig. S1). The relative expression of mCTSB (Figure 2C and D) and proCTSD (Figure 2C and E) were markedly lower in the 1-h OGD/24-h R group than in the other two groups, suggesting lysosomal dysfunction. Due to the similar relative decrease in CTSD protein levels, we examined the enzymatic activity of CTSD and lysosomal accumulation by evaluating the relative enzymatic activities of CTSD under different experimental conditions (Figure 2F). Notably, CTSD activity in neurons was significantly increased in the 1-h OGD group, but was lower in the 1-h OGD/24-h R group compared to the normoxia group or the 1-h OGD group (Figure 2G), indicating the occurrence of lysosomal dysfunction. In summary, the impaired lysosomal enzyme activity resulted in lysosomal dysfunction.
Figure 2.
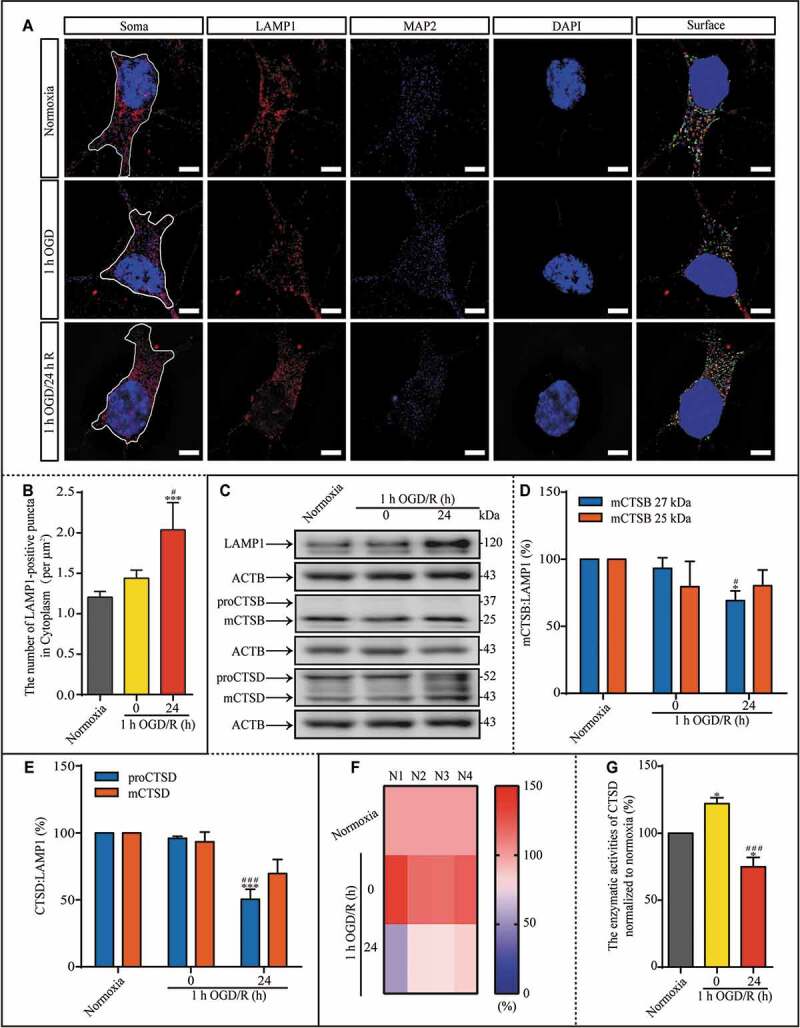
During reperfusion, lysosomal dysfunction may result from reduced CTSD activity. (A) LAMP1-positive puncta in the indicated groups were observed with SIM; scale bar: 5 μm. (B) The number of LAMP1-positive puncta per μm2 in the cytoplasm. (C-E) Representative images and analysis of western blots of LAMP1, mCTSB, proCTSD, and mCTSD in the indicated groups. (F and G) The enzymatic activity of CTSD in the neurons was evaluated with a Fluorometric Assay Kit from Abcam. N1-N4 indicated the number of replicated times. The enzymatic activities of CTSD were normalized to those of normoxia as the ratio (0–150). (B, n = 12; D and E, n = 4; G, n = 4; *p < 0.05, ***p < 0.001 vs. the normoxia group; #p < 0.05, ###p < 0.001 vs. the 1-h OGD group). Statistical comparisons were carried out with one-way ANOVA. Data are shown as the mean ± SEM. For Quantitative analysis of western blots of LAMP1, see Figure S1.
Dysfunctional lysosomal storage, characterized by increased lysosome volume and impaired autolysosomal degradation, occurs during reperfusion
Because LAMP1 is used to screen neonates for genetic LSDs [25] and CTSD dysfunction was observed during reperfusion (Figure 2), we next examined lysosomal storage during reperfusion following ischemic insult. To evaluate the autophagosomes and lysosomes simultaneously, we transfected cultured hippocampal neurons with lenti-EGFP-LC3, counterstained them with LysoTracker, and performed 3D imaging with SIM (Figure 3A). To exclude the influence of neuron size, we compared the percentage of LysoTracker-positive or LC3-positive puncta in different volumes rather than the absolute quantity. After 1 h of OGD or 24 h of reperfusion, the percentage of large LC3-positive puncta (0.1–5 μm3) was increased (Figure 3B), indicating that the number of autophagosomes was increased. The percentage of LysoTracker-positive puncta of an increased volume (> 0.5 µm3) was also increased in the 1-h OGD group and the 1-h OGD/24-h R group, suggesting a potential increase in the number of acidic organelles, such as lysosomes (Figure 3C). Furthermore, we evaluated the colocalization of LC3-positive and LysoTracker-positive puncta (Figure 3D). Compared to the normoxia group, only the 1-h OGD/24-h R group exhibited significantly higher percentages of completely and partially colocalized LysoTracker- and LC3-positive puncta and a lower percentage of non-colocalized puncta. As lysosomes are the main acidic organelles in the cytoplasm, these observations imply that the degradation capacity of the autophagosome-lysosomal machinery may be impaired during reperfusion.
Figure 3.
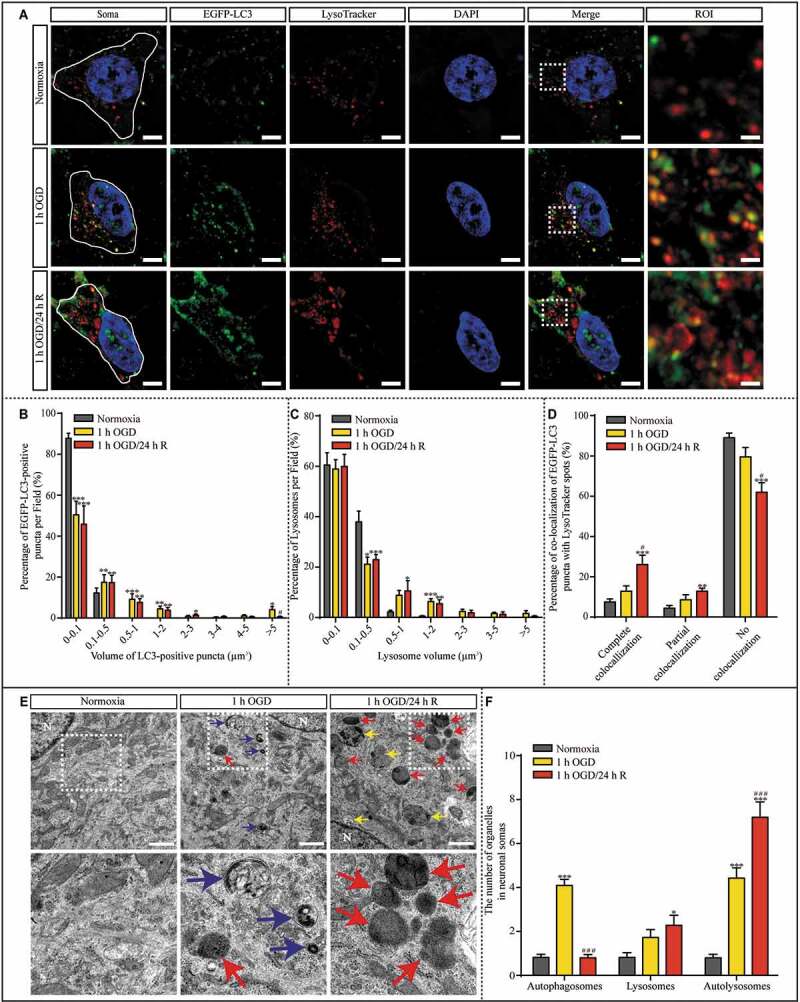
The number and volume of lysosomes increased in cultured hippocampal neurons after 1 h OGD/R 24 h exposure. (A) Representative SIM images of hippocampal neurons are shown. Lenti-EGFP-LC3-transfected neurons were counterstained with DAPI and LysoTracker; scale bar: 5 μm, High-magnification images of the boxed areas are shown in the inserts (ROI); scale bar: 1 μm. (B and C) The percentage of LC3-positive puncta and LysoTracker-positive puncta with a volume in the indicated ranges per neuron was quantified with Imaris software in randomly chosen neurons from each sample in each group. (D) The percentages of LysoTracker-positive puncta showing complete, partial, and no colocalization with LC3-positive puncta per neuron were quantified. (E) Representative TEM images of autophagosomes (blue), autolysosomes (yellow), and lysosomes (red) in the cytoplasm of neurons are presented; N: nucleus; scale bar: 1 μm and (F) quantified per TEM field from 40 randomly selected fields. (B, C and D, normoxia: n = 7; 1-h OGD: n = 8; 1-h OGD/24-h R: n = 9; F, n = 40; *p < 0.05, **p < 0.01, ***p < 0.001 vs. the normoxia group; #p < 0.05, ###p < 0.001 vs. the 1-h OGD group). Statistical comparisons were carried out with one-way ANOVA. Data are shown as the mean ± SEM
Dysfunctional lysosomal storage disrupts the housekeeping autophagic machinery
CTSD is one of the major lysosomal proteases required for the maintenance of cellular proteostasis and functions by degrading endocytic, phagocytic, and autophagic cargoes. Normally, CTSD deficiency or dysfunction significantly impairs the autophagosome-lysosomal machinery [24]. Additionally, enlarged lysosomes are associated with reduced degradative capacity [26]. Our results suggesting that the degradative capacity of lysosomes is impaired during reperfusion led us to speculate that reperfusion is associated with the occurrence of an LSD, potentially resulting in autophagosome-lysosomal dysfunction. To assess the autophagosome-lysosomal machinery, we used transmission electron microscopy (TEM) to investigate the number of autophagosomes, lysosomes, and autolysosomes in the cytoplasm of neurons and evaluated the ultra-structure of hippocampal neurons in the normoxia, 1-h OGD, and 1-h OGD/24-h R groups (Figure 3E). Compared with those in the normoxia group, neurons in the 1-h OGD group had more autophagosomes and autolysosomes in the cytoplasm (Figure 3F), whereas neurons in the 1-h OGD/24-h R group had more lysosomes and autolysosomes in the cytoplasm compared to neurons exposed only to OGD (Figure 3F). The similar quantity of autophagosomes between neurons in the normoxia group and those in the 1-h OGD/24-h R group again indicated that the degradative capacity of lysosomes may be impaired during reperfusion, resulting in lysosome and autolysosome accumulation.
Both pharmacologic and genetic disruption against OGD-induced autophagy upregulation prevent dysfunctional lysosomal storage
Because lysosomal accumulation occurred after the transient increase in autophagy, we questioned whether these processes are associated. First, the OGD-induced upregulation of autophagy was suppressed by 3-methyladenine (3-MA, 5 mM, Fig. S2), an inhibitor of autophagy (Figure 4A and B). Treatment with 3-MA significantly prevented the increase in the LAMP1 level in the 1-h OGD/24-h R group (Figure 4A and C). We also measured the number of autophagosomes, lysosomes, and autolysosomes with TEM (Figure 4D). There were increased numbers of lysosomes and autolysosomes in the cytoplasm of neurons after 24 h of reperfusion (Figure 4E); however, 3-MA treatment significantly reduced the number of lysosomes and autolysosomes, indicating that there is an association between the transient upregulation in autophagy and the subsequent accumulation of lysosomes. Finally, the hippocampal neurons were transfected with lenti-mCherry-EGFP-LC3 (Fig. S3A). Lenti-mCherry-EGFP-LC3 was used to differentiate between autophagosomes and autolysosomes. EGFP (enhanced green fluorescent protein) is acid-sensitive and will lose its fluorescence when autophagosomes fuse with lysosomes, which reduces the pH value; whereas, mCherry is acid-insensitive and positively sustained in both autophagosomes and autolysosomes. One hour of OGD increased the number of EGFP+ mCherry− puncta in the cytoplasm of neurons; however, this increase was suppressed by 3-MA treatment (Fig. S3B). The percentage of EGFP+ mCherry+ puncta was higher in the 1-h OGD group than in the normoxia group; however, this increase was also blocked by 3-MA treatment (Fig. S3C). Lysosomal accumulation was observed in neurons of the 1-h OGD/24-h R group, but 3-MA treatment prevented this accumulation (Fig. S3D).
