

Abstract
Proliferation and differentiation of preadipocytes, and other cell types, is accompanied by an increase in glucose uptake. Previous work showed that a pulse of high glucose was required during the first 3 days of differentiation in vitro, but was not required after that. The specific glucose metabolism pathways required for adipocyte differentiation are unknown. Herein, we used 3T3-L1 adipocytes as a model system to study glucose metabolism and expansion of the adipocyte metabolome during the first 3 days of differentiation. Our primary outcome measures were GLUT4 and adiponectin, key proteins associated with healthy adipocytes. Using complete media with 0 mM or 5 mM glucose, we distinguished between developmental features that were dependent on the differentiation cocktail of dexamethasone, insulin and isobutylmethylxanthine alone or the cocktail plus glucose. Cocktail alone was sufficient to activate the capacity for 2-deoxglucose uptake and glycolysis, but was unable to support the expression of GLUT4 and adiponectin in mature adipocytes. In contrast, 5 mM glucose in the media promoted a transient increase in glucose uptake and glycolysis as well as a significant expansion of the adipocyte metabolome and proteome. Using genetic and pharmacologic approaches, we found that the positive effects of 5 mM glucose on adipocyte differentiation were specifically due to increased expression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), a key regulator of glycolysis and the ancillary glucose metabolic pathways. Our data reveal a critical role for PFKFB3 activity in regulating the cellular metabolic remodeling required for adipocyte differentiation and maturation.
Keywords: Adipogenesis, glucose metabolism, glucose transport, glucose transporter 4 (GLUT4), adiponectin, Phosphofructokinase fructose bisphosphatase 3 (PFKFB3)
INTRODUCTION
Adipose tissue plays an important role in maintaining insulin sensitivity by providing both a metabolically safe place to store lipid and a source of insulin-sensitizing adipokines such as adiponectin (1, 2). The increased prevalence of obesity and obesity-related diseases has emphasized our need to understand the mechanisms that underlie adipose development both in the perinatal period and in adult obesity, with the goal of understanding the factors that promote the development of this tissue. Normal adipose tissue development occurs in the perinatal period at a time later than other organs that regulate whole body metabolism. Unlike other tissues, there is significant expansion of adipose depots in post-natal life and adult life as a result of changing energy balance (3). The development of adipose tissue begins with the recruitment and differentiation of adipocyte progenitors (APs) in response to positive energy balance. The specific signals that trigger this process in vivo are not completely known, but they are likely a combination of hormonal and nutrient signals. In vitro, transformed preadipocytes and primary APs can be differentiated to adipocyte-like cells over a period of days using a two to three-day pulse with minimal hormonal cocktail of dexamethasone, insulin, and isobutylmethylxanthine to start the process (4, 5). This supports the notion that hormone signaling is critical to adipose development, but this standard in vitro protocol does not necessarily explain the exact role that nutrients play in this process.
Our most detailed understanding of adipocyte differentiation is with the complex transcriptional cascade following hormone cocktail stimulation that leads to expression of PPAR-γ, the central regulator of adipogenesis (6–8). This transcriptional cascade is part of the intracellular response to extracellular signals transmitted to the adipocytes and APs to regulate development and function. However, extracellular signaling is not sufficient to support optimal development. Work from our lab and others have shown that the availability of glucose in combination with a hormone cocktail are needed for adipocyte development to proceed in vitro (4, 9). The specific requirement for glucose in adipocyte development is unclear because it precedes the established anabolic role of glucose to support the accumulation of lipid droplets and triglyceride storage (10).
Glucose is metabolized in preadipocytes by two main pathways, glycolysis and the pentose phosphate pathway (PPP) (11). Glycolysis generates ATP and pyruvate while the PPP generates NADPH and precursors for nucleotide synthesis. As such, both pathways play unique roles in cellular growth and development. In our previous work, we showed that a pulse of high glucose was required during the first three days of differentiation to proceed to a mature adipocyte (4). However, an unanswered question that remained from that study was to determine the mechanisms that regulated glucose metabolism over the three-day differentiation period. We hypothesize that glucose uptake and glucose metabolism would be increased during differentiation. To test this hypothesis, we have studied glucose uptake and metabolism during the early stages of differentiation. Herein, we demonstrate glucose is required for expansion of the metabolome. Using pharmacologic and genetic inhibitors of the PPP and glycolysis, our data demonstrate that the initial increase in glucose uptake supports an increase in the PPP, with upregulation of glycolysis following 24–48h later. A glucose-dependent spike in glucose uptake at day 3 of differentiation was accompanied by an increase in the expression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), a phosphofructokinase 2 (PFK2) isoform that regulates the production of fructose-2,6-bisphosphate to allosterically activate phosphofructokinase 1 (PFK1), the rate limiting step for glycolysis. PFKFB3 is one of four PFK2 isoforms known to regulate glycolysis. It is frequently overexpressed in cancer cells and has shown to be essential for growth and oncogenesis, likely due to its role for accelerating glycolysis and promoting anabolic pathways (12). This is the first study to show that PFKFB3 upregulation is an essential component of AP differentiation and metabolome expansion.
MATERIALS AND METHODS
Cell culture and animals
3T3-L1 cells (American Tissue Culture Collection, Manassas, VA; RRID:CVCL_0123) were cultured and differentiated in Minimum Essential media-alpha formulation (MEM-alpha) without ribonucleosides (12000-063,GIBCO). For experiments that varied glucose and glutamine, we used a custom MEM-alpha that did not contain either nutrient (Boca Scientific, CUSTMEDIAKIT). When using the custom media, glucose and glutamine were added as indicated to match the standard MEM-alpha formulation. Preadipocytes were grown to confluence in standard medium containing 5 mM glucose, 2 mM glutamine and supplemented with 10% calf serum. Differentiation was stimulated by changing to media containing 10% fetal bovine serum and a cocktail of 175nM insulin, 1μM dexamethasone, 0.5mM isobutyl-1-methylxanthine. After three days in this differentiation media, cells were then changed to media containing 10% fetal bovine serum without the cocktail, and harvested three days later (day 6 post-differentiation). Cells were treated at indicated times and concentrations with AZ67 (5742, Tocris), 6-aminonicotinamide (A68203, Millipore Sigma), or TEPP46 (505487, Millipore Sigma).
Primary mouse preadipocytes were cultured from the stromal vascular fraction of inguinal fat pads from male C57BL6 mice. The stromal vascular fraction was isolated by differential centrifugation following collagenase digestion of adipose pads as previously described (13). Stromal vascular cells were counted, and 150,000 cells were plated on 35 mm Pur Col™ collagen-coated dishes (#5073, Advanced BioMatrix) and grown to confluence in alpha-MEM media supplemented with 10% FBS. Two days post-confluence, cells were switched to differentiation media as described for 3T3-L1 preadipocytes. Three biological replicates were studied by isolating SVF from 3 mice/ biological replicate for a total of 9 mice. For each biological replicate, 5 35-mm dishes were plated to obtain samples for day 0, 3 and 6 post-differentiation without and with AZ67 treatment as indicated.
Five-month-old male C57BL6 mice were used for fasting and refeeding studies. Mice group housed with 2–4 mice per cage and maintained on standard chow diet with a 12-hour dark cycle from 6 pm to 6 am each day. Food was removed at 5 pm until 9 am the next morning. At that time, half of the mice were killed by CO2 asphyxiation to harvest tissues, and half were fed standard chow diet for the next 4 hours, after which they were killed. Tissues were weighed and snap frozen in liquid nitrogen and stored at −80C until further analysis. All procedures using animals were approved by the Institutional Animal Care and Use Committee at the University of Oklahoma Health Sciences Center.
Whole cell lysates and immunoblots
Whole cell extracts were prepared in lysis buffer containing 20mM HEPES, 1% NP40, 2mM EDTA, 10mM sodium fluoride, 10mM sodium pyrophosphate, 1mM sodium orthovanadate, 1mM molybdate, protease inhibitor cocktail (complete Mini EDTA-free Protease Inhibitor Cocktail, Roche), 1 mM PMSF and 50 mM DTT. Protein concentrations were determined by using a Coomassie Plus (Bradford) Assay Kit (Pierce). Lysates were fractionated using SDS-PAGE, and proteins transferred to Immobilon-FL polyvinylidene fluoride membrane (EMD Millipore Corporation, Billerica, MA, US.) Membranes were stained with anti-GLUT4 antibody (ab33780, ABCAM; RRID:AB_2191441), anti-adiponectin antibody (a gift from Philipp Scherer, UT Southwestern Medical Center, Dallas TX), anti-GLUT1 antibody (ab196357, Abcam; RRID:AB_2832207), , anti-MCT4 antibody (ab180699, Abcam), anti-HKII antibody (ab104836, Abcam), anti-PFKFB3 (ab181861, Abcam), anti-PFKFB4 (ab71622, Abcam; RRID:AB_1269727) or anti-α/β-tubulin antibody (2148, Cell Signaling) and visualized with appropriate secondary antibodies conjugated with AlexaFluor 680 (Invitrogen). Fluorescence was quantified with a Li-Cor Odyssey imager (Li-Cor Biosciences, USA). Protein expression data were compared to tubulin as a loading control.
RNA extraction and real time quantitative PCR
Total RNA was isolated from snap frozen fat pads using guanidinium isothiocyanate extraction followed by CsCl purification and samples were stored at −20°C as ethanol precipitates as described previously (14). Real-time reverse transcription PCR (qRT-PCR) analysis was performed using the following primers: mouse pfkfb3 5’ AGAACTTCCACTCTCCCACCCAAA-3’ (forward), 5’-AGGGTAGTGCCCATTGTTGAAGGA-3’ (reverse); mouse 36B4, 5’-CTGAGTGATGTGCAGCTGAT-3’ (forward), 5’-AGAAGGGGGAGATGTTCAG-3’ (reverse) and mouse actin, 5’-CCTCACTGACTACCTGATGA-3’ (forward), 5’-AGCTCATAGCTCTTCTCCAG- 3’ (reverse). All qRT-PCR were run using a CFX96 real-time PCR detection system thermal cycler (Bio-Rad). 36B4 and actin were used as housekeeping genes for ddCt calculations.
Glucose uptake assay, ATP levels and total protein
Basal glucose uptake, ATP concentration and total protein were measured in the same cell cultures propagated in 12-well culture dishes under conditions described. When indicated AZ67 was added either 39 hours (chronic) prior to the assay or 2 hours (acute) prior to the assay Basal glucose uptake assay was performed in day 3 or day 6 adipocytes using the Glucose Uptake-Glo bioluminescent assay kit (Promega Corporation, Madison WI). For basal and insulin-dependent glucose uptake, cells were serum starved in 0 mM glucose media containing 0.2% BSA for 2 hours at 37°C. Insulin (100 nM) was added during the final 30 minutes of starvation prior to beginning the glucose uptake assay. Cell number was determined by counting trypan blue excluded cells in a parallel. ATP content was determined using the CellTiter Glo Cell viability assay (Promega Corporation). Total protein was determined using the Pierce Ionic Detergent Compatibility Reagent added to the Pierce 66 nm Protein Assay Reagent (Thermoscientific Co.)
Analysis of Extracellular acidification rate (ECAR) for glycolytic capacity
ECAR measurements were performed using a Seahorse XFe24 Analyzer running Wave 2.5.0.6 (Agilent) as previously described with minor modifications (15, 16). Day 3 or day 6 cells were differentiated as indicated. When indicated, AZ67 was added either 39 hours (chronic) prior to the assay or 2 hours (acute) prior to the assay. The basal glycolysis rate was defined as the difference between ECAR following the glucose (10mM) injection and basal (glucose independent) ECAR. Glycolytic capacity was defined as the difference between ECAR following the injection of 1μM oligomycin (Sigma, 75351) and the basal ECAR reading. All experiments were performed at 37°C. Each measurement cycle consisted of a mixing time of 2 minutes, an equilibrating phase of 2 minutes, and a data acquisition period of 2 minutes (12 data points). After each XFe24 Analyzer experiment, the assay media was removed and cells were digested in 50μL of RIPA buffer (Sigma, R0278) at 4°C for 10min. BCA protein determination of each well was performed for normalization. ECAR data points refer to the average rates during the measurement cycles and each treatment group consisted of 5 wells. ECAR are reported as absolute rates (mpH/min for ECAR) after protein normalization, or expressed as a percentage of the control ECAR values.
