

Abstract
Metallo-β-lactamases (MBLs) can efficiently catalyze the hydrolysis of all classes of β-lactam antibiotics except monobactams. While serine-β-lactamase (SBL) inhibitors (e.g., clavulanic acid, avibactam) are established for clinical use, no such MBL inhibitors are available. We report on the synthesis and mechanism of inhibition of N-sulfamoylpyrrole-2-carboxylates (NSPCs) which are potent inhibitors of clinically relevant B1 subclass MBLs, including NDM-1. Crystallography reveals that the N-sulfamoyl NH2 group displaces the dizinc bridging hydroxide/water of the B1 MBLs. Comparison of crystal structures of an NSPC and taniborbactam (VRNX-5133), presently in Phase III clinical trials, shows similar binding modes for the NSPC and the cyclic boronate ring systems. The presence of an NSPC restores meropenem efficacy in clinically derived E. coli and K. pneumoniae blaNDM-1. The results support the potential of NSPCs and related compounds as efficient MBL inhibitors, though further optimization is required for their clinical development.
Keywords: antimicrobial resistance, sulfonamide, metallo-β-lactamase, taniborbactam, NDM-1
The β-lactams are one of the most important antibacterial classes;1 however, their efficacy is increasingly being eroded by resistance, most importantly by β-lactamases.2 Even carbapenems, often used as “last resort” antibiotics, are often no longer effective due to production of carbapenemases by Enterobacteriaceae strains.3 Ambler class A, C, and D β-lactamases are nucleophilic serine enzymes (serine-β-lactamases, SBLs), whereas class B are metallo-β-lactamases (MBLs).4 Combinations of a β-lactam antibiotic and a β-lactam containing SBL inhibitor have long been used as a treatment option for bacterial infections; however, no MBL combinations are clinically approved.
Presently, only a few subclasses of SBLs,5,6 e.g. Class A KPCs and Class D OXAs, are reported to efficiently hydrolyze carbapenems, and these can be countered by clinically available SBL inhibitors, e.g. avibactam.7,8 Class B MBLs, however, can hydrolyze all carbapenems and β-lactam containing SBL inhibitors.9−11 MBL inhibition is challenging in part because of the need to obtain activity against a range of relevant enzymes, which vary in their active site details.12,13 Subclass B1 and B3 MBLs are dizinc ion enzymes, whereas B2 MBLs employ one zinc ion. The B1 MBLs are the most important from a clinical perspective and include the IMP (imipenemase), NDM (New Delhi MBL), and VIM (Verona integron-encoded MBL) MBL subfamilies.14
Reported MBL inhibitors include bicyclic boronates, thiols, and succinate derivatives (Figure 1).15,16 Recently, substituted pyrroles and related compounds have been described as MBL inhibitors in the patent and scientific literature.17−20 Wachino et al. have reported that substituted pyrroles and furans bearing α-carboxylic acid and N-sulfamoyl functional groups are effective MBL inhibitors, in particular of the B1 subclass.21N-1 Sulfamoylpyrrole-2-carboxylates have also been reported as B1 MBL inhibitors,22 though no structures of them in complex with MBLs are reported. Structurally related sulfonamide-based inhibitors of metalloenzymes are used therapeutically, e.g. as carbonic anhydrase inhibitors with broad clinical utility.23−25 The N-sulfamoylpyrrole compounds are of mechanistic interest because the (initial) binding mode of some classes of potent β-lactamase inhibitors can mimic that of substrates (e.g., clavulanic acid) or tetrahedral intermediates (boronates).26 We envisaged that the approximately tetrahedral geometry about the sulfamoyl sulfur27,28 may mimic the tetrahedral intermediate formed during β-lactam hydrolysis, and the Lewis basicity of the oxygen and nitrogen atoms may enable effective coordination to the zinc ion. Here, we report on the mechanism of action and B1 MBL potencies of N-sulfamoyl-substituted pyrrole-2-carboxylic acids (NSPCs).
Figure 1.

(a) Classes of β-lactam antibiotics. (b) Outline of the MBL hydrolysis mechanism. Ligands around the zinc ions are not shown for clarity. (c) Representative SBL inhibitors. (d) Representative MBL inhibitors29−31 with sulfonamide ANT268119 and related sulfonamide and sulfamoyl inhibitors.21,22,32 Zinc-chelating functional groups are highlighted in blue.
