

ABSTRACT
Coupling of Atg8 to phosphatidylethanolamine is crucial for the expansion of the crescent-shaped phagophore during cargo engulfment. Atg21, a PtdIns3P-binding beta-propeller protein, scaffolds Atg8 and its E3-like complex Atg12–Atg5-Atg16 during lipidation. The crystal structure of Atg21, in complex with the Atg16 coiled-coil domain, showed its binding at the bottom side of the Atg21 beta-propeller. Our structure allowed detailed analyses of the complex formation of Atg21 with Atg16 and uncovered the orientation of the Atg16 coiled-coil domain with respect to the membrane. We further found that Atg21 was restricted to the phagophore edge, near the vacuole, known as the vacuole isolation membrane contact site (VICS). We identified a specialized vacuolar subdomain at the VICS, typical of organellar contact sites, where the membrane protein Vph1 was excluded, while Vac8 was concentrated. Furthermore, Vac8 was required for VICS formation. Our results support a specialized organellar contact involved in controlling phagophore elongation.
Abbreviations: FCCS: fluorescence cross correlation spectroscopy; NVJ: nucleus-vacuole junction; PAS: phagophore assembly site; PE: phosphatidylethanolamine; PROPPIN: beta-propeller that binds phosphoinositides; PtdIns3P: phosphatidylinositol- 3-phosphate; VICS: vacuole isolation membrane contact site.
KEYWORDS: Atg16, Atg21, Atg8 lipidation, organellar contact site, phagophore elongation, VICS
Introduction
Macroautophagy (hereafter autophagy) is an evolutionarily conserved, homeostatic mechanism that eliminates defective or unnecessary cellular components via lysosomal degradation. It is involved in many essential processes, including protein and organellar quality control, cell differentiation, development, and survival during starvation. Autophagy has also gained substantial attention due to its role in diverse diseases, including cancer and neurodegeneration [1,2]. Thus, understanding the autophagy mechanism has a wide impact on many physiological and pathological processes.
Autophagy in S. cerevisiae involves over 40 autophagy-related (Atg) proteins, with diverse functions, orchestrating a complex series of membrane trafficking events. The process begins at the phagophore assembly site (PAS), where the assembly of a crescent-shaped double-layered membrane, called the phagophore, occurs. The phagophore then expands and closes to engulf its cytosolic cargo, thereby forming an autophagosome. After closure, its outer membrane fuses with the vacuole for degradation [1,3]. We here focus on understanding the underlying mechanisms of phagophore elongation. It has been shown that the ER tethers the phagophore during elongation [4–6] and that Atg2 here mediates the transfer of phospholipids [7,8]. Furthermore, the crucial role of the ubiquitin-like Atg8 for phagophore expansion is known [9]. The E1-like enzyme Atg7 first activates Atg8, which then transfers Atg8 to the E2-like enzyme Atg3, and finally, couples Atg8 covalently to the membrane phospholipid phosphatidylethanolamine (PE) [10,11]. In a second conjugation system, Atg12 covalently links to Atg5. The Atg12–Atg5 conjugate then forms a complex with Atg16, which acts as an E3-like complex during Atg8 lipidation [10,12–14]. Atg8 lipidation requires the recruitment of the E3 complex Atg12–Atg5-Atg16 to the phagophore. Two pathways can mediate this process either by the interaction of Atg12 with the Atg1-kinase complex, including Atg17 and Atg13 [15], or by the PtdIns3P-dependent Atg21 [16].
In brief, the PtdIns3-kinase complex generates PtdIns3P at the phagophore [9,17,18]. It recruits a conserved family of WD40 repeat-containing PtdIns3P receptor proteins known as beta-propellers that bind phosphoinositides (PROPPINs) in yeast and WIPIs (WD40 repeat-containing proteins that interact with phosphoinositides) in mammals. Yeast contains three homologous PROPPINs, Atg18, Atg21, and Hsv2, while mammals have four orthologs: WIPI1, WIPI2, WDR45B/WIPI3, and WDR45/WIPI4. Atg21 scaffolds the PtdIns3P-dependent Atg8 lipidation by binding to Atg16 of the E3 complex and Atg8 [16]. Although Atg21 has no clear mammalian ortholog, the splice variant WIPI2B exhibits a similar scaffolding function [19]. Previously, one edge of the phagophore was found in proximity to the vacuole and was termed VICS for vacuole isolation membrane contact site [20]. We now identified Atg21 at the VICS, which suggests that Atg21-dependent Atg8 lipidation occurs here. We further characterized the VICS as an organellar contact site, where vacuolar Vph1 is excluded, but Vac8 is concentrated. Our work thus puts the phagophore into a network of organellar contacts with the vacuole on one phagophore edge and the ER at the other. Our crystal structure of Atg21 in complex with the Atg16 coiled-coil domain further shows the geometry of the complex and allowed us to uncover the important role of three salt bridges and three hydrophobic residues for its formation. Our structural data further suggested a high membrane avidity of the complex, well in agreement with our studies of the assembly of the complex.
Results
Crystal structure of Atg21 in complex with the Atg16 coiled-coil domain
To further understand the role of Atg21 in the Atg8 lipidation complex, we acquired structural information. Crystals of K. lactis Atg21 in complex with the coiled-coil domain of A. gossypii Atg16(40–124) diffracted to a resolution of 3.7 Å (Table 1). The dimeric Atg16 coiled-coil domain interacts with the bottom sides of two Atg21 beta-propellers (Figure 1A,B), with the amino terminus of AgAtg16 oriented toward the PtdIns3P-binding FRRG motif of KlAtg21. Putative contacts between KlAtg21 and AgAtg16 included three salt bridges between (I) KlAtg21 K103 and AgAtg16 D78 (corresponding to ScAtg21 R151 and ScAtg16 D101), (II) KlAtg21 K355 and AgAtg16 E74 (ScAtg21 D460 and ScAtg16 E97), (III) KlAtg21 K82 and AgAtg16 E79 (ScAtg21 K130 and ScAtg16 E102), (Figure 1C–F). The structure further showed that I81, I85, and L89, which are in three consecutive alpha-helical turns of AgAtg16, are bound by a hydrophobic cleft of KlAtg21 between blades 2 and 3 (Figure 1G,H).
Table 1.
Crystallographic data collection and refinement statistics
| |
KlAtg21-AgAtg16(40–124) |
|---|---|
| Crystallographic data | |
| Beamline | X06SA (Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland) |
| Wavelength (Å) | 0.97918 |
| Resolution range (Å)a | 46.29–3.70 (4.05–3.70) |
| Unique reflections | 17,917 |
| Redundancy | 14.2 (14.4) |
| Completeness (%) | 99.6 (99.9) |
| Space group | P3(2)21 |
| a = b (Å) | 123.45 |
| c (Å) | 185.16 |
| Rmerge (%) | 23.0 (169.7) |
| Rpim (%) | 6.2 (45.2) |
| Wilson B-factor (Å2) | 119.2 |
| I/ (I) | 9.4 (2.1) |
| CC1/2 | 99.9 (47.6) |
| Refinement statistics | |
| Rwork/Rfree | 0.2845/0.3065 |
| No. of atoms | 5698 |
| Average B factor (Å2) | 139.4 |
| Root mean square deviation | |
| Bonds Å | 0.010 |
| Angles (degree) | 1.514 |
| Ramachandran plot | |
| Favored (%) | 91.84 |
| Allowed (%) | 7.88 |
| Outlier (%) | 0.28 |
| PDB code | |
| 6RGO |
aValues for the data in highest resolution shell are shown in parentheses
Figure 1.
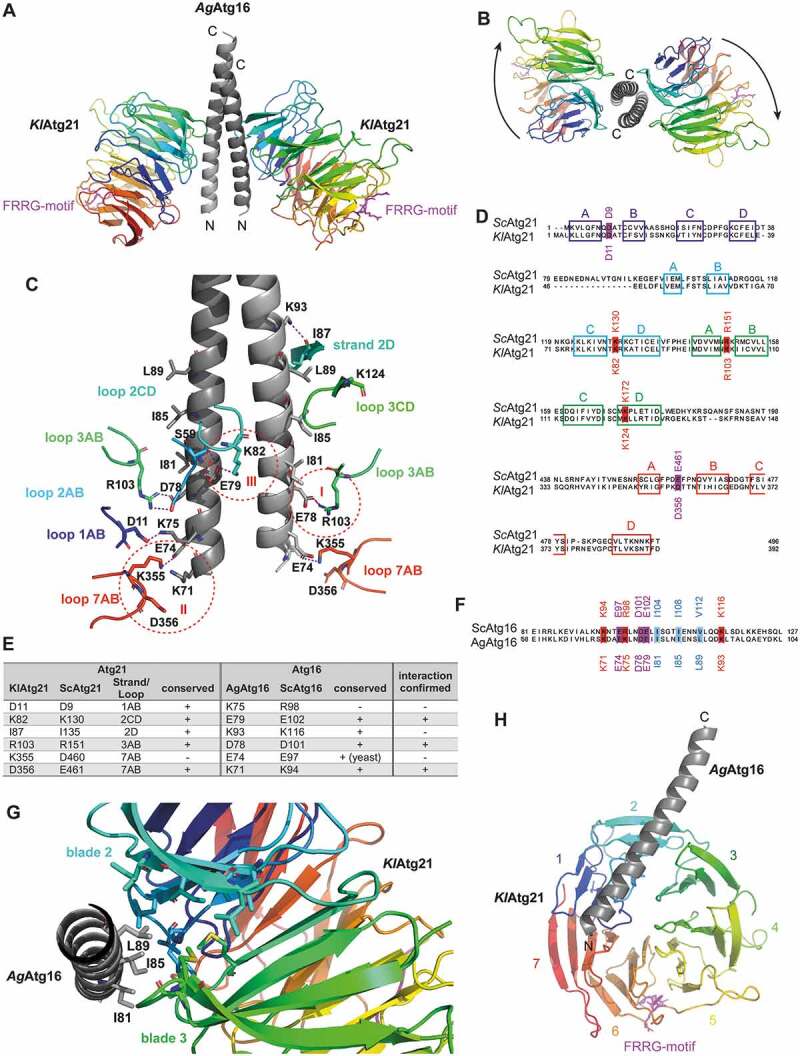
Structure of the Atg16-Atg21 complex. (A) Crystal structure of K. lactis Atg21 (rainbow-colored: blue to red from amino to carboxy terminus) in complex with the coiled-coil domain of A. gossypii Atg16(40–124) (light gray). The crystal structure was solved up to a resolution of 3.7 Å. The conserved lipid-binding motif FRRG of KlAtg21 is labeled in pink. (B) View from the top on the carboxy-terminus of the AgAtg16 coiled-coil domain and the KlAtg21 beta-propellers. Arrows indicate the orientation of the propeller blades. (C) Putative polar contacts between the AgAtg16 coiled-coil domain and KlAtg21 were predicted using PyMOL. Red dashed circles highlight three potential salt bridges (I–III). (D and F) Sequence alignment of Sc and KlAtg21 (D) or Sc and AgAtg16 (F), respectively, using Jalview and MAFFT (Clustal color code [76]). The conservation of the potentially interacting residues of Atg21 and Atg16 is shown. Colored boxes represent the respective strands of Atg21. (E) Summary of the potential polar contacts between AgAtg16 and KlAtg21, as well as their conservation and counterparts in S. cerevisiae. (G) View from the carboxy-terminus of the monomeric AgAtg16 alpha-helical domain. Residues I81, I85, and L89 of AgAtg16, which are located in three consecutive alpha-helical turns form a hydrophobic patch that inserts into a hydrophobic cleft of KlAtg21 formed between blades 2 and 3. (H) View on the bottom side of the KlAtg21 beta-propeller that shows the position of the monomeric Atg16 coiled-coil domain at the beta-propeller
To evaluate putative interactions, we first reversed the charges of the residues forming the potential salt bridges by site-directed mutagenesis and analyzed prApe1 maturation. The strongest effects were observed for mutants of the salt bridge I (ScAtg21R151E or ScAtg16D101 R) with only half of the normal prApe1 maturation (Figure 2A,B). Furthermore, both mutations almost abolished the binding of ScAtg16 to ScAtg21 in co-immunoprecipitations and reduced the number of Atg16-GFP puncta and its colocalization with mCherry-Atg21 (Figure 2C–G). Remarkably, the co-expression of ScAtg21R151E and ScAtg16D101 R recovered prApe1 maturation almost completely (Figure 2A,B). Also, the co-immunoprecipitation and localization of Atg16 were partially restored (Figure 2C–G), confirming direct interaction.
Figure 2.
