

ABSTRACT
Malignant melanoma is characterized by mutations in a number of driver genes, most notably BRAF and NRAS. Recent genomic analyses revealed that 4-9% of sun-exposed melanomas bear activating mutations in RAC1, which encodes a small GTPase that is known to play key roles in cell proliferation, survival, and migration. The RAC1 protein activates several effector pathways, including Group A p21-activated kinases (PAKs), phosphoinositol-3-kinases (PI3Ks), in particular the beta isoform, and the serum-response factor/myocardin-related transcription factor (SRF/MRTF). Having previously shown that inhibition of Group A PAKs impedes oncogenic signalling from RAC1P29S, we here extend this analysis to examine the roles of PI3Ks and SRF/MRTF in melanocytes and/or in a zebrafish model. We demonstrate that a selective Group A PAK inhibitor (Frax-1036), a pan-PI3K (BKM120), and two PI3Kβ inhibitors (TGX221, GSK2636771) impede the growth of melanoma cells driven by mutant RAC1 but not by mutant BRAF, while other PI3K selective inhibitors, including PI3Kα, δ and γ, are less effective. Using these compounds as well as an SRF/MRTF inhibitor (CCG-203,971), we observed similar results in vivo, using embryonic zebrafish development as a readout. These results suggest that targeting Group A PAKs, PI3Kβ, and/or SRF/MRTF represent a promising approach to suppress RAC1 signalling in malignant melanoma.
KEYWORDS: Melanoma, PAK, PI3K, RAC1, small GTPase, SRF/MRTF
Introduction
Malignant melanoma is a highly aggressive cancer associated with poor overall survival. Recent genomic analyses have uncovered a variety of new driver mutations in malignant melanoma including an activating mutation in RAC1 [1 2,]. RAC1 encodes a small ubiquitously expressed GTPase known to play key roles in embryonic development, immune response, cell proliferation, survival, and rearrangement of the cytoskeleton by actin filament remodelling [3–5]. In sun-exposed cutaneous melanomas, RAC1P29S is the third most common oncogenic driver mutation, following BRAFV600E and NRASQ61L/K/R [1 2,]. RAC1P29S structure analysis and biochemical studies have shown that the proline to serine substitution in the hydrophobic pocket of the switch I domain of the GTPase results in an increased cycling rate from the GDP-bound inactive state to the GTP-bound active state, triggering downstream effectors and promoting melanocyte proliferation and migration[1].
While RAC1 itself represents a challenging therapeutic target, its effectors might be more tractable. Many of RAC1’s downstream biological effects are propagated by p21-activated kinases (PAKs). PAKs are serine/threonine specific intracellular kinases that phosphorylate downstream effector substrates regulating apoptosis, cell motility, cell morphology and cytoskeleton rearrangement [6]. Overexpressed PAKs result in oncogenic effects including increased cell proliferation and cell cycle progression, evasion of apoptosis, angiogenesis, and promotion of invasion and metastasis [7]. RAC1P29S induces PAK1 activation, which in turn phosphorylates and activates MEK1 at the Serine 298 site, facilitating ERK activation and transcription of various target genes. The PAK/MEK/ERK pathway is essential for RAS-driven transformation in a mouse model of skin cancer [8] and represents a potential therapeutic target for sun-driven melanomas. Zebrafish embryos injected with RAC1P29S mRNA displayed abnormal development and PAK and ERK elevated activity [9]. Defective growth was reversed by PAK and MEK inhibitors, suggesting that inhibition of these enzymes may be useful to prevent the developmental effects of RAC1P29S mutations. [9] In addition, we and others have reported that tumours and human melanoma cell lines bearing RAC1P29S mutations are resistant to BRAF inhibitors but are sensitive to PAK and MEK inhibitors [9 10,].
In addition to PAKs, PI3Ks represent a second recognized effector for RAC1 [11]. PI3Ks are lipid signalling kinases that play key regulatory roles in cell survival, proliferation, and differentiation [12]. Class I PI3K isoforms can be divided into two families based on their regulation mode: Class IA are heterodimeric proteins composed of a p110 catalytic subunit (PI3Kα/p110α, PI3Kβ/p110β or PI3Kδ/p110δ) whose enzymatic activity depends on their binding to a regulatory p85 subunit (p85α, p55α, p50α, p85β or p55γ); and Class IB, which do not need to interact with a regulatory subunit to be active (PI3Kγ/p110γ) [12 13,]. PI3Ks transduce external signals from growth factors and cytokines into phospholipids that activate various downstream effector pathways such as the serine/threonine kinase AKT, and guanine-nucleotide exchange factors (GEFs)[14].