Results and Discussion
We targeted the synthesis of NSPC 6a, which has a para-fluorophenyl substitution at its C3 position, because this substituent has been identified as being preferred in a related series of published pyrrole inhibitors (Scheme 1).33,34 We aimed to employ mild hydrogenolytic deprotection to prepare the NSPCs because of potential competitive decarboxylation of pyrrole-2-carboxylic acids35,36 and N1-sulfonyl group cleavage under acidic or basic conditions.35−37 The efficient synthesis of 6a was readily achieved in seven steps from pyrrole (1) (12% overall yield, Scheme 1). Initial N-sulfonylation of 1 with PhSO2Cl was followed by regioselective electrophilic C3-bromination using Br2. Subsequent directed ortho-metalation and electrophilic trapping with benzylchloroformate (CbzCl) gave C2-substituted benzyl ester 3. The N-Cbz protected sulfamoyl group was installed in good yield by tetrabutylammonium fluoride (TBAF)-mediated N-sulfonyl deprotection, followed by deprotonation of the pyrrole NH with sodium hydride and then electrophilic trapping with zwitterionic sulfamoylating reagent 7.38 Subsequent Pd-catalyzed Suzuki–Miyaura cross-coupling with 4-fluorophenylboronic acid afforded the C3-aryl derivative 5 as a sodium sulfonylazanide salt which, upon hydrogenation, gave sodium carboxylate 6b (93%). The free acid 6a was obtained from 5 by acidification with aqueous HCl, followed by global deprotection with Pd/C/H2 and purification by reverse phase HPLC in 69% yield.
Scheme 1. Synthesis of 6a and NSPCs 8-14.

(a) NaH, then PhSO2Cl, DMF, 0 °C, 2 h; (b) Br2, AcOH, reflux, 1 h, 54% yield over two steps; (c) i-Pr2NLi, then CbzCl, THF, −78 to 0 °C; (d) 1 M TBAF, THF, rt, 2 h, 63% over two steps; (e) NaH, then 7, THF, 0 °C to reflux, 4 h, 69%; (f) 4-FC6H4B(OH)2, 5% Pd(dppf)Cl2, Na2CO3, dioxane: H2O 2:1, MW, 100 °C, 3 h, 70%; (g) 1 M aq HCl, then 10% Pd/C, H2 atmosphere, MeOH, rt, overnight, 69%; (h) 10% Pd/C, H2 atmosphere, MeOH, rt, overnight, 93%; (i) structure previously disclosed.
With a robust synthesis of 6 in hand, we synthesized seven other NSPC derivatives varying the C3-pyrrole substituent, with bromopyrrole 4 providing a convenient vector for late-stage diversification with aryl and heteroaryl groups via Suzuki–Miyaura coupling, followed by hydrogenation (8–14, see Supporting Information). Unexpectedly, under the hydrogenation conditions used for the preparation of aminopyrimidine 11, near equimolar quantities of zwitterionic cyclic guanidine 12 were also formed due to over-reduction;39 both products were separated by preparative HPLC. For the preparation of 14, it was necessary to first install the pinacol boronate ester at the C3 position of pyrrole 4 by Pd-catalyzed Miyaura borylation; the intermediate boronate ester then underwent coupling with the commercial heteroaryl bromide.
The NSPCs were screened against four of the currently most clinically relevant B1 MBLs, i.e. VIM-1, VIM-2, NDM-1, and IMP-1 (Table 1), using an assay employing the “fluorogenic” cephalosporin FC5.40 The NSPCs inhibit all 4 MBLs, with potencies in the submicromolar range (pIC50 s 6.5–8.5), though manifesting different inhibition profiles. Notably, the cyclic guanidine 12 is a highly potent VIM-1 inhibitor (pIC50 8.5), with a similar potency to the bicyclic boronate taniborbactam (formerly VNRX-5133), which is in Phase III clinical trials.41 Interestingly, the cyclic guanidine 12 showed higher activity toward VIM-1 than the unsaturated aminopyrimidine 11; however, 12 is a less potent inhibitor of NDM-1. Most of the NSPCs showed submicromolar activity (IC50) for inhibition of NDM-1 and VIM-2, and 6a, 6b, 10, and 13 showed nanomolar potency against NDM-1. Furthermore, some NSPCs are ∼150- to >1500-fold more potent (pIC50 values 7.3–9.2) than the bicyclic boronates CB2 or taniborbactam (pIC50 6.0 and 5.6, respectively) against IMP-1. Within the compound set tested by us, no stand-out compound potently inhibiting all four MBLs was identified, revealing the scope for further optimization of the NSPC scaffold.