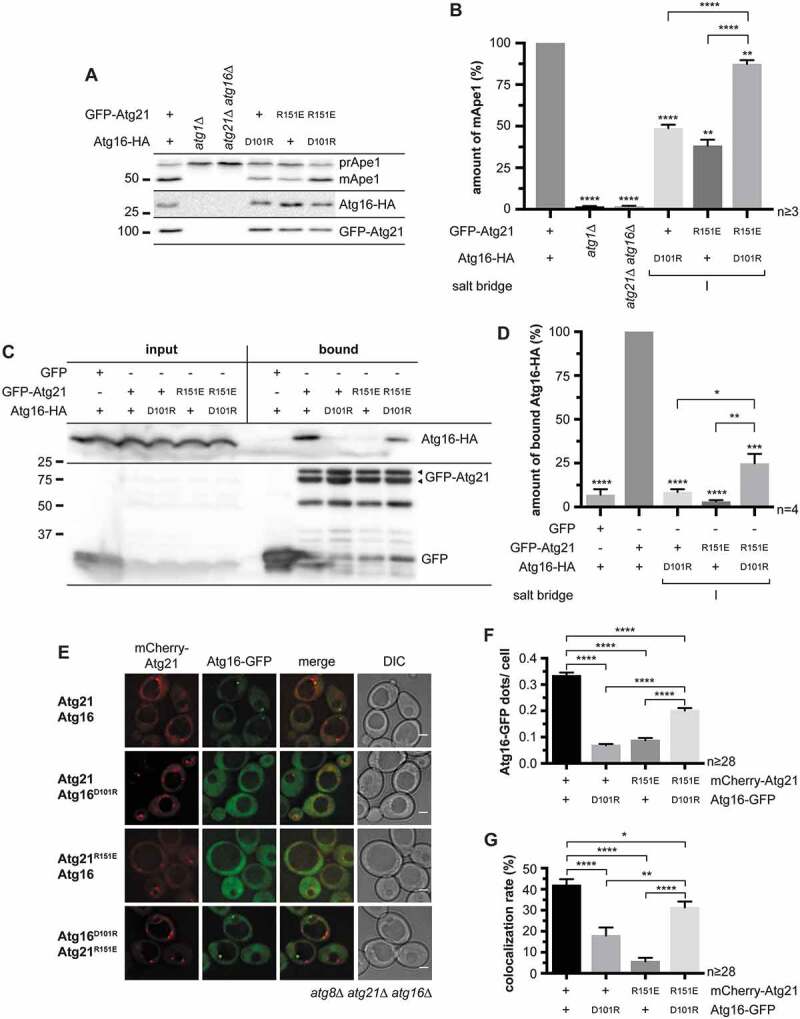
Analysis of the potential salt bridge between ScAtg21 R151 and ScAtg16 D101 (salt bridge I). (A – G) Effect of charge-change mutations of ScAtg21 R151 and ScAtg16 D101 (salt bridge I) under early stationary conditions. (A) Effect on the prApe1 maturation. (B) Quantification of the ratio of mApe1 to the total amount of Ape1 measured in (A) from ≥ three independent experiments. The amount of mApe1 of the WT was set to 100%. (C) Effect on the interaction of GFP-Atg21 and Atg16-HA using co-immunoprecipitations. (D) Quantification of (C) from ≥ four independent experiments. The amount of bound Atg16-HA was normalized to the amount of bound GFP-Atg21. The WT was set to 100%. (E) Effect on the localization of Atg16-GFP. (F and G) The number of Atg16-GFP dots per cell (F) and the number of mCherry-Atg21 dots colocalizing with Atg16-GFP (G) were determined. In four independent experiments, ≥ 28 images (n) were evaluated (≥ 1182 cells per strain). Data are presented in mean ± SEM. Statistical relevance related to the WT was determined using the one-sample t-test. Different strains were directly compared (squared bracket) using the unpaired two-tailed t-test: * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. n: number of independent experiments if not stated differently. The molecular weight marker is in kDa and scale bars represent 2 µm. DIC: differential interference contrast
KlAtg21 K355 (ScAtg21 D460) of salt bridge II is not conserved among yeast proteins (Fig. S1); thus, the interaction in the crystal might be due to a combination of proteins from different organisms for crystallization. However, the neighboring KlAtg21 D356 (ScAtg21 E461) is highly conserved and might interact with the conserved AgAtg16 K71 (ScAtg16 K94) (Figure 1C–F). Indeed, the expression of ScAtg16E97A or ScAtg21D460R did not reduce prApe1 maturation (Figure 3A,B). ScAtg21D460R even showed a slight increase, probably by creating an artificial contact with ScAtg16 E97. The stabilizing effect was further confirmed by the increased binding of ScAtg16 in co-immunoprecipitations (Fig. S2A and S2B). Instead, the expression of mutants of the neighboring residues (ScAtg21E461 R or Atg16K94E) mildly reduced prApe1 maturation and drastically affected the co-immunoprecipitation of ScAtg16 (Figure 3A,B, S2B, and S2 C). The co-expression of ScAtg21E461R and Atg16K94E again restored prApe1 maturation, co-immunoprecipitation, and localization of ScAtg16 almost to wild-type levels confirming a salt bridge between ScAtg21 E461 and ScAtg16 K94 (Figure 3A–C, S2B, S2 C, and S3). Combining mutations in the salt bridge I and II drastically decreased prApe1 maturation. ScAtg16K94E D101R alone showed about half of prApe1 maturation, while ScAtg21R151E E461R showed almost no maturation. The co-expression of both mutants restored prApe1 maturation to ~80% (Figure 3A,B). Similarly, we validated salt bridge III (Figure 3C–E, S2D, S2E, and S3). prApe1 maturation was not affected in mutants affecting other putative polar contacts (ScAtg21D9R or ScAtg16K116E) or ScAtg21K172E, which served as a control (Figs. 1C,E and 3A–D). Furthermore, ScAtg16K116E showed wild-type-like binding in co-immunoprecipitations (Fig. S2E and S2F).
Figure 3.
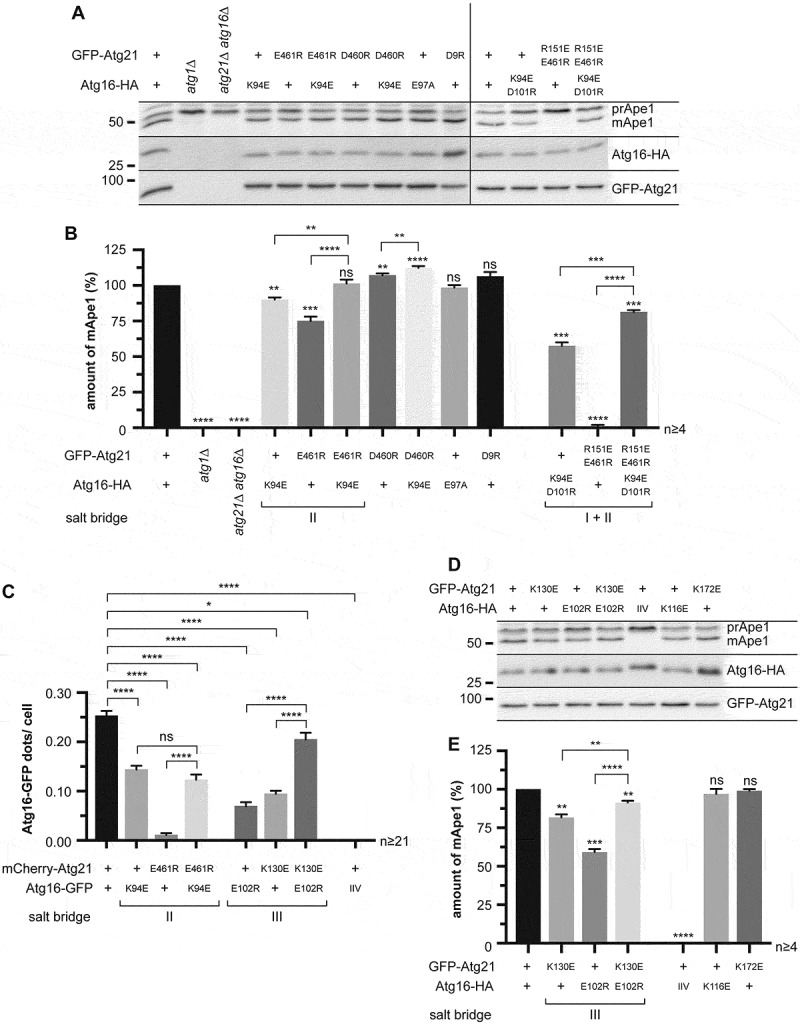
Analysis of the potential salt bridges between Atg21 E461-Atg16 K94 (salt bridge II) and Atg21 K130-Atg16 E102 (salt bridge III). (A) Effect of charge-change mutations of ScAtg21 E461 and ScAtg16 K94 (salt bridge II) together with ScAtg21D460R, ScAtg16E97A, ScAtg21D9R (control) and combinations of mutants from salt bridges I and II on the prApe1 maturation under early stationary conditions. (B) Quantification of the ratio of mApe1 to the total amount of Ape1 measured in (A) from ≥ four independent experiments. The amount of mApe1 of the WT was set to 100%. (C) Effect of the indicated mutants of salt bridge II and III on the localization of Atg16-GFP. The number of Atg16-GFP dots per cell was determined. In at least two independent experiments, ≥ 21 images (n) were analyzed, resulting in ≥ 668 counted cells per strain. (D) Effect of charge-change mutations of ScAtg21 K130 and ScAtg16 E102 (salt bridge III) with ScAtg16IIV (I104D I108D V112D) and ScAtg21K172E (control) on the prApe1 maturation under early stationary conditions. (E) Quantification of the ratio of mApe1 to the total amount of Ape1 measured in (D) from ≥ four independent experiments. The amount of mApe1 of the strain expressing the WT proteins was set to 100%. Data are presented in mean ± SEM. Statistical relevance related to the WT was determined using the one-sample t-test. Different strains were directly compared (squared brackets) using the unpaired two-tailed t-test: ns, not significant p > 0.05; * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. n: number of independent experiments if not stated differently. The molecular weight marker is in kDa
We next evaluated putative hydrophobic interactions formed by AgAtg16 I81, I85, and L89. The mutant ScAtg16I104,I108,V112D (ScAtg16IIV) of the corresponding residues of ScAtg16 blocked prApe1 maturation and the interaction with ScAtg21 (Figs. 1F, 3D,E, S2E, and S2 F). Furthermore, ScAtg16IIV-GFP shows a dispersed cytosolic localization and no colocalization with mCherry-ScAtg21 (Figure 3C and S3). Together, the Atg16-Atg21 complex is mainly stabilized by three ionic interactions and hydrophobic interactions of ScAtg16 I104, I108, and V112.
Assembly of the Atg16-Atg21 complex
To prevent Atg8 lipidation at unwanted sites, assembly of the Atg12–Atg5-Atg16 E3 complex and Atg21 must be tightly controlled. We speculated that this would occur after membrane attachment rather than in the cytosol. This notion is supported by the crystal structure, where the Atg16 coiled-coil domain binds two Atg21 molecules simultaneously, corresponding to four PtdIns3P binding sites. The increased number of PtdIns3P binding sites in the Atg16-Atg21 complex generates a high avidity to PtdIns3P-positive membranes, making the existence of a non-membrane attached complex in the cytosol unlikely. To test this, we used Atg21[FTTG], which is defective in PtdIns3P-binding. The integrity of Atg21 complexes is difficult to preserve upon cell lysis. We, therefore, used fluorescence cross-correlation spectroscopy (FCCS), which allows for the measurement of fluorescence fluctuations of individual fluorophores in intact cells. The interaction can be evaluated with the calculated cross-correlation curve between two fluorophores. As an internal control mimicking the interaction, beads carrying several fluorophores were used (Figure 4A). Atg16-GFP and mCherry-Atg21[FTTG] showed no cross-correlation, indicating no interaction in the cytosol within the detection limit, which was estimated at 1–2% interaction (Figure 4B). Since WT Atg21 is bound to the membranes, we could not measure FCCS with Atg16 in our experimental setup. As another control, we, therefore, monitored the known interaction of Atg16-GFP with mCherry-Atg5 in a strain, also expressing Atg21[FTTG]. As expected, our FCCS measurements yielded a clear cross-correlation, demonstrating binding between Atg16-GFP and mCherry-Atg5 in the cytosol (Figure 4C). We conclude that the cytosolic level of Atg21 in a complex with Atg16 is very low, suggesting that Atg21 first associates with the phagophore membrane and then binds to the E3 complex. However, we cannot exclude that the dimeric Atg21-E3 complex forms in the cytosol and very rapidly associates with membranes due to the high membrane avidity of its four PtdIns3P-binding sites.
Figure 4.