The PI3K/AKT pathway is an important regulator of normal cell physiology and is activated in 70% of sporadic melanomas including those containing the RAC1P29S mutation [15–18]. Activated AKT phosphorylates its protein targets, inhibiting apoptosis and promoting cell survival. In model systems, targeted inhibition of AKT1 and AKT2 showed little effect on melanoma cells, but AKT3 inhibition resulted in increased apoptosis, reduced cell survival and decreased tumour development, indicating that this enzyme provides a potential new therapeutic target for patients with advanced stages of melanoma [15 17].
Recent transcriptome analysis from RAC1P29S melanocytes identified an enrichment of the SRF transcription factor targets and the epithelial to mesenchymal transition genes (EMT) [11]. The nuclear transcription factor serum response factor (SRF) and its myocardin-related transcription factor (MRTF) co-factor are regulated by actin polymerization. Upon GTPase activation of the WAVE regulatory complex (WRC), the ARP2/3 complex catalyzes the polymerization of G-actin into F-actin liberating the SRT/MRTF transcription factor which may now bind to DNA and induce the transcription of epithelial to mesenchymal transition (EMT) genes. SRF/MRTF inhibitors could therefore represent an attractive approach to tackling resistance in melanoma bearing the RAC1P29S mutation [11].
Vemurafenib and dabrafenib, a BRAF and a MEK inhibitor, respectively, inhibit growth and promote tumour regression in BRAFV600E mutant melanomas [19–21]. The effect of these drugs is limited due to an eventual reactivation of the MAPK pathway and the PI3K/AKT signalling pathway that leads to drug resistance [22–28]. BRAFi and AKTi combinatorial therapies in human melanoma cells display promising effects on abatement of tumour growth. Additionally, combination of MEK, BRAF and AKT inhibitors delayed signs of drug resistance [29]. In melanoma cells, a combination of PI3Kβ and PI3Kα inhibitors is necessary to inhibit the PI3K signalling in long-term treatments [30]. BKM120, a pan-PI3K inhibitor, prevented AKT activation, cell cycle arrest in the G2-M phase, and induced apoptosis in human melanoma cells that metastasize to the brain in in vitro and in vivo assays. The combination of BKM120 with the MEK inhibitor binimetinib, further inhibited melanoma cell line proliferation [31]. Finally, PAK and MEK inhibitors have also been shown to cause tumour regression and loss of ERK and AKT activity in a RAS-mediated skin cancer mouse model, suggesting that blockade of these pathways could provide therapeutic benefit [8].
Here, we compare the results of inhibiting three distinct classes of RAC1 effectors – PAKs, PI3Ks, and SRF/MRTF – on the growth and survival of RAC1-mutant melanoma cell lines and on RAC1-driven changes in zebrafish embryonic development.
Materials and methods
Reagents
PI3K inhibitors: TGX221, GSK2636771, AS252424, GSK2269557, BKM120 and BYL719; AKT 1/2/3 inhibitor: MK 2206; and MRTF/SRF inhibitor: CCG-203,971 were purchased from Selleckchem. PAK1 inhibitor Frax-1036 was generously provided by Genentech.
Cell culture
501mel, YUROB, YUFIC, YURIF and YUHEF melanoma cell lines were generously provided by Ruth Halaban (Yale University) and maintained in OptiMEM media (Invitrogen, Carlsbad, CA, USA) supplemented with 5% FBS and Penicillin-Streptomycin. 451Lu, WM1791 and WM1960 were generously provided by Meenhard Herlyn (Wistar Institute) and maintained in 80% MCDB153, 20% Leibovitz’s L-15, supplemented with 2% FBS, 5 μg/ml insulin, and 1.68 mM CaCl2. All cells were cultured in a humidified incubator with 5% CO2 at 37° C. Cells were tested for mycoplasma and authenticated by sequencing the BRAF, PREX2, and RAC1 genes.
Cell viability (mitochondrial activity)
Melanoma cell lines were plated in 96-well plates at 5000 cells/well in the corresponding medium. After 24 h, the cells were treated with increasing concentrations of PI3K inhibitors. Cell viability was evaluated after 72-h incubation with drugs. Culture media were replaced by 100 μL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to a final concentration of 0.33 mg/mL dissolved in culture media and incubated at 37°C with 5% CO2 for 1 h in the dark. Supernatant was removed and replaced with DMSO to dissolve formazan crystals. Absorbance value was measured at 570 nm. Experiments were done in triplicate and 0.1% DMSO was used as negative control. IC50 was calculated from three independent experiments.