Table 1. Activity of N-Sulfamoyl Pyrrole Carboxylate Derivatives against Clinically Relevant MBLs.
| pIC50 |
||||
|---|---|---|---|---|
| VIM-1 | NDM-1 | VIM-2 | IMP-1 | |
| CB2 | 7.142 | 7.543 | 8.543 | 6.043 |
| taniborbactam | 8.141 | 8.041 | 8.341 | 5.641 |
| 6a | 6.9 | 8.1 | 7.7 | 9.2 |
| 6b | 6.9 | 8.2 | 7.5 | 8.9 |
| 8 | 7.1 | 7.9 | 6.8 | 8.6 |
| 9 | 6.5 | 7.9 | 6.7 | 9 |
| 10 | 7.1 | 8.1 | 7.8 | 8.9 |
| 11 | 7.4 | 7.9 | 7.3 | 8.2 |
| 12 | 8.5 | 6.5 | 7.9 | 7.3 |
| 13 | 6.6 | 8.8 | 6.8 | >9.2 |
| 14 | 7.4 | 7.9 | 8.2 | 8.3 |
| AMRC272a,c | 6.122 | 4.522 | 6.422 | |
| AMRC276a,c | 6.822 | 4.222 | 7.422 | |
| AMRC364a,c | 5.622 | 4.422 | 5.722 | |
| AMRC439a,c | 6.422 | 4.322 | 6.322 | |
| SPCb | 6.021 | 7.721 | 6.621 | |
Determined using 100 μM nitrocefin.22
Determined by measuring hydrolysis of imipenem.21
Structure in Figure 1d. Assay details are given in the Supporting Information. Enzyme concentration: 100 pM (VIM-1), 20 pM (NDM-1), 20 pM (IMP-1), and 500 pM (VIM-2); the concentration of FC5 was 5 μM. Note: inhibition data are reported as pIC50 values (pIC50 = −log10IC50) and repeated in quadruplicate.
Our pIC50 values for 9 obtained from the fluorescence-based assay are higher than the previously reported pIC50 values for AMRC272 when using a nitrocefin-based assay.22 The discrepancy likely reflects different enzymatic assay conditions, protein constructs, and enzyme purification procedures; however, it should be noted that both assays show a clear preference for inhibition of IMP-1 over VIM-2.22
Crystallography
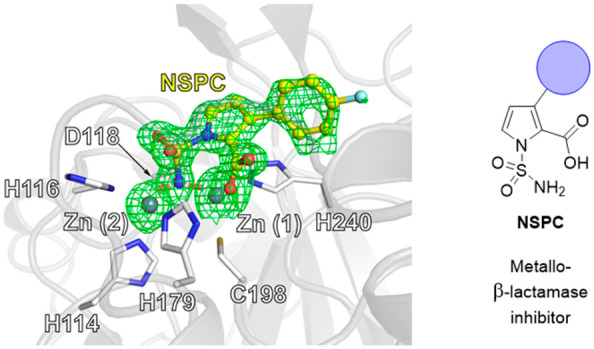
We investigated the NSPC ligand–enzyme interaction by crystallography and obtained a structure for VIM-1 complexed with 6 (space group: P121 1, 1.21 Å resolution, Figure 2). The structure was solved by molecular replacement (PDB: 5N5G),44 with iterative fitting of 6 at the active site. The two zinc ions (coordinated by H114, H116, D118 (Zn1), H179, C198, and H240(Zn2)) were refined with occupancies of 0.75 for both Zn1 and Zn2. The reduced occupancy for Zn1 is due to partial oxidation of Cys198 to a 3-sulfino alanine residue (Csd198), which was modeled and refined in a ratio of Cys (75%) to Csd (25%), as shown in previous work on VIM-1 (PDB 5FQA).45
Figure 2.

VIM-1 active site binding mode of sulfamoyl inhibitor 6 and comparison with that of a bicyclic boronate and SPC. (a) Polder omit map48 of VIM-1:Zn2:6 (PDB: 7AYJ, 1.21 Å resolution, 3.0 σ contour level) showing the NSPC sulfamoyl NH2 group replaces the dizinc bridging water. (b) Superimposition of VIM-1:Zn2:CB2 (PDB: 7AYJ, yellow) and VIM-2:Zn2:CB2 (PDB: 5FQC, teal)43 structures reveals related binding modes. Note, whereas binding of CB2 to VIM-2 has an impact on the Zn–Zn distance compared to unligated VIM-2, the effects of binding of 6 on this distance are negligible (see Supporting Information Figure S1e–g); note also that the aryl side chains of CB2 and 6 project in different directions. (c) Superimposition of VIM-1:Zn2:SPC (PDB: 7AYJ, yellow) and Vim-2:Zn2:NSPC (PDB: 6KZN, salmon)21 structures reveal the same binding mode.