Metabolic profiling
Metabolic profiling was performed on 3T3-L1 cells 3 days post-differentiation. Cells were grown on 60mm plates and differentiated as indicated. When used, AZ treatment was carried out for the final 39 hours of differentiation. Media was aspirated, the cells were quickly washed twice with pre-warmed (37°C) ultra-pure water, snap-frozen with liquid nitrogen and stored at −80°C until further extraction. Frozen cells were scraped, and metabolites were extracted using methanol:chloroform:water (2:1:1) solution as previously described (17). Prior to the extraction, 20mg/ml of ribitol was added to each sample as an internal standard. A 400μL aliquant of each extract were completely dried by SpeedVac and subjected to two-step derivatization procedure as previously described (17). Three quality controls (QCs) (prepared by pooling an equal volumes of each sample), analytical standards of malonate, lactate, glucose and leucine, and two blank samples were included to the analysis on Agilent Gas Chromatography – Mass Spectrometry instrument.
Detected peaks were annotated in Agilent MassHunter software (vsB.07.01/Build7.1.524.0) according to the integrated NIST library and external analytical standards. The relative abundance of metabolites was calculated by peak areas and normalized by the internal standard. Statistical analysis was performed using multiple Student’s t-tests using a False Discovery Rate correction set at 1%.
Oil red O staining
Oil red O staining was performed on cells that had been washed twice in PBS prior to fixation by 10% formalin for 30 minutes at room temperature. After fixation, the cells were washed twice with PBS. The cells were then stained with a filtered 0.3% solution of Oil Red O in 60% isopropanol for 15 minutes at room temperature. The Oil Red O solution was removed and cells were washed 5 times with water or until the rinse water ran clear. The Oil Red O stain was eluted with 99% isopropanol and absorbance at 500nM was recorded. The cells were washed twice with water, and counterstained with 5 mg/mL solution of crystal violet in water for 30 minutes at room temperature. Crystal violet stain was removed and cells were washed 5 times with water or until rinse water ran clear. The stain was eluted in methanol. The absorbance at 540nM was recorded. The data were presented as a ratio of oil red O absorbance/crystal violet absorbance
Statistical analysis
Data are expressed as the mean and standard deviation. Comparisons among groups were performed with GraphPad Prism software using one-way or two-way ANOVA with Dunnett’s multiple comparison tests when comparing to a single control or a Tukey’s test when making multiple comparisons within a single experiment. Two sample experiments were analyzed using Student’s t-tests.
RESULTS
Glucose is required for optimal adipocyte maturation
We have previously shown that high glucose was required for optimal maturation of 3T3-L1 adipocytes and mouse stromal vascular cells differentiated in vitro (4). In that study, standard culture conditions using Dulbecco’s modified Eagle medium (DMEM) supplemented with 25 mM glucose were compared to 5 mM glucose. In the current study, cells were propagated and differentiated using alpha-minimum essential medium (MEM), which supported adipocyte differentiation at a more physiologic glucose concentration of 5 mM glucose. To determine if glucose and glutamine, two of the most abundant soluble macronutrients in the circulation are interchangeable for adipocyte development, we differentiated preadipocytes with 0 or 2 mM glutamine and 0 or 5 mM glucose added to the base media (Fig. 1A). To confirm that serum supplementation did not contribute significant glucose or glutamine to complete media, we performed GC-MS analysis of the media with and without glucose, and glucose and glutamine standards. The results were in line with our assumption, showing no glucose in low glucose medium, and no glutamine in either media (Suppl. Fig. 1). Cells were harvested 6 days post-differentiation, and maturation was assessed by measuring expression of cell-associated GLUT4 and adiponectin as well as the ability of adiponectin to be secreted into the media. Expression of GLUT4 and adiponectin were increased only when glucose was present in the media, regardless of glutamine (Fig. 1A and B). GLUT1 expression was decreased in the presence of 5 mM glucose, but not impacted by glutamine. Taken together, we concluded that 5 mM glucose, but not 2 mM glutamine, was required for adipocyte maturation.
Figure 1.
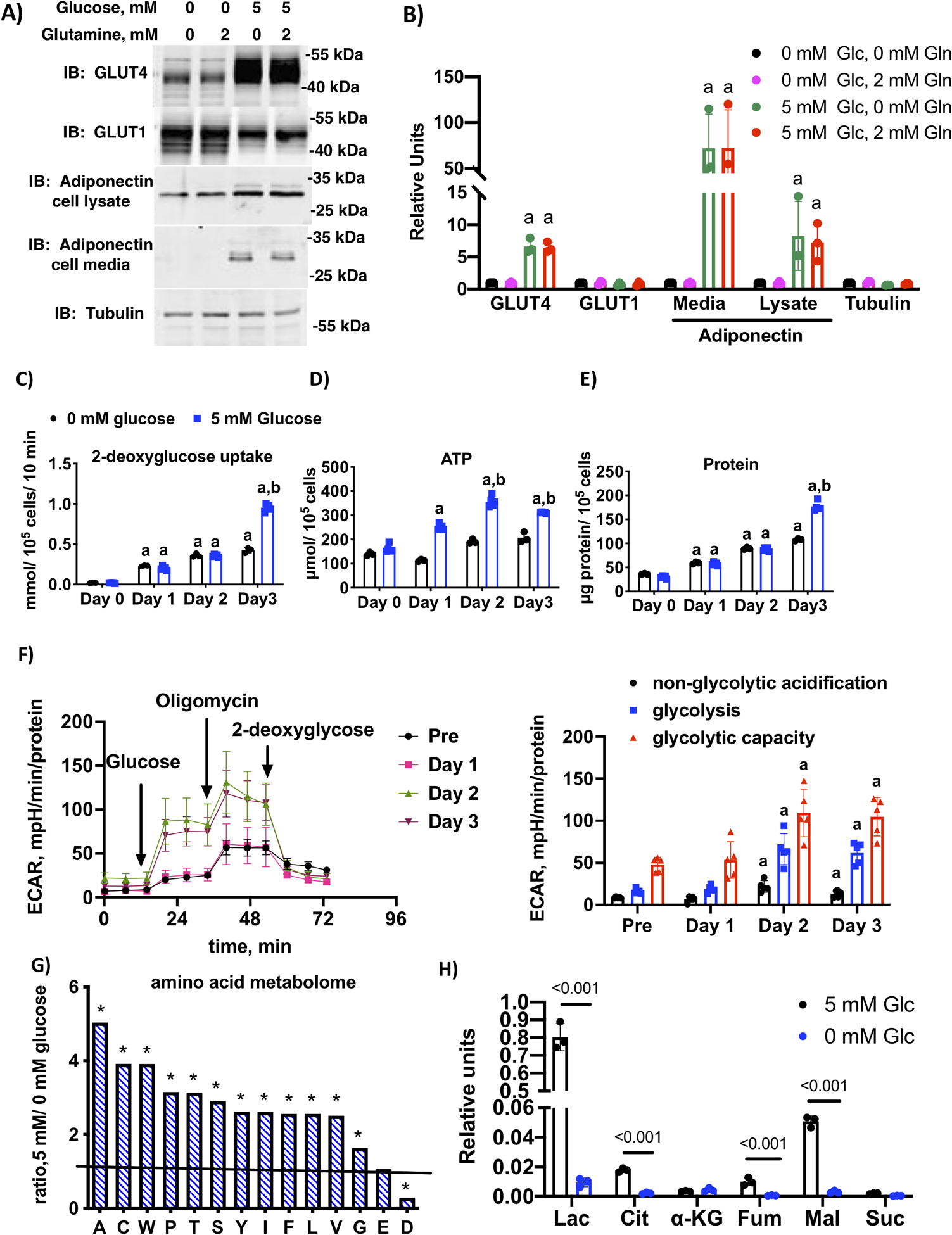
Glucose and glucose metabolism are required for 3T3-L1 adipocyte maturation. A) Immunoblotting of cell lysates differentiated in media containing either 0 or 5 mM glucose and 0 or 2 mM glutamine for the first 3 days of differentiation. All cells were switched to media plus 5 mM glucose and 2 mM glucose days 4 through 6 and harvested on day 6. Media and cell lysates were immunolabeled with indicated antibodies and a representative blot of n=3 independent trials is shown. B) Quantification of n=3 independent experiments. Letter “a” indicates significant difference compared to 0 mM glucose and 0 mM glutamine by one-way ANOVA and Dunnett’s multiple comparison test (p < 0.001). C) 2-deoxyglucose uptake, D) ATP concentration and E) total protein in day 0 (preadipocytes), day 1, day 2 and day 3 3T3-L1 adipocytes differentiated in media with 0 mM or 5 mM glucose and 2 mM glutamine. Histograms marked by different letters indicate statistical differences from all other conditions (n=5 replicates) (p < 0.01). Data were analyzed using two-way ANOVA and Tukey’s post-hoc test. F) Glycolysis stress test measuring extracellular acidification rate (ECAR) at day 0 (preadipocytes), day 1, day 2 and day 3 post-differentiation in media supplemented with 5 mM glucose and 2 mM glutamine. The first graph shows the trace for the acidification experiment with additions labeled and the second graph shows the quantification of the area under the curve for non-glycolytic acidification, glycolysis and glycolytic capacity, respectively. Letter “a” indicates statistical difference compared to the corresponding component of the stress test in preadipocytes. Data analyzed by one-way ANOVA and Dunnett’s multiple comparison test (p < 0.01). G) Ratio of relative concentration of amino acids (A alanine, C cysteine, W tryptophan, P proline, T threonine, S serine, Y tyrosine, I isoleucine, F phenylalanine, L leucine, V valine, G glycine, E glutamate, D aspartate) in day 3 adipocytes differentiated in media supplemented with 0 mM or 5 mM glucose and 2 mM glutamine. The ratio was derived from the average of n=4 replicates per condition. * indicates when the ratio was significantly different from unity using multiple Student’s t-tests as described in methods (p < 0.001). H) Key organic acids, lactate (Lac), citrate (Cit), alpha-ketoglutarate (α-KG), fumarate (Fum) Malate (mal), and succinate (Suc) from day 3 adipocytes differentiated with 0 mM or 5 mM glucose and 2 mM glutamine. Relative differences between conditions (n=3) were analyzed by Student’s t-test using a False Discovery Rate correction as described in methods and q value is indicated.
Our previous work showed that a pulse of high glucose was required for adipocyte maturation only during the first 3 days of adipocyte differentiation (D-Phase) when DII cocktail was present, and this condition also required activity of the pentose phosphate pathway (4). To clarify the role played by glucose, we carried out a time course over the first 3 days of differentiation in cells treated with 0 mM or 5 mM glucose. 2-Deoxyglucose uptake was measured on day 0, day 1, day 2 and day 3 post-differentiation. Glucose uptake was increased after day 1 of differentiation under both 0 mM and 5 mM glucose (Fig. 1C). Glucose treatment increased ATP levels beginning on day 1(Fig. 1D). On day 3, there was an additional glucose-dependent increase in glucose uptake that was mirrored by an increase in total cellular protein, a measure of increased anabolism (Fig. 1E). To determine glycolytic rates, we measured the extracellular acidification rate (ECAR) by Seahorse XF analysis on preadipocytes and differentiating adipocytes at 1, 2, and 3 days post-differentiation in 5 mM glucose containing media (Fig. 1F). This assay measures ECAR basally (non-glycolytic acidification), upon the addition of glucose (glycolysis), and maximally upon the addition of oligomycin (glycolytic capacity). ECAR was significantly elevated by days 2 and 3 post-differentiation, indicating that cells become glycolytic two days after treatment with the hormone cocktail.