Immunoblotting
Cells were grown in six-well plates until 80% confluence was reached. Inhibitors were added at a final concentration of 100 nM and cells were cultured in a humidified incubator with 5% CO2 at 37° C for 24 h. Cells were washed with Dulbecco’s PBS media and scrapped with 200 μl of TNS buffer. Protein concentration was determined by Bradford and the samples were normalized to protein content. Western blot assays were performed using standard techniques. Primary antibodies used in this study were: anti-phospho-PAK1 (pSer144) (#2606), -total PAK1 (#2602), -phospho-ERK1/2 (pThr202/pTyr204) (#9101), -total ERK1/2 (#9102), -phospho-AKT (Ser473) and –total AKT (#9272) from Cell Signalling Technology.
Proliferation assays
Proliferation was evaluated with the xCELLigence technology (Acea Bioscience, San Diego, CA, USA, distributed by Roche) in E-16-well plates. Melanoma cells were monitored during 72 h. The impedance value of each well was automatically monitored by microelectrodes placed on the bottom of plates. The impedance changes detected were proportional to the number of adhering cells and expressed as the cell index value. The experiments were conducted in triplicate. The results are shown as mean ± standard deviation (SD) of at least three independent experiments (* p < 0.05, ** p < 0.01, *** p < 0.001 versus control).
Wound healing assay
Melanoma cells were seeded on six-well plates and manually scraped with a 200 μL pipette tip. Cells were washed once with growth media, and then grown in fresh growth media with the IC50 of the inhibitors for 24 h. Images were acquired at 100x magnification using an EVOS fluorescence microscope and the number of cells that cross into the wound area from their reference point at time zero was analysed. To determine the migration rate of the cells, the wound areas were quantified using ImageJ software. The percentage of migration was quantified by measuring the size of the cell free-area. Data represent the mean (SD) of three independent experiments. (* p < 0.05, ** p < 0.01 versus Control).
Zebrafish microinjection experiments
Wild-type AB zebrafish embryos were collected and maintained in the Zebrafish Core Facility of Fox Chase Cancer Centre under standard conditions39. Capped mRNA was obtained as follows: RAC1 (P29S) cDNA was subcloned into the pSGH2 vector. Transcripts were made in vitro for antisense (HindIII and T3 RNA polymerase) or sense (SacII and SP6 RNA polymerase) full-length mRNA using the mMessage Machine kit (Ambion). Eight pairs of fish were bred and the resulting one-cell-stage embryos were injected with Phenol red or Phenol red + Rac1P29S mRNA directly in the cytoplasm using a nitrogen-powered Picospritzer III injector (Intracel) conjugated to a Nikon SMZ 1000 stereomicroscope at a final concentration of 35 ng/μl, as described previously40. Eggs were randomly separated in 50 egg-batches and kept in E3 medium at 28̊ C. 4 hpf mRNA injected embryos were incubated in E3 medium containing 1 μM of PI3K, PAK1, AKT or Rho/MRTF/SRF inhibitors for 1 h and then washed thoroughly to avoid unwanted abnormalities caused by the inhibitor. Control embryos were incubated in E3 with DMSO at the same final concentration as the small molecule inhibitors. To analyse zebrafish morphology, 24 h-dechorionated embryos were placed on a glass depression slide in 1% methylcellulose to stabilize the embryo. Morphology was assessed visually using a light transmission Nikon SMZ 1500, and representative images were recorded using a Nikon digital sight DS Fi1 camera. Embryonic phenotypes were scored as normal if they presented an elongated body axis, eye and heart development. These experiments were repeated three times using different breeders (* p < 0.05, ** p < 0.01, *** p < 0.001 vs. control).
All animal procedures were performed in accordance with IACUC guides and regulations.
Statistics
Statistical analyses were carried out using the paired Student’s t-test. All values reflect the mean (SD), with a significance cut-off of * p < 0.05, ** p < 0.01, *** p < 0.001. All statistical analyses were completed in GraphPad Prism 6.0 or 7.0 (La Jolla, CA, USA).
Results
Hotspot mutations of BRAF, NRAS and RAC1 genes have been identified as the most frequent drivers in melanoma. In this study, we analyse the effect of diverse inhibitors on melanoma cell lines that harbour different mutations. Table 1 shows the target of each small molecule inhibitor.
Table 1.