Figure 4.
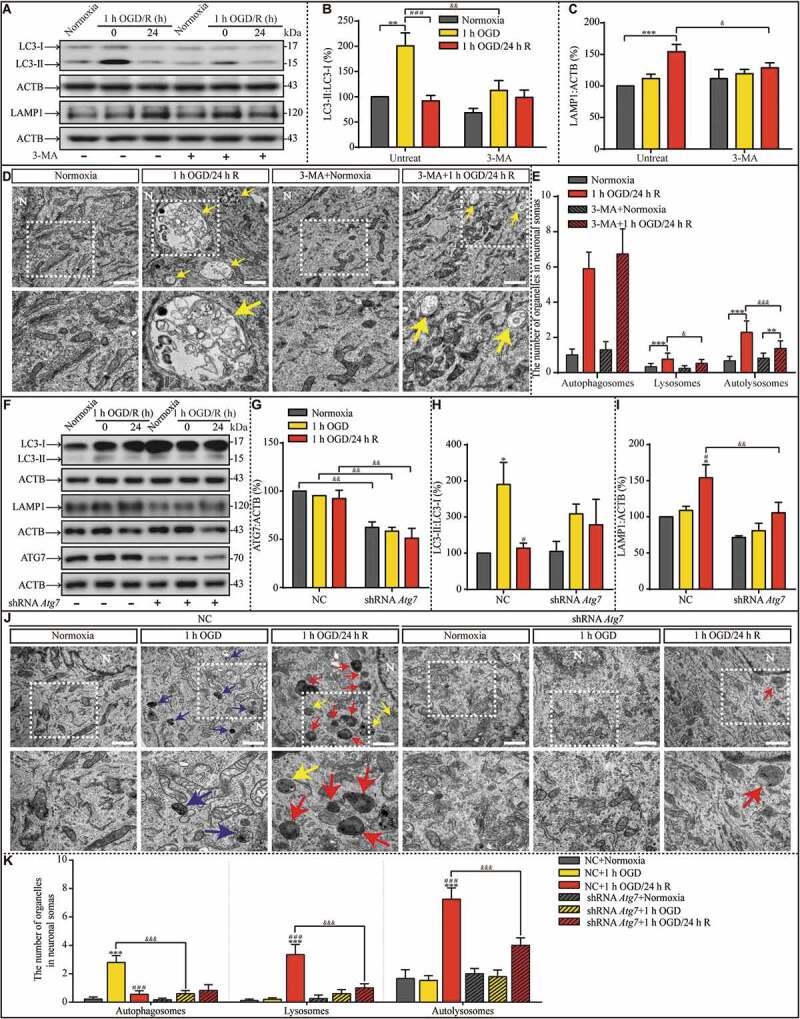
Suppression of OGD-induced autophagy upregulation by 3-MA pretreatment or ATG7 genetic knockdown prevents lysosomal accumulation during the subsequent reperfusion, respectively. (A-C) The LC3-II:LC3-I ratio and LAMP1 protein levels in indicated groups. (D and E) Representative images and quantitative analysis of autophagosomes, lysosomes, and autolysosomes in indicated groups with or without 3-MA treatment. Yellow arrows depict autolysosomes. N: nucleus; scale bar: 1 μm. (B, n = 6; C, n = 5; E, normoxia: n = 18; 1-h OGD/24-h R: n = 21; 3-MA+normoxia: n = 17; 3-MA+1-h OGD/24-h R: n = 27; B, C and E, **p < 0.01, ***p < 0.001 vs. the indicated normoxia group, respectively; ###p < 0.001 vs. the 1-h OGD group; &p < 0.05, &&p < 0.01, &&&p < 0.001 vs. the indicated condition without 3-MA). (F-I) Representative western blots and quantitative analysis of ATG7, LC3, and LAMP1 in the indicated groups. ACTB was used as the loading control. (J and K) The numbers of autophagosomes, lysosomes, and autolysosomes in the indicated groups were evaluated with TEM. Scale bar: 1 μm. NC: negative control. N: nucleus. Blue, yellow, or red arrowheads indicate representative autophagosomes, autolysosomes, or lysosomes, respectively (G, H and I, n = 3; K, NC+normoxia, n = 9; NC+1-h OGD, n = 15; NC+1-h OGD/24-h R, n = 20; shRNA Atg7+normoxia, n = 12; shRNA Atg7+1-h OGD, n = 13; shRNA Atg7+1-h OGD/24-h R, n = 17; G, H, I and K, *p < 0.05, ***p < 0.001 vs. the NC+normoxia group; #p < 0.05 or ###p < 0.001 vs. the NC+1-h OGD group; &&p < 0.01 or &&&p < 0.001 vs. the indicated condition with NC). Statistical comparisons were carried out with one-way ANOVA. Data are shown as the mean ± SEM
To further explore the effects of early autophagy upregulation on lysosomal accumulation, we knocked down ATG7 (autophagy related 7) with a lenti-shRNA Atg7. The attachment of phosphatidylethanolamine to LC3 by the E1-like enzymes that ATG7 contributes to LC3 lipidation is the vital step for autophagosome formation [27]. ATG7 knockdown (Fig. S4; Figure 4F and G) suppressed the OGD-induced increase in the LC3-II:LC3-I ratio (Figure 4F and H). Notably, ATG7 knockdown also suppressed LAMP1 protein accumulation in the neurons exposed to OGD and reperfusion (Figure 4F and I). Moreover, TEM images showed more autophagosomes in the cytoplasm of neurons in the 1-h OGD group than in the normoxia group (Figure 4J and K), and more lysosomes and autolysosomes in the cytoplasm of neurons in the 1-h OGD/24-h R group than in the 1-h OGD group (Figure 4K). However, when ATG7 was knocked down, the early increase in autophagy did not occur after OGD, nor did the late accumulation of lysosomes and autolysosomes after reperfusion. Moreover, 3-MA pretreatment can alleviate OGD-induced neuronal injuries, and RAPA pretreatment increased OGD-induced injuries (Fig. S5A and B). As TFEB (transcription factor EB) is vital for autophagosome formation and the biogenesis of lysosomes [28,29], to further explore the basis of lysosomal accumulation, we measured TFEB protein levels and found them to be reduced by OGD exposure and increased after 24 h of reperfusion (Fig. S6). TFEB knockdown (Fig. S7A) before OGD prevented the increase of LAMP1 (Fig. S7B-E). In summary, these results suggest that the early increased production of autophagosomes facilitates late lysosomal accumulation in a TFEB-dependent manner.
RAPA-induced autophagy upregulation is followed by dysfunctional lysosomal storage
The previous results suggest that OGD-induced autophagy influences lysosomal biogenesis, function, and accumulation. To verify this, we used 2 h rapamycin (RAPA, 50 nM) treatment to activate autophagy (Fig. S8) and assessed its effect by measuring the change in the LC3-II:LC3-I ratio. Two hours of RAPA treatment successfully induced autophagy, which then returned to basal level (Figure 5A and B). Similar to OGD exposure, the increase in autophagy was transient, suggesting that maintenance of upregulation of autophagy for neurons is limited. However, in contrast to OGD exposure, 24 h of reperfusion following RAPA withdrawal resulted in another increase in autophagy. Notably, LAMP1 protein levels increased after 24 h of continuous culture following RAPA withdrawal (Figure 5A and C). To observe the condition of the autophagosome-lysosomal machinery after RAPA treatment, we used TEM to investigate the number of autophagosomes, lysosomes, and autolysosomes in the cytoplasm of hippocampal neurons under control conditions, after 2 h RAPA exposure, and 2 h RAPA exposure followed by subsequent culture without RAPA (Figure 5D). The number of autophagosomes increased significantly after 2 h of RAPA exposure and returned to basal level during the subsequent culture period (Figure 5E). The number of lysosomes increased in the culture period following the removal of RAPA. The number of autolysosomes increased after RAPA exposure and further increased in the subsequent reperfusion period (Figure 5E). Furthermore, RAPA treatment did not influence CTSD activity, which subsequently decreased after the removal of RAPA (Figure 5F and G). Based on these results and the results shown in Figure 4 and Figure 5, we propose that the transient increase in autophagy is the onset of the autophagosomal-lysosomal dysfunction, including lysosomal activity and storage dysfunction.
Figure 5.
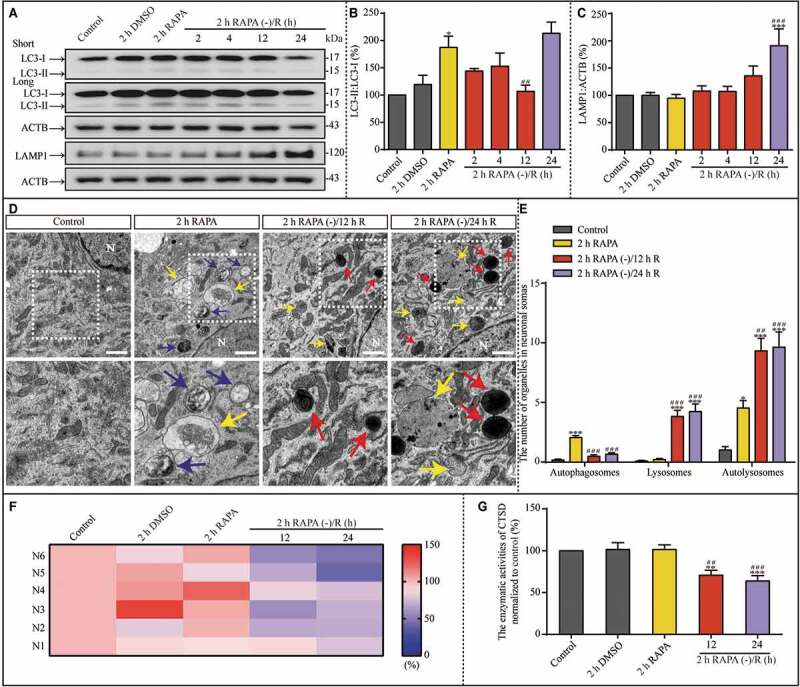
RAPA-induced autophagy upregulation is followed by lysosomal accumulation in the subsequent culture without RAPA. (A-C) Representative western blots and the quantitative analysis of LC3 and LAMP1 in the indicated groups. ACTB was used as the loading control. (D and E) Representative TEM images and the quantitative analysis of autophagosomes, lysosomes, and autolysosomes in the indicated groups. N: nucleus; scale bar: 1 μm. (F and G) The enzymatic activity of CTSD in the indicated groups was evaluated with a Fluorometric Assay Kit from Abcam. N1-N6 indicated that this experiment was replicated 6 times. The activities of enzymatic activity of CTSD were normalized to those of normoxia and shown as the ratio (0–150). Blue, yellow, or red arrowheads indicate representative autophagosomes, autolysosomes, or lysosomes, respectively. (B, n = 5; C, n = 6; E, control, n = 24; 2 h RAPA, n = 34; 2 h RAPA (-)/12 h R, n = 38; 2 h RAPA (-)/24 h R, n = 35; G, n = 6; *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control group; ##p < 0.01, ###p < 0.001 vs. the 2 h RAPA group). Statistical comparisons were carried out with one-way ANOVA. Data are shown as the mean ± SEM
The effects of autophagy and lysosome dysfunction on synaptic plasticity during reperfusion are similar to those of genetic LSDs
The lysosomal dysfunction characteristics in the reperfusion period, including lysosome accumulation, enlargement, and dysfunction, are indicative of dysfunctional lysosomal storage. In genetic LSDs, defects in lysosomal degradation cause progressive lysosomal dysfunction and neurodegeneration; therefore, next we sought to explore the significance of dysfunctional lysosomal storage on synaptic function and autophagic machinery during reperfusion. To this end, we evaluated the architecture of synaptic terminals in hippocampal neurons under the 1 h OGD and 1 h OGD/24 h R using TEM ultra-structure analysis (Figure 6A). The number of autophagosomes in the neuronal processes increased significantly after 24 h of reperfusion (Figure 6B). Furthermore, the number of autolysosomes in neuronal processes increased significantly after 1 h of OGD and further increased after 24 h of reperfusion (Figure 6B). Again, the accumulation of autophagosomes and autolysosomes in these neuronal processes suggested that the autophagosome-lysosomal machinery was impaired and that synaptic functions may be influenced by autophagosome-lysosomal dysregulation. Next, we found that the inhibition of early autophagy activation with 3-MA prevented the accumulation of both autolysosomes and lysosomes (Figure 6C and D). Notably, autophagosomes still accumulated in the neuronal processes during reperfusion (Figure 6D), again indicating that the impairment in autophagic flux results from lysosomal dysfunction. However, RAPA exposure (Figure 6E) resulted in a greater increase in autolysosomes after 24 h reperfusion than after 1 h OGD (Figure 6F), partially mimicking the effects of OGD exposure.