The increase in ATP and total protein prompted an investigation of the role of glucose in development of the adipocyte metabolome, in particular the glucose dependent changes in amino acids and glucose metabolic intermediates. Soluble extracts from three biological replicates each of day 3 adipocytes differentiated with 0 or 5 mM glucose were analyzed by GC-mass spectrometry to determine relative concentrations of metabolites. Thirty-seven peaks were identified as unique metabolites with the high probability using NIST library. Lactate, glucose, and leucine were confirmed by external standards. Two out of 37 metabolites were detected only in 1 group: lysine in 0 mM glucose, and fructose-6-phosphate in 5 mM glucose (Suppl Tables 1 and 2). The global scale analysis of the metabolic profiling data was performed by principal component analysis (Suppl. Figure 2). Consistent with an increase in total protein, a majority of the amino acid metabolome, both essential and non-essential amino acids, was significantly increased by 5 mM glucose in the media (Fig. 1G). The exceptions were glutamine, which was not different between media, and aspartate, which was significantly reduced in 5 mM glucose media. As expected, 5 mM glucose increased lactate, alanine, citrate, fumarate and malate (Fig. 1H).
To begin to understand the molecular underpinnings of the glucose-dependent increase in glucose uptake on day 3 of differentiation, we carried out a time-course from day 0 through day 3 of cells differentiated in 0 or 5 mM glucose. The lysates were probed for some key regulators of glucose uptake and glycolysis. We observed that PFKFB3 and GLUT1 were the only proteins that showed a time-dependent pattern of expression that was also dependent on the carbohydrate content of the media (Fig. 2A and B). Interestingly, GLUT1 was increased by 0 mM glucose media while PFKFB3 was increased in 5 mM glucose media. The mono-carboxylate transporter 4 (MCT4), which serves as a lactate transporter, and pyruvate carboxylase (PC), an anaplerotic enzyme, also increased in a time-dependent fashion, independent of media glucose concentration (Figures 2A and B). Hexokinase II (HKII) and pyruvate dehydrogenase (PDH) trended up as a function of time, but they did not reach statistical significance under these conditions. To determine if 0 mM glucose was inhibiting initiation of differentiation, we immunoblotted lysates for Pref-1, a transmembrane protein expressed in preadipocytes, which is downregulated as cells progress through differentiation (18). Down-regulation of Pref-1 was dependent on DII-cocktail, but not dependent on the media glucose concentration (Figure 2A and B).
Figure 2.
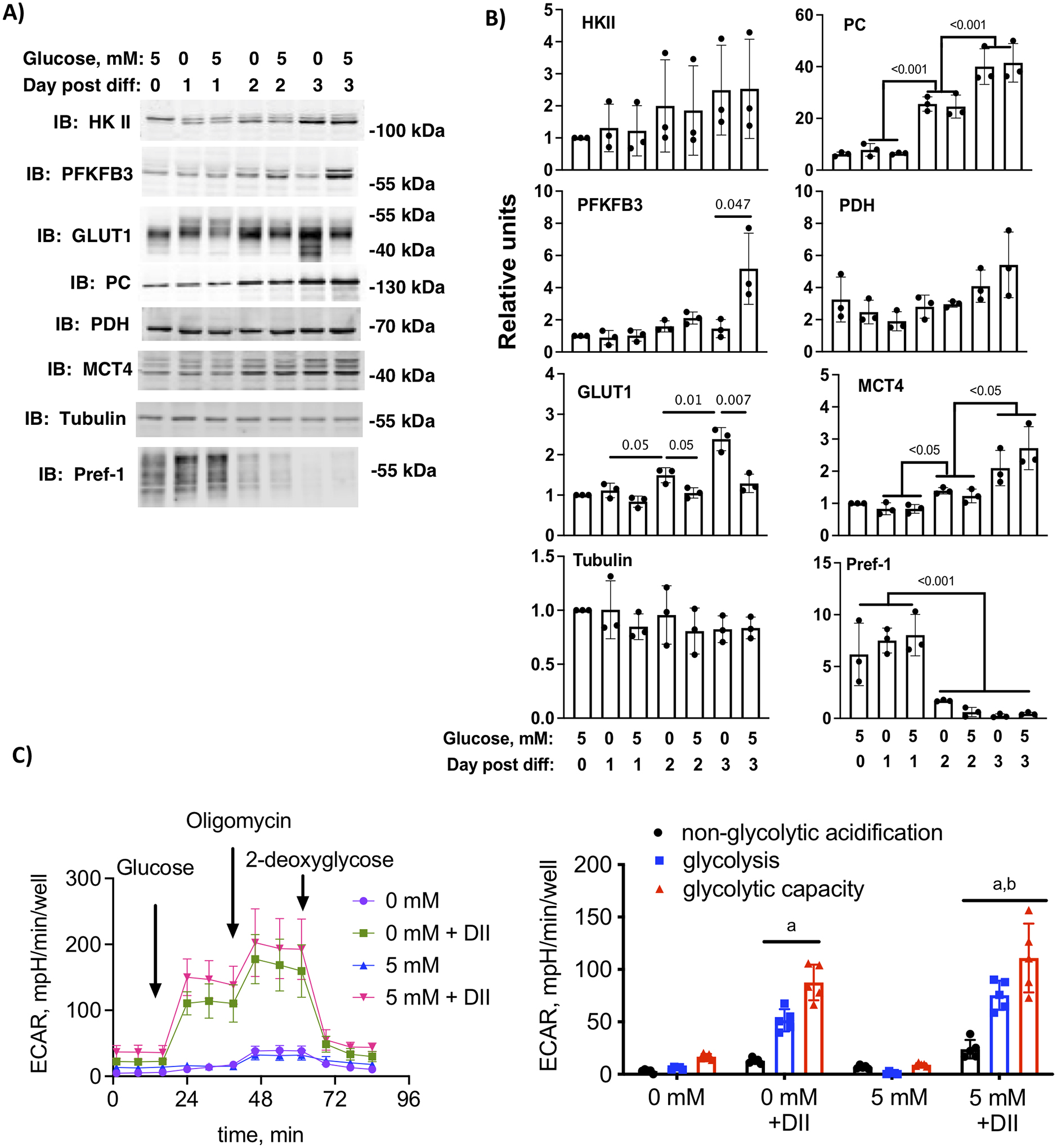
Glucose and hormone dependent expression of key glycolytic metabolites. A) 3T3-L1 cells differentiated in 0 mM or 5 mM glucose and 2 mM glutamine were harvested on day 0 (preadipocytes), day 1, day 2 or day 3 post-differentiation. Cell lysates were immunolabeled as indicated. B) Quantification of n=3 independent experiments. Statistical differences between days and conditions were determined by two-way ANOVA and Tukey’s post-hoc test. The p-values are shown. C) Glycolysis stress test measuring extracellular acidification rate (ECAR) at day 3 post-differentiation in media supplemented with 0 mM 5 mM glucose and 2 mM glutamine with or without added differentiation hormone cocktail (DII). The first graph shows the trace for the acidification experiment with additions labeled and the second graph shows the quantification of the area under the curve for non-glycolytic acidification, glycolysis and glycolytic capacity, respectively. Letters indicate statistical differences compared to the corresponding component of the stress test in 0 mM glucose cells without DII. Data analyzed by one-way ANOVA and a Tukey’s post hoc test (p < 0.01).
The differential response of key glycolytic enzymes prompted us to ask if glucose exposure alone impacted the development of glycolytic capacity in differentiating adipocytes. We therefore measured ECAR in cells differentiated with 0 or 5 mM glucose. A parallel set of cells were cultured with 0 or 5 mM glucose without the DII cocktail. DII cocktail alone upregulated the capacity for cells to carry out glycolysis even in cells differentiated in 0 mM glucose, and 5 mM glucose in the media further increased the capacity of the cells for glycolysis (Fig. 2C). The upregulation in glycolysis is consistent with the differentiation-dependent changes of key regulators of glycolysis such as MCT4, which was upregulated independently of glucose. Importantly, differentiation in 5 mM glucose further increased the glycolytic capacity and is consistent with the increase in total (phosphorylated and non-phosphorylated) PFKFB3 protein expression in the 5 mM glucose cells. The PFKFB3 antibody is directed toward the C-terminal end of the protein, allowing identification of all known PFKFB3 splice variants.
Glucose uptake and glycolysis is transiently up-regulated on day 3 differentiation.
A requirement for glucose during early differentiation implies that glucose uptake and/or glycolysis is increased specifically during D-phase. To test this hypothesis, we measured basal 2-deoxyglucose uptake in cells on day 3 and day 6 post differentiation and found that basal glucose uptake was highest on day 3 (Fig. 3A). Increased glucose uptake was accompanied by an increase in ATP and total protein (Fig. 3B and C). Glycolysis, as measured by ECAR, was significantly upregulated in day 3 cells, and reduced in day 6 cells (Fig. 3D). To determine enzymes or transporters that might be responsible for transient increase in glucose uptake and glycolysis, we probed protein extracts from 3T3-L1 cells at days 3 and 6 post-differentiation for key regulators of these processes (19). The only molecule that was specifically upregulated in day 3 cells compared to day 6 was PFKFB3 (Fig. 3E), potentially accounting for the transient increase in glucose uptake and glycolysis. To quantify the changes in expression between day 3 and day 6, we determined the ratio of proteins for each independent experiment. Only PFKFB3 was significantly lower on day 6 compared to day 3 (Fig. 3F). In contrast, GLUT4 and MCT4 were significantly increased in day 6 compared to day 3 cells, while GLUT1 and HK II were not significantly changed.
Figure 3.
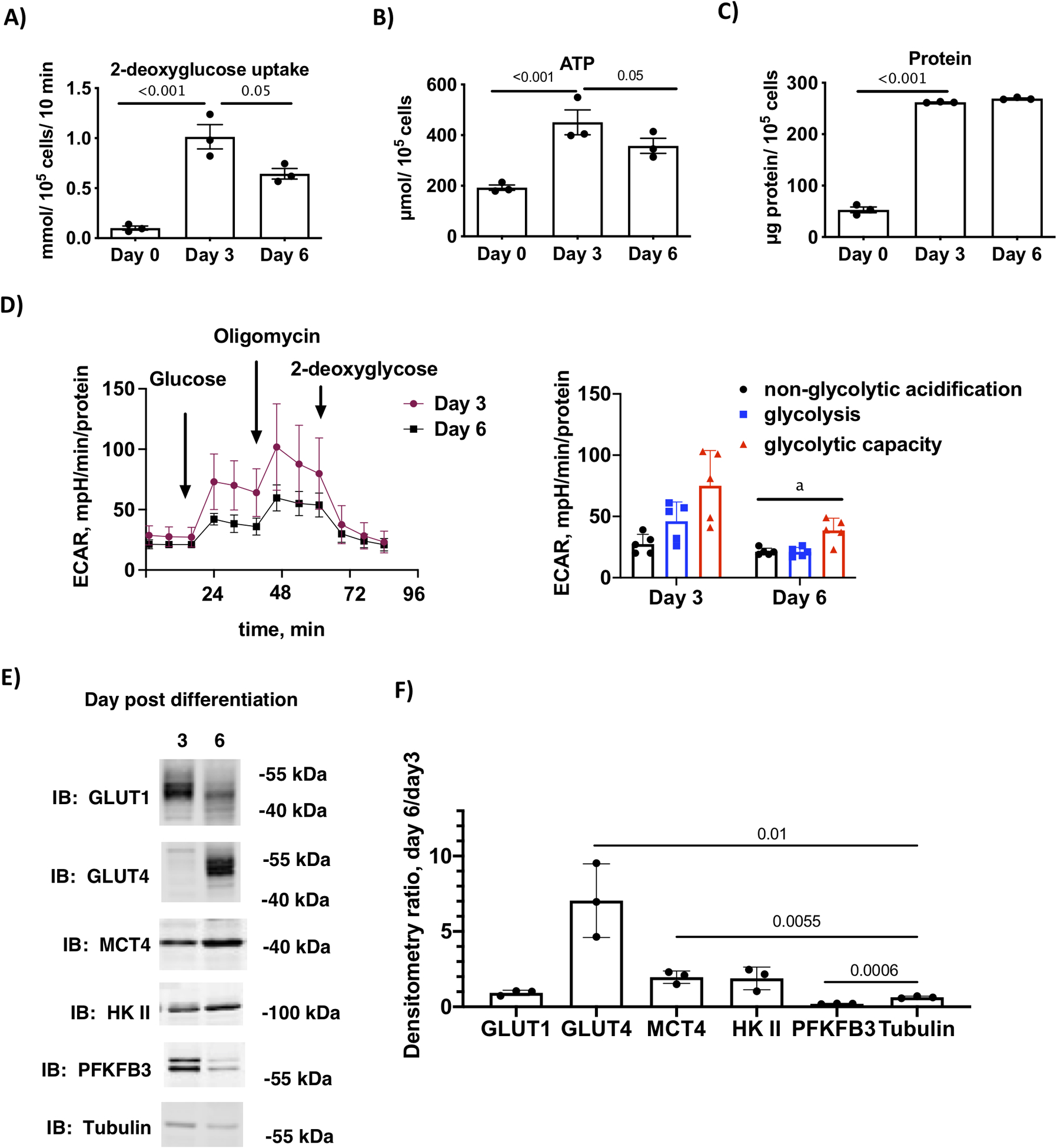
Glucose uptake and glycolysis are transiently upregulated on day 3 of differentiation. A) 2-deoxyglucose uptake, B) ATP concentration and C) total protein in day 0 (preadipocytes) and day 3 and day 6 3T3-L1 adipocytes differentiated in media with 5 mM glucose and 2 mM glutamine. Statistical differences (n=3 replicates) are one-way ANOVA and Tukey’s post-hos test. D) Glycolysis stress test measuring extracellular acidification rate (ECAR) at day 3 and day 6 post-differentiation as described above. The first graph shows the trace for the acidification experiment with additions labeled and the second graph shows the quantification of the area under the curve for non-glycolytic acidification, glycolysis and glycolytic capacity, respectively. Differences between day 3 and day 6 (n=5 replicates) were compared for each component of the stress test using Student’s t-test and indicated by the letter “a” (p < 0.01). E) Immunoblot analysis of key proteins that regulated glycolysis in cells at day 3 and day 6 post-differentiation. F) Quantification the ratio of expression of key proteins on day6/day 3 of n=3 independent experiments. The ratios for each protein were compared to the ratio for our loading control, tubulin. The p values are given for ratios that differed from tubulin.