Uncomplexed VIM-1:Zn2 (PDB: 5N5G)44 has a dizinc bridging hydroxide/water, as do other B1MBls.26 The NH2- of sulfamoyl group of 6 replaces this “hydrolytic” water, probably in its deprotonated form, though this cannot be discerned from the crystal structure. The C2-carboxylate of 6 ligates to Zn2, as observed in substrate derived complexes and those of inhibitors with analogously placed carboxylates, including CB2/VNRX-5133 (Figure 1).41−43
The binding mode of 6 is related to that of bicyclic boronate MBL inhibitors, (e.g., VIM-2:Zn2:CB2 (PDB: 5FQC, superimposition Figure 2b)),43 in which the binding of the two boron-bound oxygens mimics the binding modes proposed for the two oxygens in the oxyanion intermediate in MBL catalysis. However, in the NSPC complex, one of these oxygens is “replaced” by the tetrahedral sulfamoyl amino group (NR2-S-NH2 108°; NR2SO(1) 105°; NR2–S-O(2) 114°, O(1)-S-NH2 111°, see Figure S1a). Comparison of the Zn(1)–Zn(2) distances in the VIM-1:Zn2:6 and the VIM-2:Zn2:CB2 complex reveals differences. The Zn(1)–Zn(2) distance is increased in the VIM-2:Zn2:CB2 complex to 4.34 Å compared to 3.47 and 3.62 Å44 in the unligated VIM-2:Zn2 (PDB: 5N5G) and VIM-1:Zn2 (PDB: 4NQ2) complexes, respectively46 (see Supporting Information Figure S1e). Binding of 6, however, does not substantially increase the Zn(1)–Zn(2) distance, i.e. it is 3.60 Å compared to the unligated VIM-1/VIM-2. This distance is similar to those reported for a hydrolyzed VIM-1:Zn2:meropenem complex (PDB: 5N5I, Zn(1)–Zn(2): 3.50 Å, Figure S1d)44 and other unligated VIM family members.47
Antimicrobial susceptibility testing of the NSPCs in combination with Meropenem, following CLSI guidelines,49,50 was performed in a minimum inhibition concentration (MIC) antimicrobial assay format with 4 clinically relevant NDM-1 producing strains of Escherichia coli and Klebsiella pneumoniae (Table 2). At a fixed concentration of 8 μg mL–1, the N-sulfamoyl pyrroles reduce the meropenem MIC from 64 to ∼0.375 μg mL–1. In all cases, the MICs for the NSPCs were better than those for VNRX-5133. At a concentration of 0.5 μg mL–1 of the NSPC, the analogue potencies can be compared; 6 and 8–10 exhibit greater potency compared to the higher mass compounds 13 and 14, bearing 4-morpholinophenyl or bicyclic heteroaromatic groups at C3, respectively. These differences may reflect differences in uptake as the pIC50s of 13/14 against NDM-1 are consistent with the other analogues (Table 1). Furthermore, the comparative data for aminopyrimidine 11 and its saturated analogue 12 show the latter is significantly less active at 0.5 μg mL–1, in particular with the B68-1, S117, and IR47 strains, likely reflecting their relative NDM-1 pIC50 values (Table 1). These data correlate with the submicromolar/nanomolar enzymatic inhibition for NDM-1 and show the ability of the series to penetrate the cell membrane, at least in the studied Enterobacteriaceae clinical isolates.
Table 2. MIC Values of Meropenem (MEM)-SPC Combination against NDM-1 Producing Enterobacteriaceae.
| strain | MIC
(μg mL–1) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| species, genotype | MEM | [I] (μg mL–1) | MEM VNRXa | MEM 6a | MEM 6b | MEM 8 | MEM 9 | MEM 10 | MEM 11 | MEM 12 | MEM 13 | MEM 14 | |
| IR57 | E. coli | 64 | 0.5 | 1 | 1 | 0.5 | 1 | 1 | 1.5 | 4 | 4 | 1.5 | |
| blaNDM-1 | 2 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | 0.375 | ≤0.25 | 0.5 | ≤0.25 | |||
| 8 | 0.5 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | 0.375 | 0.5 | ≤0.25 | |||
| B68-1 | K. pneumoniae | 64 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 1 | 1 | 16 | 1 | 3 | |
| blaNDM-1 | 2 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | |||
| 8 | 0.5 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | 0.5 | 0.5 | 0.5 | ≤0.25 | |||
| S117 | E. coli | 256 | 0.5 | 4 | 2 | 1 | 1 | 1.5 | 1.5 | 32 | 8 | 4 | |
| blaNDM-1 | 2 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | 0.5 | ≤0.25 | |||
| 8 | 1 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | 0.5 | 0.5 | 0.5 | ≤0.25 | |||
| IR43 | K. pneumoniae | 128 | 0.5 | ≤0.25 | ≤0.25 | 0.375 | 0.5 | 0.5 | 0.5 | 16 | 0.5 | 1.5 | |
| blaNDM-1 | 2 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | |||
| 8 | 0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | ≤0.25 | 0.375 | 0.5 | ≤0.25 | |||
MIC values of meropenem (MEM) and VRNX were at 10 μg mL–1.41 MIC experiments were repeated in triplicate.