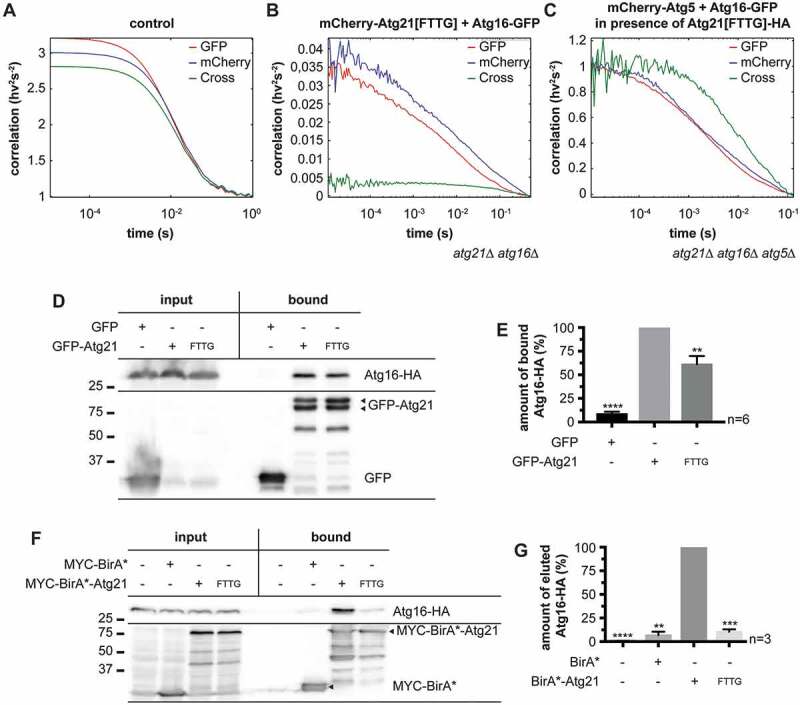
The Atg16-Atg21 complex is preferentially associated with membranes. (A) FCCS measurement of an in vitro control with fluorescent beads containing dyes emitting in four wavelength channels to establish the overlap volume of the two color-foci as well as to determine the quality of the alignment in the detection channels. (B and C) FCCS measurements of living yeast cells in early stationary phase containing Atg16-GFP together with mCherry-Atg21[FTTG] (B) or together with mCherry-Atg5 and Atg21[FTTG]-HA (C). Autocorrelation curves are shown in red (GFP-fused molecules) and blue (mCherry-fused molecules) and cross-correlation curves are shown in green. Correlations are plotted logarithmically. (D-G) Analysis of the interaction between the lipid-binding-deficient Atg21[FTTG] mutant and Atg16. (D) Co-immunoprecipitations of GFP-Atg21[FTTG] and Atg16-HA. (E) Quantification of (D) from six independent experiments. The amount of bound Atg16-HA was normalized to the amount of bound GFP-Atg21 or GFP-Atg21[FTTG]. The normalized amount of Atg16-HA bound to WT GFP-Atg21 was set to 100%. (F) BioID assay. BirA* alone or the empty vector pUG36 (-) served as controls for unspecific or background biotinylation in yeast cells. BirA* and its fusion proteins were detected using anti-MYC antibodies. (G) Quantification of (F) from three independent experiments. The amount of eluted Atg16-HA was normalized to its amount in the input fraction. The normalized amount of Atg16-HA eluted from the BirA*-ATG21 expressing strain was set to 100%. Data are presented in mean ± SEM. Statistical relevance was determined using the one-sample t-test: ** p < 0.01; *** p < 0.001; **** p < 0.0001. n: number of independent experiments. The molecular weight marker is in kDa
We further assessed complex formation between Atg16 and Atg21 using standard methods. Compared to GFP-Atg21, GFP-Atg21[FTTG] co-immunoprecipitated only 61% of Atg16-HA (Figure 4D,E), suggesting that there was still binding but less. We also used the split-ubiquitin system to analyze binding. Here, the N-terminal half of ubiquitin (Nub) was fused with Atg21 and the C-terminal domain (Cub) with Atg16. The interaction of the proteins restores ubiquitin, which is cleaved by ubiquitin proteases and finally leads to the degradation of a proteolytically sensitive Ura3 protein. Cells with interacting proteins thus are unable to grow without uracil but are not sensitive to 5-fluoroorotic acid (5-FOA) [21]. Similar to the co-immunoprecipitation results, the split-ubiquitin system demonstrated an interaction between Atg21[FTTG] and Atg16, thus in the absence of Atg21 membrane binding (Fig. S4). In contrast to the other methods used, this interaction seemed to be even stronger than that of the WT proteins. However, it must be considered that both immunoprecipitation and the split-ubiquitin system also detect weak and transient interactions. In our immunoprecipitations, only a low percentage of Atg16-HA from the input fraction was precipitated. Furthermore, the concentration of the bait protein on beads might favor interaction. In addition, in the split-ubiquitin system, both ubiquitin-halves already possess a binding affinity, which might stabilize otherwise weak interactions. Also, the membrane association of the Atg16-Atg21 complex might sterically hinder the association of the ubiquitin halves, leading to the underestimation of their interaction. All these points limit the reliability of both immunoprecipitation and the split-ubiquitin system.
Thus, as a final approach, we looked for a method to quantitatively monitor protein interactions in intact cells. To this end, we attached the unselective E. coli biotin ligase BirA* to the bait protein Atg21. Proteins in close proximity are then biotinylated and can be isolated with biotin-affinity columns. For BirA*-Atg21, we set the amount of biotinylated Atg16-HA to 100%. For BirA*-Atg21[FTTG], we detected only 11% and for the BirA* control 7% of biotinylated Atg16-HA (Figure 4F,G). This result corresponds to the lack of cross-correlation in the FCCS measurements. We conclude that in the cytosol, only minor amounts of Atg21[FTTG] and Atg16 are in a complex. Together, our quantitative analyses suggest that the Atg16-Atg21 complex preferentially occurs when associated with membranes.
Localization of Atg21 at the phagophore
Remarkably, in the Atg16-Atg21 crystal structure, the PtdIns3P binding sites of the two Atg21 molecules are in a conformation, which suggests preferential binding to concave membranes (Figure 1A). Assuming that the Atg16-Atg21 complex is preferentially associated with membranes, we were wondering where Atg21 is located at the phagophore.
To allow spatial resolution of the Atg-machinery during the early stages of autophagosome biogenesis when Atg8 lipidation mediates elongation of the crescent-shaped phagophore, we overexpressed the selective autophagic cargo prApe1. This approach results in the formation of a giant cargo, which can be monitored by fluorescence microscopy [20]. Using GFP-Atg8 or Atg16-GFP, we first established conditions allowing for the detection of the different stages of phagophore elongation around prApe1-RFP (Fig. S5). The quantification showed that elongated and crescent-shaped phagophores were best detectable in wild-type cells starved for 1–2 h in nitrogen-free SD-N medium (Fig. S5B and S5 C). To validate our approach, we further localized two phagophore-associated proteins with known localization: Atg14, which is a specific component of the PtdIns3-kinase complex at phagophores, and one of the PROPPINs, Atg18. As published, Atg14-3xGFP was restricted to the phagophore edge proximal to the vacuole (VICS) (Fig. S6A), while Atg18-GFP was detected at both edges of the elongating phagophore (Fig. S6B) [20]. We also recapitulated the localization of Atg3, the E2-like enzyme during Atg8 lipidation. As amino- or carboxy-terminal tagging of Atg3 affects its function, we inserted GFP between D265 and G266. As published, Atg3 was dispersed over the phagophore (Fig. S6C) [22,23]. Thus, our system recapitulates known molecular events of phagophore biogenesis.
Next, we analyzed the localization of Atg21-YFP in nitrogen-starved cells. The majority (~80%) of the Atg21-YFP puncta colocalized with the phagophore edge in contact with the vacuole (Figure 5A,B). The residual Atg21 puncta were also in contact with the vacuole, but within the crescent-shaped phagophore (Figure 5A). Thus, the phagophore can either grow out of the Atg21 punctum in different directions, or more than one phagophore is formed. 3-dimensional reconstruction of mCherry-Atg8 in these cells indeed showed a single phagophore growing in different directions (Figure 5C). A large part of Atg8 lipidation depends on Atg21 [15,16], we thus expect that Atg21-dependent Atg8 lipidation occurs at the VICS.
Figure 5.
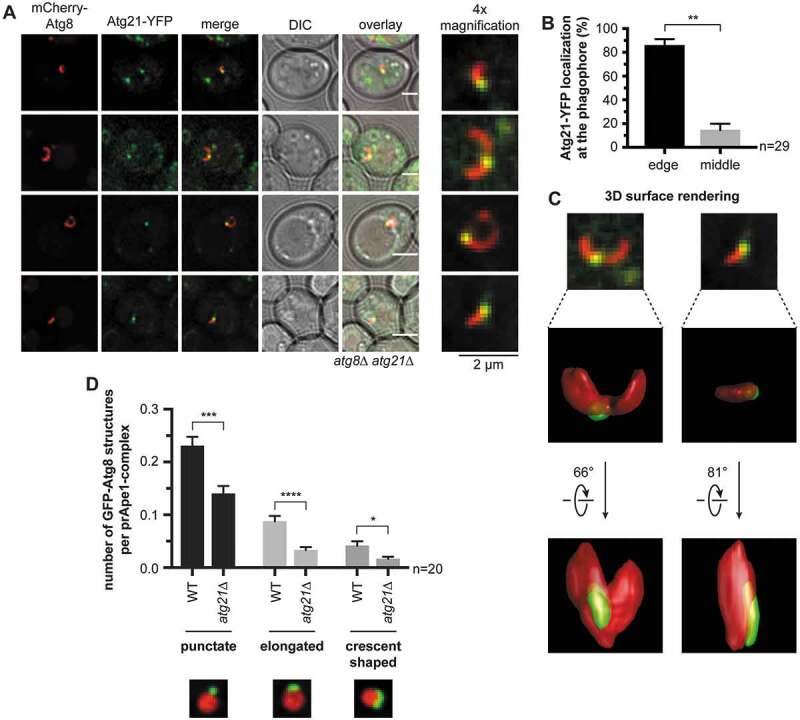
Atg21 localizes to the VICS. (A) Distribution of Atg21 at the phagophore. In an atg8∆ atg21∆ strain, plasmid-encoded mCherry-ATG8 and ATG21-YFP were expressed using their endogenous promoters. The APE1-overexpressing cells were analyzed after 1 h of starvation in SD-N. Scale bars: 2 µm. DIC: differential interference contrast. (B) Quantification of the localization of the Atg21-YFP puncta at the mCherry-Atg8-positive phagophores in (A). In three independent experiments, 29 images (n) were analyzed, resulting in 54 counted phagophores. (C) 3-dimensional reconstruction from the collected z-stacks of the respective mCherry-Atg8-positive phagophores of (A) using Huygens Professional. Arrows indicate rotation angles between the depicted views. (D) Effect of ATG21 deletion on the formation of the phagophore in APE1-overexpressing cells after 1 h starvation. In the atg8∆ APE1-RFP (WT) or the atg8∆ APE1-RFP atg21∆ (atg21∆) strain, plasmid-encoded GFP-ATG8 was expressed using its endogenous promoter. In two independent experiments with ≥ 1001 counted cells per strain, the colocalization of GFP-Atg8-positive structures with prApe1 complexes was determined. The shape of the colocalizing GFP-Atg8 structures was grouped into punctate, slightly elongated, and crescent-shaped. The colocalization rate of each group per prApe1 complex was calculated. Data are presented in mean ± SEM. Statistical relevance was determined using the unpaired two-tailed t-test: * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. n: number of analyzed images per strain
We further analyzed the effect of ATG21 deletion on PAS formation and phagophore elongation. In line with former reports [16], the recruitment of GFP-Atg8 to prApe1-RFP complexes was significantly reduced. We now also found decreased phagophore growth (Figure 5D), well in agreement with the role of the lipidated Atg8 in phagophore expansion [9].
The membrane association of Atg21 requires its binding to PtdIns3P [16]. Thus, we also monitored the distribution of PtdIns3P using mRFP-2xFYVE (a fusion protein containing the PtdIns3P-binding FYVE domain) (Fig. S6D). In agreement with published data [24,25], PtdIns3P was dispersed across the phagophore. This result suggests additional mechanisms that restrict the localization of Atg21 and Atg18 at the phagophore.
The vacuole forms a specialized Vac8-positive domain near the phagophore
The proximity of the phagophore to the vacuolar membrane has been reported [20]. However, it has not been clarified if a contact site is formed and if it affects autophagy. Typically, the protein composition at organellar contacts is distinct from the residual membrane. For example, at the nucleus-vacuole junction (NVJ), which is formed during piecemeal microautophagy of the nucleus, Vph1, a component of the vacuolar H+-ATPase is excluded, while the phosphatase Pho8 is evenly distributed at the vacuole [26]. We analyzed the distribution of these vacuolar proteins in our APE1-overexpressing system. 3xtagBFP-Pho8 was dispersed all over the vacuolar membrane (Fig. S7A). Also, fluorescence intensity profiling showed no alterations in the vicinity of the phagophore. Vph1-GFP, however, was clearly excluded from the vacuolar membrane near the prApe1 complex and in the vicinity of about 90% of GFP-Atg8-positive puncta and crescent-shaped structures (Figure 6A–C). Vph1-GFP was further excluded from the vacuolar membrane proximal to the Hoechst 33342-stained nucleus, according to the presence of NVJs (Figure 6D,E). To further evaluate the spatial organization, we also co-expressed Vph1-mCherry with Atg21-YFP and mTagBFP2-Atg8. As expected, the Atg21-YFP puncta colocalized with the Vph1-exclusion zone (Fig. S7 C and S7D).
Figure 6.