Inhibition of PFKFB3 inhibits adipocyte maturation
PFKFB3 was the only key regulator of glycolysis that was upregulated (data shown in Fig. 2A and B and summarized in Figure 4A) in a glucose-dependent manner causing us to hypothesize that the increase in glucose-uptake and glycolysis were dependent on this enzyme. PFK-1 protein, the target of PFKFB3 activity, was not measured. To test the hypothesis, we used both genetic and pharmacological approaches to determine if PFKFB3 activity was required for glucose uptake and adipocyte maturation. Genetic knockdown of PFKFB3 was accomplished using pooled PFKFB3 siRNAs transfected into preadipocytes followed by treatment with DII in 5 mM glucose media adipocytes (Figure 4B). At day 3 post-differentiation, the gene knockdown resulted in an 80% decrease in immunostaining of PFKFB3 (Fig. 4C and B). On day 3, the PFKFB3 knockdown resulted in a 23% decrease in glucose uptake, presumably due to the decrease in glycolysis (Fig. 4D). At day 6 post-differentiation, the gene knockdown resulted in reduced expression of GLUT4, cell-associated adiponectin and secreted adiponectin (Fig. 4E and F). These data support the hypothesis that PFKFB3 protein is playing a role in adipocyte maturation.
Figure 4.
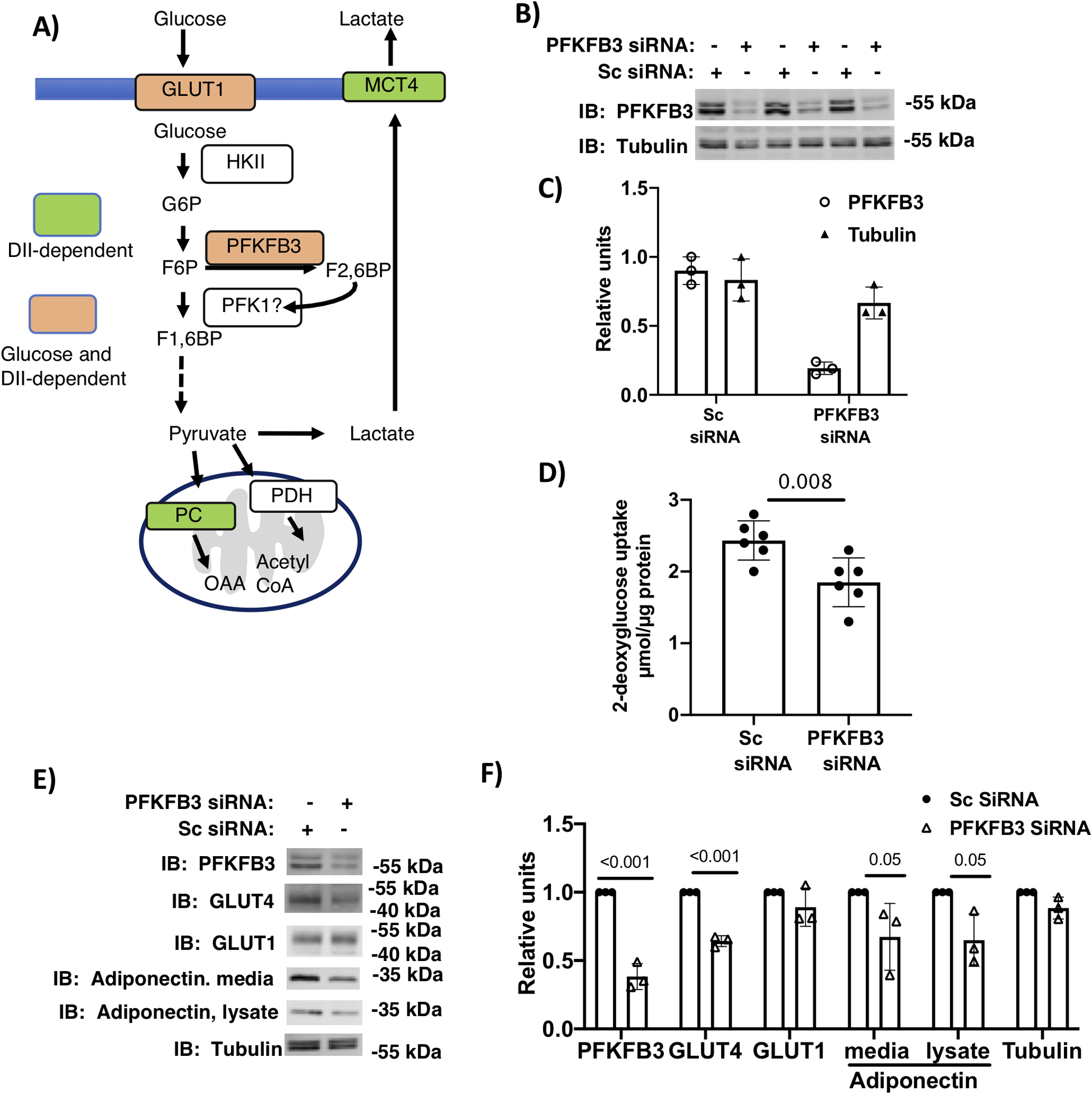
Genetic knockdown of PFKFB3 inhibited glucose uptake and adipocyte maturation. A) schematic summary of key regulators of glycolysis during the first 3-days of differentiation 3T3-L1 cells. B) Transfection of adipocytes on day 1 of differentiation (in 5 mM glucose and 2 mM glutamine) with PFKFB3 siRNA or scrambled (Sc) siRNA were harvested on day 3. Lysates were immunolabeled for PFKFB3 and tubulin. B) Relative densitometry of the 3 replicates shown. D) Basal 2-deoxy glucose uptake in day 3 adipocytes transfected on day 1 with PFKFB3 siRNA or Sc siRNA. Analysis of n=6 replicates was carried out using a Student’s t-test. E) 3T3-L1 adipocytes differentiated in media supplemented with 5 mM glucose and 2 mM glutamine and transfected with PFKFB3 siRNA or Sc siRNA on day 1 were harvested on day 6. Lysates and media were immunoblotted as indicated F) Quantification of immunoblots. Differences were determined using Student t-tests and p values are shown.
To further confirm the importance of PFKFB3 in adipocyte maturation, we employed pharmacologic inhibition using AZ67, a compound that reduces fructose 2,6-bisphosphate concentrations in a dose dependent manner and with minimal off-target effects (20). Because we determined that PFKFB3 was transiently upregulated by day 3 of differentiation, AZ67 treatment was pulsed for different periods during D-phase to identify when PFKFB3 activity was most important. Three different treatment protocols were used for this study (Fig. 5A). Cells were treated with 30 μM AZ67 in DII media containing 5 mM glucose for the first 33 hours of DII treatment (Pulse A); last 39 hours of DII treatment (Pulse B); or the full DII treatment period (Pulse C). Differentiation media was replaced with 5 mM glucose media containing 10% FBS and cells were harvested on day 6. We first determined that AZ67-treatment did not inhibit initiation of differentiation by probing cell lysates for Pref-1 (Figure 2B and C). Cell lysates were then probed for GLUT4, PFKFB3 and tubulin, while both lysates and cell medium were probed for adiponectin (Fig. 5D and E). Pulse A of AZ67did not alter expression of GLUT4 or adiponectin, our key markers of adipocyte maturation. In contrast, treatment by pulse B and pulse C both inhibited expression of both GLUT4 and adiponectin, indicating that expression of these markers is dependent on PFKFB3 activity in the last 39 hours of DII treatment. PFKFB3 expression itself was not inhibited by AZ67 treatment.
Figure 5.