Conclusions
Our biochemical and microbiological results combined with recently published data34,22 reveal the NSPCs as promising MBL inhibitors with particularly potent (low nM) activity against NDM-1 and IMP-1 and submicromolar activity against VIM-1 and VIM-2 enzymes. The structural studies presented here define an NSPC binding mode very similar to that of the α-carboxylate- and N-sulfamoyl-substituted furans and pyrroles as MBL inhibitors described by Wachino et al.21 Thus, the likely deprotonated amino group of the tetrahedral sulfamoyl group bridges the two zinc ions replacing the hydrolytic water in a manner reflecting a tetrahedral intermediate in catalysis. The structural analyses suggest that the relatively low activity against VIM-1/VIM-2 may in part reflect differences in inhibitor (and substrate) carboxylate binding mode. Dynamics at the dizinc center of the protein, which are not observable with cryo-temperature crystal structures, may also account for the observed differences in inhibition data between the tested MBL enzymes. Indeed, the overall highly conserved active site architecture of the MBL superfamily enzymes supports a wide range of reactions, including nucleic acid hydrolysis and redox reactions, and in some cases, human MBLs are being pursued as drug targets.51,52 The compact and polar nature of the NSPCs and related scaffolds suggests that they may have wide utility as inhibitors of MBL superfamily enzymes. However, further derivatization of the NSPCs is warranted to increase potency and spectrum of activity toward the most abundant resistance causing MBLs to restore utility of important β-lactamase antibacterials.
Methods
The experimental procedures describing the synthesis and characterization of the compounds, the evaluation of their biological activity (enzyme assays, in vitro antibacterial susceptibility testing), and X-ray crystallography studies are fully described in the Supporting Information.
General Information
Commercially available reagents and solvents were from Merck or Fluorochem and were used as received. All manipulations with air- and moisture-sensitive compounds were carried out under a positive pressure of argon in flame-dried glassware. Reactions under microwave conditions were carried out in Biotage Initiator EXP microwave reactor with Robot Sixty sample processor.
Chromatographic separations/purifications were performed either using manually packed columns with Silica gel 60 (Merck, 15–40 μm) for dry column vacuum chromatography (DCVC)) or using Reveleris X2 Flash Chromatography Purification System (BÜCHI) with FlashPure Silica prepacked columns. Reactions were monitored by TLC on silica gel 60 F254 plates (Merck).
NMR spectra were acquired using a 600 MHz Bruker Avance III HD machine equipped with a 5 mm DCH cryoprobe and a 400 MHz Bruker Avance II equipped with a 5 mm BBFO probe. Chemical shifts were referenced to residual protio- and perdeuterio-solvent resonances (δH 7.26 and δC 77.16 for CDCl3; δH 2.50 and δC 39.52 for DMSO-d6) as internal standards for 1H NMR and 13C NMR spectra, respectively. 19F NMR spectra were referenced indirectly via the 2H signal of the lock substance (CDCl3 or DMSO-d6) and the Ξ(19F) value. All NMR spectra were processed with MestReNova software v. 14.1.
Low resolution mass spectrometry (LRMS) data were obtained using a Waters Acquity H-class UPLC with a Sample Manager FTN and a TUV dual wavelength detector coupled to a QDa single quadrupole analyzer using electrospray ionization (ESI). UPLC separation was achieved with a C18 reversed-phase column (Acquity UPLC BEH C18, 2.1 × 50 mm, 1.7 μm) operated at 40 °C, using a linear gradient of the binary solvent system of buffer A (H2O:MeCN:formic acid, 95:5:0.1 v/v/v%) to buffer B (MeCN:formic acid, 100:0.1 v/v%) from 0 to 100% B in 3.5 min, then 1 min at 100% B, maintaining a flow rate of 0.8 mL/min. High resolution mass spectra were recorded using a Bruker μTOF (ESI) spectrometer. The m/z values are reported in Daltons.
Analytical HPLC was carried out using an Ultimate HPLC system (Thermo Scientific) consisting of a LPG-3400A pump (1 mL/min), a WPS-3000SL autosampler, and a DAD-3000D diode array detector (220 and 254 nm) using a Gemini-NX C18 column (4.6 × 250 mm, 3 μm, 110 Å, Phenomenex); gradient elution 0 to 100% B (MeCN-H2O-TFA 90:10:0.1 v/v/v%) in solvent A (H2O-TFA 100:0.1 v/v%) over 15 min.