PI3K, AKT and SRF/MRTF selective inhibitors and biological activity
| Inhibitor | Target |
|---|---|
| BKM120 (Buparlisib) | PI3Kα/β/δ/γ |
| BYL719 (Alpelisib) | PI3Kα,minimal effect on PI3Kβ/γ/δ |
| TGC221 | PI3Kβ, minimal effect on PI3Kα |
| GSK2636771 | PI3Kβ |
| AS-252424 | PI3Kγ, minimal effect on PI3Kβ/δ |
| GSK2269557 (Nemiralisib) | PI3Kδ |
| MK-2206 2HCL | AKT1/2/3 |
| CCG-203971 | Rho/MRTF/SRF |
Viability, proliferation, migration and signalling assays were performed in eight different cell lines: YUROB (WT), 501mel (BRAFV600E), YURIF (BRAFV600K/RAC1P29S), 451Lu (BRAFV600E), YUFIC (NRASQ61R), YUHEF (RAC1P29S), WM1791 (K-RasG12D/RAC1P29S) and WM1960 (N-RASQ61K/RAC1P29S). Table 2 displays the genotype of each melanoma cell line used in this study.
Table 2.
Genetic profiles of melanoma cell lines
| Cell line | Mutation |
|---|---|
| YUROB | WT |
| 501mel | BRAFV600E |
| YURIF | BRAFV600E/Rac1P29S |
| 451Lu | BRAFV600E |
| YUFIC | NrasQ61K |
| YUHEF | Rac1P29S |
| WM1791C | KrasG12D/Rac1P29S |
| WM1960 | NrasQ61K/Rac1P29S |
Cytotoxic effects of PI3K inhibitors on melanoma cell lines
We evaluated the viability of melanoma cell lines when exposed to increasing concentrations of the isoform-selective and the pan-PI3K inhibitors using the reduction of MTT to formazan at 72 h as readout. Viability was significantly reduced in all the melanoma cell lines exposed to 0.1 μM of the pan-PI3K inhibitor BKM120, and when exposed to higher concentrations of this drug (0.5 μM), cell survival dropped below 50% (Figure 1a). When cell lines were exposed to 0.01 μM of the specific PI3Kα isoform inhibitor (BYL719), viability of cells that harbour mutations in BRAF and NRAS decreased to 60%. Cell viability was reduced as the concentration of BYL719 was increased. In contrast, PI3Kα inhibitors had no significant effect on cells that harbour RAC1 mutations (Figure 1b). When adding the isoform PI3Kβ selective inhibitors (TGX221, GSK2636771) cell lines containing RAC1 mutations were preferentially affected (Figure 1 c,d). When exposed to 0.05 μM of TGX221, RAC1 mutant cells survival was reduced to less than 50%. When increasing the concentration to 0.1 μM the survival rate decreased to 20%. The viability of cells that harbour mutations in BRAF and NRAS was reduced to 70% survival with the highest concentration tested (Figure 1c); following the same behaviour pattern, when cells were exposed to 0.1 μM of the GSK2636771 inhibitor, RAC1 mutant cell survival was reduced to less than 40%, but when cells that harbour mutations in BRAF and NRAS were treated with this drug, no significant viability effects were observed (Figure 1d). The PI3Kγ inhibitor AS252424 slightly reduced the survival of all cell lines to at least 80% at a concentration of 0.05 μM, control cell line viability at a concentration of 0.05 μM was around 40% (Figure 1e). Finally, treating the cells with the PI3Kδ inhibitor GSK2269557, had no effects in any of the melanoma cell lines (Figure 1f). The calculated IC50 values for each cell line treated with the tested drugs are shown in Supplementary Table 1.
Figure 1.
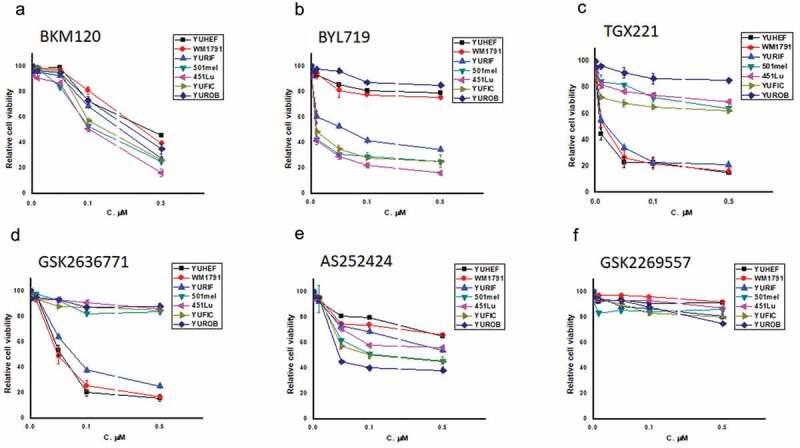
Effect of selective PI3K inhibitors on melanoma cell viability. Melanoma cells were treated for 72 h with concentrations ranging from 0.01 µM to 0.5 µM of (a) pan-PI3K (BKM120), (b) PI3Kα (BYL719), (c) PI3Kβ (TGX221), (d) PI3Kβ (GSK2636771), (e) PI3Kγ (AS252424), (f) PI3Kδ (GSK2269557). Cell viability was determined by an MTT assay. Data represent the mean (SD) of three independent experiments
Cell proliferation and migration in response to PI3K inhibitors
We next evaluated the consequences of PI3K inhibition on cell proliferation and migration. Proliferation was studied in three melanoma cell lines with different genotypes that have only one mutation: YUROB (WT), YUHEF (RAC1P29S), and 501mel (BRAFV600E). Consistent with our previous results, WT cell proliferation was not significantly affected by the exposure to any of the inhibitors used in this study (Figure 2a). Treatment of RAC1P29S mutant cell lines with the Group A PAK inhibitor Frax-1036, nearly abolished cell proliferation (Figure 2b) which is consistent with data previously reported by our group [9]. PI3Kβ inhibitors (TGX221, GSK2636771) also reduced RAC1P29S mutant cell proliferation, while all the other drugs tested caused a milder decrease in cell proliferation. 501mel cells bearing the BRAFV600E mutation displayed a significant reduced proliferation rate by the addition of the pan-PI3K and the PI3Kα inhibitor, and a milder reduction when exposed to PI3Kβ inhibitors (Figure 2c).