Figure 6.
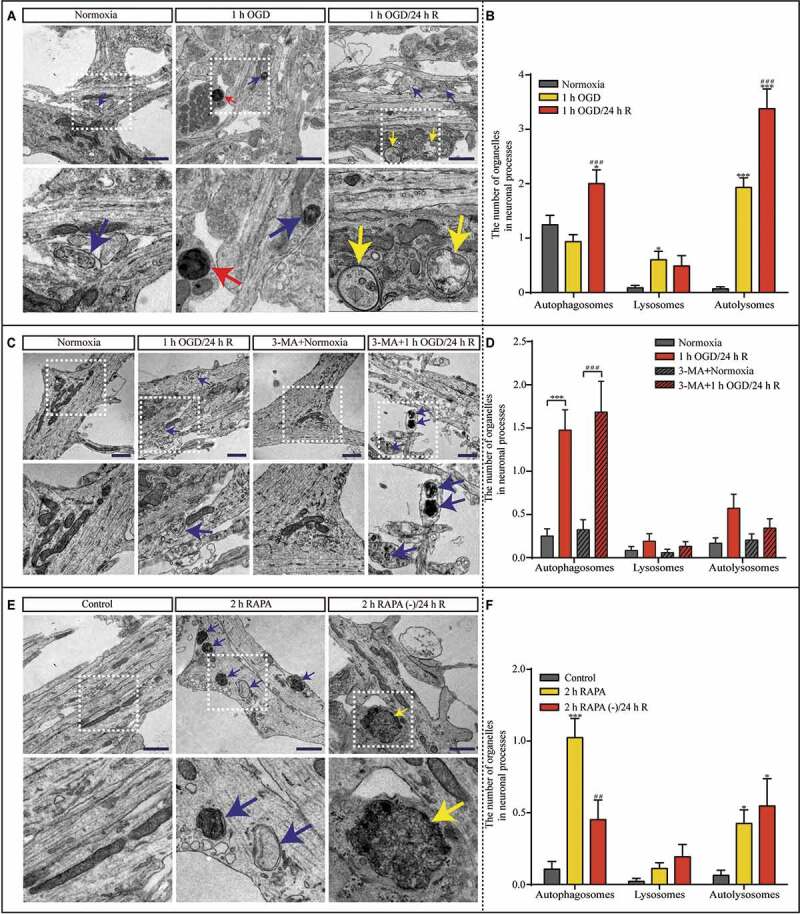
Hippocampal neuronal processes show dysfunctional autophagy after OGD and subsequent reperfusion, which can be disrupted by 3-MA pretreatment and mimicked by RAPA treatment. (A and B) Representative TEM images and quantitative analysis of the autophagosomes, lysosomes, and autolysosomes in neuronal processes after normoxia, OGD, or OGD/reperfusion treatment (n = 45, *p < 0.05, ***p < 0.001 vs. the normoxia group; ###p < 0.001 vs. the 1-h OGD group). (C and D) Representative TEM images and quantitative analysis of the number of autophagosomes, lysosomes, and autolysosomes in neuronal processes in the indicated groups (normoxia, n = 36; 1-h OGD/24-h R, n = 21; 3-MA+normoxia, n = 34; 3-MA+1-h OGD/24-h R, n = 38; ***p < 0.001 vs. the normoxia group; ###p < 0.001 vs. the 3-MA+normoxia group). (E and F) Representative TEM images and quantitative analysis of the number of the autophagosomes, lysosomes, and autolysosomes in the neuronal processes exposed to RAPA (control, n = 47; 2 h RAPA, n = 80; 2 h RAPA (-)/24 h R, n = 31; *p < 0.05, ***p < 0.001 vs. the control group; ##p < 0.01 vs. the RAPA group); scale bar: 1 μm. Blue, yellow, or red arrowheads indicate representative autophagosomes, autolysosomes, or lysosomes, respectively. Statistical comparisons were carried out with one-way ANOVA. Data are shown as the mean ± SEM
The average size of synaptic vesicles of the readily releasable pool (RRP) and thickness of the postsynaptic density (PSD) were significantly lower, and synaptic space width was increased in the 1-h OGD/24-h R groups compared to the normoxia group (Fig. S9A-D). However, postsynaptic membrane length was decreased only after OGD exposure. Notably, the average postsynaptic membrane length in the 24 h reperfusion group was significantly greater than that in the 1-h OGD group (Fig. S9E). Structural changes of synaptic sites in hippocampal neurons normally occur at the onset of a worsening phenotype and are concomitant with lysosomal enlargement, which is characteristic of overt lysosomal dysfunction [30–33]. Because of the effect of the observed lysosomal dysfunction on synaptic structure and synaptic function, we examined the ultra-structural changes in the synaptic machinery to identify the relevant structures impacted by the lysosomal dysfunction. Here, TEM analysis showed structural alterations at synaptic sites similar to those observed with lysosomal-dysfunction-associated neurodegeneration. Considering that ischemic conditions are associated with many stressors, we blocked the lysosomal dysfunction in hippocampal neurons by suppressing the initial upregulation of autophagy via 3-MA treatment to test which synaptic dysfunction phenotype is primarily associated with lysosomal dysfunction. Along with the reduction in lysosome accumulation, the inhibition of OGD-induced autophagy via 3-MA treatment (Figure 7B) prevented the decrease in RRP size and PSD thickness, and increase in synaptic space width (Figure 7C-E). However, there was no significant difference in postsynaptic membrane length among the experimental groups (Figure 7F). Moreover, 2 h RAPA treatment followed by subculturing produced ultra-structural changes in synaptic structures (Figure 7G). RRP size and PSD thickness were reduced after 2 h RAPA treatment and after 2 h RAPA treatment followed by 24 h of reperfusion (Figure 7H and I). Similar to OGD exposure, RAPA treatment produced an increase in synaptic space width (Figure 7J). Similar to 1 h OGD followed by 24 h reperfusion, RAPA treatment had no effect on postsynaptic membrane length (Figure 7K). Because the ultra-structural changes in synaptic structures followed those of autophagic impairment, it can be concluded that synaptic dysfunction was the inevitable consequence of autophagic dysfunction in neurons.
Figure 7.
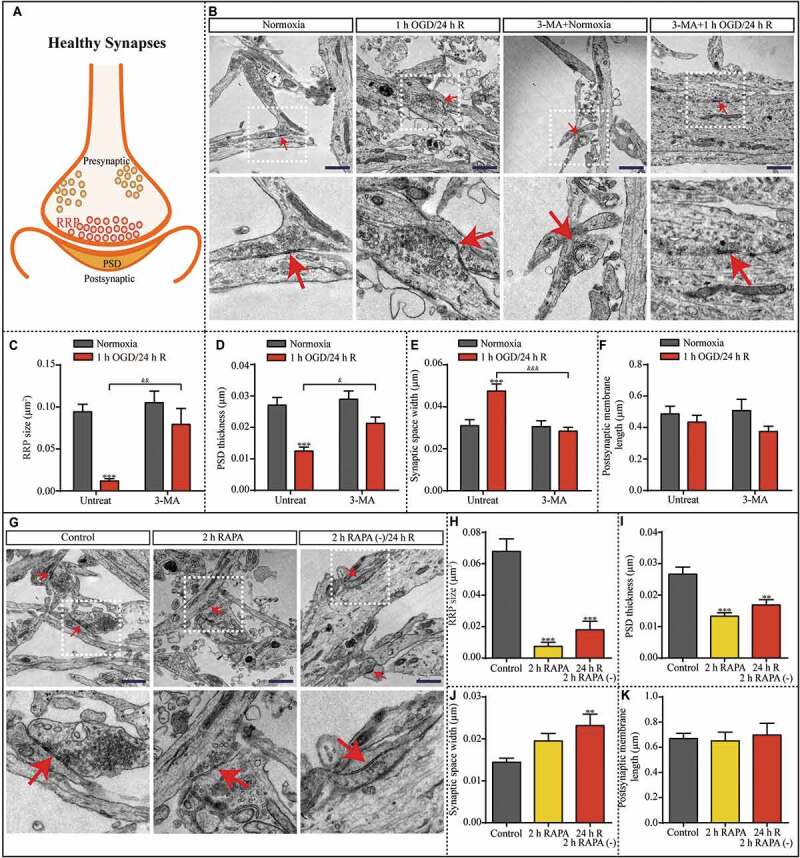
Synaptic structures are altered after 1 h OGD/24 h reperfusion; these changes are prevented by 3-MA treatment and partially mimicked by RAPA treatment. (A) Illustration of healthy synaptic structure under normoxic condition. (B) Representative TEM images showing the synapses of neurons in the indicated groups with or without 3-MA. RRP size (C), PSD thickness (D), synaptic space width (E), and postsynaptic membrane length (F) in the neurons were evaluated in the indicated groups (normoxia, n = 31; 1-h OGD/24-h R, n = 24; 3-MA+normoxia, n = 19; 3-MA+1-h OGD/24-h R, n = 31; ***p < 0.001 vs. the normoxia group; &p < 0.05, &&p < 0.01, &&&p < 0.001 vs. 1-h OGD/24-h R without 3-MA). (G) Representative TEM images showing the synapses of neurons exposed to RAPA treatment. (H) RRP size, (I) PSD thickness, (J) synaptic space width, and (K) postsynaptic membrane length in the neurons were evaluated in the indicated groups (control, n = 27; 2 h RAPA, n = 21; 2 h RAPA (-)/24 h R, n = 19; **p < 0.01, ***p < 0.001 vs. the control group); scale bar: 1 μm. Red arrowheads indicate representative synapses. Statistical comparisons were carried out with one-way ANOVA. Data are shown as the mean ± SEM
The dysfunction in turnover of synaptic proteins may contribute to the impairment in synaptic plasticity during reperfusion
To explore the possible mechanisms contributing to synaptic dysfunction in reperfusion, RNA sequencing was performed to investigate the differentially expressed genes (DEGs). Using the Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO) and pathway enrichment analyses were carried out to determine the functions and pathways of DEGs. We gathered 214 differentially expressed mRNAs in hippocampal neurons between the 1-h OGD and normoxia groups, and 251 differentially expressed mRNAs between the 1-h OGD/24-h R and normoxia groups. Among these, 98 DEGs were determined to be involved in the functions of the nervous system. Moreover, KEGG and GO analyses indicate these DEGs are greatly enriched in several biological processes and pathways. A functional pathway cluster analysis using the Clusters of Orthologous Groups (COG) database was performed and most neuronal DEGs were associated with long-term potentiation, long-term depression, and Alzheimer disease (Figure 8A). In addition, the related biological processes mainly include synaptic vesicle membranes, exocytic vesicle membranes, excitatory synapses, and the axon terminus (Figure 8B). Of note, these pathways and processes suggest these DEGs are closely related to synaptic plasticity. In addition, there are many plasticity-associated DEGs in the neurons after 1-h OGD/24-h R, such as Snap25 (synaptosomal-associated protein 25), Syt1 (synaptotagmin I), Syn1 (synapsin I), Hpca (hippocalcin), and Tubb3 (tubulin, beta 3 class III) (Figure 8C).
Figure 8.