Preparative HPLC (prepHPLC) was carried out using an Ultimate HPLC system (Thermo Scientific) consisting of a LPG-3200BX pump (20 mL/min), a Rheodyne 9725i injector, a 10 mL loop, a MWD-300SD detector (220 and 254 nm), and an AFC-3000SD automated fraction collector using a Gemini-NX C18 column (21.2 × 250 mm, 5 μm, 110 Å, Phenomenex); gradient elution 0 to 100% B (MeCN-H2O-formic acid 90:10:0.1 v/v/v%) in solvent A (H2O-formic acid 100:0.1 v/v%) over 15 min (unless noted otherwise).
Data for both analytical and preparative HPLC were acquired and processed using Chromeleon software v. 6.80.
3-Bromo-1-(phenylsulfonyl)-1H-pyrrole (2)
N1-Sulfonylation53
Sodium hydride (2.20 g, 55.0 mmol, 60 wt %, 1.1 equiv) was added portionwise to the solution of pyrrole (3.47 mL, 50.0 mmol, 1.0 equiv) in dry DMF (150 mL) at 0 °C. The obtained mixture was stirred for 1 h at the same temperature (note for this step: a constant flow of nitrogen gas was used to reduce foaming). Benzenesulfonyl chloride (7.66 mL, 66.0 mmol, 1.2 equiv) was added slowly over 5 min at 0 °C; the cooling bath was removed, and the reaction was further stirred for 0.5 h at room temperature (starting pyrrole was consumed, TLC). The reaction mixture was carefully quenched with half-saturated NH4Cl (200 mL) at 0 °C and diluted with 200 mL of EtOAc. The organic phase was washed with water (4 × 150 mL), brine (150 mL), dried over Na2SO4, and concentrated under reduced pressure, providing 10.64 g of crude 1-(phenylsulfonyl)-1H-pyrrole as a beige solid which was taken through to the next step without further purification.
Bromination54
A solution of bromine (2.57 mL, 50 mmol, 1 equiv) in acetic acid (40 mL) was added dropwise to the solution of 1-(phenylsulfonyl)-1H-pyrrole (10.64 g, ∼50 mmol, 1 equiv) in AcOH (90 mL). The mixture was refluxed for 1 h, then cooled to rt, concentrated and coevaporated with toluene (2 × 150 mL). Purification by DCVC (5% EtOAc - heptane) afforded 11.53 g of the desired product as a purple oil which solidified upon standing. Further purification by crystallization from MeOH (20 mL) gave 7.69 g (54% from pyrrole) of 2 as a white crystalline solid.
1H NMR (400 MHz, CDCl3) δ 7.90–7.83 (m, 2H), 7.67–7.59 (m, 1H), 7.58–7.46 (m, 2H), 7.19–7.15 (m, 1H), 7.09 (t, J = 2.9 Hz, 1H), 6.29 (dd, J = 3.4, 1.6 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 138.6, 134.4, 129.7, 127.1, 121.4, 119.9, 116.5, 102.4. The analytical data are consistent with those reported in the literature.54
Benzyl 3-Bromo-1-(phenylsulfonyl)-1H-pyrrole-2-carboxylate (3)
According to a modified version of the reported procedure,34 a 1.6 M solution of n-BuLi in cyclohexane (25.0 mL, 40.0 mmol, 1.25 equiv) was slowly added to a precooled −78 °C stirred solution of i-Pr2NH (5.87 mL, 41.6 mmol, 1.3 equiv) in anhydrous THF (24 mL) under argon atmosphere. After addition was complete, the reaction mixture was stirred for 10 min at −10 °C, then recooled back to −78 °C. A solution of pyrrole derivative 2 (9.15 g, 32.0 mmol, 1 equiv) in THF (30 mL) was added over 20 min at −78 °C. The reaction flask was stirred at the same temperature for 1 h, followed by dropwise addition (∼15 min) of benzyl chloroformate (CbzCl) (8.22 mL, 57.6 mmol, 1.8 equiv) in 10 mL THF. (Note: the traces of CO2 from the CbzCl solution in THF were removed with the stream of argon before use.) The reaction mixture was stirred for 30 min at −78 °C, then slowly warmed to 0 °C (∼2 h), quenched with 50 mL of NH4Clsat, and diluted with EtOAc (100 mL) and H2O (100 mL). The aqueous layer was extracted with EtOAc (2 × 100 mL), dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by DCVC (2%, then 10% EtOAc/Hept) to give 12.16 g of crude product as an orange oil; impure fractions containing the desired compound were repurified by DCVC (2%, then 10% EtOAc/Hept) and afforded ester 3 (10.94 g, purity >80% by 1H NMR) as a pale orange oil which solidified upon storage in the refrigerator.