Figure 2.
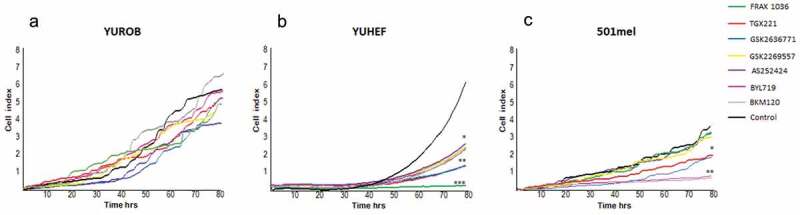
Proliferation of WT, BRAF and RAC1-mutant melanoma cell lines in presence of targeted inhibitors. (a) WT cells (YUROB), (b) RAC1 mutant (YUHEF) and (c) BRAF mutant (501mel) were treated with 100 nM of pan-PI3K (BKM120), PI3Kα (BYL719), PI3Kβ (TGX221, GSK2636771), PI3Kγ (AS252424), PI3Kδ (GSK2269557) and PAK1 (Frax-1036) small molecule inhibitors. Cell number was measured during 72 h using an XCELLigence device. Representative data of three independent experiments. The results are shown as mean (SD) of at least three independent experiments (* p < 0.05, ** p < 0.01, *** p < 0.001 versus control)
RAC1 is well known as a regulator of cell motility through its effects on the actin cytoskeleton. A wound-healing assay was used to test whether PI3K inhibitors would differentially affect the migration of melanoma cells depending on their driver mutation(s). None of the inhibitors impeded migration in WT cell lines (YUROB). The migration of RAC1P29S mutant cells (YUHEF) was severely diminished by the pan-PI3K and the PI3Kβ inhibitors, and partially reduced by the PI3Kα inhibitor. The BRAFV600E mutant cell (501mel) migration was reduced by exposure to the pan-PI3K and the PI3Kα inhibitor, and partially reduced by the PI3Kβ inhibitors. When cell lines had both RAC1 and BRAF mutations (YURIF), cell migration was strongly diminished by the pan-PI3K, the PI3Kα, and the PI3Kβ inhibitors. PI3Kγ and δ inhibitors had only a mild inhibitory effect in YURIF cell line (Figure 3 a,b).
Figure 3.