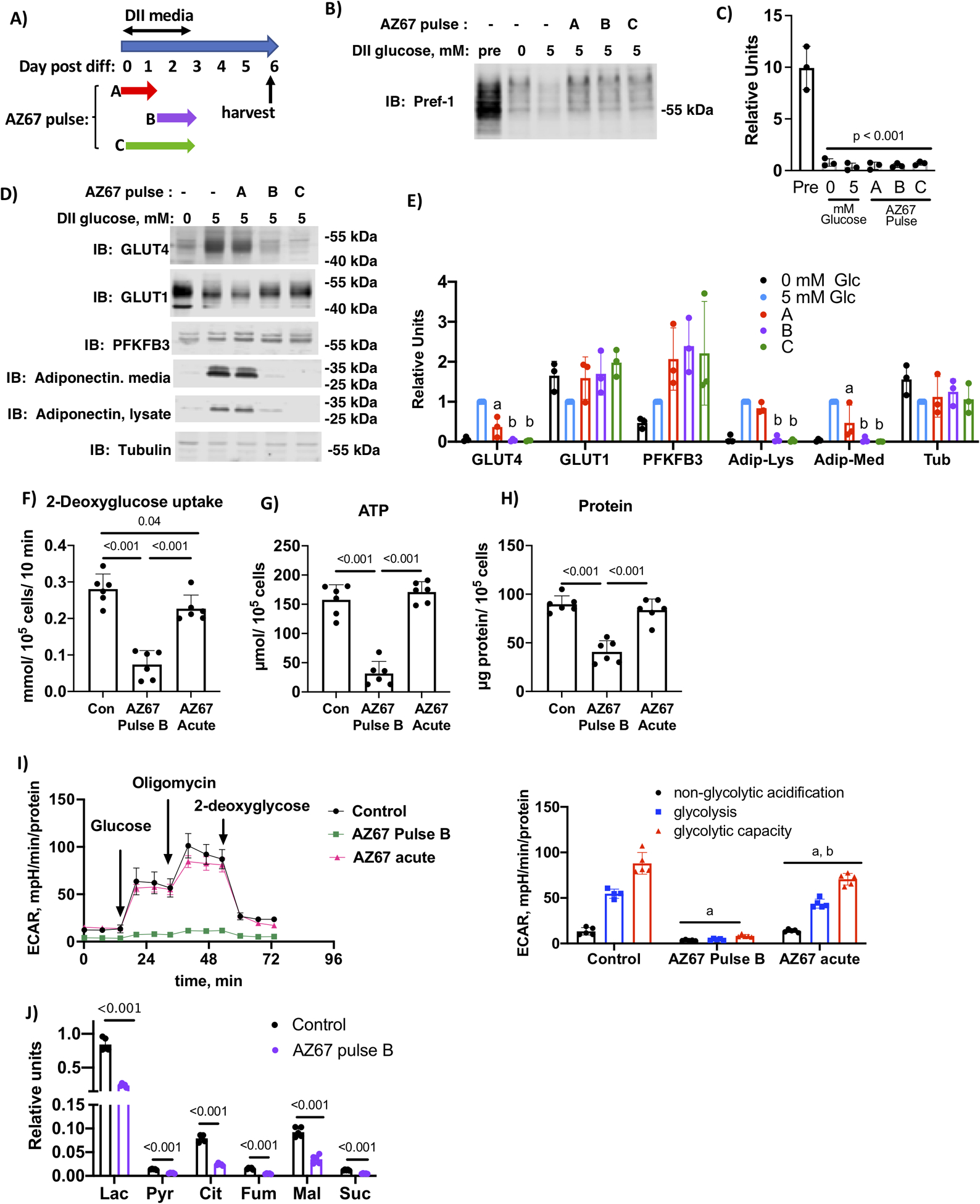
Chronic inhibition of PFKFB3 during the final 39 hours of D-phase prevented adipocyte maturation. A) Schematic depiction of the experimental design. B) 3T3-L1 cells differentiated in media supplemented with 0 or 5 mM glucose and 2 mM glutamine were treated with AZ67 as indicated. Cell lysates were prepared on day 6 post-differentiation and lysates and media were immunoblotted as indicated. C) Quantification of immunoblots of n=3 independent experiments were analyzed by one-way ANOVA using a Tukey’s post-hoc test to compare 5 mM glucose with different AZ67 pulses, p value is indicated. D) 3T3-L1 cells differentiated in media supplemented with 0 or 5 mM glucose and 2 mM glutamine were treated with AZ67 as indicated. Cell lysates were prepared on day 6 post-differentiation and lysates and media were immunoblotted as indicated. E) Quantification of immunoblots of n=3 independent experiments were analyzed by one-way ANOVA using a Tukey’s post-hoc test to compare 5 mM glucose with different AZ67 pulses. Histograms marked with different letters are significantly different from all other conditions for a given immunolabel. F) 2-deoxyglucose uptake, G) ATP and H) total protein for day 3 adipocytes differentiated with media containing 5 mM glucose and 2 mM glutamine and treated with AZ67 as indicated. Analysis of n=6 independent replications was analyzed by one-way ANOVA and tukey’s post-hoc test. I) Glycolysis stress test performed in day 3 adipocytes as described for F-H. The first graph shows the trace for the acidification experiment with additions labeled and the second graph shows the quantification of the area under the curve for non-glycolytic acidification, glycolysis and glycolytic capacity, respectively. Letters indicate statistical differences compared to the corresponding component of the stress test. Data analyzed by one-way ANOVA and a Tukey’s post hoc test (p < 0.01). H) key metabolites in day 3 adipocytes differentiated as described for F-H. Differences are indicated as determined by multiple t-tests. J) Key organic acids, lactate (Lac), pyruvate (Pyr), citrate (Cit), fumarate (Fum) Malate (mal), and succinate (Suc) from day 3 adipocytes differentiated with 5 mM glucose and 2 mM glutamine and treated with AZ67 Pulse B protocol. Relative differences between conditions (n=5) were analyzed by Student’s t-test using a False Discovery Rate correction as described in methods and q value is indicated.
To determine how PFKFB3 inhibition altered glucose uptake, we measured 2-deoxyglucose uptake on day 3 of DII treatment in cells treated with 30 μM AZ67 chronically (Pulse B) or acutely for 1 hour prior to the assay. As expected, chronic AZ67 treatment reduced 2-deoxyglucose uptake 3.8-fold, while the acute treatment reduced glucose uptake by 20% (Fig. 5F). Chronic, but not acute treatment with AZ67 decreased both ATP levels and protein concentration (Fig. 5G and H), which supports the notion that PFKFB3 activity is required specifically between day 2 and 3 to upregulate the capacity for cells to expand the metabolome. Consistent with the 2-deoxyglucose uptake, chronic exposure to AZ67 prevented upregulation of the glycolytic machinery, as determined by ECAR. Chronic treatment completely ablated glycolysis while acute treatment inhibited by approximately 20% (Fig. 5I). To determine if chronic AZ67 treatment altered the adipocyte metabolome, soluble extracts from five biological replicates each of day 3 adipocytes treated without or with 30 μM AZ67 chronically (Pulse B) were analyzed by GC-mass spectrometry to determine relative concentrations of metabolites. Forty-four peaks were identified as unique metabolites with high probability using NIST library. Lactate, glucose, pyruvate, alanine and leucine were confirmed by external standards (Suppl Tables 3 and 4). The global scale analysis of the metabolic profiling data was performed by principal component analysis (Suppl. Figure 3). Chronic exposure of cells to AZ67 decreased the glycolytic products pyruvate and lactate, as well as other TCA intermediates (Fig. 5J). This is consistent with a role of PFKFB3 in accelerating glycolysis and anaplerosis.
Previously, we showed that inhibiting glucose metabolism through the PPP also diminished adipocyte development (4). To determine if inhibition of PPP followed the same time course as PFKFB3 inhibition, we treated cells with 30 μM 6-aminonicotinamide (6-AN), an inhibitor of both 6-phosphogluconate dehydrogenase and glucose-6-phosphate dehydrogenase, using the same timing strategy as for PFKFB3 inhibition (Fig. 6A). We first determined that 6-AN-treatment did not inhibit initiation of differentiation by probing cell lysates for Pref-1 (Figure 3B and C). In contrast to AZ67 treatment, 6-AN treatment inhibited adipocyte maturation when given both in the first 33 hours of differentiation or the final 39 hours of differentiation (Fig. 6D and E). Cell-associated adiponectin appeared to increase with 6-AN treatment, but this was likely a result of decreased secretion of adiponectin into the media. These data indicate that the requirement for PPP-dependent glucose metabolism precedes the requirement for PKFFB3-dependent glucose metabolism.
Figure 6.
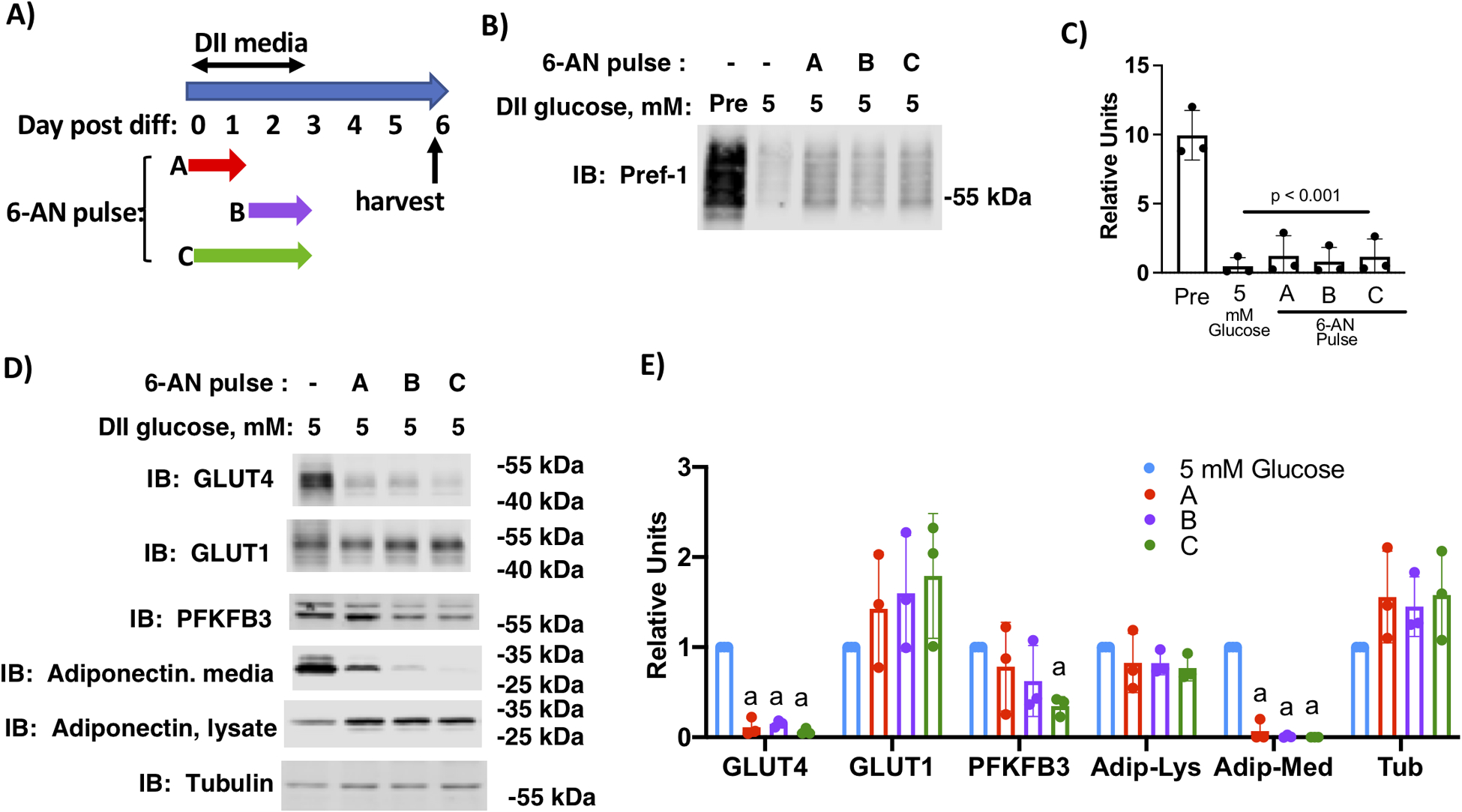
Inhibition of pentose phosphate pathway inhibited adipocyte maturation. A) Schematic depiction of the experimental design. B) 3T3-L1 cells differentiated in media supplemented with 0 or 5 mM glucose and 2 mM glutamine were treated with 6-AN as indicated. Cell lysates were prepared on day 6 post-differentiation and lysates and media were immunoblotted as indicated. C) Quantification of immunoblots of n=3 independent experiments were analyzed by one-way ANOVA using a Tukey’s post-hoc test to compare 5 mM glucose with different AZ67 pulses, p value is indicated.D) 3T3-L1 cells differentiated in media supplemented with 5 mM glucose and 2 mM glutamine were treated with 6-AN as indicated. Cell lysates were prepared on day 6 post-differentiation and lysates and media were immunolabeled as indicated. E) Quantification of immunoblots of n=3 independent experiments were analyzed by one-way ANOVA using a Tukey’s post-hoc test to compare 5 mM glucose with different 6-AN pulses. Histograms marked with different letters are significantly different from all other conditions for a given immunolabel.
To determine if PFKFKB3 activity is required for differentiation of non-transformed mouse preadipocytes, we cultured preadipocytes derived from the stromal vascular fraction of mouse inguinal fat pads. Unlike 3T3-L1 preadipocytes which are grown to confluence in media supplemented with 10% calf serum, primary preadipocytes were grown to confluence in media supplemented with 10% fetal bovine serum. Once the cells were two days past confluence, differentiation was initiated with DII media for the next 72 hours. Cells were treated without or with 30 μM AZ67 for the final 39 hours of DII treatment (Pulse B) (Figure 7A). Cells were harvested at days 0, 3 and 6 post-differentiation and immunolabeled for Pref-1, PFKFB3, Glut4, adiponectin and tubulin (Fig. 7B and C). Media from day 6 cells was immunolabeled for adiponectin. Chronic treatment with AZ67 for the final 39 hours of differentiation inhibited adipocyte maturation as was observed for differentiation of 3T3-L1 cells. Day 0 primary preadipocytes did not express high levels of Pref-1, reflecting a potential difference in the specific population of preadipocytes in the confluent primary adipocytes compared to 3T3-L1 preadipocytes. PFKFB3 expression was induced in the differentiated primary adipocytes, and expression was highest at day 6 in both control and AZ67 treated cells.