1H NMR (400 MHz, CDCl3) δ 7.93–7.88 (m, 2H), 7.64–7.57 (m, 1H), 7.55 (d, J = 3.4 Hz, 1H), 7.51–7.45 (m, 2H), 7.41–7.31 (m, 5H), 6.40 (d, J = 3.5 Hz, 1H), 5.28 (s, 2H). The analytical data are consistent with those reported in the literature.34
Benzyl 3-Bromo-1H-pyrrole-2-carboxylate (S1)
A 1 M solution of TBAF in THF (28.6 mL, 28.6 mmol, 1.1 equiv) was added dropwise to a solution of N-sulfonylpyrrole 3 (10.94 g, ∼26 mmol, 1 equiv) in dry THF (75 mL) at room temperature. The obtained reaction mixture was stirred for 2 h, and then H2O (100 mL) was added. The aqueous layer was extracted with EtOAc (3 × 75 mL); the organic extracts were combined, washed with water (2 × 100 mL), brine (100 mL), dried over Na2SO4, and concentrated to dryness. Purification by DCVC (5%, then 20% EtOAc/Hept) afforded two fractions containing the product:
-
1.
The less polar fraction (1.32 g) was repurified by column chromatography on SiO2 (Reveleris, 0→15% EtOAc/Hept gradient), providing 942 mg of S1 as a white solid.
-
2.
The more polar fraction (5.87 g) was crystallized from 30 mL of a 20% EtOAc/Hept mixture. The precipitate (1.08 g) was discarded, and the mother liquor was concentrated to give 4.69 g of S1 as a colorless oil, quickly solidifying upon standing.
The repurified product obtained from both fractions was of analogous purity (1H NMR) and equally acceptable for the next chemical step. Total yield −5.64 g (63% over 2 steps from 2).
1H NMR (400 MHz, CDCl3) δ 9.35 (br s, 1H), 7.50–7.44 (m, 2H), 7.43–7.31 (m, 3H), 6.85 (t, J = 3.1 Hz, 1H), 6.35 (t, J = 2.9 Hz, 1H), 5.36 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 160.1, 135.9, 128.7, 128.3, 122.9, 120.1, 115.1, 104.2, 66.5. The analytical data are consistent with those reported in the literature.34
((Benzyloxy)carbonyl)((4-(dimethyliminio)pyridin-1(4H)-yl)sulfonyl)azanide (7)
A solution of benzyl alcohol (6.28 mL, 60.6 mmol, 1.0 equiv) in CH2Cl2 (100 mL) was cooled to 0 °C followed by the dropwise addition of chlorosulfonyl isocyanate (5.21 mL, 60 mmol, 1.0 equiv). After stirring at 0 °C for 10 min, 4-(dimethylamino)pyridine (14.7 g, 120 mmol, 2 equiv) was added portionwise, and the reaction mixture allowed to warm to room temperature and stirred overnight. The resulting mixture was diluted with CH2Cl2 (100 mL), washed with water (3 × 100 mL), dried over MgSO4, filtered, and concentrated to dryness under reduced pressure to give the desired product as a white solid (18.0 g, 90%).
1H NMR (400 MHz, DMSO-d6) δ 8.51–8.43 (m, 2H), 7.37–7.27 (m, 3H), 7.27–7.22 (m, 2H), 6.98–6.90 (m, 2H), 4.87 (s, 2H), 3.22 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 157.5, 156.6, 138.4, 136.9, 128.2, 127.6, 127.6, 106.3, 65.9, 40.0. The analytical data are consistent with those reported in the literature.55
Benzyl 1-(N-((Benzyloxy)carbonyl)sulfamoyl)-3-bromo-1H-pyrrole-2-carboxylate (4)
Sodium hydride (60% in mineral oil, 822 mg, 20.6 mmol, 1.5 equiv) was added portionwise to a precooled 0 °C solution of benzyl 3-bromo-1-pyrrole-2-carboxylate (S1) (3.84 g, 13.7 mmol, 1 equiv) in dry THF (41 mL). After stirring at 0 °C for 30 min, sulfamoylating reagent 7 (5.06 g, 15.1 mmol, 1.1 equiv) was added, and the reaction mixture was heated to reflux for 4 h. After cooling to 0 °C, the reaction was quenched by the dropwise addition of water (40 mL), concentrated under reduced pressure to remove organic solvents, and extracted into ethyl acetate (3 × 100 mL). The combined organic phases were washed with brine (40 mL), dried over Na2SO4, and concentrated to dryness under reduced pressure. The residue was purified by column chromatography on SiO2 (Reveleris, 20→100% EtOAc/Hept, then 0→20% MeOH/EtOAc gradients) to give 7.03 g of the product 4 as the sodium salt as a beige foam. The residue was taken up in EtOAc (40 mL), sequentially washed with 0.5 M HClaq (2 × 40 mL), brine (40 mL), dried over Na2SO4, and evaporated to dryness in vacuo and purified by column chromatography (Reveleris, 10→50% EtOAc/heptane gradient) to afford 4.68 g (69%) of the desired compound 4 as a yellowish oil.