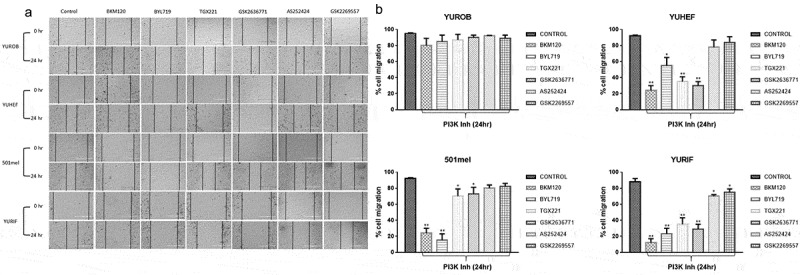
Consequences of PI3K inhibitors on cell migration. A cell culture wound-healing assay was performed in six-well confluent plates. After creating a gap by scratching a confluent plate of cells, WT (YUROB), RAC1 (YUHEF), BRAF (501 mel) and BRAF/RAC1 (YURIF) cell lines were treated with 100 nM pan-PI3K (BKM120), PI3Kα (BYL719), PI3Kβ (TGX221, GSK2636771), PI3Kγ (AS252424) and PI3Kδ (GSK2269557) inhibitors. (a) Images were taken in an inverted microscope at 0 and 24 h. Scale bar 400 μm for all images. (b) Migration was quantified by measuring the length of the cell-free area. Maximum (100%) migration was determined by the control. Percentage of migration of each treated cell line was measured against untreated cells (control). The results are shown as mean (SD) of at least three independent experiments (* p < 0.05, ** p < 0.01 versus control)
Rac1-mutant signalling response to PI3K inhibitors
To assess the functional role of PI3K inhibition, we examined RAC1 downstream effector proteins signalling in response to PI3K inhibitors in melanoma cell lines. Western blot analysis showed that treatment with specific PI3K drugs (α, β, γ, δ, and pan-PI3K inhibitor) did not affect signalling in the WT cell line, but modified signalling in the mutant cell lines (Figure 4). In non-treated cells, PAK and AKT activity was increased in RAC1 and NRAS mutant cells while ERK activity remained constant in all the cell lines. The phosphorylation of PAK and AKT, which indicates their activity, was significantly decreased in RAC1 mutants when exposed to the pan-PI3K inhibitor (BKM120) and to the PI3Kβ inhibitors (TGX221 and GSK2636771). The PI3Kα inhibitor (BYL719) significantly reduced the activation of AKT in all cell lines. No effects were observed when cells were treated with PI3Kγ or δ inhibitors. ERK activity was nearly abolished in all mutant cell lines by the pan-PI3K inhibitor, and was suppressed by the PI3Kα inhibitor (BYL719) in cell lines that have only a BRAF or an NRAS mutation. PI3Kβ-inhibitors (TGX221 and GSK2636771) reduced ERK phosphorylation in RAC1-mutant cell lines. These results suggest that different isoforms of PI3K regulate signalling of AKT, PAK and ERK (Figure 4).
Figure 4.
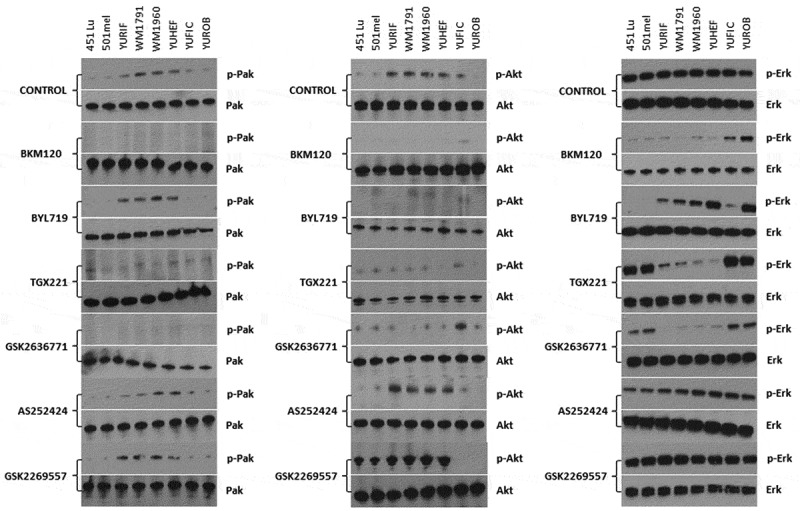
PI3K inhibitors cell signalling modification in melanoma cell lines bearing RAC1 and BRAF mutations. Melanoma cell lines with different genotypes were grown under standard conditions. Cells were treated with vehicle (control) or 100 nM of the indicated PI3K inhibitors for 24 h. Lysates were analysed by Western blot for PAK, AKT, and ERK phosphorylation. Representative data of 3 independent experiments are shown
Effect of RAC1 signalling pathway inhibitors on zebrafish embryonic development
To examine in vivo signalling roles for RAC1 effectors, we employed a zebrafish embryonic development assay. This system has been deployed by us and others in previous studies to elucidate signalling pathways from human oncogenes such as NRAS, BRAF, and RAC1. Overexpression of RAC1P29S hinders zebrafish embryonic development and activates ERK signalling [22]. We previously showed that these defects were blocked by inhibiting components of the RAS/MAPK pathway with small molecule PAK or MEK inhibitors [22]. Therefore, we asked whether these developmental defects could be suppressed by small molecule inhibitors of additional RAC1 effectors such as PI3K and SRF/MRTF. To determine if these inhibitors affect development, we introduced mRNA encoding RAC1P29S into one-cell stage zebrafish embryos. The l RAC1P29S phenotype is characterized by pericardial oedema, small/absent eyes, and reduced head size in ~97% of the embryos (Figure 5). Specific small molecule inhibitors of PI3K, SRF/MRTF, AKT and PAK were diluted in the embryonic water during an hour at 4 hpf, and development was followed for 24 h. We demonstrated that a Group 1 PAK inhibitor (Frax-1036: 40/38 normal, 40/2 abnormal), a PI3Kα inhibitor (BYL71: 40/35 normal, 40/5 abnormal), an MRTF/SRF inhibitor (CCG-203,971: 40/33 normal, 40/7 abnormal), a pan-PI3K inhibitor (BKM120: 40/33 normal, 40/7 abnormal), and a PI3Kβ inhibitor (TGX221: 40/31 normal, 40/9 abnormal) almost completely prevented the Rasophaty like phenotype induced by activated RAC1. In contrast, a second PI3Kβ inhibitor (GSK2636771:40/25 normal, 40/15 abnormal) and an AKT inhibitor (MK2206: 40/24 normal, 40/16 abnormal), did not successfully prevent the Rasopathy-like phenotype (Figure 5 a,b). These results suggest that, of the three main groups of RAC1 effectors, Group A PAKs and/or SRF/MRTF represent the most effective targets to antagonize the developmental defects induced by RAC1P29S.