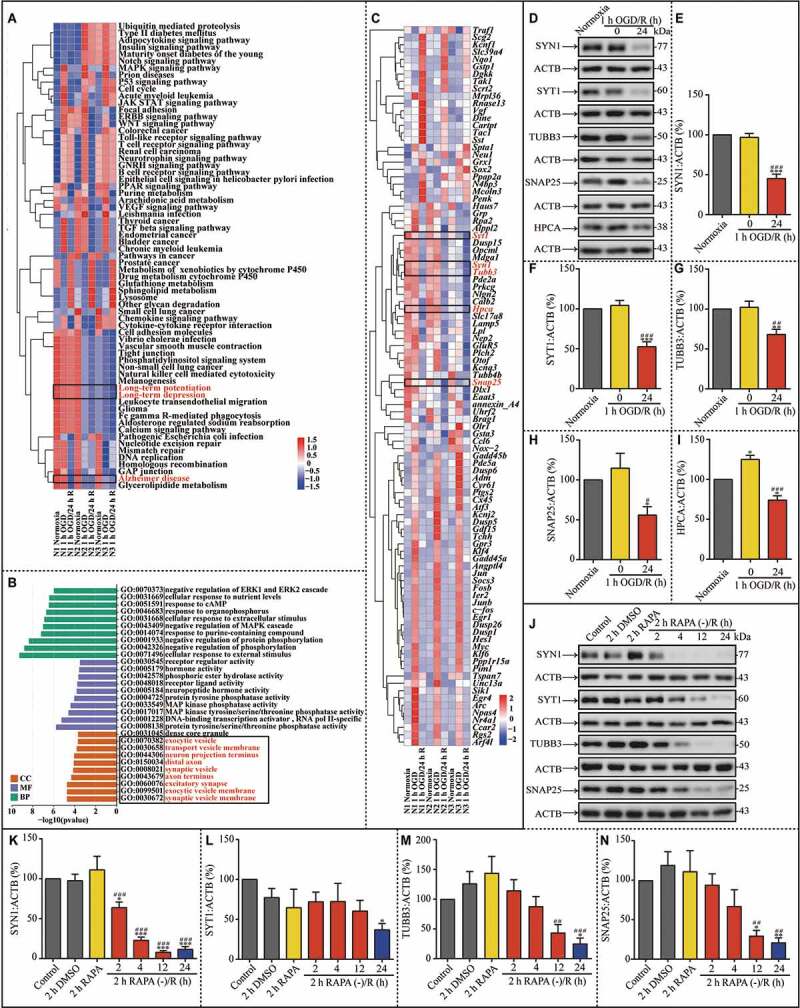
The turnover of synaptic proteins is impaired after 1 h OGD/24 h reperfusion; these changes are partially mimicked by RAPA treatment. (A) Heat map of key KEGG pathways of the DEGs in the indicated groups. The rows are samples in different groups and the columns are the KEGG pathways. N1-N3 indicated the number of replicated times. (B) Results of GO enrichment analysis of DEGs are presented in the indicated groups. Abscissa is the enriched GO, and ordinate is the number and ratio of DEGs. Different colors indicate different GO classes, namely molecular function, biological process, and cellular component. (C) Heat map of the selected 98 DEGs in the indicated groups. Each row indicates a sample and each column a gene. Red represents upregulated genes and blue downregulated ones. (D-I) Representative western blots and quantitative analysis of SYN1, SYT1, TUBB3, SNAP25 and HPCA in the indicated groups (E, n = 10; F, n = 9; G, n = 5; H, n = 5; I, n = 3; *p < 0.05, **p < 0.01, ***p < 0.001 vs the normoxia group; #p < 0.05, ##p < 0.01, ###p < 0.001 vs 1-h OGD group). (J-N) Representative western blots and quantitative analysis of SYN1, SYT1, TUBB3 and SNAP25 in neurons after RAPA treatment or after the subsequent culture without RAPA (K, n = 6; L, n = 4; M, n = 5; N, n = 5; *p < 0.05, **p < 0.01, ***p < 0.001 vs the control group; ##p < 0.01, ###p < 0.001 vs rapa group). ACTB was used as the loading control. Statistical comparisons were carried out with paired t-tests in the DEGs selected experiment, and with one-way ANOVA in the western blotting experiment. Data are shown as the mean ± SEM
Synapses must maintain a balance between protein biosynthesis and degradation for homeostasis and synaptic plasticity [34]. The disruption of proteostasis, a cellular network determining protein life from synthesis to degradation, will lead to the deterioration of functions and ultra-structures [35]. Synaptic homeostasis depends on efficient autophagy function for the turnover of synaptic proteins [36,37]. To further explore the results of RNA sequencing and the molecular basis of the aforementioned impairment in synaptic ultra-structures, several synaptic proteins implied by RNA sequencing were evaluated for their expression level in the hippocampal neurons of the experimental groups. Western blot assays showed that SYN1, SYT1, TUBB3, SNAP25 and HPCA were significantly down-regulated in hippocampal neurons during 1-h OGD/24-h R compared to those of the normoxia group (Figure 8D-I). These results were consistent with the RNA sequencing of these 5 genes (Figure 8C). Similarly, reperfusion following RAPA withdrawal resulted in the downregulation of SYN1, SYT1, TUBB3 and SNAP25 (Figure 8J-N). Based on the lysosomal dysfunction, downregulated synaptic proteins and sequential occurrence of disruption of proteostasis after transient upregulated autophagy, it can be concluded that the autophagy-dependent impairment in the dynamic turnover of synaptic proteins may be responsible for the ultra-structural changes at synaptic sites.
LAMP1 accumulation and impaired synaptic ultra-structures occurred after acute ischemia in MCAO mice
We observed significant autophagic dysfunction in synaptic ultra-structures in cultured hippocampal neurons after OGD exposure. Therefore, we wondered what would happen in vivo in an ischemic mouse model. Transient middle cerebral artery occlusion (tMCAO)-induced focal cerebral ischemia was performed as 1 h occlusion and 6 h (tMCAO-Rep 6 h) or 72 h reperfusion (tMCAO-Rep 72 h) (Figure 9A), which was assessed by the infarct volume, cerebral edema of the ischemic cortex, and neurobehavioral scores (Figure 9B-D). The infarct volume increased significantly after tMCAO-Rep 6 h; moreover, the neurobehavioral scores increased both after tMCAO-Rep 6 h and tMCAO-Rep 72 h. Furthermore, the LC3-II:LC3-I ratio and LAMP1 protein levels were analyzed with western blot assays in the contralateral and ipsilateral cortex (Figure 9E). Both the LC3-II:LC3-I ratio and the levels of LAMP1 were not influenced in the sham group mice (Figure 9F and G). However, the LC3-II:LC3-I ratio increased in the ipsilateral cortex of the MCAO mice upon tMCAO-Rep 6 h compared to that of the contralateral cortex (Figure 9F). The levels of LAMP1 increased significantly in the ipsilateral cortex of tMCAO-Rep 72 h mice (Figure 9G). Moreover, the ultra-structures in cytoplasm of the cortex of MCAO mice were evaluated with TEM assays (Figure 9H). The number of autophagosomes in the cytoplasm increased distinctively upon tMCAO-Rep 6 h; however, the number of lysosomes and autolysosomes in the cytoplasm increased distinctively upon tMCAO-Rep 72 h (Figure 9I), which were consistent with the results from the western blot assays.
Figure 9.
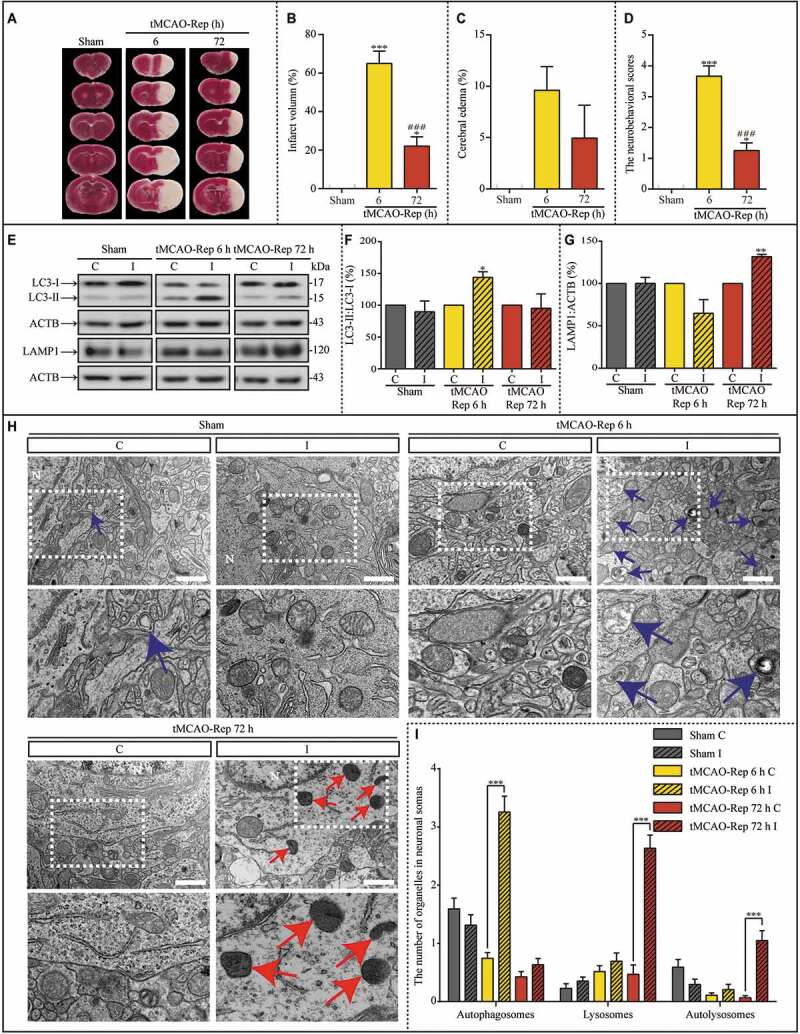
Lysosome dysfunction is transiently induced by 1 h MCAO insult during subsequent reperfusion in vivo. (A) Representative images of TTC-stained cerebral coronal sections of mice in sham, 1 h MCAO/6 h reperfusion (tMCAO-Rep 6 h), and 1 h MCAO/72 h reperfusion (tMCAO-Rep 72 h). Quantification of brain infarct volume (B), cerebral edema (C), and neurobehavioral scores (D) in tMCAO-Rep 6 h and tMCAO-Rep 72 h mice (B-D, n = 3; *p < 0.05, ***p < 0.001 vs. the sham group; ###p < 0.001 vs. tMCAO-Rep 6 h group). Statistical comparisons were carried out with one-way ANOVA. (E-G) Representative western blots and the quantitative analysis of the LC3-II:LC3-I ratio and LAMP1 in the indicated groups (F and G, n = 3; *p < 0.05 and **p < 0.01 vs. the contralateral group). I: ipsilateral, C: contralateral. Statistical comparisons were carried out with paired t-tests. (H and I) Representative TEM images and quantitative analysis of the number of autophagosomes, lysosomes, and autolysosomes in the cytoplasm of neurons exposed to sham, tMCAO-Rep 6 h, and tMCAO-Rep 72 h (sham C/I, n = 44/51; tMCAO-Rep 6 h C/I, n = 74/39; tMCAO-Rep 72 h C/I, n = 47/63; ***p < 0.001 vs. the contralateral group); scale bar: 1 μm. Blue or red arrowheads indicate representative autophagosomes or lysosomes, respectively. Statistical comparisons were carried out with unpaired t-tests. Data are shown as the mean ± SEM
We then asked whether in vivo upregulated autophagy is the onset of the subsequent lysosomal accumulation. After intracerebroventricular (i.c.v) injection of 3-MA, the infarct volume, cerebral edema, and the neurobehavioral scores in the contralateral cortex and ipsilateral cortex were compared (Figure 10A-D). The infarct volume and the neurobehavioral scores upon tMCAO-Rep 6 h were salvaged against insults in the duration of reperfusion by 3-MA. Furthermore, the LC3-II:LC3-I ratio and LAMP1 protein levels were evaluated with western blot assay with or without 3-MA pretreatment (Figure 10E-G). After the transient upregulation of autophagy (Figure 10F), the following lysosomal accumulation was significantly alleviated (Figure 10G). In addition, the ultra-structures in the cytoplasm of the cortex of MCAO mice were assessed with TEM assays (Figure 10H). After the increased number of autophagososomes was reduced by 3-MA, both the number of lysosomes and autolysosomes were reduced correspondingly (Fig. S10). Moreover, the synaptic ultra-structures in the cortex of sham and MCAO mice was assessed with TEM assays (Fig. S11A). Synaptic space width increased and PSD thickness decreased significantly after both tMCAO-Rep 6 h and tMCAO-Rep 72 h, and RRP size decreased only after tMCAO-Rep 72 h (Fig. S11B-D). Meanwhile, there is no significant difference in the postsynaptic membrane length between different groups (Fig. S11E). Furthermore, PSD thickness reversed to the basal level after the 3-MA pretreatment (Fig. S11C). This phenomenon was similar to what occurred in the in vitro experiments, indicating that dysfunctional lysosomal storage and associated synaptic dysfunction are common after both in vivo and in vitro ischemia.
Figure 10.