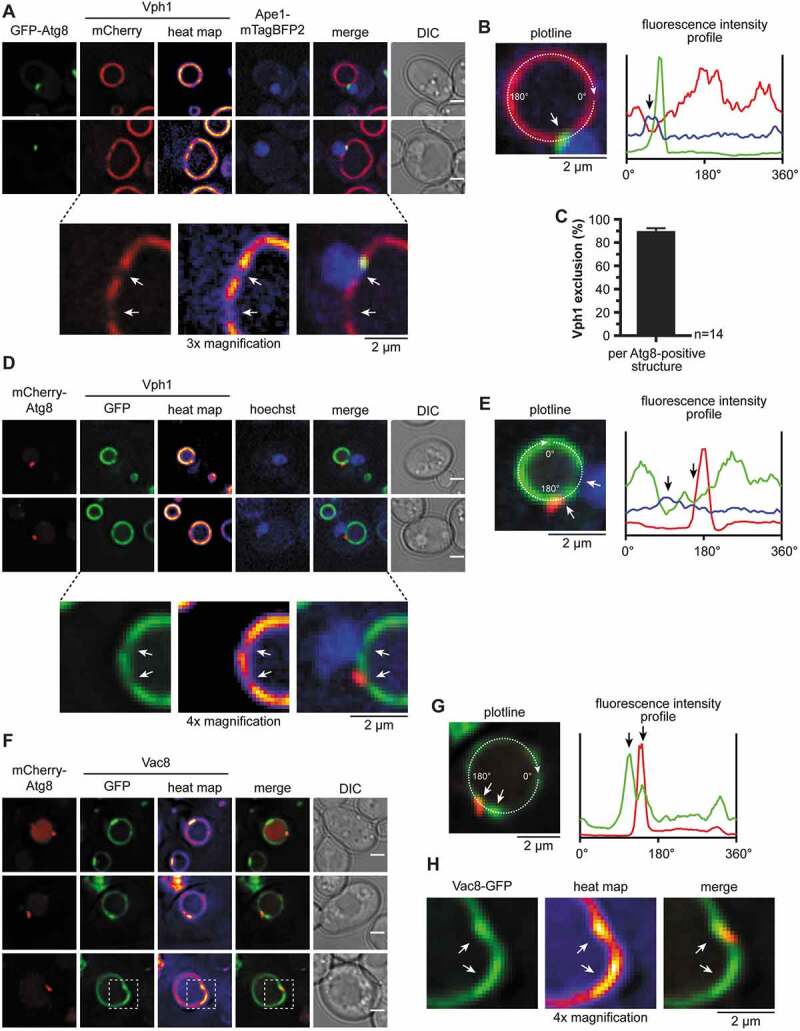
The vacuolar membrane is Vac8-enriched and Vph1-depleted at the VICS. (A-E) Analysis of the distribution of Vph1 at the vacuolar membrane in relation to the growing phagophore and the nucleus in APE1-overexpressing cells after 1 h starvation in SD-N. White and black arrows highlight the exclusion zones of Vph1 in proximity with the phagophore and the nucleus. (A) In an atg8∆ VPH1-mCherry APE1-mTagBFP2 strain, plasmid-encoded GFP-ATG8 was expressed from its endogenous promoter. (B) Fluorescence profile of the Vph1-mCherry-positive vacuole from the respective cell of (A). Fluorescence intensities of Vph1-mCherry (red), GFP-Atg8 (green), and Ape1-mTagBFP2 (blue) were measured along the depicted circular plotline and plotted against the circular line distance from 0°-360°. (C) Quantification of the number of Vph1 exclusions in proximity with GFP-Atg8-positive structures in (A). 14 images (n) were analyzed with a total of 602 counted cells. Data are presented in mean ± SEM. (D) In an atg8∆ VPH1-GFP strain, plasmid-encoded mCherry-ATG8 was expressed from its endogenous promoter. The nucleus was visualized using Hoechst 33324. (E) Fluorescence profile of the Vph1-GFP-positive vacuole from the respective cell of (D). Fluorescence intensities of Vph1-GFP (green), mCherry-Atg8 (red), and Hoechst 33,324 (blue) were measured along the depicted circular plotline and plotted against the circular line distance from 0°-360°. (F-H) Distribution of Vac8-GFP at the vacuolar membrane in relation to the growing phagophore. White and black arrows highlight Vac8-enriched regions. (F) In an atg8∆ vac8∆ strain, plasmid-encoded VAC8-GFP and mCherry-ATG8 were expressed using their endogenous promoters. The APE1-overexpressing cells were analyzed after 1–2 h starvation in SD-N. (G) Fluorescence profile of the Vac8-GFP-positive vacuole from the respective cell of (F). Fluorescence intensities of Vac8-GFP (green) and mCherry-Atg8 (red) were measured along the depicted circular plotline and plotted against the circular line distance from 0°-360°. (H) 4x Magnification of the white dashed boxes in (F). Scale bars: 2 µm. DIC: differential interference contrast
We expected that vacuolar proteins involved in VICS formation accumulate at this site. A good candidate is the vacuolar protein Vac8, which is involved in autophagy [27] and is crucial for the formation of NVJs [28]. Indeed, Vac8-GFP accumulated at the VICS in proximity with mCherry-Atg8 (Figure 6F–H). Together, the protein composition of the vacuolar membrane in the vicinity of the phagophore is distinct, indicating an organellar contact.
Vac8 is required for the formation of the phagophore vacuole contact site and phagophore elongation
Next, we analyzed the role of Vac8 and Atg21 for VICS formation in cells expressing GFP-Atg8, prApe1-RFP, and BFP-Pho8 (Figure 7A–E). While the deletion of ATG21 only had a minor effect on VICS formation (Figure 7A–C), the deletion of VAC8 caused a drastic increase in phagophores without vacuolar contact. This observation points to a crucial role of Vac8 in establishing phagophore vacuole contacts.
Figure 7.
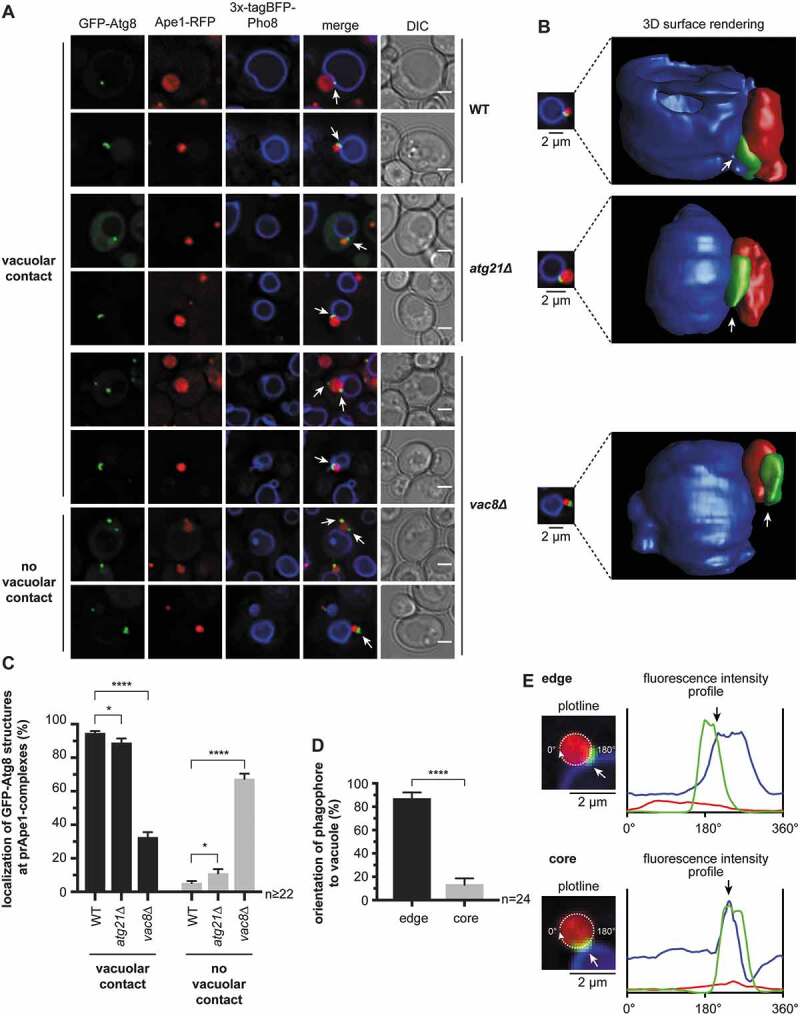
Vac8 is required for the contact between the vacuole and phagophore. (A-C) Analysis of the deletion of ATG21 and VAC8 on the vacuolar contact with the phagophore. (A) In the atg8∆ APE1-RFP 3xtagBFP-PHO8 (WT) strain, either ATG21 (atg21∆) or VAC8 (vac8∆) was deleted. Plasmid-encoded GFP-ATG8 was expressed using its endogenous promoter. Analysis was done in APE1-overexpressing cells after 1 h of starvation. White arrows highlight the position of the GFP-Atg8-positive structures. (B) 3-dimensional reconstruction from the collected z-stacks of the respective cells from (A) using Huygens Professional. White arrows highlight the position of the GFP-Atg8-positive phagophores. (C) Quantification of the number of GFP-Atg8-positive structures of each strain in (A) with and without contact with the vacuolar membrane. In two independent experiments ≥ 22 images (n) with ≥ 935 cells per strain were analyzed. (D) The orientation of the GFP-Atg8-positive phagophores toward the vacuolar membrane was quantified in the WT strain of (A). Phagophores were grouped into those that contact the vacuole with one edge (edge) and those that contact the vacuole at their circumference (core). In total, 24 images (n) from two independent experiments were analyzed with 50 counted phagophores. (E) Fluorescence profiles were measured around prApe1-RFP complexes from WT cells in (A) to support the grouping in (D). Fluorescence intensities of prApe1-RFP (red), GFP-Atg8 (green), and 3x-tagBFP-Pho8 (blue) were measured along the depicted circular plotline and plotted against the circular line distance from 0°-360°. The contact of the phagophore edge (upper panel) and of the circumference of the phagophore (lower panel) with the vacuole can be distinguished by the distance between the GFP-Atg8 and the 3x-tagBFP-Pho8 intensity peaks. White and black arrows highlight the contact site of phagophore and vacuole. Data are presented in mean ± SEM. Statistical relevance was determined using the unpaired two-tailed t-test: * p < 0.05; **** p < 0.0001. Scale bars: 2 µm. DIC: differential interference contrast
The deletion of VAC8 has several effects, such as vacuole fragmentation. Thus, we further wanted to generate a Vac8 mutant protein with reduced binding to Atg13. Vac8 uses R276, R317, and R359 to interact with either Atg13 during autophagy or with Nvj1 at the NVJ [29]. We thus generated a Vac8R276,317,359E (Vac8[EEE]) triple mutant. As published, Vac8[EEE]-GFP did not accumulate near the nucleus, consistent with its inability to interact with Nvj1 and to form NVJs (Figure 8A) [29]. However, under the required starvation conditions, Vac8[EEE]-GFP was still enriched in the vicinity of the Atg8-positive structures and prApe1-mTagBFP2 complexes (Figure 8A–C). So, Vac8 is either interacting with additional components of the Atg machinery or the Vac8[EEE] mutant possesses residual interaction with Atg13. Indeed, in co-immunoprecipitations of Vac8[EEE]-GFP, compared to the wild-type Vac8-GFP, we observed a significant reduction to ~41%, but no loss of, binding to Atg13-HA (Figure 8D,E). The remaining interaction might be sufficient to microscopically detect its enrichment near phagophores. Accordingly, the number of GFP-Atg8-positive structures without vacuolar contact was only slightly increased in Vac8[EEE]-HA-expressing cells (Figure 8F). However, phagophore elongation was significantly decreased with Vac8[EEE]-HA or in vac8∆ cells (Figure 8G). We, thus, conclude that the VICS plays a role in phagophore elongation (Figure 8F,G, and S8).
Figure 8.
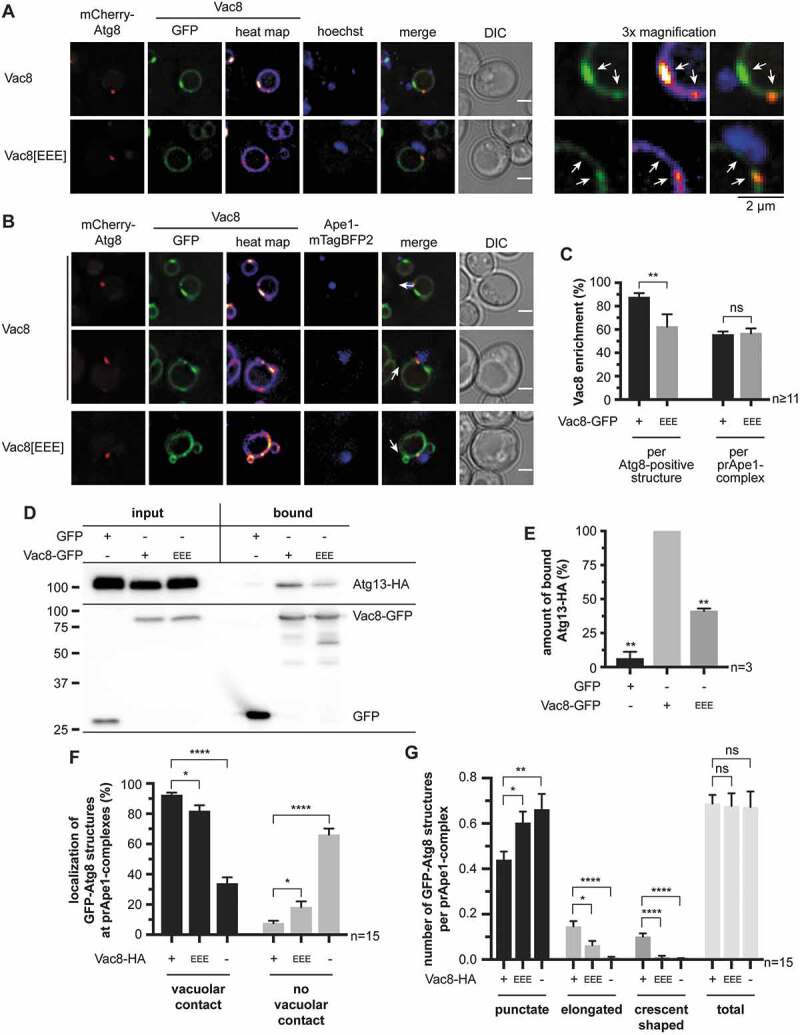
Efficient phagophore growth requires Vac8 and its interaction with Atg13. (A-C) Effect of charge-change mutation of R276 R317 R359 in the Atg13/Nvj1-binding region of Vac8 (Vac8[EEE]) on the distribution of Vac8-GFP at the vacuolar membrane. Analysis was done in APE1-overexpressing cells after 1 h of starvation. White arrows highlight Vac8-enriched regions. (A) Distribution of Vac8-GFP in relation to the nucleus. In an atg8∆ vac8∆ strain, plasmid-encoded mCherry-ATG8 and VAC8-GFP or VAC8[EEE]-GFP were expressed using their endogenous promoters. The nucleus was visualized using Hoechst 33324. (B) Distribution of Vac8-GFP in relation to the phagophore and the prApe1 complex. In an atg8∆ vac8∆ APE1-mTagBFP2 strain, plasmid-encoded mCherry-ATG8 and VAC8-GFP or VAC8[EEE]-GFP were expressed using their endogenous promoters. (C) Quantification of the number of Vac8-enriched regions at the vacuolar membrane in proximity with the mCherry-Atg8-positive phagophores or the prApe1-mTagBFP2 complexes of (B). ≥ 11 images (n) with ≥ 24 mCherry-Atg8-positive structures per strain were analyzed. (D) Effect of the Vac8[EEE]-GFP mutant on the interaction with Atg13-HA using co-immunoprecipitations. (E) Quantification of (D) in three independent experiments (n). The amount of bound Atg13-HA was normalized to its amount in the input fraction and to the amount of bound GFP, Vac8-GFP, or Vac8[EEE]-GFP, respectively. The WT was set to 100%. (F and G) Effect of the Vac8[EEE] mutant on the vacuolar contact (F) and the formation of the phagophore (G). In an atg8∆ vac8∆ APE1-mTagBFP2 strain plasmid-encoded GFP-ATG8 and VAC8-HA or VAC8[EEE]-HA were expressed using their endogenous promoters. Furthermore, an atg8∆ vac8∆ APE1-mTagBFP2 strain carrying an empty vector (-) was analyzed. The vacuolar membrane was visualized using FM 4–64. Analysis was done in APE1-overexpressing cells after 1 h of starvation. In total, 15 images (n) with ≥ 390 cells per strain were analyzed. (F) Quantification of the number of GFP-Atg8-positive structures of each strain with and without contact with the vacuolar membrane. (G) Quantification of the phagophore growth by grouping the GFP-Atg8-positive structures into punctate, slightly elongated, and crescent-shaped and calculating the colocalization rate of each group per prApe1 complex. Data information: data are presented in mean ± SEM. Statistical relevance related to the WT was determined using the one-sample t-test. Different strains were directly compared (squared brackets) using the unpaired two-tailed t-test: ns, not significant p > 0.05; * p < 0.05; ** p < 0.01; **** p < 0.0001. Scale bars: 2 µm. DIC: differential interference contrast
We next stained the vacuole with the styryl dye FM 4–64 [30] and monitored phagophore formation as before. FM 4–64 was absent from growing phagophores, suggesting a diffusion barrier that prevents unregulated lipid-mixing, which is typical for organellar contact sites (Figure 9A,B). However, this does not exclude a regulated lipid-transfer from vacuole to phagophore reminiscent to the function of Atg2 at the ER-phagophore contact site [5,8]. To examine the relationship between the ER-phagophore and the vacuole-phagophore contact, we analyzed cells co-expressing mCherry-Atg8, 3xtagBFP-Pho8, and the ER-marker Sec63-GFP. Contacts between mCherry-Atg8-positive phagophores and Sec63-GFP were mainly detected at the phagophore edge most distant from the vacuole (Figure 9C,D). In vac8∆ cells, the mCherry-Atg8-positive structures still contacted the ER, but the vacuolar contact was often absent (Figs. 7D,E and 9C,D), suggesting the independent existence of both contacts. We, thus, expect that growing phagophores are spanned between the vacuole and ER.