Figure 5.
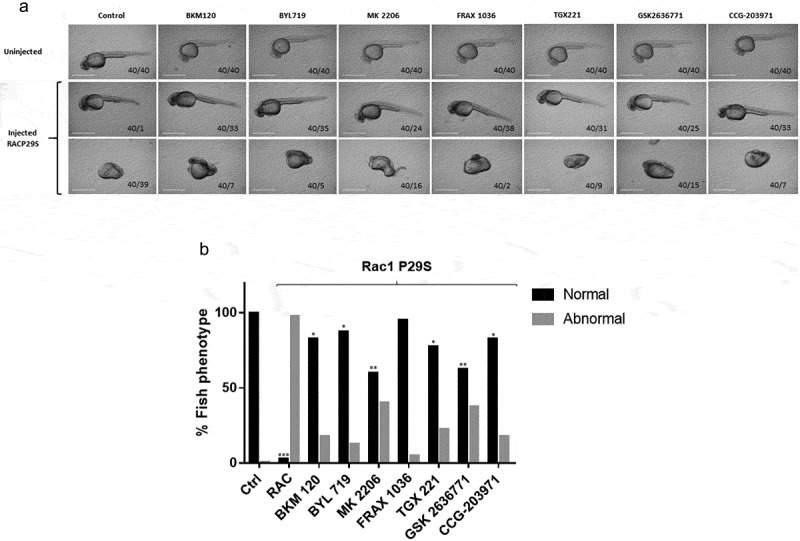
RAC1 overexpression in zebrafish embryonic development and its treatment with PI3K, AKT and Rho/MRTF/SRF inhibitors. Eight pairs of fish were bred and the resulting embryos were injected with Phenol red or Phenol red + RAC1P29S mRNA during the one-cell stage. 1 μM inhibitors were added at 4 hpf, then removed after 1 h and washed thoroughly. Embryonic morphology was scored at 24 hpf by a blinded observer. (a) Representative images of developmental abnormalities were performed with a Nikon digital sight DS Fi1 camera. Scale bar 100 μm for all images. (b) Embryo phenotypes were scored as normal if they presented an elongated body axis, eye and heart development. The results are shown as mean (SD) of at least three independent experiments (* p < 0.05, ** p < 0.01 versus control)
Discussion
Melanoma is the most aggressive form of skin cancer and it is strongly associated with poor prognosis, low overall survival and drug resistance. RAC1P29S is the third most common mutation found in sun-exposed melanoma. We and others have shown that the RAC1P29S protein activates downstream effector proteins such as PAK, PI3Kβ and SRF/MRTF, among others. Furthermore, PI3Ks activate AKT, which have a positive feedback effect on RAC1 that may in turn also stimulate certain carcinogenic properties in cells [14].
As RAC1 mutations confer poor prognoses, the inhibition of RAC1P29S and its downstream effectors might provide new therapeutic targets for appropriately-selected drug-resistant tumours and advanced staged melanoma. To date, few effective targets for RAC1-mutated melanoma other than PAK have been reported, prompting us to examine and compare the effect of PAK, PI3K, and SRF/MRTF inhibition in melanocyte signalling and zebrafish development.
Cell viability and migration assays revealed that, among all PI3K inhibitors tested, selective inhibitors for PI3Kβ were the most effective against RAC1 mutants, whereas selective PI3Kα inhibitors were most effective in the setting of BRAFV600K mutations. As expected, the PI3Kδ and γ inhibitors had fewer effects on melanocyte viability since these isoforms are mainly expressed in leukocytes [12]. The pan-PI3K inhibitor showed a non-selective decline in cell viability, growth and migration in all cell lines tested. These results are consistent with the idea that the main PI3K isoform engaged by RAC1 is PI3Kβ, and that targeting other PI3K isoforms does not add benefit in this setting. As a selective PI3K inhibitor would be expected to have less toxicity to cells, this finding could have therapeutic implications.