Figure 7.
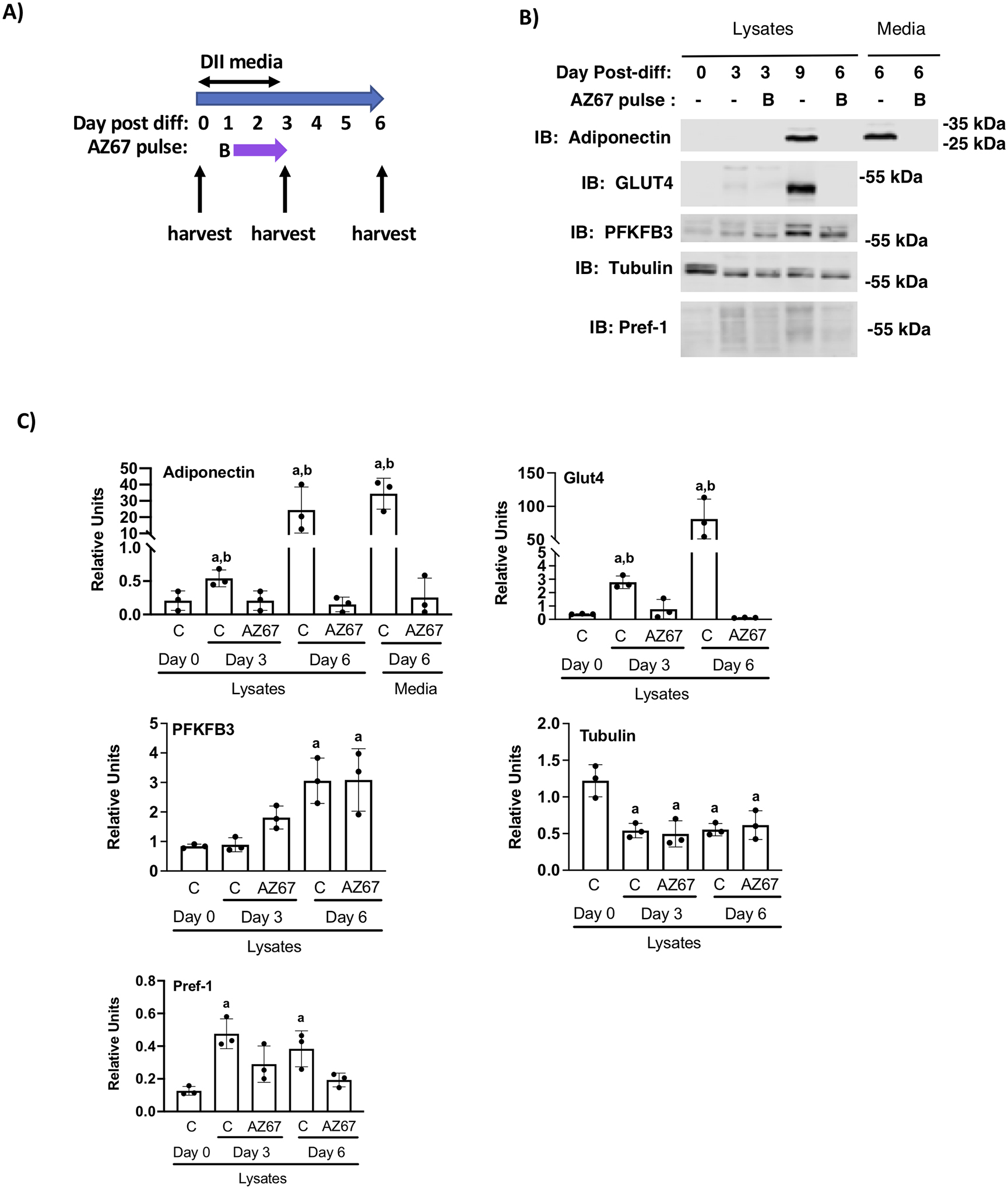
Chronic inhibition of PFKFB3 during the final 39 hours of D-phase prevented adipocyte maturation of primary inguinal preadipocytes. A) Schematic depiction of the experimental design. B) Inguinal adipose tissue preadipocytes were differentiated in media supplemented with 5 mM glucose and 2 mM glutamine were treated with AZ67 as indicated. Cell lysates were prepared on day 0, 3 and 6 post-differentiation and lysates and media were immunoblotted as indicated. C) Quantification of immunoblots of n=3 independent experiments were analyzed by one-way ANOVA using a Tukey’s post-hoc test to compare control cells and AZ67 treated cells over the time-course. Bars indicated by the letter “a” indicate a significant difference compared to day 0 cells (p < 0.01). The letter “b” indicates a significant difference between control and AZ67 cells (p < 0.01).
PFKFB3 regulates adipocyte function in mature adipocytes
Interestingly, in adult mice, the highest levels of PFKFB3 mRNA are found in adipose tissue (21). This indicates a role for PFKFB3 in glucose metabolism in mature adipocytes. To test this, we treated 3T3-L1 adipocytes beginning on day 4 of differentiation with either 0 or 30 μM AZ67 for the next 48 hours. Cells were harvested on day 6 post-differentiation and cell lysates were probed for GLUT4, PFKFB3, and tubulin, while both lysates and cell medium were probed for adiponectin (Fig. 8A and B). AZ67 treatment significantly decreased GLUT4 expression. Intracellular adiponectin was unchanged but was significantly decreased in the media. PFKFB3 content was increased, suggestive of a compensatory mechanism. Oil red O staining showed that neutral lipid staining on day 6 adipocytes treated with AZ67 was reduced 10% compared to controls (Fig. 8C). This is likely due to decreased glyceroneogenesis for glycerol backbones given that branch chain amino acids are the main carbon source for de novo lipogenesis (22). To determine if AZ67 inhibited either basal or insulin-dependent glucose uptake in the mature cells, we carried out 2-deoxyglucose uptake in day 6 adipocytes that were treated without or with 30 μM AZ67 since day 4. Basal 2-deoxyglucose uptake was increased by AZ67 treatment, while the insulin-mediated glucose uptake was completely inhibited (Fig. 8D). Oil red O-staining was measured in basal cells, therefore, the increase basal 2-deoxyglucose uptake in the treated cells was not likely supporting triacylglycerol synthesis in the AZ67-treated cells.
Figure 8.
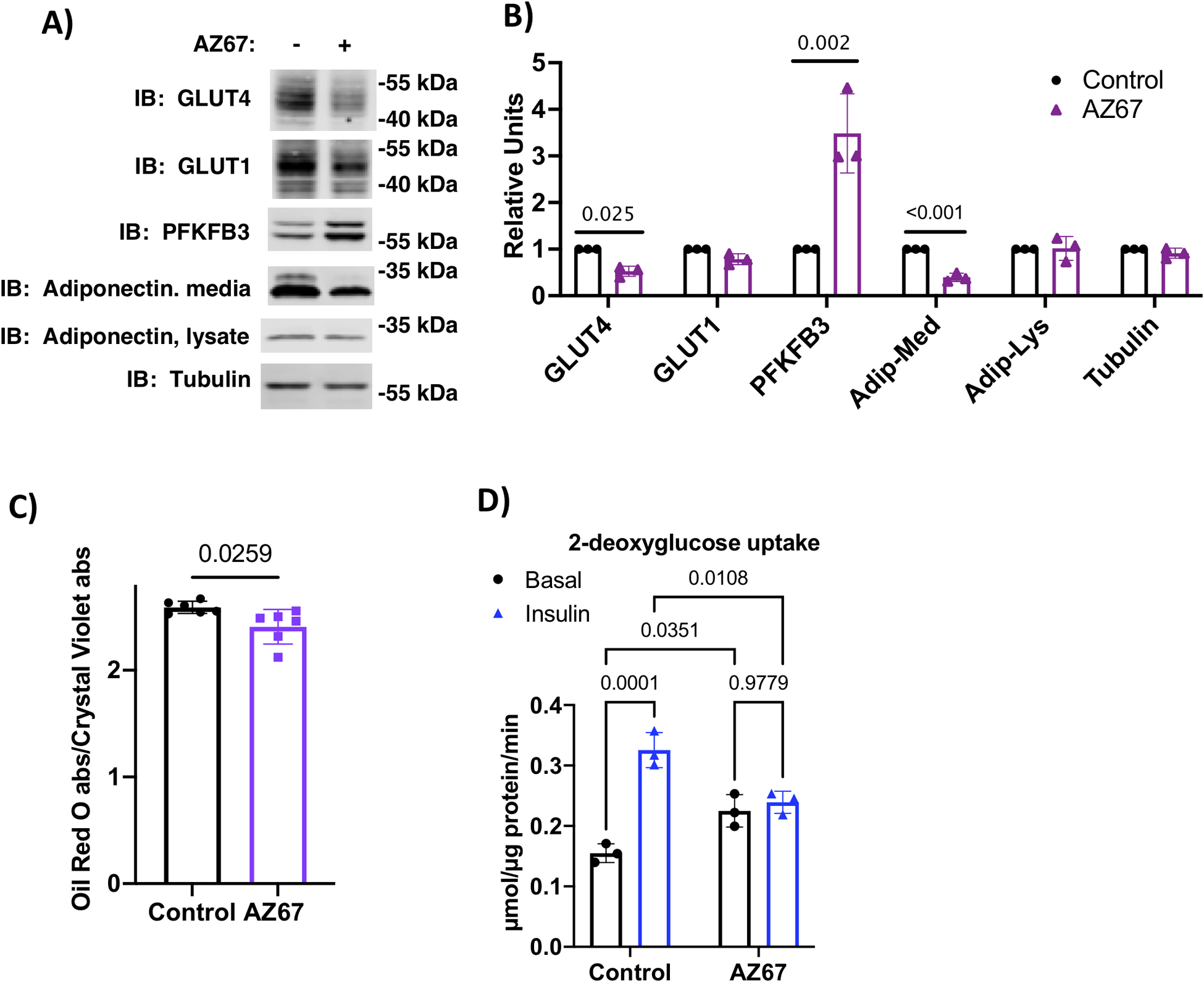
PFKFB3 activity regulates mature adipocyte function. A) Immunoblot analysis of indicated proteins from day 6 adipocytes treated for the previous 48 hrs without or with 30 μM AZ67. B) Quantification of immunoblots from n=3 independent experiments analyzed by Student’s t-test. C) Oil red O staining in day 6 adipocytes treated for the previous 48 hrs without or with 30 μM AZ67. Data are presented as the ratio of absorbance of oil red O/ crystal violet eluates. Data from n=6 independent replicates were analyzed by Student’s t-test.
These findings suggest that PFKFB3 may also play a role in the insulin/fed state dependent increase in adipocyte glycolysis and glyceroneogenesis in vivo (23). To begin testing this possibility, we measured PFKFB3 protein expression in mice during the fed and fasted states in epididymal (EPI) and inguinal (SQ) fat. We found that PFKFB3 was significantly lower in both fat pads of fasted animals and higher after 4 hours of refeeding fasted mice (Fig. 9A and B). A small, but significant difference was observed for PFKFB4 in the EPI, but not SQ fat. The changes in PFKFB3 protein expression were not due to changes in pfkfb3 mRNA, indicating that a translational or post-translational mechanism regulated protein expression in the fasted and refed conditions (Fig. 9C). In conclusion, our results indicate PFKFB3 plays an essential role in glucose mediated adipocyte differentiation and in regulating glucose metabolism in mature adipocytes in vitro. Changes in PFKFB3 protein levels in fasted and refed fat predict that glucose metabolism in vivo may be regulated, in part, by PFKFB3.
Figure 9.