Figure 9.
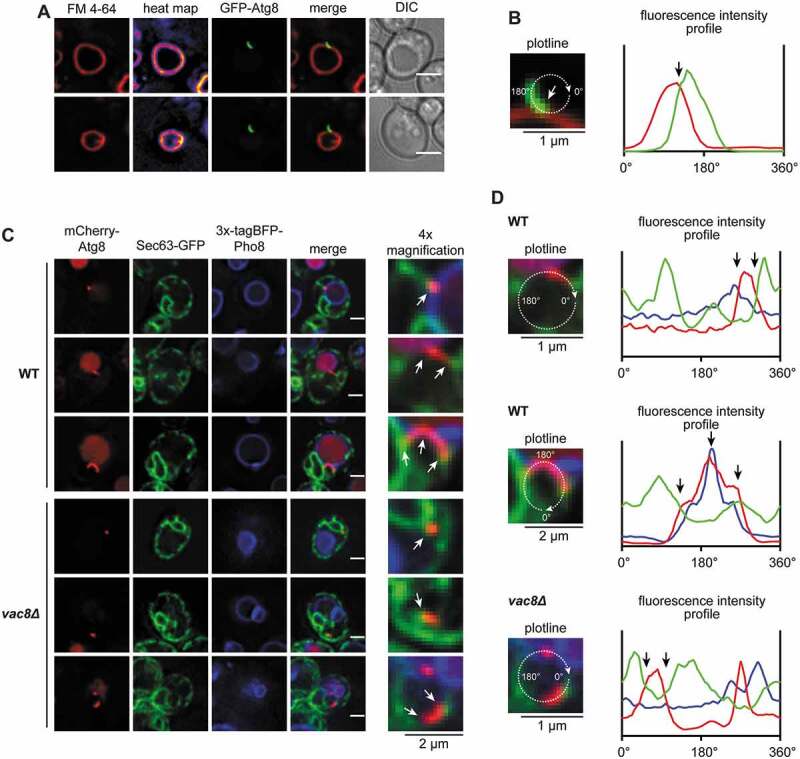
The phagophore is arranged within a network of organellar contacts with the vacuole on one edge and the ER at the other. (A) Analysis of the distribution of FM 4–64 at the vacuolar membrane and the phagophore. In an atg8∆ strain, plasmid-encoded GFP-ATG8 was expressed using its endogenous promoter. Analysis was done in APE1-overexpressing cells after 1 h of starvation. Heat maps represent fluorescence intensities of FM 4–64. (B) Fluorescence profile of the GFP-Atg8-positive phagophore from the respective cell of (A). Fluorescence intensities of GFP-Atg8 (green) and FM 4–64 (red) were measured along the depicted circular plotline and plotted against the circular line distance from 0°-360°. The white and black arrow highlights the contact site of vacuole and phagophore. (C) Analysis of the localization of Sec63-GFP in relation to the phagophore and the vacuole-phagophore contact site. In an atg8∆ 3xtagBFP-PHO8 (WT) or an atg8∆ 3xtagBFP-PHO8 vac8∆ (vac8∆) strain, plasmid-encoded SEC63-GFP and mCherry-ATG8 were expressed using their endogenous promoters. White arrows highlight the contacts of phagophore and ER or vacuole. (D) Fluorescence profiles of the mCherry-Atg8-positive phagophores from the respective cells of (C). Fluorescence intensities of Sec63-GFP (green), mCherry-Atg8 (red) and 3xtagBFP-Pho8 (blue) were measured along the depicted circular/oval plotline and plotted against the circular line distance from 0°-360°. Black arrows highlight the contact sites of phagophore and ER or vacuole. White scale bars: 2 µm. DIC: differential interference contrast
Discussion
We solved the crystal structure of Atg21 in complex with the Atg16 coiled-coil domain, which points with its amino terminus toward the PtdIns3P-binding sites of Atg21 and thus the membrane (Figure 1). Combined with published structural and functional data of Atg12–Atg5 in complex with the Atg16 amino-terminal domain [31–33], we predict that Atg12–Atg5 is oriented near the membrane around the circumference of Atg21 toward its top side. This orientation might be stabilized by membrane binding of Atg5 via K160 and R171 [34]. Furthermore, a membrane-binding amphipathic alpha-helix was identified in the amino-terminal part of human ATG16L1 [35], which might also be present in the yeast homolog and could contribute to membrane association.
The Atg16-Atg21 complex is stabilized by ionic interactions (Figs. 2 and 3). Interestingly, the mammalian ATG16L1 complex with WIPI2B is similarly stabilized by a salt bridge between WIPI2B R108 and ATG16L1 E230 [19]. WIPI2B R108 directly corresponds to ScAtg21 R151 (salt bridge I), highlighting the conservation of functional principles. However, despite the conservation of ScAtg16 D101 in the mammalian complex, not the corresponding residue but ATG16L1 E230 is used for the salt bridge. ATG16L1 E230 is part of a carboxy-terminal extension of the coiled-coil domain that continues into a WD40 beta-propeller domain, which are both missing in the yeast protein [36]. In our structure, the carboxy-terminus of Atg16 is oriented away from the membrane above the beta-propeller of Atg21 (Figure 1). In a similar orientation, the WD40 domain of the mammalian ATG16L1 would be exposed away from the lipidation site. This orientation might explain why this WD40 domain is only required for non-canonical lipidation at the phagosomes after the uptake of bacteria, but not for normal autophagy [37,38]. A study further identified a WIPI2 mutation, which is associated with a complex developmental disorder, that affects the interaction of WIPI2B with ATG16L1, highlighting the medical relevance of this interaction [39].
We also showed the essential role of Atg16 I104, I108, and V112 for binding to Atg21 (Figure 3C,D, S2E, and S2F). Interestingly, there are reports that these residues mediate the formation of a coat-like structure of Atg12–Atg5-Atg16, with Atg8-PE, on the convex, outside of the phagophore [14,40]. This observation would suggest that Atg16 can either be in complex with Atg21 or part of a proposed coat-like complex.
In the crystal, the Atg16 coiled-coil domain binds to two Atg21 molecules simultaneously, corresponding to four PtdIns3P binding sites. This arrangement generates high avidity to PtdIns3P-positive membranes and makes the existence of a non-membrane-attached complex in the cytosol unlikely. Recent studies also highlighted the membrane affinity of human ATG16L1 [35,41]. In line, our analyses on complex assembly (Figure 4) uncovered that PtdIns3P-binding-deficient Atg21[FTTG] only interacts with Atg16 to a limited extent in the cytosol.
In fluorescence microscopy, Atg21-YFP was restricted to the phagophore edge close to the vacuole, the VICS (Figure 5). Remarkably, Vac8 was enriched at the VICS, while Vph1 was excluded (Figs. 6 and 8). This observation underlines the formation of a specialized subdomain at the vacuolar membrane. Vph1 exclusion is reminiscent of NVJs and the phase separation at vacuolar membranes observed after glucose-limitation [42]. Here sterol-rich, liquid-ordered domains devoid of Vph1 and liquid-disordered domains, including Vph1, are formed. While this work was under revision, the vacuolar tethering of phagophores by Vac8 was also detected in another study [43]. Interestingly, Pho8 was evenly distributed at NVJs, which is consistent with its access to VICS (Fig. S7A) [26]. The lack of FM 4–64 transfer from the vacuole to the phagophore suggests that no free lipid mixing occurs between the vacuole and the phagophore membrane (Figure 9A,B). Vac8 interacts with Atg13 [44], and their binding sites have been characterized [29,45,46]. We observed a significant loss of VICS formation in the absence of Vac8, or when mutating the Atg13-binding site of Vac8 (Figs. 7 and 8). The VICS, thus, fulfills the criteria of an organellar contact site, which is typically involved in non-vesicular lipid transfer [47]. It is, thus, tempting to speculate that lipids targeted to the vacuole by fusion of the autophagosomal outer membrane might here be recycled to novel phagophores. This scenario would ensure vacuolar homeostasis and explain the involvement of Vac8 into phagophore elongation (Figure 8). In this scenario, additional membranes could be supplied by the ER-phagophore contact site [5,7,8] and via vesicular transport.
The localization of Atg21 to the VICS (Figure 5A) indicates that the part of Atg8 lipidation, which depends on its presence, might be restricted to this site. This restriction of Atg8 would open an easy way to deliver Atg8-PE to both the inner and outer phagophore membrane to mediate its elongation. Indeed, the deletion of ATG21 affected phagophore elongation (Figure 5D). However, in contrast to Vac8, the absence of Atg21 had only mild effects on VICS formation (Figure 7). Atg21 and Vac8 are both essential for selective autophagy. Unselective starvation-induced autophagy can proceed in their absence, albeit at a significantly reduced level [44,48]. Indeed, the formation of aberrantly small autophagosomes were observed even in the absence of Atg8 [49].
PtdIns3P mediates membrane binding of Atg21, but since it is dispersed over the whole phagophore (Fig. S6D) [24], additional mechanisms must restrict Atg21 localization. Probably, preferential binding to curved membranes contributes to this restriction. Indeed, loop 6CD of yeast PROPPINs partially penetrates membranes and contributes to membrane binding and bending [50–53]. Regulated membrane association of Atg18 by phosphorylation of this loop underlines this function [54]. Furthermore, our structure of KlAtg21 in complex with the AgAtg16 coiled-coil domain shows the two Atg21 molecules in a twisted orientation (Figure 1A). Most likely, yet unknown protein-protein interactions of Atg21 at the VICS are involved, which might also explain why the complex does not assemble on other PtdIns3P-positive membranes, such as the endosomes.
Materials and methods
Yeast strains, media and growth conditions
All Saccharomyces cerevisiae strains used in this study are listed in Table 2. They are derivatives of the WT strain WCG4a [55] if not stated differently. The indicated knockouts were generated as described by using pFA6a-hphNT1 or pFA6a-natNT2 as templates [56]. Chromosomal tagging with yeGFP, mCherry, RFP, 6xHA, mTagBFP2 or 3xGFP was done by following previous reports and using pYM25, pYM25-mCherry, pYM25-RFP, pYM17, pFA6a-TagBFP2-natNT2 or pFA6a-3xGFP-HIS3MX6 as templates, respectively [56,57]. 3xtagBFP-PHO8 was integrated into the LEU2 locus by cutting pRS305-PGK1p-3xtagBFP-PHO8 [6] with BstEII. The resulting fragment was transformed into the indicated strains. Generated strains were subsequently verified by PCR of the genomic DNA.
Table 2.