In the embryonic zebrafish development assays, the rasopathy-like phenotype induced by mutant RAC1 was most successfully reversed when using PAK or SRF/MRTF inhibitors, while PI3Kβ and AKT inhibitors were in general less effective, with some isoform specificity. As the zebrafish developmental assay is driven by high level, transient expression of transgenes, it may represent a more stringent assay for drug studies than in vitro studies using melanoma cell lines in culture, with the proviso that the zebrafish readout represents organismal development as opposed to cancer development. However, in most aspects both the developmental and in vitro data are in general agreement, arguing that both systems can be used to evaluate RAC1 signalling. Using these systems, our results, like those recently reported in a Rac1P29S mouse model, [11] suggest that targeting Group A PAKs and/or SRF/MRTF, and possibly also PI3K/AKT signalling, could become a promising approach to suppress RAC1 signalling in malignant melanoma.
Supplementary Material
Acknowledgments
We thank Drs. Ruth Halaban and Meenhard Herlyn for providing melanoma cell lines, Genentech for providing Frax-1036, and the Fox Chase Cancer Center Animal Facility for assistance with zebrafish experiments.
Funding Statement
This work was supported by grants from the NIH (JC), the Melanoma Research Foundation (JC), and Presupuesto Interno del Instituto de Química/UNAM (DAO) as well as Cátedras CONACYT and NCI Core Grant P30 CA06927 (to Fox Chase Cancer Center).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Didsbury J, Weber RF, Bokoch GM, et al. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. 1989;264:16378–16382. [PubMed] [Google Scholar]
- [4].Haataja L, Groffen J, Heisterkamp N.. Characterization of RAC3, a novel member of the Rho family. J Biol Chem. 1997;272:20384–20388. [DOI] [PubMed] [Google Scholar]
- [5].Marei H, Malliri A. GEFs: dual regulation of Rac1 signaling. Small GTPases. 2017;8:90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ye DZ, Field J. PAK signaling in cancer. Cell Logist. 2012;2:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Radu M, Semenova G, Kosoff R, et al. PAK signalling during the development and progression of cancer. Nat Rev Cancer. 2014;14:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chow HY, Jubb AM, Koch JN, et al. p21-activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012;72:5966–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Araiza-Olivera D, Feng Y, Semenova G, et al. Suppression of RAC1-driven malignant melanoma by group A PAK inhibitors. Oncogene. 2018;37:944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Watson IR, Li L, Cabeceiras PK, et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 2014;74:4845–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lionarons DA, Hancock DC, Rana S, et al. RAC1(P29S) induces a mesenchymal phenotypic switch via serum response factor to promote melanoma development and therapy resistance. Cancer Cell. 2019;36:68–83 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu S, Knapp S, Ahmed AA. The structural basis of PI3K cancer mutations: from mechanism to therapy. Cancer Res. 2014;74:641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Amzel LM, Huang CH, Mandelker D, et al. Structural comparisons of class I phosphoinositide 3-kinases. Nat Rev Cancer. 2008;8:665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu P, Cheng H, Roberts TM, et al. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stahl JM, Sharma A, Cheung M, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–7010. [DOI] [PubMed] [Google Scholar]
- [16].Cheung M, Sharma A, Madhunapantula SV, et al. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 2008;68:3429–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Davies MA, Stemke-Hale K, Tellez C, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer. 2008;99:1265–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010;70:6670–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sondergaard JN, Nazarian R, Wang Q, et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med. 2010;8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. [DOI] [PubMed] [Google Scholar]
- [22].Fedorenko IV, Paraiso KH, Smalley KS. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem Pharmacol. 2011;82:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–20416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Su F, Bradley WD, Wang Q, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969–978. [DOI] [PubMed] [Google Scholar]
- [29].Lassen A, Atefi M, Robert L, et al. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol Cancer. 2014;13:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Herkert B, Kauffmann A, Molle S, et al. Maximizing the efficacy of MAPK-targeted treatment in PTENLOF/BRAFMUT melanoma through PI3K and IGF1R inhibition. Cancer Res. 2016;76:390–402. [DOI] [PubMed] [Google Scholar]
- [31].Niessner H, Schmitz J, Tabatabai G, et al. PI3K pathway inhibition achieves potent antitumor activity in melanoma brain metastases in vitro and in vivo. Clin Cancer Res. 2016;22:5818–5828. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.