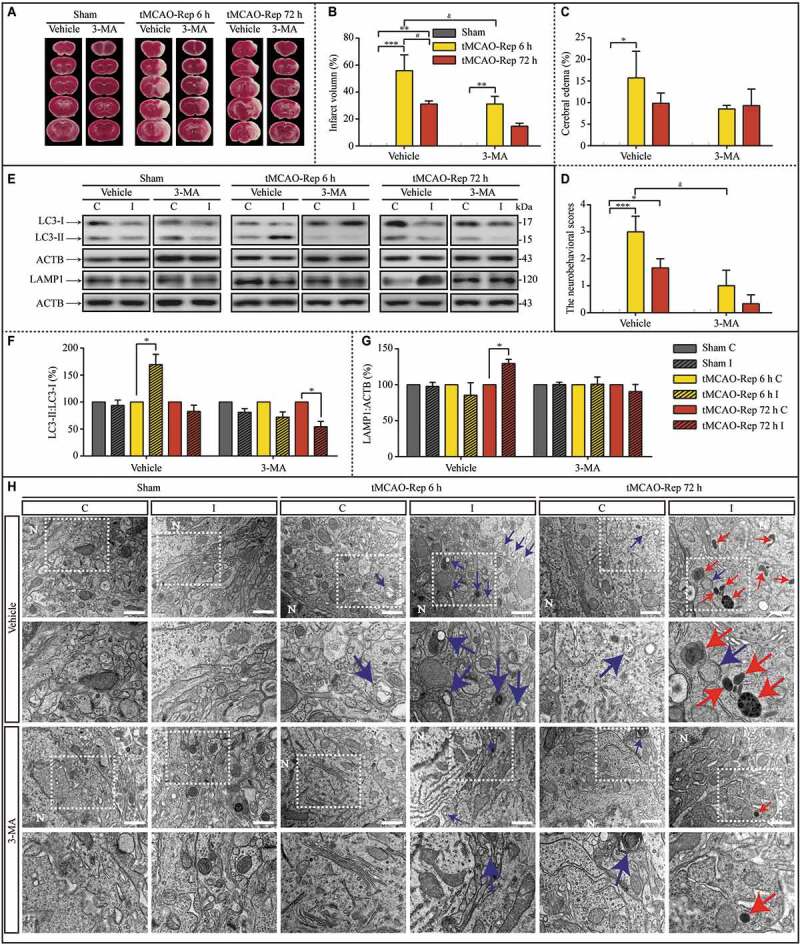
3-MA pretreatment via the intracerebroventricular (i.c.v) injection alleviated ischemic injuries induced by 1 h MCAO insults during subsequent reperfusion. (A) Representative images of TTC-stained cerebral coronal sections of mice in sham, tMCAO-Rep 6 h, and tMCAO-Rep 72 h with or without 3-MA. Quantification of brain infarct volume (B), cerebral edema (C), and neurobehavioral scores (D) in tMCAO-Rep 6 h and tMCAO-Rep 72 h mouse (B-D, n = 3; *p < 0.05, **p < 0.01, ***p < 0.001 vs. the indicated sham group; #p < 0.05 vs. tMCAO-Rep 6 h group; &p < 0.05 vs. vehicle+tMCAO-Rep 6 h group). Statistical comparisons were carried out with one-way ANOVA. (E-G) Representative western blots and the quantitative analysis of the LC3-II:LC3-I ratio and LAMP1 with or without 3-MA in the indicated groups. I: ipsilateral, C: contralateral. (F, Vehicle+tMCAO-Rep 6 h, n = 4 and n = 3 in other groups; *p < 0.05 vs. the contralateral group). Statistical comparisons were carried out with paired t-tests. (H) Representative TEM images of the number of autophagosomes, lysosomes, and autolysosomes in the cytoplasm of neurons exposed to sham, tMCAO-Rep 6 h, and tMCAO-Rep 72 h; scale bar: 1 μm. Blue or red arrowheads indicate representative autophagosomes or lysosomes, respectively. (vehicle Sham C/I, n = 49/43; 3-MA+Sham C/I, n = 41/38; Vehicle+tMCAO-Rep 6 h C/I, n = 24/40; 3-MA+tMCAO-Rep 6 h C/I, n = 44/37; Vehicle+tMCAO-Rep 72 h C/I, n = 41/45; 3-MA+tMCAO-Rep 72 h C/I, n = 40/48 each groups; *p < 0.05, ***p < 0.001 vs. the contralateral group). Statistical comparisons were carried out with unpaired t-tests. For quantitative analysis of the TEM images, see Figure S10. Data are shown as the mean ± SEM
Lysosomal dysfunction in reperfusion may result from unmatched activation levels of MTOR with increased requirements for lysosome-dependent degradation
In response to ischemic stress, MTOR is inhibited, resulting in the induction of autophagy [38]. MTOR (mechanistic target of rapamycin kinase) is usually reactivated after prolonged stimulation. Autophagic lysosome reformation (ALR), a vital process for maintaining lysosome homeostasis, depends on MTOR reactivation, wherein autolysosome tubulation occurs and increases vesicles that finally develop into functional lysosomes [39]. To explore the possible mechanisms contributing to dysfunctional lysosomal storage, relative levels of PRKAA/AMPKα (protein kinase AMP-activated catalytic subunit alpha), phospho-PRKAA (p-PRKAA), RPS6KB/p70S6K (ribosomal protein S6 kinase), and phospho-RPS6KB (p-RPS6KB) were evaluated with western blot assays (Figure 11A). It is well-known that p-RPS6KB levels are phosphorylated by the rapamycin-sensitive MTORC1 (MTOR complex 1), responsible for regulating autophagy [40]. In addition, it is postulated that PRKAA stimulates autophagy by suppressing the activation of MTORC1 [41]. Thus, in the current study, the phosphorylated levels of PRKAA and RPS6KB were investigated for activating levels of MTOR. The phosphorylated levels of RPS6KB depend on the activation levels of MTOR. p-PRKAA levels were increased and p-RPS6KB levels were reduced by OGD insults (Figure 11B and C). The phosphorylated levels of PRKAA and RPS6KB returned to basal levels in the subsequent 24 h reperfusion. 3-MA-pretreatment salvaged phosphorylated levels of RPS6KB increased with OGD, but not those of PRKAA (Figure 11B and C). Similar phenomenon was observed after the intervention of autophagy with Atg7 shRNAs (Figure 11D-F). Furthermore, the phosphorylated levels of PRKAA and RPS6KB were assessed with western blot assay in tMCAO-Rep 6 h and tMCAO-Rep 72 h mice (Figure 11G). p-PRKAA levels were increased and p-RPS6KB levels were reduced after tMCAO-Rep 6 h in MCAO mice (Figure 11H and I). After tMCAO-Rep 72 h, the phosphorylated levels of PRKAA and RPS6KB returned to basal levels (Figure 11H and I). Based on the current results, the activation levels of MTOR did not match with the requirement of degradation of accumulated lysosomes and autolysosomes in the reperfusion duration.
Figure 11.
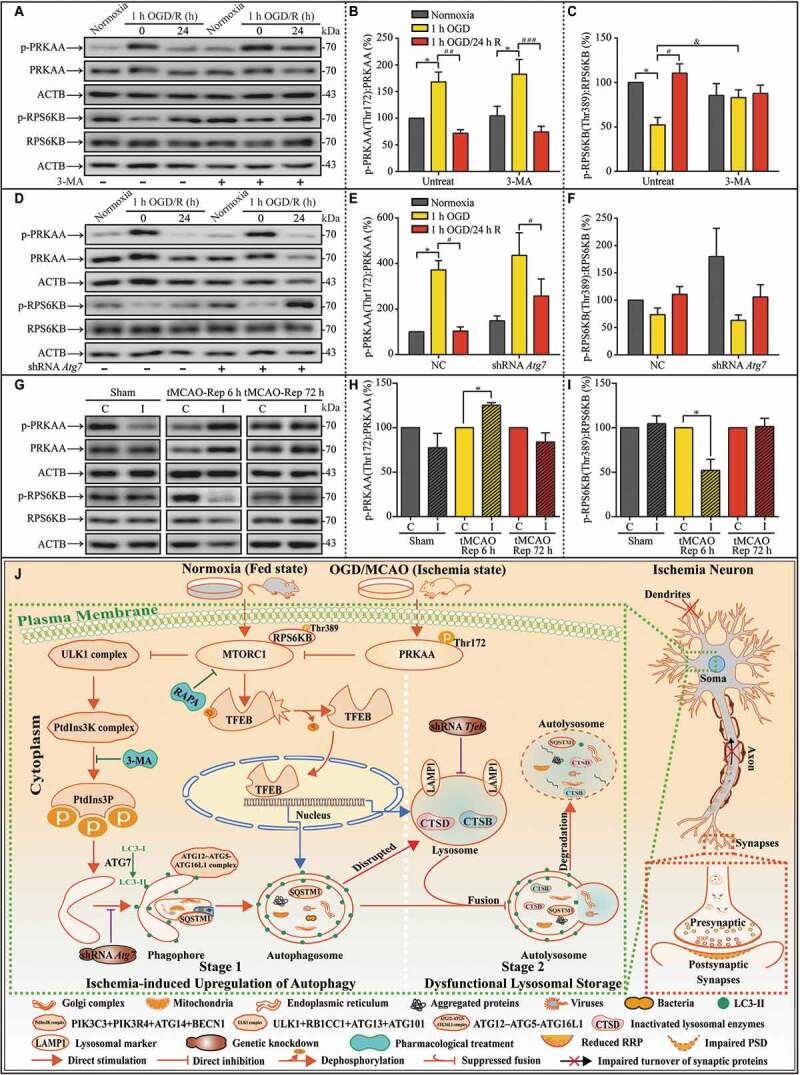
Unmatched activation levels of MTOR may contribute to lysosomal accumulation during subsequent reperfusion in in vitro and in vivo experiments. (A-C) Representative western blots and quantitative analysis of p-PRKAA, PRKAA, p-RPS6KB, and RPS6KB with or without 3-MA in cultured neurons of the indicated groups (B, n = 5; C, n = 4; *p < 0.05 vs. the indicated normoxia group; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. the indicated 1-h OGD group; &p < 0.05 vs. OGD group without 3-MA). Statistical comparisons were carried out with one-way ANOVA. (D-F) Representative western blots and quantitative analysis of p-PRKAA, PRKAA, p-RPS6KB, and RPS6KB with or without ATG7 knockdown in cultured neurons of the indicated groups (n = 4; *p < 0.05 vs. NC + the normoxia group; #p < 0.05 vs. the indicated OGD group). Statistical comparisons were carried out with one-way ANOVA. (G-I) Representative western blots and quantitative analysis of p-PRKAA, PRKAA, p-RPS6KB, and RPS6KB of MCAO mice in the indicated groups (tMCAO-Rep 6 h, n = 4; n = 3 in each of the other groups; *p < 0.05 vs. the contralateral group). Statistical comparisons were carried out with paired t-tests. Data are shown as the mean ± SEM. (J) Proposed model for the scenario of upregulated autophagy by ischemic stress, subsequent lysosomal dysfunction, and synaptic impairment in the ultra-structures. Here, we highlight the central role of neuronal autophagy in influencing lysosome functions and synaptic injuries. In this model, in the early stages of ischemia, the level of intracellular autophagy is upregulated. In the subsequent reperfusion, lysosome function regulated by MTOR signaling does not meet the requirement for the clearance of remnants of the transient autophagy burst, which leads to functional lysosomal accumulation. Finally, the impairment in turnover of synaptic proteins results in the aggravation of ischemic synaptic function damage, especially in the later stages of reperfusion. This model suggests that neuron-targeted-regulation of the lysosomal biogenesis gene MTOR may rescue dysfunctional lysosomes and improve synaptic function in later stages of reperfusion
Discussion
In the current study, we found that autophagy activation after ischemic insult is only transient, as the increased number of autophagosomes quickly return to basal levels. Furthermore, the early activation of autophagy may be the initiating factor that contributes to late-stage lysosomal dysfunction (Figure 11J). As lysosomes are vital for autophagosome degradation [42], lysosomal dysfunction may be considered a disorder of the autophagosome-lysosomal machinery. It was an interesting phenomenon that autophagy upregulation was followed by the dysfunction of the endosome-lysosomal system, which resulted in the impairment of autophagic machinery by the injured degradation of autophagosome contents. Because the early autophagy upregulation was transient, the subsequent dysfunction remained in a long-term and autophagosome-formation-independent manner. Our results suggest that in addition to the protein level of SQSTM1 and the ratio of LC3-II:LC3-I, other parameters should be evaluated to assess the status of the autophagosome-lysosomal machinery and provide a clearer understanding of autophagic machinery dysregulation. The initial activation or inhibition of autophagy only functions as a prelude to a prolonged dysfunction of autophagic machinery that has a greater impact than the earlier changes. Furthermore, we propose that too much emphasis has been placed on autophagosome formation, which is only one stage of the autophagy machinery; therefore, the other stages of autophagy and the relationship between them should be fully addressed in future studies, including the functions of lysosomes. We are not challenging the previous studies, but only provide a possible prospect in autophagy study.
In the acute ischemia stage, transient upregulation of autophagy with increased autophagosomes and greater ongoing autophagic flux occurs, followed by lysosomal accumulation. Further investigation revealed that lysosome enlargement and lysosome dysfunction resulted from decreased CTSD activity, indicating dysfunctional lysosomal storage. Moreover, there was accumulation of autophagosomes in neuronal processes, which cannot be transported from the terminals to the cytoplasm of neurons. Based on these phenomena, it can be concluded that the response capacity to stressors is different for the individual stages of autophagy. Autophagy is normally regulated by upregulation or suppression via the formation of autophagosomes or the fusion between autophagosomes and lysosomes [43]. However, in the current study, significant accumulation of undegraded autolysosomes was observed, suggesting that additional methods targeting autophagy should be considered, such as increased activity of lysosomal enzymes.