List of S. cerevisiae strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| WCG4a (WT) | MATα his2-11,15 leu2-3,112 ura3 | [55] |
| atg1∆ | WCG4a atg1∆::KAN | [77] |
| atg8∆ | WCG4a atg8∆::KAN | [78] |
| atg8∆ atg3∆ | WCG4a atg8∆::KAN atg3∆::hphNT1 | This study |
| atg8∆ ATG14-3xGFP | WCG4a atg8∆::KAN ATG14-3xGFP(S65 T)::HIS3MX6 |
This study |
| atg8∆ atg18∆ | WCG4a atg8∆::KAN atg18∆::natNT2 | This study |
| atg8∆ atg21∆ | WCG4a atg8Δ::KAN atg21∆::natNT2 | [16] |
| atg8∆ atg21∆ atg16∆ | WCG4a atg8∆::KAN atg21∆::hphNT1 atg16∆::natNT2 |
[16] |
| atg8Δ atg21Δ VPH1-mCherry | WCG4a atg8∆::KAN atg21∆::natNT2 VPH1-mCherry::hphNT1 |
This study |
| atg8Δ APE1-RFP | WCG4a atg8∆::KAN APE1-mRFP::hphNT1 |
This study |
| atg8Δ APE1-RFP atg21∆ | WCG4a atg8∆::KAN APE1-mRFP::hphNT1 atg21∆::natNT2 | This study |
| atg8Δ APE1-RFP 3xtagBFP-PHO8 | WCG4a atg8∆::KAN APE1-mRFP::hphNT1 pRS305-PGK1p- 3xtagBFP-PHO8 |
This study |
|
atg8Δ APE1-RFP atg21Δ 3xtagBFP-PHO8 |
WCG4a atg8∆::KAN APE1-mRFP::hphNT1 atg21∆::natNT2 pRS305-PGK1p-3xtagBFP-PHO8 |
This study |
| atg8Δ APE1-RFP vac8Δ 3xtagBFP-PHO8 | WCG4a atg8∆::KAN APE1-mRFP::hphNT1 vac8∆::natNT2 pRS305-PGK1p-3xtagBFP-PHO8 |
This study |
| atg8∆ vac8∆ | WCG4a atg8∆::KAN vac8∆::hphNT1 | This study |
| atg8∆ vac8∆ APE1-mTagBFP2 | WCG4a atg8∆::KAN vac8∆::hphNT1 APE1-mTagBFP2::natNT2 |
This study |
| atg8∆ 3xtagBFP-PHO8 | WCG4a atg8∆::KAN pRS305-PGK1p- 3xtagBFP-PHO8 LEU2::natNT2 |
This study |
| atg8∆ 3xtagBFP-PHO8 vac8∆ | WCG4a atg8∆::KAN pRS305-PGK1p- 3xtagBFP-PHO8 LEU2::natNT2 vac8∆::hphNT1 |
This study |
| atg8∆ VPH1-GFP | WCG4a atg8∆::KAN VPH1-yeGFP::hphNT1 |
This study |
| atg8∆ VPH1-mCherry APE1-mTagBFP2 | WCG4a atg8∆::KAN VPH1-mCherry::hphNT1 APE1-mTagBFP2::natNT2 | This study |
| atg16Δ APE1-RFP | WCG4a atg16∆::natNT2 APE1-RFP::hphNT1 |
This study |
| atg21∆ atg16∆ | WCG4a atg21∆::KAN atg16∆::natNT2 | [16] |
| atg21∆ atg16∆ atg5∆ | WCG4a atg21∆::KAN atg16∆::natNT2 atg5∆::hphNT1 |
This study |
| SEY6210 (SEY WT) | Sey6210 MATa ura3-52 leu2-3,112 his3-∆200 lys 2–801 trp1-∆901 suc2-∆9 mel GAL |
[79] |
| SEY6210 atg16∆ | Sey6210 atg16∆::natNT2 | [16] |
| SEY6210 atg16∆ atg21∆ | Sey6210 atg16∆::natNT2 atg21∆::hphNT1 |
This study |
| vac8∆ ATG13-6xHA | WCG4a vac8∆::hphNT1 ATG13-6xHA::natNT2 |
This study |
Strains were grown at 30°C in complete minimal medium (0.67% [w:v] yeast nitrogen base w/o amino acids [Becton Dickinson, 291920], 2% [w:v] glucose [Roth, 6780.2], pH 5.6, supplemented with appropriate amino acids) for auxotrophic selection. Autophagy was induced by incubating stationary cells (OD600 4–5) for 1–2 h in nitrogen-free SD-N medium (0.17% [w:v] yeast nitrogen base w/o amino acids and ammonium sulfate [Becton Dickinson, 233520], 2% glucose).
Plasmids
All used plasmids are listed in Table 3. The mutated and MYC-tagged gene of the biotin protein ligase (BirA*) from E. coli was donated by H. D. Schmitt (Dep. of Neurobiology, Max Planck Institute for Biophysical Chemistry, Goettingen). It was cloned into pUG36 using the XbaI/XhoI restriction sites. The plasmids Nub-Atg21, Nub-Ubc6, and Ste14-Cub were a kind gift of F. Reggiori (Dep. of Cell Biology, University of Groningen). The yomTagBFP2 gene derived from the plasmid pFA6a-link-yomTagBFP2-Kan, which was a gift from Wendell Lim and Kurt Thorn (Addgene, 44899) [58].
Table 3.
List of plasmids used in this study
| Name | Characteristics | Source |
|---|---|---|
| AgAtg16(40-124)-6xHis | pET-28a AgATG16(40-124)-6xHis KANR | This study |
| Atg3-GFP | pRS313 ATG3p-ATG3-ATG3D265-yeGFP-ATG3G266-ATG3 t | This study |
| Atg16-Cub | pRS313 MET17p-ATG16-Cub-RURA3 | [16] |
| Atg16-HA | pRS313 CUP1p-ATG16-6xHA | [16] |
| Atg16E97A-HA | pRS313 CUP1p-ATG16E97A-6xHA | This study |
| Atg16K94E-HA | pRS313 CUP1p-ATG16K94E-6xHA | This study |
| Atg16K94E D101R-HA | pRS313 CUP1p-ATG16K94E D10R-6xHA | This study |
| Atg16D101R-HA | pRS313 CUP1p-ATG16D101R-6xHA | This study |
| Atg16E102R-HA | pRS313 CUP1p-ATG16E102R-6xHA | This study |
| Atg16K116E-HA | pRS313 CUP1p-ATG16K116E-6xHA | This study |
| Atg16IIV-HA | pRS313 CUP1p-ATG16I104,I108,V112D-6xHA | This study |
| Atg16-GFP | pRS313 ATG16p-ATG16-GFP | [16] |
| Atg16K94E-GFP | pRS313 ATG16p-ATG16K94E-GFP | This study |
| Atg16D101R-GFP | pRS313 ATG16p-ATG16D101R-GFP | This study |
| Atg16E102R-GFP | pRS313 ATG16p-ATG16E102R-GFP | This study |
| Atg16IIV-GFP | pRS313 ATG16p-ATG16I104,I108,V112D-GFP | This study |
| Atg18-GFP | pUG23 ATG18p-ATG18-yeGFP-CYC1 t | This study |
| Atg21[FTTG]-HA | pRS315 ATG21p-ATG21R343,R344 T-His6- 3xHA |
This study |
| Atg21-YFP | pRS313 ATG21p-ATG21-EYFP | This study |
| Atg21-YFP | pRS315 ATG21p-ATG21-EYFP | This study |
| BirA* | pUG36 MET17p-MYC-BirAR118 G-CYC1 t | This study |
| BirA*-Atg21 | pUG36 MET17p-MYC-BirAR118 G-ATG21- CYC1 t |
This study |
| BirA*- Atg21[FTTG] | pUG36 MET17p-MYC-BirAR118 G-ATG21R343,R344T-CYC1 t | This study |
| GFP-Atg8 | pRS313 ATG8p-GFP-ATG8-ATG8 t | This study |
| GFP-Atg8 | pRS315 ATG8p-GFP-ATG8-ATG8 t | [80] |
| GFP-Atg21 | pUG36 MET17p-yeGFP-ATG21-CYC1 t | [81] |
| GFP-Atg21D9R | pUG36 MET17p-yeGFP-ATG21D9R-CYC1 t | This study |
| GFP-Atg21D460R | pUG36 MET17p-yeGFP-ATG21D460R-CYC1 t | This study |
| GFP-Atg21E461R | pUG36 MET17p-yeGFP-ATG21E461R-CYC1 t | This study |
| GFP-Atg21[FTTG] | pUG36 MET17p-yeGFP-ATG21R343,R344T-CYC1 t | This study |
| GFP-Atg21K130E | pUG36 MET17p-yeGFP-ATG21K130E-CYC1 t | This study |
| GFP-Atg21R151E | pUG36 MET17p-yeGFP-ATG21R151E-CYC1 t | This study |
| GFP-Atg21R151E E461R | pUG36 MET17p-yeGFP-ATG21R151E E461R-CYC1 t | This study |
| GFP-Atg21K172E | pUG36 MET17p-yeGFP-ATG21K172E-CYC1 t | This study |
| KlAtg21-6xHis | pET-28a KlATG21-6xHis KANR | This study |
| mCherry-Atg5 | pUG36 MET17p-mCherry-ATG5-CYC1 t | This study |
| mCherry-Atg8 | pRS315 ATG8p-mCherry-ATG8-ATG8 t | This study |
| mCherry-Atg21 | pUG36 MET17p-mCherry-ATG21-CYC1 t | [16] |
| mCherry-Atg21E461R | pUG36 MET17p-mCherry-ATG21E461R-CYC1 t | This study |
| mCherry- Atg21[FTTG] | pUG36 MET17p-mCherry-ATG21R343,R344 T-CYC1 t | [16] |
| mCherry-Atg21K130E | pUG36 MET17p-mCherry-ATG21K130E-CYC1 t | This study |
| mCherry-Atg21R151E | pUG36 MET17p-mCherry-ATG21R151E-CYC1 t | This study |
| mRFP-2xFYVE | pRS315 AgTEF1p-mRFP-2xFYVE(Hrs)- CYC1 t | [81] |
| Nub-Atg5 | pRS314 CUP1p-Nub-ATG5-ATG5 t | [16] |
| Nub-Atg21 | pRS314 CUP1p-Nub-ATG21 | F. Reggiori |
| Nub- Atg21[FTTG] | pRS314 CUP1p-Nub- ATG21R343,R344 T | This study |
| Nub-Ubc6 | pRS314 CUP1p-Nub-UBC6 | F. Reggiori |
| pFA6a-3xGFP- HIS3MX6 |
3xGFPS65 T-ADH1 t AgTEF1p-HIS3MX6-AgTEF1 t | This study |
| pFA6a-hphNT1 | AgTEF1p-HygR-CYC1 t | [56] |
| pFA6a-natNT2 | AgTEF1p-NrsR-ADH1 t | [56] |
| pFA6a-TagBFP2-natNT2 | TagBFP2 AgTEF1p-NrsR-ADH1 t | This study |
| pRS305-PGK1p-3xtagBFP-PHO8 | PGK1p-3xtagBFP-PHO8 LEU2 | [6] |
| pRS313 | CEN/ARS AmpR ori lacZ’ HIS3 | [82] |
| pRS314 | CEN/ARS AmpR ori lacZ’ TRP1 | [82] |
| pRS423-CUP1p-Ape1 | pRS423 2μ HIS3 CUP1p-APE1 | This study |
| pUG36 | CEN/ARS AmpR ori URA3 MET17p-yeGFP-lacZ’/MCS-CYC1 t | [83] |
| PVac8-GFP | pUG23 VAC8p-yeGFP-CYC1 t | This Study |
| pYEX-BX(Ape1) | pYEX-BX 2μ URA3 CUP1p-APE1 | [20] |
| pYM17 | 6xHA AgTEF1p-NrsR-ADH1 t | [56] |
| pYM25 | yeGFP AgTEF1p-HygR-CYC1 t | [56] |
| pYM25-mCherry | mCherry AgTEF1p-HygR-CYC1 t | This study |
| pYM25-RFP | mRFP1 AgTEF1p-HygR-CYC1 t | This study |
| Sec63-GFP | pRS316 SEC63p-SEC63-GFPS65 T V163A | [84] |
| Ste14-Cub | pRS313 MET17p-STE14-Cub-RURA3 | F. Reggiori |
| Vac8-GFP | pUG23 VAC8p-VAC8-yeGFP-CYC1 t | This study |
| Vac8[EEE]-GFP | pUG23 VAC8p-VAC8R276,R317,R359E-yeGFP-CYC1 t | This study |
| Vac8-HA | pRS313 VAC8p-VAC8-6xHA | This study |
| Vac8[EEE]-HA | pRS313 VAC8p-VAC8R276,R317,R359E-6xHA | This study |
| yomTagBFP2-Atg8 | pRS313 ATG8p-yomTagBFP2-ATG8-ATG8 t | This study |
Indicated mutations of Atg21, Atg16, and Vac8 were introduced into the plasmids Atg21-GFP, mCherry-Atg21, BirA*-Atg21, Nub-Atg21, Atg21-HA, Atg16-HA, Atg16-GFP, Vac8-GFP, and Vac8-HA, respectively, using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, 210518).
Antibodies
The anti-Ape1 antibody was described previously [59]. The anti-MYC antibody (cell supernatant precipitated with ammonium sulfate) was from S. Callegari and P. Rehling (Dep. of Cellular Biochemistry, University of Goettingen). Other antibodies were anti-GFP (from mouse IgG1κ; Roche, 11814460001), HA-probe antibody (F-7, mouse monoclonal IgG2a; Santa Cruz Biotechnology, sc-7392), horseradish peroxidase-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific, G-21234) and horseradish peroxidase-conjugated goat anti-mouse IgG (Dianova, 115–035-166).
Fluorescence microscopy
For the generation of giant prApe1 complexes in the cell, APE1 was overexpressed using high copy plasmids pYEX-BX(Ape1) or pRS423-CUP1p-Ape1 as described previously [20]. Respective cells were grown to stationary phase in complete minimal medium supplemented with 100 µM CuSO4 and subsequently starved for 1–2 h in SD-N medium. To visualize the nucleus, 12.5 μg/ml Hoechst 33324 (Thermo Fisher Scientific, H1399) were added to the culture 15 min before analysis by fluorescence microscopy. For the FM™ 4–64 staining, stationary cells were incubated for 30 min with 20 µg/ml FM™ 4–64 dye (Thermo Fisher Scientific, T3166) prior to starvation in SD-N medium.
For localization studies of Atg16 and Atg21, atg8∆ atg16∆ atg21∆ cells carrying mCherry-Atg21 and Atg16-GFP or their variants as indicated were grown to early stationary phase (OD600 2) in complete minimal medium supplemented with 0.3 mM methionine.