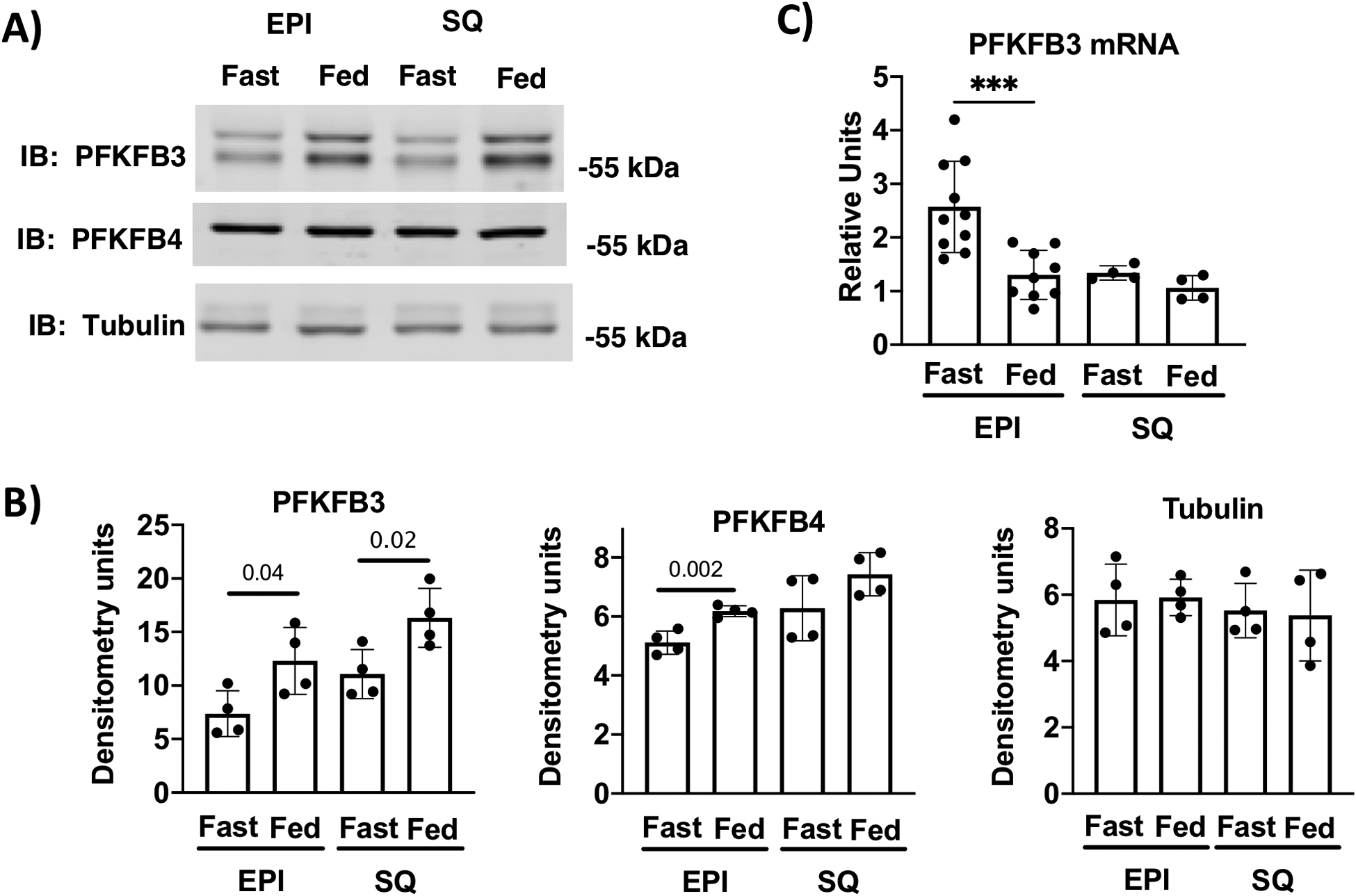
PFKFB3 expression is dependent on fasting/refeeding cycle. A) immunoblot analysis of cell lysates from fasted and 4 hr refed epididymal (EPI) and inguinal subcutaneous fat (SQ). B) Quantification of n=4 independent experiments are shown. Differences between fasted a refed for EPI and SQ were analyzed using Student’s t-tests. C) PFKFB3 mRNA from fasted and 4 hour refed male mice n=4 to 10 replicates. Differences between fasted a refed for EPI and SQ were analyzed using Student’s t-tests.
DISCUSSION
Our data reveal an important interaction between hormone signaling and glucose availability in development of mature 3T3-L1 adipocytes. Glucose, but not glutamine, was specifically required for development indicating that the glucose effect is different than the Warburg effect often observed in cancer cells (24) Within the first day of differentiation, we observed a 20-fold increase in the capacity for glucose uptake that was instigated by the addition of the hormone cocktail. 2-Deoxyglucose uptake was further enhanced when 5 mM glucose was present in the media (Fig 2A). Differentiation hormone cocktail with no glucose added to the media was able to increase expression of several key proteins that are known to regulate glucose uptake and glycolysis, including hexokinase II and the lactate transporter MCT4 (19). Hormone signaling alone was also able to suppress expression of Pref-1, a marker of very early stage preadipocytes, which is highly expressed in 3T3-L1 preadipocytes indicating that differentiation was activated (Fig. 2) (25, 26). Hormonal signaling was also sufficient to produce some of the cellular machinery to support glycolysis when substrate becomes available. Importantly, hormone-dependent and glucose-dependent signals were required for increased expression of PFKFB3, an isoform of the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase family that plays a key role in regulating cellular fructose 2,6-bisphosphate (Fru-2,6-BP), an important allosteric activator of phosphofructokinse-1 (PFK-1). These data support the notion that, following the initiation of the differentiation, preadipocytes require a carbohydrate signal to proceed through the differentiation program.
PFKFB3 expression was transiently upregulated on day three of differentiation, a time that coincided with both an increase in basal glucose uptake and a significant increase in the cellular proteome and the amino acid content of the maturing adipocytes. These data support the notion that a transient acceleration in glucose uptake and glycolysis may serve to enhance amino acid uptake and/or decrease amino acid catabolism, thus increasing protein synthesis. While the mechanism for these functions is not known, it is possible that epigenetic changes in gene expression, differential posttranslational acetylation, and/or glycosylation of key proteins may be responsible for these changes (27).
3T3-L1 cells are a transformed cell line representing a very early clonal line of preadipocytes that were derived from Day 17–19-day-old Swiss 3T3 mouse embryos and may be metabolically different than preadipocytes from adult mouse adipose depots (28). In previous work from our lab, we confirmed that differentiation of adult mouse primary preadipocytes were also dependent on glucose supplementation for differentiation (4). In the current paper, we confirmed that differentiation of the adult mouse preadipocytes were also dependent on PFKFB3 activity for maturation. There were some notable differences between the 3T3-L1 adipocytes and mouse preadipocytes. Mouse preadipocytes expressed lower levels of Pref-1 compared to 3T3-L1 preadipocytes (Fig. 7). This may be due to the fact that 3T3-L1 cells are embryonic and the inguinal preadipocytes were from adult mice. The temporal pattern of PFKFB3 expression was also different during development of the mouse inguinal adipocytes. In mouse preadipocytes cells, the highest level of expression was seen at day 6 post differentiation (compare Fig. 3E and Fig, 7). This temporal pattern of PFKFB3 expression was also reported several years ago by Atsumi et al, who found PFKFB3 protein was upregulated in 3T3-L1 adipocytes reaching its highest level at day 6 (29). The glucose concentration used to culture 3T3-L1 adipocytes was not specified and other markers of differentiation were not shown. For this reason, it is difficult to compare our differentiation time course with the previous work.
PFKFB3, like other members of the PFKFB family, is a bifunctional enzyme that can catalyze both the production and consumption of Fru-2,6,-BP. The kinase activity of PFKFB3 is dominant over the phosphatase activity which means that it favors the production of Fru-2,6-BP content (30). This shunts glucose catabolism towards glycolysis and away from other ancillary glucose metabolic pathways. We observed a glucose-dependent increase in ATP concentration on day 1 of differentiation. It is likely that this increase in ATP concentration was due to the expansion of the adenine nucleotide pool, a product of the pentose phosphate pathway, rather than a change in the energy charge of the cells. Our previous study demonstrated that the energy charge of the cells differentiated in low glucose was the same as cells differentiated in high glucose (4). In that study, we also observed that the total pool size of nicotinamide adenine dinucleotide (NAD) was glucose-dependent.
The expansion of the nucleotide pool prior to the upregulation of PFKFB3 suggests that the initial increase in glucose uptake is directed towards the pentose phosphate pathway. This is further supported by our observation that a pulse of 6-AN, which inhibits the pentose phosphate pathway, completely blocked differentiation when applied during the first 33 hours of differentiation. In contrast, a similarly-timed pulse of AZ67, which inhibits PFKFB3 activity, had no effect on differentiation. AZ67 treatment inhibited differentiation when added during hours 34–72 of the differentiation period, consistent with the timing of the increased PFKFB3 expression. Taken together, the temporal difference in the effects of glucose metabolism by 6-AN and AZ67 suggest that flux through the pentose phosphate pathway is more important at the onset of differentiation. The upregulation of glycolysis plays a critical role between day 2 and day 3 of differentiation, leading to production of additional metabolites, proteins, or cellular structures that are important for the mature adipocyte.
PFKFB3 is highly upregulated in proliferating cells and in many types of cancer (31). Human epidermal growth factor receptor-2 (HER2) positive breast cancer cells express high levels of PFKFB3 protein. The expression of PFKFB3 can be decreased by inhibiting HER2 with a neutralizing antibody, indicating that PFKFB3 is a target of tyrosine kinase signaling (32). In adipocyte differentiation, PKFKB3 expression requires both hormone cocktail stimulation and glucose in the media, indicating hormone stimulation of glucose uptake may be the key to the upregulation of PFKFB3 as differentiation proceeds. When glucose is not added to the media, hormone signaling is not able to increase glucose uptake into the cell, providing the needed signal to increase expression of PFKFB3. It is unclear if the requirement for carbohydrate is also present for PFKFB3 expression in cancer cells.
In adult mice, pfkfb3 mRNA and protein is very highly expressed in white adipose tissue and appears to play an important role in adipocyte development and adipocyte inflammation (Fig. 9) (21, 33). Whole body heterozygous knockout of PFKFB3 in mice resulted in reduced adipocyte cell size, increased adipocyte inflammation and decreased glucose tolerance under high fat feeding (21). Adipocyte-specific overexpression of PFKFB3 under the control of the AP2 promoter increased lipid storage in adipose tissue, improved insulin resistance and reduced inflammation in high fat fed mice (33). These data demonstrate a key role for PFKFB3 in adipose tissue development, but do not specifically address the pathway(s). Our findings support multiple roles that PFKFB3 may play in adipocyte metabolism. During early stages of adipocyte development, we demonstrated that PFKFB3 was associated with increased glucose uptake and glycolysis. In mouse adipose tissue, we observed that PFKFB3 expression appeared to be regulated by nutritional state. PFKFB3 protein was lower in fasted mice compared to when the fasted mice were refed for 4 hours, consistent with changes in glucose uptake expected with the transition between fasting and refeeding. The decrease of PFKFB3 content in response to fasting is similar to what we have previously reported for PFKFB2 expression in the heart (34). In that case, we found that PFKFB2 is rapidly degraded by multiple pathways in the absence of insulin, presumably as part of a glucose sparing program. The data presented here are consistent with the observation that pharmacologic inhibition of PFKFB3 activity in mature 3T3-L1 adipocytes inhibited AKT signaling and insulin-dependent GLUT4 translocation (35). While it is clear that increased translocation of GLUT4 to the plasma membrane increases glucose uptake, these data when combined with our observation that PFKFB3 is upregulated within 4 hours of refeeding suggest that PFKFB3 enzyme activity coordinates with GLUT4 translocation to increase glucose uptake and glycolysis.
A specific role that PFKFB3 might play in fed state regulation of glucose metabolism could be to divert glucose away from pentose phosphate pathway to promote glycolysis and glyceroneogenesis for triglyceride synthesis (23). We show that chronic inhibition of PFKFB3 in 3T3-L1 adipocytes from day 4 to day 6 post-differentiation reduced accumulation of triacylglycerol, presumably through decreased glyceroneogenesis as this in the major pathway through which glucose contributes to lipogenesis (22, 36). Importantly, the oil Red O staining was carried out in mature cells that were stimulated with 10% fetal bovine serum, but not exogenous insulin between day 4 and day 6. We showed that basal glucose uptake was increased on day 6 in AZ67-treated mature adipocytes (Fig. 8). This further supports the notion that PFKFB3-dependent glycolysis plays a significant in regulating normal adipocyte metabolism.