LSDs are generally genetic and often fatal diseases induced by mutations in lysosomal-function-associated proteins, including lysosomal enzymes, non-enzymatic lysosomal proteins, and non-lysosomal proteins [44]. Genetic LSDs are characterized by the accumulation of undegraded cargoes in lysosomes and lysosomal dysfunction. Although multiple forms of multi-systemic dysfunction are found in LSDs, most LSDs exhibit neurologic symptoms and neurodegeneration [45]. Interest in these diseases has increased because some LSDs share signaling pathways with neurodegenerative diseases, including AD and PD [30,46,47]. For neurons, lysosomal disorders have been associated with increased susceptibility to ischemic injury. Feldt-Rasmussen U reported that Fabry disease, an X-linked lysosomal storage disorder, has the classic phenotypes of acroparesthesia, hypohidrosis, and corneal opacity in childhood [48]. Some of these patients suffer from strokes in the third to fifth decades of life, indicating that lysosomal disorders play a role in the occurrence of stroke. Based on the synaptic dysfunction observed in genetic LSDs, we examined the synaptic structure and found that synaptic dysfunction accompanied this change in lysosome function. In addition, we found that this lysosomal accumulation may be the inevitable consequence after autophagy dysfunction, regardless if autophagy upregulation is induced by OGD or rapamycin exposure. Vascular cognitive impairment has long been characterized by a common profile that differs from other causes of cognitive impairment and dementia, such as AD [49]. Based on the prevalence of autophagy dysfunctions in these disorders, they may share a similar pathogenic pathway.
For neurons, the processes are more complicated because their highly polarized structure causes the spatial separation of various components [50]. However, recent reports suggest that autophagosomes formed locally can be degraded via distal axonal lysosomal machinery [51]. The abnormal formation or retrograde transport of autophagosomes disrupts the clearance of excitotoxins, resulting in synaptic disorders [52]. Furthermore, the rate-limiting step of cargo degradation is considered to be the fusion of autophagosomes and lysosomes [53]. In the current study, dysfunctions of the autophagy-lysosomal system occurred together with synaptic dysfunction, again suggesting the vital role of lysosomes. Furthermore, the inhibition of lysosomal proteolysis damages the axonal transport of degradative organelles, suggesting that lysosomes can have a profound influence on neuronal function [54].
A series of abnormalities in autophagic pathways are associated with dysfunction of the lysosomal system and are associated with the onset of AD [55]. Autophagic vacuoles with partly digested proteins specifically accumulate in focal axonal swellings, resulting in the neuritic dystrophy that is characteristic of AD [56]. Therefore, because disorders of both retrograde transport and autophagic systems are associated with lysosomal function, furthering our understanding of lysosome function to improve treatment options should be a priority. Although some reports suggest that dysfunctional autophagy induces disorders of some synaptic proteins contributing to synaptic dysfunction [57], lysosomal dysfunction is actually the molecular basis of this dysfunction based on our current findings. Thus, we propose that lysosomal dysfunction may be the cause of synaptic disorders under certain stress conditions, such as ischemic stress. In the current study, a number of synaptic-function-associated proteins, including SYN1, SYT1, TUBB3, SNAP25, and HPCA, which are important for the hemostasis of synaptic functions and the maintenance of the synaptic ultra-structures, decreased during the reperfusion. Of note, the total protein levels of PRKAA and RPS6KB did not change in the neurons in different experimental conditions as the synaptic proteins did. Because these processes involve highly specialized structures that are remote from the cytoplasm, the efficiency of the autophagic machinery, which contributes to the dynamic turnover of synaptic proteins, may not be reversed sufficiently. Our observation of dysfunctional lysosomal storage during reperfusion following acute ischemia suggests that the maintenance of lysosomal function may be a good target for achieving synaptic plasticity after acute ischemia.
In neurons, MTOR is distributed at both presynaptic and postsynaptic sites (or to lysosomes) where it acts as surveillance on autophagy [58,59]. ALR is a vital process for maintaining lysosome homeostasis, which acts as an evolutionarily conserved lysosome regeneration cycle [39]. In addition to clathrin and PtdIns(4,5)P2 were also identified as potential regulators for ALR and KIF5B and necessary for autolysosome tubulation, MTOR reactivation is the onset of the occurrence of ALR. Furthermore, MTOR may affect the degradation capacity of autolysosomes by the regulation of lysosomal pH [60]. Generally, after MTOR is reactivated, autolysosome tubulation occurs and increases vesicles that finally develop into functional lysosomes [39]. Inhibition of MTOR reactivation results in the accumulation of enlarged autolysosomes and impaired lysosomal functions. In the current study, MTOR activation after 1 h OGD/24 h reperfusion or tMCAO-Rep 72 h, addressed by phosphorylated levels of PRKAA and RPS6KB, was not significantly changed compared to that of the normoxia or sham group in both in vivo and in vitro models, although there is a large accumulation of lysosomes and autolysosomes. Thus, it can be concluded that the biogenesis of lysosomes did not change with the requirement of autophagy. Furthermore, production of some lysosomal enzymes, such as CTSD, did not synchronize with the other components of lysosomes. Obviously, although the activation of MTOR reversed to basal levels, the basal level of MTOR is not sufficient for the high demand for lysosomal function after ischemic stroke. In the near future, an MTOR function-related strategy against the accumulation of lysosomes following ischemic stress may be a promising gateway for alleviating lysosomal dysfunction.
Unlike lysosomal function, lysosome transport and axonal morphology were not affected by autophagic activation with RAPA [54]. Autophagy activation has been shown to coincide with axonal swelling during nerve growth factor deprivation and excitotoxicity in PC12 cells [61,62]. Autophagy activation is involved in neurodegeneration [63,64]. In our study, autophagy activation with RAPA induced LAMP1 accumulation and disrupted dendritic morphology. We made three conclusions based upon these observations. First, the entire autophagy process has 5 stages, which may differ in their capacity to adapt to stress. Therefore, the activation of autophagosome formation alone may cause a signaling cascade, such as a higher requirement for lysosomal function, because 3-MA pretreatment partially prevents the accumulation of LAMP1 and changes in synaptic morphology. Second, dysfunctional lysosomal storage accompanied by the reduced biogenesis of lysosomal hydrolases may be another cause of lysosomal dysfunction. Third, lysosomal dysfunction is the molecular basis of these synaptic plasticity disorders because several synaptic proteins decrease simultaneously.
In summary, an early transient upregulation of autophagy may result in undegraded cargoes, especially in the neurites of neurons. The following MTOR-dependent biogenesis of lysosomes and lysosomal functions are not capable of clearing these undegraded materials which cause impairments in the dynamic turnover of synaptic proteins. Finally, the synaptic ultra-structures are modified. Based on these results, we suggest that therapeutic strategies for synaptic dysfunction after ischemia should include lysosome-targeting compounds, such as those affecting lysosomal degradation efficiency. Our current study provides a model of dysfunctional lysosomal storage. Although the lysosomal storage induced by ischemia and reperfusion are moderate compared with those caused by genetic mutations, their epigenetic significance may provide insights into the mechanisms underlying the effects of the genetic mutations. Additionally, ischemia-induced functional lysosomal storage plays a role in the maintenance of synaptic morphology; therefore, its influence on the synaptic network and cognitive function deserves further exploration. To our knowledge, this is the first report on dysfunctional lysosomal storage after ischemic stress, which sheds light on a novel treatment target for post-ischemia synaptic dysfunction, even post-stroke cognitive impairment.
Materials and methods
Animals
For development of a MCAO model, 6- to 8-week-old (22.5–24 g) male C57BL/6 mice were purchased from Charles River Laboratories (213; Beijing, China). Animals were housed 4–5 per cage under standard housing conditions (room temperature [RT] 20 ± 2°C with a 12-h light–dark cycle) with free access to food and water. All procedures and treatments were conducted in accordance with the ethical regulations set by the Animal Experimentation Committee of Capital Medical University.
For development of an OGD model, Newborn Wistar rats were used within 24 h of birth and were purchased from Charles River Laboratories (102; Beijing, China). All procedures and treatments were conducted in accordance with the ethical regulations set by the Animal Experimentation Committee of Capital Medical University.
Agents and antibodies
Hanks’ balanced salt solution (HBSS; 14175095), 0.25% trypsin-EDTA (25200072), Dulbecco’s modified Eagle’s medium (DMEM; 10569010), Neurobasal-A Medium (10888022), Glucose-free DMEM (11966025), B27 (17,504,044), and GlutaMAX (35050061) were all obtained from Gibco. Poly-D-lysine hydrobromide (27964994) was obtained from Sigma-Aldrich. Penicillin-streptomycin (PSL02) was obtained from Caisson Labs. 3-MA (HY-19312), and RAPA (HY-10219) were obtained from Med Chem Express.
Primary antibodies were as follows: anti-LC3B (Abcam, ab63817; Cell Signaling Technology, 3868; dilution ratio: 1:750 for western blot), anti-SQSTM1 (Proteintech, 18420-1-AP and 55274-1-AP; dilution ratio: 1:1000 for western blot), anti-LAMP1 (Abcam, ab24170 and ab25630; dilution ratio: 1:1000 for western blot, 1:100 for immunofluorescence staining), anti-TFEB (Proteintech, 13372-1-AP; dilution ratio: 1:500 for western blot, 1:100 for immunofluorescence staining), anti-CTSD (Proteintech, 21327-1-AP; dilution ratio: 1:1500 for western blot), anti-CTSB (Abcam, ab214428; dilution ratio: 1:750 for western blot), anti-MAP2 (Abcam, ab75713; dilution ratio: 1:100 for immunofluorescence staining), anti-ATG7 (Cell Signaling Technology, 8558; dilution ratio: 1:750 for western blot), anti-PRKAA/AMPKα (Cell Signaling Technology, 2603; dilution ratio: 1:750 for western blot), anti-p-PRKAA/AMPKα (Thr172; Cell Signaling Technology, 2535; dilution ratio: 1:750 for western blot), anti-RPS6KB/p70S6K (Cell Signaling Technology, 2708; dilution ratio: 1:750 for western blot), anti-p-RPS6KB/p70S6K (Thr389; Cell Signaling Technology, 9234; dilution ratio: 1:750 for western blot), anti-SYN1 (Cell Signaling Technology, 5297; dilution ratio: 1:750 for western blot), anti-SYT1 (Cell Signaling Technology, 14558; dilution ratio: 1:750 for western blot), anti-SNAP25 (Cell Signaling Technology, 5309; dilution ratio: 1:750 for western blot), anti-HPCA (Abcam, ab126740; dilution ratio: 1:750 for western blot), anti-TUBB3 (Abcam, ab41489; dilution ratio: 1:750 for western blot), anti-ACTB/β-actin (Proteintech, 60008-1-Ig; dilution ratio: 1:5000 for western blot), and anti-TUBB/β-tubulin (Proteintech, 10068-1-AP; dilution ratio: 1:1000 for western blot).
Secondary antibodies for western blot were from Cell Signaling Technology as follows: anti-mouse IgG, HRP-linked antibody (7076s), anti-rabbit IgG, HRP-linked antibody (7074s). These antibodies were used at 1:5000 dilution ratio. Secondary antibodies for immunofluorescence staining were from Thermo Fisher Scientific as follows: goat anti-chicken IgY (H + L) Alexa Fluor 647 (A-21449), goat anti-mouse IgG (H + L) Alexa Fluor 594 (A-11005), and goat anti-rabbit IgG (H + L) Alexa Fluor 488 (A-11034). These antibodies were used at 1:200 dilution ratio.
Mouse middle cerebral artery occlusion model (MCAO)
A MCAO mouse model was developed as follows. Briefly, mice were anesthetized with an intraperitoneal injection of 1.25% tribromoethanol (Sigma-Aldrich, T48402). Through a midline skin incision, the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were isolated. The proximal CCA and distal ECA were ligated. A MCAO suture (0.23 ± 0.02 mm diameter of head end, 0.104 mm diameter of suture body, 3 cm Length; RWD Life Science, MSMC23B104PK50), with its head end coated with soft silicone, was inserted into the right ICA through the broken end of the ECA to block the origin of the right middle cerebral artery. Meanwhile, a sham-operated mouse received the same surgical procedures without inserting MCAO sutures. Body temperature was maintained at 37°C by a water circulation heat preservation system (RWD Life Science, Shenzhen, China) during surgery and for 2 h after the start of reperfusion. MCAO sutures were removed from the mice after 1 h of occlusion, followed by reperfusion.
For the i.c.v injection, anesthetized mice were placed in a stereotaxic apparatus (RWD Life Science, Shenzhen, China) and a burr hole was drilled 0.5 mm posterior to bregma, 1.0 mm lateral to the sagittal suture, and 1.75 mm in depth. Mice were given an i.c.v injection of 15 µg 3-MA before 30 min in MCAO with a microinjector. 3-MA was dissolved in normal saline by heating the solution to 60°C immediately before injection. Vehicle mice were injected with the same volume of normal saline solution (5 µL). The dosages of 3-MA were selected according to previous studies [65].