In vivo fluorescence microscopy was performed using a DeltaVision® Core setup, based on an inverted IX71 microscope (Olympus), equipped with the UPlanSApo x100, 1.4 numerical aperture, oil immersion objective (Olympus) and a CoolSNAPHQ2™ couple-charged device camera (Roper Scientific). For the detection of fluorescent proteins and dyes, the following built-in filter sets were used, DAPI (excitation wavelength: 390/18 nm; emission wavelength: 435/48 nm), FITC (excitation wavelength: 475/28 nm; emission wavelength: 523/36 nm), TRITC (excitation wavelength: 542/27 nm; emission wavelength: 594/45 nm), GFP (excitation wavelength: 475/28 nm; emission wavelength: 525/50 nm) and mCherry (excitation wavelength: 575/25 nm; emission wavelength: 632/69 nm). The DeltaVision® Core setup was operated using softWoRx® (Applied Precision), which was also used for the deconvolution of the images. The cells were imaged taking z-stacks of at least 20 focal planes with a distance of 0.2 µm. The resulting images were further processed using Fiji [60]. Fluorescence intensities were measured along with circular/oval or linear plotlines using Fiji. Resulting profiles were plotted in arbitrary units against the line distance of the plotline (in µm or degree). Heat maps of the fluorescence intensity were generated by coloring the indicated channel with the look-up-table fire (bright [yellow; high intensity] to dark [deep blue; low intensity]) using Fiji. 3-dimensional reconstructions were generated by processing the imaged focal planes with the surface rendering function of Huygens Professional (Scientific Volume Imaging). Images were reduced to the area of interest prior to processing.
Dual-color fluorescence cross-correlation spectroscopy (FCCS)
(I) Preparation of cultures for FCCS of living yeast cells: atg21∆ atg16∆ cells carrying Atg16-GFP and mCherry-Atg21[FTTG] or atg21∆ atg16∆ atg5∆ cells carrying Atg16-GFP, mCherry-Atg5, and Atg21[FTTG]-HA were grown in complete minimal medium to early stationary phase (OD600 1–2).
(II) Microscope: A modified Olympus IX-71 (Olympus Deutschland GmbH, Hamburg, Germany) microscope with an accessible standard side port on the right side was used. The samples were scanned at a speed of 2–4 ms per 100 nm pixel using a 3-axis piezo-stage (P-562.3 CD, Physical Instruments), which was driven with a digital piezo controller (E-710, Physical Instruments).
(III) Excitation system: A pulsed interleaved excitation was achieved with a white light laser (Fianium) filtered with a 560 clean-up filter (FF01-560/14, Semrock) and a 485 nm pulsed diode laser equipped with a clean-up filter (FF01-488/10). The inter-pulse duration was 25 ns. The two lasers were coupled through a polarization-maintaining single-mode optical fiber (PMC-400-4.2-NA010-3-APC-250-V, Schaefter and Kirchhoff GmbH, Germany).
(IV) Main Optical Unit: The fiber output was collimated to a beam of 9 mm diameter by coupling it to an infinity-corrected 4X objective (UPLSAPO4X, Olympus). The excitation beam was then reflected by a dichroic mirror (FITC/CY5 [51008bs], Chroma Technology Corporation), guiding it into the side port of the microscope. The excitation power used for both wavelengths was around 10–25 kW/cm2. A high N.A. objective was used for exciting the samples (UPLSAPO 60XW, 1.2 N.A., Olympus) and for collecting the fluorescence emission. The emission light was then passed through the dichroic mirror and focused onto a 50 µm pinhole for confocal imaging and FCS measurements. After the pinhole, the fluorescence photons were split according to their wavelength with the help of an emission dichroic mirror (HC BS 560, AHF Analysentechnik AG, Germany). These two separated streams of photons were then focused on the active area (~ 175 µm) of two single-photon counting modules (SPCM–AQRH-14, Excellitas). Additional band pass filters (FF01-525/45, FF01-590/36, Semrock) were used to block any undesired back-scattered laser light. The dark count rate of the detector was less than 150 counts per s.
(V) Data acquisition and processing: The transistor-transistor-logic (TTL) pulses from the SPCM were recorded with a 2 ps time resolution by a multichannel picosecond event timer and Time-Correlated Single Photon Counting (TCSPC) module (HydraHarp 400, PicoQuant, GmbH). Due to the perfectly synchronized lasers with a fixed time delay between them (interleaved) the photons arising from direct emission of the green and red fluorophores are temporally separated from the spectral crosstalk of the green fluorescence into the red emission channel. By setting defined time gates, we select the photons that correspond to the direct emissions of the fluorophores collected on the respective detectors. Auto-correlations and spectral cross-correlations are performed on these photon streams. The data analysis was performed with the help of custom-written Matlab (MathWorks®) routines.
Expression and purification of KlAtg21 and AgAtg16(40-124)
Full-length KlAtg21 or the truncated AgAtg16(40–124) construct were cloned in a pET-28a vector using NdeI/XhoI restriction sites and transformed in E. coli BL21-Gold(DE3) (Agilent Technologies, 230132) competent cells. Proteins were expressed individually overnight at 25°C in the ZYM-5052 auto-inducing medium [61]. Cells were harvested and resuspended in 50 mM HEPES (GERBO Biotech, 1009) pH 7.5, 250 mM NaCl, 30 mM imidazole (Sigma-Aldrich, 56750), 1 mM TCEP (Sigma-Aldrich, 75259) and stored frozen at −20°C. For complex purification thawed cell pellets of KlAtg21 and AgAtg16(40–124) were mixed in a 2:1 ratio, supplemented with one protease inhibitor tablet (complete™, EDTA-free Protease Inhibitor Cocktail, Roche, 05056489001), DNase I (AppliChem, A3778), lysozyme (Roth, 8259) and 1 mM MgCl2. Cell suspensions were stirred for 15 min at room temperature and then homogenized with a homogenizer and lysed with three repetitions in a microfluidizer M-110 L (Microfluidics Corporation) according to the manufacturer’s instructions. Cell debris was pelleted at 30597 x g and 4°C for 45 min. The supernatant was applied to a 5 ml HisTrap column (GE Healthcare) connected to the Äkta Prime FPLC system at 4°C using 50 mM HEPES pH 7.5, 250 mM NaCl, 30 mM imidazole, 1 mM TCEP. The column was washed with 12 column volumes. His-tagged proteins were eluted with a gradient over 10 column volumes to 50 mM HEPES pH 7.5, 250 mM NaCl, 400 mM imidazole, 1 mM TCEP. Eluted proteins were concentrated and then applied to a Superdex 200 16/60 HiLoad column (GE Healthcare) and ran with 30 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP. Fractions were pooled and concentrated to 10–30 mg/ml. For complex purification, only fractions with complexed AgAtg16-KlAtg21 were pooled. Purified proteins were aliquoted, flash cooled with liquid nitrogen and stored at −80°C.
Crystallization and structure determination of the AgAtg16-KlAtg21 complex
The AgAtg16(40-124)-KlAtg21 complex was crystallized using a dropped vapor diffusion setup at 20°C. The complex was subjected to in-situ proteolysis with 1:1000 clostripain (Proti-AceTM 2, Hampton Research, HR2-432) and a precipitant composed of 15% (w:v) PEG 4000 (Sigma-Aldrich, 817006) and 0.1 M HEPES pH 7.0. Crystals were soaked in crystallization solution supplemented with 30% ethylene glycol (Sigma-Aldrich, 102466) and 3% PEG 4000 before flash cooling in liquid nitrogen. A complete 3.7 Å resolution native data set (Table 1) was collected at 100 K on a beamline X06SA equipped with Pilatus 6 M detector (Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland).
Data processing
Diffraction images were integrated using MOSFLM [62] and scaled with AIMLESS [63] from the CCP4 [64] suite. The estimation of the overall highest resolution limit (3.7 Å) were performed using default settings as implemented in AIMLESS: half-dataset correlation CC1/2 > 0.3 and mean I:σ(I) > 1.50. Data collection statistics are presented in Table 1.
Structure solution and refinement
The HHPRED [65] server was used to search the PDB for KlAtg21 homologous structures and revealed Atg18 from Pichia angusta (PDB id: 5LTG) as the most suited model for Molecular Replacement (MR) search (corresponding sequence identity and similarity are 32% and 56%, respectively). The initial model preparation, utilizing the HHPRED sequence alignment, was performed using phenix.sculptor [66], and involved the removal of unaligned residues and stripping all non-identical side chains to the gamma atom. The MR search, performed with PHASER [67], revealed a clear solution for two KlAtg21 monomers occupying the asymmetric unit (corresponding TFZ scores for the 1st and the 2nd molecule were 9.7 and 19.0, respectively). In order to overcome the difficulties in manual model rebuilding related to the limited resolution of experimental data, one KlAtg21 monomer was subjected to energy and density-guided refinement in Rosetta [68,69] utilizing constraints from an averaged 2mFo-DFc electron density map calculated with PHENIX [70]. One thousand generated models with the highest ROSETTA score were rescored using PHASER (Refine and Phase protocol). The best model was subjected to the next round of modeling using ROSETTA employing a newly calculated averaged electron density map. Prior to modeling, the template was manually inspected in order to verify the correctness of the employed sequence alignment. This routine was repeated until the convergence has been reached (three times). The subsequent ROSETTA-based refinement was used to rebuild regions around insertions and up to seventeen amino acid long gaps in the verified sequence alignment. During this step, the ROSETTA loop modeling protocol [71] combined with cyclic coordinate descent closure [72] was employed. From a few thousand decoys, the best 100 models obtained from the PHASER rescore step were subjected to reciprocal-space refinement in PHENIX using strong non-crystallographic symmetry (NCS) restraints between two KlAtg21 monomers and grouped B-factor refinement. The best model was refined at 3.7 Å to crystallographic R and Rfree factors of 0.3566 and 0.3833, respectively (mean FOM = 0.61). Manual model inspection in Coot [73] revealed a clear mFo-DFc electron density map corresponding to a fragment of a dimeric AgAtg16(40–124) located between two KlAtg21 molecules. Two dimeric AgAtg16(40–124) coiled-coil assemblies were modeled as poly-A using Coot in both “forward” and “reversed” polypeptide directions. The assemblies were added to the two KlAtg21 monomers and subjected to refinement in PHENIX employing an identical protocol. The “forward” KlAtg21-AgAtg16(40–124) model revealed significantly lower crystallographic R factors (R = 0.3119, Rfree 0.3378) then the “reversed” one (R = 0.3142, Rfree 0.3427). From this step on, the refinement was continued only with the “forward” KlAtg21-AgAtg16(40–124) model. A detailed inspection of the electron density map showed a clearly visible KlAtg21 R103 side chain pointing toward a residue of AgAtg16(40–124) being most likely a negatively charged amino-acid – preferably D over E. The further analysis revealed that the subsequently following three alpha-helical turns of AgAtg16(40–124) were bound in a hydrophobic cleft formed between two beta-blades of KlAtg21. Due to a limited depth of this cleft, the potential hydrophobic residues of AgAtg16(40–124) could be either A, V, I, L or G. Searching for an amino acid sequence patch in the form of E/D-X-X-A/V/I/L/G-X-X-X-A/V/I/L/G-X-X-X-A/V/I/L/G present in AgAtg16(40–124) revealed only one possibility leading to assignment of the negatively charged amino acid interacting with KlAtg21 R103 to D78. Hence, the hydrophobic AgAtg16(40–124) residues bound in a cleft are: I81, I85 and L89.
The model was manually verified against simulated annealing (SA) omit maps in Coot and further refined with PHENIX in both real and reciprocal space. Grouped B-factor refinement and strong NCS restrains were used to reduce the risk of overfitting. The final model consisting of residues 1 to 392 for each KlAtg21 monomer (missing loops: 136–150, 285–334) and residues 70–104/107 for AgAtg16(40–124) dimer, has been refined at 3.7 Å resolution to crystallographic R and Rfree factors of 28.45% and 30.65%, respectively). Details are presented in Table 1. Atomic coordinates and structure factors were deposited in the Protein Data Bank (PDB, http://www.rcsb.org/) under PDB id code 6RGO. PISA software was used to analyze macromolecular interfaces. Figures were prepared using PyMol (http://www.pymol.org).
prApe1 maturation assay
atg21∆ atg16∆ cells carrying Atg21-GFP and Atg16-HA or their variants as indicated were grown to early stationary phase (OD600 2–3) in complete minimal medium without methionine. atg1∆ and atg21∆ atg16∆ cells carrying the empty vectors pUG36 and pRS313 were included into the analysis as negative controls. The Ape1 maturation assay was performed according to [74].
Immunoprecipitation of Atg21-GFP
atg16∆ atg21∆ cells carrying pUG36 and Atg16-HA (control), Atg21-GFP and Atg16-HA or their variants as indicated were grown to early stationary phase (OD600 2) in complete minimal medium without methionine. Immunoprecipitations were performed as described before [16].
Immunoprecipitation of Vac8-GFP
vac8∆ ATG13-6xHA cells carrying VAC8p-GFP (VAC8 promoter control), Vac8-GFP or Vac8[EEE]-GFP were grown to stationary phase (OD600 3–4). 200 OD600 of cells were harvested (720 x g, 5 min), washed once with 20 ml SD-N medium (720 x g, 5 min), and starved for 2 h in 20 ml of fresh SD-N medium. After starvation, cells were harvested (720 x g, 5 min, 4°C) and washed once with cold 50 mM Tris-HCl pH 7.5 (720 x g, 5 min, 4°C). Immunoprecipitations were performed according to the manufacturer’s recommendations using the µMACS GFP Isolation Kit (Miltenyi Biotec, 130–091-125) together with the µ Columns (Miltenyi Biotec, 130–042-701). Instead of the supplied lysis buffer, the following buffer was used for cell lysis and washing of the columns: 50 mM Tris-HCl pH 7.5, 1 mM EDTA (Roth, 8043.1), 0.5% (w:v) TWEEN® 20 (Sigma-Aldrich, P7949), 1 mM PMSF (Roth, 6367.1), 1x cOmplete™ (EDTA-free Protease Inhibitor Cocktail, Roche, 05056489001) and 1x PhosSTOP (Phosphatase Inhibitor Cocktail, Roche, 04906837001). Cells were lysed using glass beads. For this, the cell pellet was resuspended in 750 µl lysis buffer and added to 400 µl of glass beads. After 30 min harsh vortexing at 4°C the cell lysate was centrifuged at 3000 x g and 4°C for 5 min. The supernatant was used for immunoprecipitation. Input and bound fractions were analyzed by western blot.