While we have specifically studied the role of glucose metabolism in adipocyte differentiation, it is likely that transient upregulation of glucose metabolism and glycolysis is part of the differentiation program in other tissues and in some cancers. For example, T cell activation and immunologic activity was accompanied by increased energy demand and cellular biosynthesis which was fueled by a rapid increase in glucose and glucose metabolism (37, 38). Increased glucose metabolism in activated T-cells is accompanied by increased expression of PFKFB3 that is induced by growth factor activation (39); however, the role of exogenous glucose in PFKFB3 upregulation was not studied.
In conclusion, we have shown that there is an important developmental metabolic program that supports the transition from a preadipocyte to a mature adipocyte. The initial hormonal switch upregulates glucose uptake that first serves the pentose phosphate pathway to build up the nucleotide pool to support the clonal expansion of preadipocytes that occurs during the first 24 hours of 3T3-L1 differentiation (11). The capacity for cells to undergo glycolysis develops by day 2 of differentiation. Our data support the notion that interventions that blunt the upregulation of glycolysis or deprive glycolytic substrate prevent the further development into mature adipocytes, at least with regard to expression of key adipocyte proteins. While our previous work supported the role of the pentose phosphate pathway during the early stages of adipocyte differentiation, the current work shows that both pentose phosphate pathway and glycolysis are each required at different, times during the differentiation program.
Supplementary Material
Acknowledgements
The work was supported by grants from the Oklahoma Center for Adult Stem Cell Research (OCASCR) and the Oklahoma Center for advancement of Science and Technology (OCAST, HR17-018) to ALO, the National Institutes of Health (HL125625) to KMH, the National Institutes of Health (AG049058) to TMG, and the National Institutes of Health (P30GM114731) in support of AB. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Benjamin S. Harris for technical assistance.
Abbreviations:
- 6-AN
6-amino-3-pyridinecarboxamide
- AZ67
(2S)-N-[4-[[3-Cyano-1-[(3,5-dimethyl-4-isoxazolyl)methyl]-1H-indol-5-yl]oxy]phenyl]-2-pyrrolidinecarboxamide
- DII
dexamethasone, isobutylmethylxanthine and insulin hormone cocktail
- ECAR
extracellular acidification rate
- EPI
epididymal adipose pad
- SQ
subcutaneous adipose pad
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
LITERATURE CITED
- 1.Ye J-M, Dzamko N, Cleasby ME, Hegarty BD, Furler SM, Cooney GJ, and Kraegen EW (2004) Direct demonstration of lipid sequestration as a mechanism by which rosiglitazone prevents fatty-acid-induced insulin resistance in the rat: comparison with metformin. Diabetologia 47, 1306–1313 [DOI] [PubMed] [Google Scholar]
- 2.Ye R, and Scherer PE (2013) Adiponectin, driver or passenger on the road to insulin sensitivity? Molecular Metabolism 2, 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holtrup B, Church CD, Berry R, Colman L, Jeffery E, Bober J, and Rodeheffer MS (2017) Puberty is an important developmental period for the establishment of adipose tissue mass and metabolic homeostasis. Adipocyte 6, 224–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson RM, Griesel BA, Gurley JM, Szweda LI, and Olson AL (2017) Glucose availability controls adipogenesis in mouse 3T3-L1 adipocytes via up-regulation of nicotinamide metabolism. J Biol. Chem 292, 18556–18564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubin CS, Hirsch A, Fung C, and Rosen OM (1978) Development of hormone receptors and hormone responsiveness in vitro. J. Biol. Chem 253, 7570–7578 [PubMed] [Google Scholar]
- 6.Rosen ED, and MacDougald OA (2006) Adipocyte differentiation from the inside out. Nature Rev. Mol. Cell. Biol 7, 885–896 [DOI] [PubMed] [Google Scholar]
- 7.Rosen ED, and Speigelman BM (2014) What we talk about when we talk about fat. Cell 156, 20–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosen ED, Walkley CJ, Puigserver P, and Spiegelman BM (2000) Transcriptional regulation of adipogenesis. Genes Dev. 14, 1293–1307 [PubMed] [Google Scholar]
- 9.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CR (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krycer JR, Quel LE, Francis D, Zadoorian A, Weiss FC, Cooke KC, Nelson ME, Diaz-Vegas A, Humphrey SJ, Scalzo R, Hirayama A, Ikeda S, Shoji F, Suzukie K, Huynh K, Giles C, Varney B, Nagarajan SR, Hoy AJ, Soga T, Meiklle PJ, Cooney GJ, Fazakerley DJ, and James DE (2020) Insulin signaling reqires glucose to promote lipid anabolism in adipocytes. J. Biol. Chem 295, 13250–13266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu L-B, Shah S, Fan J, Park JO, Wellen KE, and Rabinowitz JD (2016) Malic enzyme tacers reveal hypoxia-induced switch in adipocyte NADPH pathway usage. Nature Chem. Biol 12, 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yi M, Ban Y, Tan YX,W, Li G, and Xiang B. (2019) 6-phosphofructo-2-kinase/2,6-biphosphatase 3 and 4: A pair of valves fro fine-tuning of glucose metabolism in human cancer. Mol. Metab 20, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Church CD, Berry R, and Rodeheffer MS (2014) Isolation and study of adipocyte precursors. Methods Enzymol 537, 31–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olson AL, Perlman S, and Robillard JE (1990) Developmental regulation of angiotensinogen gene expression in sheep. Pediatric research 28, 183–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz, and Ferrick DA (2007) Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic funciton and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol 292, C125–C136 [DOI] [PubMed] [Google Scholar]
- 16.Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood CC, Hales LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, and Rathmell JC (2015) Metabolic programming and PDHK1 control CD4+ T-cell subsets and inflammation. J. Clin. Invest 125, 194–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Batushansky A, Lopes EBP, Zhu S, Humphries KM, and Griffin TM (2019) GC-MS method for metabolic profiling of mouse femoral head articular cartilage reveals distinct effects of tissue culture and development. Osteoarthritis and Carilage 27, 1361–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smas CM, Kachinskas D, Liu CM, Xie X, Dircks LK, and Sul HS (1998) Transcriptional control of the pref-1 gene in 3T3-L1 adipocyte differentiation. J. Biol. Chem 48, 31751–31758 [DOI] [PubMed] [Google Scholar]
- 19.Tanner LB, Goglia AG, Wei MH, Sehgal T, Parsons LR, Park JO, White E, Toettcher JE, and Rabinowitz JD (2018) Four key steps control glycolytic flux in mammalian cells. Cell Systems 7, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burmistrova O, Olias-Arjona A, Lapresa R, Jimenez-Blasco D, Eremeeva T, Shishov D, Romanov S, Zakurdaeva K, Almeida A, Fedichev PO, and Boanos JP (2019) Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice. Sci. Reports 9, 11670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huo Y, Guo X, Li H, Wang H, Zhang W, Wang Y, Shou H, Gao Z, Telang S, Chesney J, Chen YE,Y,J, Chapkin RS, and Wu C. (2010) Disruption of inducible 6-phosphofructo-2-kinse ameliorates diet-induced adiposity but exacerbastes systemic insulin resistance and adipose tissue inflammatory reponse. J Biol. Chem 285, 3713–3721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciarldi TP, and Metallo CM (2016) Branched-chain amino acid catabolims fuels adipocyte differentiation and lipogenesis. Nature Chem. Biol 12, 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nye CK, Hanson RW, and Kalhan SC (2008) Glyceroneogenesis is the dominant pathway for triglyceride glycerol synthesis in vivo in the rat. J. Biol. Chem 283, 27565–27574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hensley CT, Wasti AT, and BDeBerardinis RJ (2013) Glutamine and cancdr: cell bioogy, physiology, and clinical opportunities. J. Clin. Invest 123, 3678–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smas CM, and Sul HS (1993) Pref-1, a protein containin EGF-like repeats, inhibits adipocyte differentiation. Cell 73, 725–734 [DOI] [PubMed] [Google Scholar]
- 26.Hudak CS, Gulyaeva O, Wang Y-T, Park SM, Lee L, Kang C, and Sul HS (2014) Pref-1 marks very early mesenchymal precursors required for adipose tissue development and expansion. Cell Reports 8, 678–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wellen KE, and Thompson CB (2012) A two-way street: Reciprocal regulation of metabolism and signaling. Nature Reviews 13, 270–276 [DOI] [PubMed] [Google Scholar]
- 28.Green H, and Meuth M (1974) An established pre-adipose cell line and its differentiation in culture. Cell 3, 127–133 [DOI] [PubMed] [Google Scholar]
- 29.T. A, Nishio, Niwa H, Takeuchi J, Bando H, Shimizu C, Yosioka N, Bucala R, and Koike T (2005) Expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase/PFKFB3 isoforms in adipocytes and their potenial role in glycolytic regulation. Diabetes 54, 3349–3357 [DOI] [PubMed] [Google Scholar]
- 30.Sakakibara R, Kato M, Okamura N, Nakagawa T, Komada Y, Tominage N, Shimojo M, and Fukasawa M (1997) Characterization of a human placental fructose—6-phoshphate, 2-kinase/Frictose-2, 6-bisphosphatase. J Biochem 122, 122–128 [DOI] [PubMed] [Google Scholar]
- 31.Shi L, Pan H, Liu Z, Xie J, and Han W (2017) Roles of PFKFB3 in cancer. Singal Transduction and Targeted Therapy 2, e17044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Neal J, Clem A, Reynolds L, Dougherty S, Imbert-Fernandez Y, Telang S, Chesney J, and Clem BF (2016) Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res. Treat 160, 29–40 [DOI] [PubMed] [Google Scholar]
- 33.Huo Y, Guo X, Li H, Xu H, Halim V, Zhang W, Wang H, Fan Y,Y, Ong KT, Woo SL, Chapkin RS, Mashek DG, Chen Y, Dong H, Lu F, Wei L, and Wu C (2012) Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects aginst diet-induced insulin resistance and inflammatory responses. J Biol. Chem 287, 21492–21500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bockus LB, Matsuzaki S, Vadvalkar S, Young ZT, Giorgione JR, Newhardt MF, Kinter M, and Humphries KM (2017) Cardiac insulin signaling regulates glycolysis through phosphofructokinse 2 content and activity. J. Am. Heart Assoc 6, e007159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trefely S, Khoo PS, Krycer JR, Chaudhuri R, Fazakerley DJ, Parker BL, Sultani G, Lee JW, Stephan JP, Torres E, Jung K, Kuijl C, James DE, Junutula JR, and Stockli J (2015) Kinome screen identifies PFKFB3 and glucose metabolism as important regulators of the insulin/insulin-like growth factor (IGF)-1 signaling pathway. J Biol. Chem 290, 25834–25846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rotondo F, Ho-Palma AC, Remesar X, Fernandez-Lopez JA, del Mar Romero M, and Alemany M (2017) Glycerol is synthesized adn secreted by adipocytes to dispose fo excess glucose, via glycerogenesis and increased acyl-glycerol turnover. Scientific Reports 7, 8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobs SR, Herman CE, MacIver NJ, Wofford JA, Wieman HL, Hammen JJ, and Rathmell JC (2008) Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol 180, 4476–4486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beckermann KE, Hongo R, Ye X, Carbonell K, Contreras-Healey DC, Siska PJ, Barone S, Roe CE, Smith CC, Vincent BG, Mason FM, Irish JM, Rathmell WK, and Rathmell JC (2020) CD28 costimulation drives tumor-infiltrating T cell glycolysis to promote inflammation. JCI Insight 5, e138729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simon-Molas H, Arnedo-Pac C, Fontova P, Vidal-Alabro A, Castano E, Rodriguez-Garcia A, Navarro-Sabate A, Lloberas N, Manzano A, and Bartrons R (2018) PI3K-Akt signaling controls PFKFB3 expression during human T-lymphocyte activation. Mol. Cell. Biochem 448, 187–197 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.