Measurement of motor deficits, cerebral infarct volume, and cerebral edema
The motor deficits in mice subjected to sham or MCAO surgery were evaluated using Zea-Longa scoring criteria as follows: 0 point, mice behave normally; 1 point, mice can’t fully stretch their left front legs; 2 points, mice turn around in a circle; 3 points, mice fall down to the left side; 4 points, mice cannot move by themselves, losing consciousness. The motor deficits were evaluated by an examiner without knowing experimental conditions.
To determine the infarct volume, mice were anesthetized with an intraperitoneal injection of 1.25% tribromoethanol and sacrificed by decapitation at the indicated time point after reperfusion. The coronal brain slices at 1-mm intervals were incubated in 2% 2, 3,5-triphenyltetrazolium chloride (TTC; Solarbio, T8170) for 30 min at 37°C. Then, the slices were fixed through the immersion in 4% paraformaldehyde (Solarbio, P1110) and were taken by photographic image. Cerebral infarct areas were measured by an examiner without knowing experimental conditions using Image-Pro Plus 6.0. The percentage of the infarct volume was calculated using the following equation: Ipsilateral Infarct volume/Contralateral hemispheric volume × 100%. The percentage of cerebral edema was determined as an indicator of cerebral edema using the following equation: (Ipsilateral hemispheric volume – Contralateral hemispheric volume)/Contralateral hemispheric volume × 100%.
Primary hippocampal neuron culture
Hippocampal neurons were prepared from newborn Wistar rats. The hippocampi were isolated in ice-cold HBSS, digested in 0.125% trypsin-EDTA for 12 min at 37°C, triturated with a siliconized pipette, and plated on 12-mm diameter coverslips (for SIM or Immunofluorescent staining) at a density of 3.5 × 105 cells/mL, in 12-mm plastic dishes at a density of 7.5 × 105 cells/mL (for TEM), or in 6-well plastic dishes, which were pre-coated with 0.01% poly D-lysine for at least 12 h, at a density of 5 × 105 cells/mL. Plating medium (DMEM containing 4.5 g/L glucose and 100 mg/L sodium pyruvate) supplemented with 100 U/mL penicillin/streptomycin, 10% fetal bovine serum, and 10% horse serum was replaced with Neurobasal-A Medium containing 0.3 g/L GlutaMAX, 2% B27 supplement, and 100 U/mL penicillin/streptomycin 4 h after plating. Neurons were incubated in 5% CO2 at 37°C for one week before experiments, and one-third of the medium was replaced every 3 days.
Establishment of the oxygen-glucose deprivation/reperfusion (OGD/R) model
Cells were randomly assigned to different experimental groups. To establish the OGD model, the hippocampal neuron culture medium was changed to serum- and glucose-free DMEM on day 8 of in vitro culture. The neuron cultures were placed in a closed hypoxia incubator chamber (Stemcell Technologies, 27,310) filled with 2% O2, 5% CO2 and 93% N2 at 37°C. After 1 h, the cells were removed from the chamber. The medium was replaced with fresh neurobasal culture medium supplemented with 2% B27, GlutaMAX, and 100 U/mL penicillin/streptomycin. Then, the cultured neurons were incubated in 5% CO2 at 37°C to achieve reperfusion. At the indicated time point following reperfusion, the neurons were used for experiments.
RNA sequencing
Total messenger RNA (mRNA) was extracted with Trizol (Life Ambion, 15596018) from the hippocampal neurons of normoxia, 1-h OGD, and 1-h OGD/24-h R groups. Then, the obtained RNAs were evaluated with Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA) and Invitrogen Qubit Fluorometer. Total RNA samples that met the following requirements were used in subsequent experiments: RNA integrity number (RIN) > 7.0 and a 28S:18S ratio > 1.8. High-throughput RNA sequencing (RNA-seq) was conducted by Illumina HiSeq 2500 (Illumina, San Diego, CA, USA) at CapitalBio Corporation (Beijing, China). The raw sequencing data were aligned to the rat preference genome. We used a p < 0.05 and fold changes cutoff as |log2 ratio|≥0.5 for addressing the differentially expressed genes in the RNA-seq analysis. If there were several transcripts in a gene, sequencing depth and expression were calculated using the longest one. GSVA (https://doi.org/10.1186/1471-2105-14-7) was used to assess KEGG gene set enrichment in gene expression RNA-seq data [66]. GO enrichment analysis (p value<0.05) against genes was conducted using clusterProfiler [67]. Heatmap was plotted using pheatmap R package (version 1.0.12).
Pharmacological treatments of cultured neurons
To determine the effects of the autophagy inhibitor, 3-MA on oxygen-glucose deprivation (OGD)-reperfusion-induced autophagy, 3-MA (5 mM) was dissolved in normal culture medium and pre-incubated with primary neurons for 1 h. Following, the medium was replaced with glucose-free DMEM for 1-h OGD treatment. To activate autophagy, RAPA (50 nM) was dissolved in dimethyl sulfoxide (DMSO), diluted with normal culture medium, and pre-incubated with primary neurons for 2 h. The medium was then replaced with fresh Neurobasal culture medium supplemented with 2% B27, GlutaMAX, and 100 U/mL penicillin-streptomycin, and incubated under 5% CO2 at 37°C to achieve reperfusion.
Western blot assay
After MCAO, the ipsilateral parietal cortex (penumbra of middle cerebral artery) and the corresponding area of the contralateral cortex were rigorously homogenized with FastPrep-24 (MP Biomedicals, USA). For in vitro samples, after subjection to the corresponding experimental treatments, culture mediums of these cells were removed and cells were washed 3 times with pre-cooled phosphate-buffered saline (PBS; Applygen Technology, C1053). Then, the in vivo tissue samples and in vitro cell samples were lysed with RIPA Lysis buffer (Applygen Technology, C1053) supplemented with PMSF (Beyotime Biotechnology, ST506), Complete Protease Inhibitor EASYpacks EDTA-Free (Roche, 04693132001), and Phosphatase Inhibitor Cocktail 2 (Sigma-Aldrich, P5726) for 60 min at 4°C. Samples were collected and centrifuged at 15000 x g for 20 min at 4°C. The supernatant was transferred to a new tube. Protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225). Supernatants were boiled in Protein SDS-PAGE Loading Buffered Solution (ROBY, RBU114-2). Equal amounts of protein samples were loaded and separated in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (8%-12% SDS-PAGE depending on the molecular weight of the protein of interest) with SDS-PAGE running buffer (Applygen Technology, B1005). Proteins were then transferred onto a PVDF membrane (Millipore, IPVH00010) with WB transfer buffer (Solarbio, D1060). Subsequently, the membrane was blocked in blocking solution containing 10% nonfat powdered milk (Solarbio, D8340) for 1 h. After being washed 3 times with Tris buffered saline plus tween-20 (TBST; Solarbio, T1082), samples were probed with indicated primary antibodies for overnight incubation at 4°C. On the next day, membranes were washed 3 times with TBST and incubated with HRP-linked secondary antibody for 1.5 h at RT. After washing 3 times with TBST, samples were detected with Enhanced Chemiluminescent (NCM Biotech, P10300) and visualized by using Amersham imager 600 (General Electric Company, USA). The expression of the target band relative to the loading control, ACTB or TUBB, was quantified with ImageJ software.
CTSD activity
CTSD activity was measured using a Fluorometric Assay Kit from Abcam (ab65302). The kit is a fluorescence-based assay that utilizes the preferred CTSD substrate sequence (GKPILFFRLK(Dnp)D-R-NH2) labeled with methyl coumaryl amide (MCA). Samples were prepared according to the manufacturers’ instructions. Briefly, cells (5 × 105) were harvested for each condition, washed with ice-cold PBS, resuspended in 100 µL of PBS, and homogenized quickly by pipetting up and down multiple times. Samples were centrifuged at 100 x g for 5 min at 4°C, and the supernatant was decanted without disturbing the cell pellet. The cell pellet was transferred to a new tube in 200 µL of chilled CD Cell Lysis Buffer and then incubated on ice for 10 min. Samples were centrifuged at 21,000 x g for 5 min at 4°C to remove any insoluble material. The cleared cell lysate was transferred to a new tube. Then, 50 µL of cell lysate and 52 µL of reaction mix (50 µL of reaction buffer and 2 µL of substrate) were added to each well of a black 96-well plate (CORNING, 3916), and the samples were incubated at 37°C for 30 min. The fluorescence released upon substrate cleavage was measured using a SynergyH1 microplate reader (Biocompare, USA) at Ex/Em = 328/460 nm.
Plasmids and lentiviral vectors
Autophagosomes were measured by analyzing EGFP-MAP1LC3B puncta. Cultured hippocampal neurons expressing EGFP-MAP1LC3B were generated by the transfection of pLenti-UBC-EGFP-3 FLAG-MAP1LC3B (Obio Technology, Shanghai, China), with pLenti-UBC-EGFP-3 FLAG used as the negative control (NC).
For short hairpin RNAs (shRNAs) transfection, three shRNAs targeting Atg7 and negative control (NC) with lentiviruses particles were purchased from Obio Technology (Shanghai, China). Cultured hippocampal neurons were transfected according to the following procedure. In brief, neurons (5 × 105 per well) were seeded in 6-well plates for up to 3 days. Then, lentivirus expressing the respective shRNA plasmid was added to the medium. After 24 h of incubation in a 5% CO2 incubator, the medium was removed, and 2 mL of fresh medium was added to each well. A multiplicity of infection (MOI) of 10 was used as the optimal concentration for shRNA transduction in our experiment. The transfected neurons were cultured for 96 h and harvested for the subsequent experiments. The knockdown efficiency of three shRNAs targeting Atg7 was determined by western blotting. We chose three different shRNAs against the target gene (Atg7) as follows: shRNA Atg7-#10,651: CCTGTGGGCTTGGATCAAA; shRNA Atg7-#10,652: GCAAGCGAAAGCTGGTCAT; shRNA Atg7-#10,653: GGTGTTTAACTCTTCACAT; shRNA NC-#10,657: TTCTCCGAACGTGTCACGT.
Immunofluorescent staining
Hippocampal neurons were plated onto 12-mm coverslips at a density of 3.5 × 105 cells/mL. At the indicated time points, the cells were subjected to OGD/R establishment. To detect the LAMP1 signals, neurons were washed 3 times with cold PBS and then fixed with 4% paraformaldehyde at RT for 15 min. The samples were then washed 3 times with PBS, permeabilized with 0.25% Triton X-100 (AMRESCO, 9,002,931) in PBS for 10 min, blocked with 5% normal goat serum (ZSGB-bio, ZLI-9056) in PBS for 60 min, and incubated with anti-LAMP1, and anti-MAP2 antibodies overnight at 4°C, followed by Alexa Fluor 594-conjugated goat anti-mouse IgG, Alexa Fluor 488-conjugated anti-rabbit IgG, and Alexa Fluor 647-conjugated goat anti-chicken IgY for 1.5 h at RT. The samples were washed with PBS containing 0.05% Triton X-100 (PBST) between and after antibody incubation. The samples were mounted on a microscope slide containing DAPI-Fluoromount-G (Southern Biotech, 010020). The super resolution images were taken using A1 N-SIM STORM with a 100× oil immersion objective. The super resolution images were analyzed by Imaris 9.3.1 (Bitplane, Switzerland).
The super resolution imaging of autophagosomes and lysosomes
Hippocampal neurons were plated onto 12-mm diameter coverslips (Thermo Fisher Scientific, 1,254,580) at a density of 3.5 × 105 cells/mL. At the indicated time points, the cells were subjected to RNA interference, pharmacological treatment, and OGD/R establishment. To detect the number of autophagosomes and lysosomes, hippocampal neurons were first transfected with pLenti-UBC-EGFP-3FLAG-MAP1LC3B/LC3 at a multiplicity of infection (MOI) of 10 for 96 h and then incubated in neurobasal culture medium containing LysoTracker Red DND-99 (Thermo Fisher Scientific, L7528) for 30 min. The neurons were then washed 3 times with cold PBS and fixed with 4% paraformaldehyde at RT for 15 min. The samples were mounted on a microscope slide containing DAPI-Fluoromount-G. The super resolution images were acquired using an A1 N-SIM STORM microscope with a 100× oil immersion objective (Nikon, Japan). The volume and size of autophagosomes and lysosomes were determined with Nikon Instruments Software-Elements Advanced Research (NIS-Elements AR; Nikon, Japan) and Imaris software. For evaluation of colocalization of LC3-positive and lyso-tracker-positive puncta, taking the distance from the center of the signal to the maximum as a statistic, we take the sum of the radius of the two signals as the reference, greater than the sum of the radius as non-colocalization, less than or equal to any radius as the complete colocalization, and between greater than any half of the radius and less than the sum of the two signal radius as partial colocalization.
To calculate the number of LAMP1-positive puncta per μm2 in the cytoplasm, the neurons were stained with LAMP1 and MAP2 using immunofluorescence staining assay and counterstained with DAPI. The boundary and size of neuronal cytoplasm were determined with MAP2 staining using the function “surface” of Imaris software. The LAMP1-positive puncta within the range of the neuronal cytoplasm were addressed with the function “spot” of Imaris software. After that, the number of LAMP1-positive puncta per μm2 in the cytoplasm was calculated.