Split-ubiquitin assay
The split-ubiquitin assay was performed according to a previous report [16].
Biotin identification (BioID) assay
atg21∆ atg16∆ cells carrying the empty vector pUG36, BirA*, BirA*-Atg21 or BirA*- Atg21[FTTG] together with Atg16-HA were grown to early stationary phase (OD600 2–3) in complete minimal medium without methionine which was supplemented with 10 µM biotin (Sigma-Aldrich, B4501). Cell lysis and isolation of biotinylated proteins was performed according to [75] using 0.2 ml gravity flow strep-tactin sepharose columns (iba, 2–1202-501). Input and elution fractions were analyzed by western blot. The MYC-tagged BirA* and its fusion proteins were detected using the anti-MYC antibody.
Quantification and statistical analysis
Western blots were quantified using Fiji [60]. To plot and statistically evaluate the data obtained from quantification or fluorescence microscopy, Prism 7.0 c for Mac OSX (GraphPad Software) was used. Graphs were plotted using the mean value and error bars indicate the standard error of the mean (SEM). The number (n) of independent experiments is indicated for each graph. Statistical evaluations of prApe1 maturation, co-immunoprecipitations or the BioID assay were performed using the one-sample t-test. For this, the WT strain/protein was used as a 100% reference. Two datasets were directly compared using the two-tailed t-test. The statistical relevance is indicated in the graphs as follows: not significant (ns) for P > 0.05, * for P < 0.05, ** for P < 0.01, *** for P < 0.001 and **** for P < 0.0001.
Supplementary Material
Acknowledgments
We are very grateful to K. Kuehnel for preparation of the Atg16-Atg21 crystals. We further thank P. Schlotterhose for technical support, and H.D. Schmitt and P. Rehling for plasmids and antibodies.
Funding Statement
This work was supported by the “Deutsche Forschungsgemeinschaft” [SFB860 B04 for LM, JMS and MT, SFB1190 P06 for FO and MT, SFB1190 P12 for MM and SFB803 B09 for BK].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Wen X, Klionsky DJ.. An overview of macroautophagy in yeast. J Mol Biol. 2016;428:1681–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 2014;26C:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Reggiori F, Klionsky DJ. Autophagic processes in yeast: mechanism, machinery and regulation. Genetics. 2013;194:341–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gomez-Sanchez R, Rose J, Guimarães R, et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J Cell Biol. 2018;217:2743–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kotani T, Kirisako H, Koizumi M, et al. The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation. Proc Natl Acad Sci U S A. 2018;115:10363–10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Graef M, Friedman JR, Graham C, et al. ER exit sites are physical and functional core autophagosome biogenesis components. Mol Biol Cell. 2013;24:2918–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Valverde DP, Yu S, Boggavarapu V, et al. ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol. 2019;14:jcb.201811139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Osawa T, Kotani T, Kawaoka T, et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat Struct Mol Biol. 2019;26:281–288. [DOI] [PubMed] [Google Scholar]
- [9].Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19:3290–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. [DOI] [PubMed] [Google Scholar]
- [11].Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. [DOI] [PubMed] [Google Scholar]
- [12].Kuma A, Mizushima N, Ishihara N, et al. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. The. J Biol Chem. 2002;277:18619–18625. [DOI] [PubMed] [Google Scholar]
- [13].Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. [DOI] [PubMed] [Google Scholar]
- [14].Fujioka Y, Noda NN, Nakatogawa H, et al. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J Biol Chem. 2010;285:1508–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Harada K, Kotani T, Kirisako H, et al. Two distinct mechanisms target the autophagy-related E3 complex to the pre-autophagosomal structure. eLife. 2019;8:685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Juris L, Montino M, Rube P, et al. PI3P binding by Atg21 organises Atg8 lipidation. Embo J. 2015;34:955–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Suzuki K, Kirisako T, Kamada Y, et al. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. Embo J. 2001;20:5971–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Obara K, Sekito T, Ohsumi Y. Assortment of phosphatidylinositol 3-kinase complexes–Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell. 2006;17:1527–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dooley HC, Razi M, Polson HEJ, et al. WIPI2 Links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014;55:238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Suzuki K, Akioka M, Kondo-Kakuta C, et al. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J Cell Sci. 2013;126:2534–2544. [DOI] [PubMed] [Google Scholar]
- [21].Wittke S, Lewke N, Muller S, et al. Probing the molecular environment of membrane proteins in vivo. Mol Biol Cell. 1999;10:2519–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ngu MP, Hirata E, Suzuki K. Visualization of Atg3 during Autophagosome Formation in Saccharomyces cerevisiae. J Biol Chem. 2015;290:8146–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sakoh-Nakatogawa M, Kirisako H, Nakatogawa H, et al. Localization of Atg3 to autophagy-related membranes and its enhancement by the Atg8-family interacting motif to promote expansion of the membranes. FEBS Lett. 2015;589:744–749. [DOI] [PubMed] [Google Scholar]
- [24].Cheng J, Fujita A, Yamamoto H, et al. Yeast and mammalian autophagosomes exhibit distinct phosphatidylinositol 3-phosphate asymmetries. Nat Commun. 2014;5:3207. [DOI] [PubMed] [Google Scholar]
- [25].Obara K, Noda T, Niimi K, et al. Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes Cells. 2008;13:537–547. [DOI] [PubMed] [Google Scholar]
- [26].Dawaliby R, Mayer A. Microautophagy of the nucleus coincides with a vacuolar diffusion barrier at nuclear-vacuolar junctions. Mol Biol Cell. 2010;21:4173–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang Y, Catlett N, Weisman L. Vac8p, a vacuolar protein with armadillo repeats, functions in both vacuole inheritance and protein targeting from the cytoplasm to vacuole. J Cell Biol. 1998;140:1063–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kvam E, Goldfarb D. Nucleus-vacuole junctions and piecemeal microautophagy of the nucleus in S. cerevisiae. Autophagy. 2007;3:85–92. [DOI] [PubMed] [Google Scholar]
- [29].Jeong H, Park J, Kim H-I, et al. Mechanistic insight into the nucleus-vacuole junction based on the Vac8p-Nvj1p crystal structure. Proc Natl Acad Sci U S A. 2017;114:E4539–E48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vida T, Emr S. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Noda NN, Fujioka Y, Hanada T, et al. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013;14:206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sakoh-Nakatogawa M, Matoba K, Asai E, et al. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat Struct Mol Biol. 2013;20:433–439. [DOI] [PubMed] [Google Scholar]
- [33].Metlagel Z, Otomo C, Takaesu G, et al. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proc Natl Acad Sci U S A. 2013;110:18844–18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Romanov J, Walczak M, Ibiricu I, et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. Embo J. 2012;31:4304–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lystad AH, Carlsson SR, de la Ballina LR, et al. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat Cell Biol. 2019;21:372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wilson MI, Dooley HC, Tooze SA. WIPI2b and Atg16L1: setting the stage for autophagosome formation. Biochem Soc Trans. 2014;42:1327–1334. [DOI] [PubMed] [Google Scholar]
- [37].Fletcher K, Ulferts R, Jacquin E, et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. Embo J. 2018;37:e97840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rai S, Arasteh M, Jefferson M, et al. The ATG5-binding and coiled coil domains of ATG16L1 maintain autophagy and tissue homeostasis in mice independently of the WD domain required for LC3-associated phagocytosis. Autophagy. 2019;15(4):599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jelani M, Dooley HC, Gubas A, et al. A mutation in the major autophagy gene, WIPI2, associated with global developmental abnormalities. Brain. 2019;142:1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kaufmann A, Beier V, Franquelim HG, et al. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell. 2014;156:469–481. [DOI] [PubMed] [Google Scholar]
- [41].Dudley LJ, Cabodevilla AG, Makar AN, et al. Intrinsic lipid binding activity of ATG16L1 supports efficient membrane anchoring and autophagy. Embo J. 2019;38:e100554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Toulmay A, Prinz WA. Direct imaging reveals stable, micrometer-scale lipid domains that segregate proteins in live cells. J Cell Biol. 2013;202:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hollenstein DM, Gomez-Sanchez R, Ciftci A, et al. Vac8 spatially confines autophagosome formation at the vacuole in S. cerevisiae. J Cell Sci. 2019;132:jcs235002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Scott SV, Nice DC, Nau JJ, et al. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J Biol Chem. 2000;275:25840–25849. [DOI] [PubMed] [Google Scholar]
- [45].Park J, Kim H-I, Jeong H, et al. Quaternary structures of Vac8 differentially regulate the Cvt and PMN pathways. Autophagy. 2019;00:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gatica D, Damasio A, Pascual C, et al. The carboxy terminus of yeast Atg13 binds phospholipid membrane via motifs that overlap with the Vac8-interacting domain. Autophagy. 2019;8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Molino D, Nascimbeni AC, Giordano F, et al. ER-driven membrane contact sites: evolutionary conserved machineries for stress response and autophagy regulation? Commun Integr Biol. 2017;10:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Meiling-Wesse K, Barth H, Voss C, et al. Atg21 is required for effective recruitment of Atg8 to the preautophagosomal structure during the Cvt pathway. J Biol Chem. 2004;279:37741–37750. [DOI] [PubMed] [Google Scholar]
- [49].Abeliovich H, Dunn WA, Kim J, et al. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol. 2000;151:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Krick R, Busse RA, Scacioc A, et al. Structural and functional characterization of the two phosphoinositide binding sites of PROPPINs, a β-propeller protein family. Proc Natl Acad Sci U S A. 2012;109:E2042–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Baskaran S, Ragusa MJ, Boura E, et al. Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy. Mol Cell. 2012;47:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Watanabe Y, Kobayashi T, Yamamoto H, et al. Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J Biol Chem. 2012;287:31681–31690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Busse RA, Scacioc A, Krick R, et al. Characterization of PROPPIN-phosphoinositide binding and role of loop 6CD in PROPPIN-membrane binding. Biophys J. 2015;108:2223–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tamura N, Oku M, Ito M, et al. Atg18 phosphoregulation controls organellar dynamics by modulating its phosphoinositide-binding activity. J Cell Biol. 2013;202:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Thumm M, Egner R, Koch B, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349:275–280. [DOI] [PubMed] [Google Scholar]
- [56].Janke C, Magiera M, Rathfelder N, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. [DOI] [PubMed] [Google Scholar]
- [57].Longtine M, McKenzie A, Demarini D, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. [DOI] [PubMed] [Google Scholar]
- [58].Lee S, Lim WA, Thorn KS. Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS ONE. 2013;8:e67902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Barth H, Thumm M. A genomic screen identifies AUT8 as a novel gene essential for autophagy in the yeast Saccharomyces cerevisiae. Gene. 2001;274:151–156. [DOI] [PubMed] [Google Scholar]
- [60].Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. [DOI] [PubMed] [Google Scholar]
- [62].Battye TGG, Kontogiannis L, Johnson O, et al. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zimmermann L, Stephens A, Nam S-Z, et al. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J Mol Biol. 2018;430:2237–2243. [DOI] [PubMed] [Google Scholar]
- [66].Bunkóczi G, Read RJ. Improvement of molecular-replacement models with Sculptor. Acta Crystallogr D Biol Crystallogr. 2011;67:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].McCoy AJ, Grosse-Kunstleve RW, Adams PD, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].DiMaio F, Terwilliger TC, Read RJ, et al. Improved molecular replacement by density- and energy-guided protein structure optimization. Nature. 2011;473:540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bender BJ, Cisneros A, Duran AM, et al. Protocols for molecular modeling with rosetta3 and rosettascripts. Biochemistry. 2016;55:4748–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Qian B, Raman S, Das R, et al. High-resolution structure prediction and the crystallographic phase problem. Nature. 2007;450:259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Canutescu AA, Dunbrack RL. Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Emsley P, Lohkamp B, Scott WG, et al. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Barth H, Meiling-Wesse K, Epple UD, et al. Autophagy and the cytoplasm to vacuole targeting pathway both require Aut10p. FEBS Lett. 2001;508:23–28. [DOI] [PubMed] [Google Scholar]
- [75].Opitz N, Schmitt K, Hofer-Pretz V, et al. Capturing the Asc1p/Receptor for Activated C Kinase 1 (RACK1) microenvironment at the head region of the 40s ribosome with quantitative BioID in yeast. Mol Cell Proteomics. 2017;16:2199–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Waterhouse AM, Procter JB, Martin DMA, et al.. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Straub M, Bredschneider M, Thumm M. AUT3, a serine/threonine kinase gene, is essential for autophagocytosis in Saccharomyces cerevisiae. J Bacteriol. 1997;179:3875–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Lang T, Schaeffeler E, Bernreuther D, et al. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. Embo J. 1998;17:3597–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Harding TM, Hefner-Gravink A, Thumm M, et al. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J Biol Chem. 1996;271:17621–17624. [DOI] [PubMed] [Google Scholar]
- [80].Krick R, Bremer S, Welter E, et al. Cdc48/p97 and Shp1/p47 regulate autophagosome biogenesis in concert with ubiquitin-like Atg8. J Cell Biol. 2010;190:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Krick R, Henke S, Tolstrup J, et al. Dissecting the localization and function of Atg18, Atg21 and Ygr223c. Autophagy. 2008;4:896–910. [DOI] [PubMed] [Google Scholar]
- [82].Sikorski R, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Niedenthal R, Riles L, Johnston M, et al. Green fluorescent protein as a marker for gene expression and subcellular localization in budding yeast. Yeast. 1996;12:773–786. [DOI] [PubMed] [Google Scholar]
- [84].Prinz W, Grzyb L, Veenhuis M, et al. Mutants affecting the structure of the cortical endoplasmic reticulum in Saccharomyces cerevisiae. J Cell Biol. 2000;150:461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.