Transmission electron microscopy (TEM)
Following subjection to MCAO surgery, mice were subjected to transcardial perfusion with 10 mL of pre-cooled 0.9% NaCl, followed by 40 mL of 4% paraformaldehyde. The ipsilateral parietal cortex (I) and the corresponding area of the contralateral cortex (C) were obtained and kept overnight in 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4).
Cell samples in vitro were prefixed in a mixture of 2% paraformaldehyde and 2.5% glutaraldehyde for 2 h at RT. After the samples were washed 3 times with 0.1 M phosphate buffer (PB, 1 min each), they were postfixed in a mixture of 1% osmic acid and 1.5% potassium ferricyanide for another 1 h at 4°C. The cells were then washed 3 times with 0.1 M PB (15 min per wash) and stained with 1% uranyl acetate for 1 h at RT, followed by three washes with ddH2O (15 min each). Samples were dehydrated in a gradient of ethanol solutions ranging from 30% to 100% ethanol (30%, 50%, 70%, 80%, and 90% for eight min; 100% for eight min 3 times), followed by propylene oxide (two 10 min washes). Samples were gradually embedded with 3 mixtures of EPON812 and propylene oxide (ratios of 1:1, 2:1, and 3:1 for 2 h at 4°C) and subsequently embedded in pure EPON812 overnight. Following, the samples were incubated with fresh EPON812 for 8 h. The cell samples were embedded for 4 h at RT, incubated overnight at 37°C, and cured in the oven at 60°C for 24 h. The polymerized cell samples were sectioned and stained with 1% uranyl acetate for 20 min followed by lead citrate for 5 min. 70 nm-thick ultra-thin sections were prepared, and images in vitro were acquired using a Tecnai Spirit 120 kV TEM (FEI, USA) at an acceleration voltage of 80 kV. Images in vivo were acquired using a JEM-2100 200 kV TEM (JEOL, Japan) at an acceleration voltage of 100 kV. The ultra-structures in the cytoplasm and processes of neurons were measured manually using ImageJ software analysis. To make an accurate TEM quantitation in our experimental condition, we differentiated the structures between autophagosomes, lysosomes, and autolysosomes according to the following criteria. Vesicles with double-membrane structures engulfing cytoplasmic material were defined as autophagosomes. Monolayer organelles with high electron density were called lysosomes. A monolayer structure with significantly enlarged volume and abundant undegraded materials (autophagosomes and other membrane components) were defined as enlarged autolysosomes.
Supplementary Material
Funding Statement
This work is supported by grants from the National Natural Science Foundation of China (31771292 and 31871147), and Beijing Natural Science Foundation (7202006).
Disclosure statement
The authors declare no competing financial interests.
Statistical analysis
Statistical analyses were performed by one-way ANOVA or t-tests using GraphPad Prism 8.0.1 software. The results are presented as the mean ± standard error of the mean (SEM). Details for each analysis can be found in the figure legends.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Sun JH, Tan L, Yu JT.. Post-stroke cognitive impairment: epidemiology, mechanisms and management. Ann Transl Med. 2014;2(8):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333(24):1581–1587. [DOI] [PubMed] [Google Scholar]
- [3].Yang Y, Lv SY, Lyu SK, et al. The protective effect of apelin on ischemia/reperfusion injury. Peptides. 2015;63:43–46. [DOI] [PubMed] [Google Scholar]
- [4].Wei K, Wang P, Miao CY. A double-edged sword with therapeutic potential: an updated role of autophagy in ischemic cerebral injury. CNS Neurosci Ther. 2012;18(11):879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang P, Shao BZ, Deng Z, et al. Autophagy in ischemic stroke. Prog Neurobiol. 2018;163-164:98–117. [DOI] [PubMed] [Google Scholar]
- [6].Hou K, Xu D, Li F, et al. The progress of neuronal autophagy in cerebral ischemia stroke: mechanisms, roles and research methods. J Neurol Sci. 2019;400:72–82. [DOI] [PubMed] [Google Scholar]
- [7].Pei X, Li Y, Zhu L, et al. Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic stroke. Exp Cell Res. 2019;382(2):111474. [DOI] [PubMed] [Google Scholar]
- [8].Soreng K, Neufeld TP, Simonsen A. Membrane trafficking in autophagy. Int Rev Cell Mol Biol. 2018;336:1–92. [DOI] [PubMed] [Google Scholar]
- [9].Wu M, Lao YZ, Tan HS, et al. Oblongifolin C suppresses lysosomal function independently of TFEB nuclear translocation. Acta Pharmacol Sin. 2019;40(7):929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Koerver L, Papadopoulos C, Liu B, et al. The ubiquitin-conjugating enzyme UBE2QL1 coordinates lysophagy in response to endolysosomal damage. EMBO Rep. 2019;20(10):e48014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang DM, Zhang T, Wang MM, et al. TIGAR alleviates ischemia/reperfusion-induced autophagy and ischemic brain injury. Free Radic Biol Med. 2019;137:13–23. [DOI] [PubMed] [Google Scholar]
- [13].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tang G, Gudsnuk K, Kuo SH, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83(5):1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13(7):805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. [DOI] [PubMed] [Google Scholar]
- [17].Kaye EM. Lysosomal storage diseases. Curr Treat Options Neurol. 2001;3(3):249–256. [DOI] [PubMed] [Google Scholar]
- [18].Ferreira CR, Gahl WA. Lysosomal storage diseases. Trans Sci Rare Dis. 2017;2(1–2):1–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Markello TC, Bernardini IM, Gahl WA. Improved renal function in children with cystinosis treated with cysteamine. N Engl J Med. 1993;328(16):1157–1162. [DOI] [PubMed] [Google Scholar]
- [20].Ahrens-Nicklas RC, Tecedor L, Hall A, et al. Neuronal network dysfunction precedes storage and neurodegeneration in a lysosomal storage disorder. JCI Insight. 2019;4(21):e131961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Noda T. Regulation of Autophagy through TORC1 and mTORC1. Biomolecules. 2017;7(3):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gresham GE, Phillips TF, Wolf PA, et al. Epidemiologic profile of long-term stroke disability: the Framingham study. Arch Phys Med Rehabil. 1979;60(11):487–491. [PubMed] [Google Scholar]
- [23].Sensenbrenner B, Rouaud O, Graule-Petot A, et al. High prevalence of social cognition disorders and mild cognitive impairment long term after stroke. Alzheimer Dis Assoc Disord. 2020;34(1):72–78. [DOI] [PubMed] [Google Scholar]
- [24].Marques ARA, Di Spiezio A, Thiessen N, et al. Enzyme replacement therapy with recombinant pro-CTSD (cathepsin D) corrects defective proteolysis and autophagy in neuronal ceroid lipofuscinosis. Autophagy. 2020;16(5):811–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Meikle PJ, Brooks DA, Ravenscroft EM, et al. Diagnosis of lysosomal storage disorders: evaluation of lysosome-associated membrane protein LAMP-1 as a diagnostic marker. Clin Chem. 1997;43(8Pt 1):1325–1335. [PubMed] [Google Scholar]
- [26].Henry AG, Aghamohammadzadeh S, Samaroo H, et al. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet. 2015;24(21):6013–6028. [DOI] [PubMed] [Google Scholar]
- [27].Tanida I, Yamasaki M, Komatsu M, et al. The FAP motif within human ATG7, an autophagy-related E1-like enzyme, is essential for the E2-substrate reaction of LC3 lipidation. Autophagy. 2012;8(1):88–97. [DOI] [PubMed] [Google Scholar]
- [28].Zhao E, Czaja MJ. Transcription factor EB: a central regulator of both the autophagosome and lysosome. Hepatology. 2012;55(5):1632–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lloyd-Evans E, Haslett LJ. The lysosomal storage disease continuum with ageing-related neurodegenerative disease. Ageing Res Rev. 2016;32:104–121. [DOI] [PubMed] [Google Scholar]
- [31].Matsushita K, Numakawa T, Odaka H, et al. Presynaptic dysfunction in neurons derived from tay-sachs iPSCs. Neuroscience. 2019;414:128–140. [DOI] [PubMed] [Google Scholar]
- [32].Sambri I, D’Alessio R, Ezhova Y, et al. Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol Med. 2017;9(1):112–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wolfe DM, Lee JH, Kumar A, et al. Autophagy failure in alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013;37(12):1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bingol B, Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron. 2011;69(1):22–32. [DOI] [PubMed] [Google Scholar]
- [35].Douglas PM, Dillin A. Protein homeostasis and aging in neurodegeneration. J Cell Biol. 2010;190(5):719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. [DOI] [PubMed] [Google Scholar]
- [37].Komatsu M, Wang QJ, Holstein GR, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104(36):14489–14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hwang JY, Gertner M, Pontarelli F, et al. Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ. 2017;24(2):317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Saiki S, Sasazawa Y, Imamichi Y, et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2011;7(2):176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jia R, Guardia CM, Pu J, et al. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy. 2017;13(10):1648–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shi B, Ma M, Zheng Y, et al. mTOR and Beclin1: two key autophagy-related molecules and their roles in myocardial ischemia/ reperfusion injury. J Cell Physiol. 2019;234(8):12562–12568. [DOI] [PubMed] [Google Scholar]
- [44].Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199(5):723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Verity C, Winstone AM, Stellitano L, et al. The epidemiology of progressive intellectual and neurological deterioration in childhood. Arch Dis Child. 2010;95(5):361–364. [DOI] [PubMed] [Google Scholar]
- [46].Keilani S, Lun Y, Stevens AC, et al. Lysosomal dysfunction in a mouse model of sandhoff disease leads to accumulation of ganglioside-bound amyloid-beta peptide. J Neurosci. 2012;32(15):5223–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kresojevic N, Dobricic V, Svetel M, et al. Mutations in niemann pick type C gene are risk factor for alzheimer’s disease. Med Hypotheses. 2014;83(5):559–562. [DOI] [PubMed] [Google Scholar]
- [48].Feldt-Rasmussen U. Fabry disease and early stroke. Stroke Res Treat. 2011;2011:615218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sachdev P, Kalaria R, O’Brien J, et al. Internationlal Society for Vascular, D. Cognitive, Diagnostic criteria for vascular cognitive disorders: a VASCOG statement. Alzheimer Dis Assoc Disord. 2014;28(3):206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shen DN, Zhang LH, Wei EQ, et al. Autophagy in synaptic development, function, and pathology. Neurosci Bull. 2015;31(4):416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Farfel-Becker T, Roney JC, Cheng XT, et al. The secret life of degradative lysosomes in axons: delivery from the soma, enzymatic activity, and local autophagic clearance. Autophagy. 2020;16(1):167–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Katsumata K, Nishiyama J, Inoue T, et al. Dynein- and activity-dependent retrograde transport of autophagosomes in neuronal axons. Autophagy. 2010;6(3):378–385. [DOI] [PubMed] [Google Scholar]
- [53].Tanida I, Minematsu-Ikeguchi N, Ueno T, et al. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1(2):84–91. [DOI] [PubMed] [Google Scholar]
- [54].Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci. 2011;31(21):7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer’s disease. J Alzheimers Dis. 2006;9(3 Suppl):277–289. [DOI] [PubMed] [Google Scholar]
- [56].Uddin MS, Mamun AA, Labu ZK, et al. Autophagic dysfunction in Alzheimer’s disease: cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis. J Cell Physiol. 2019;234(6):8094–8112. [DOI] [PubMed] [Google Scholar]
- [57].Hwang JY, Yan J, Zukin RS. Autophagy and synaptic plasticity: epigenetic regulation. Curr Opin Neurobiol. 2019;59:207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–1945. [DOI] [PubMed] [Google Scholar]
- [59].Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33(2):67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rong Y, McPhee CK, Deng S, et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci U S A. 2011;108(19):7826–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Batistatou A, Greene LA. Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: correlation with suppression of endonuclease activity. J Cell Biol. 1991;115(2):461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yang Y, Xu K, Koike T, et al. Transport of autophagosomes in neurites of PC12 cells during serum deprivation. Autophagy. 2008;4(2):243–245. [DOI] [PubMed] [Google Scholar]
- [63].Yue Z, Horton A, Bravin M, et al. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35(5):921–933. [DOI] [PubMed] [Google Scholar]
- [64].Wang QJ, Ding Y, Kohtz DS, et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26(31):8057–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang X, Yan H, Yuan Y, et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9(9):1321–1333. [DOI] [PubMed] [Google Scholar]
- [66].Reiman M, Laan M, Rull K, et al. Effects of RNA integrity on transcript quantification by total RNA sequencing of clinically collected human placental samples. Faseb J. 2017;31(8):3298–3308. [DOI] [PubMed] [Google Scholar]
- [67].Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.