1H NMR (400 MHz, CDCl3) δ 7.50–7.44 (m, 3H), 7.41–7.24 (m, 9H), 6.32 (d, J = 3.4 Hz, 1H), 5.35 (s, 2H), 5.14 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 159.5, 149.6, 134.7, 134.1, 129.7, 129.1, 128.9, 128.8, 128.8, 128.7, 128.6, 121.6, 114.9, 112.4, 69.5, 68.0. The analytical data are consistent with those reported in the literature.34
General Procedure A: Suzuki–Miyaura Cross-Coupling Reaction
A microwave vial charged with bromide 4 (197 mg, 0.40 mmol, 1 equiv), the corresponding boronate (0.52 mmol, 1.3 equiv), Na2CO3 (127 mg, 1.20 mmol, 3 equiv), Pd(dppf)Cl2·DCM (16.3 mg, 0.02 mmol, 0.05 equiv) and a degassed dioxane/H2O mixture (2:1, 2 mL) was purged with argon, sealed, and stirred under microwave irradiation at 100 °C for 3 h. After cooling to rt, the reaction mixture was diluted with 2 mL of EtOAc and 2 mL of H2O; the organic phase was separated, and the aqueous layer was extracted with EtOAc (2 × 2 mL). The combined organic phases were filtered through a pad of Na2SO4 with Celite on top, and the filtrate was concentrated under reduced pressure and further purified by column chromatography on SiO2 (Reveleris purification system) to afford the desired C3 substituted pyrrole.
Sodium ((Benzyloxy)carbonyl)((2-((benzyloxy)carbonyl)-3-(4-fluorophenyl)-1H-pyrrol-1-yl)sulfonyl)azanide (5)
Use of General Procedure A with 4-fluorophenyl)boronic acid as the coupling partner gave, after purification by column chromatography (Reveleris, 20→90% EtOAc/Hept gradient), the desired compound 5 as a yellow amorphous solid in 70% yield (149 mg).
1H NMR (600 MHz, CDCl3) δ 7.55 (d, J = 3.1 Hz, 1H), 7.13 (t, J = 7.4 Hz, 1H), 7.10–7.03 (m, 5H), 7.00 (t, J = 7.6 Hz, 2H), 6.86 (dd, J = 8.4, 5.3 Hz, 2H), 6.69–6.61 (m, 4H), 5.84 (d, J = 3.1 Hz, 1H), 4.89 (s, 2H), 4.81 (s, 2H); 13C NMR (151 MHz, CDCl3) δ 162.2 (d, J = 246.5 Hz), 161.8, 158.4 (br), 136.5, 136.1, 134.5, 131.2 (d, J = 3.3 Hz), 130.9 (d, J = 8.1 Hz), 129.7, 128.44, 128.40, 128.3, 128.1, 127.9, 127.8, 118.9, 114.6 (d, J = 21.5 Hz), 110.9, 67.9, 67.2; 19F NMR (376 MHz, CDCl3) δ −115.4; LRMS (ESI) m/z: [M-Na]− calcd for C26H20FN2NaO6S 507.1, found 507.2.
General Procedure B: Hydrogenolysis of N-Cbz and O-Bn Protected Pyrrole-2-carboxylates
A one-necked round-bottom flask containing a 0.025 M methanol solution of the corresponding O-Bn- and N-Cbz-protected pyrrole (1 equiv) was charged with Pd on carbon (10% w/w, 0.2 equiv), sealed, and evacuated/backfilled with dihydrogen gas (3 times). The reaction mixture was hydrogenated at atmospheric pressure (H2 balloon) overnight under vigorous stirring, then filtered through a pad of Celite, and the filter cake was washed with methanol. The combined filtrates were concentrated to dryness under reduced pressure.
3-(4-Fluorophenyl)-1-sulfamoyl-1H-pyrrole-2-carboxylic acid (6a)
The N-Cbz sodium salt 5 (72.7 mg, 0.137 mmol) was dissolved in 10 mL of EtOAc, transferred to a separatory funnel, and successively washed with 1 M HClaq (2 × 5 mL), brine (5 mL), and dried over Na2SO4. The solvent was removed in vacuo to give S8 as a yellow oil in 96% yield (67.2 mg).
1H NMR (600 MHz, CDCl3) δ 8.69 (br s, 1H), 7.55 (d, J = 3.3 Hz, 1H), 7.38–7.34 (m, 3H), 7.34–7.28 (m, 3H), 7.27–7.22 (m, 4H, overlapped with solvent peak), 7.04–7.00 (m, 2H), 6.94–6.87 (m, 2H), 6.23 (d, J = 3.3 Hz, 1H), 5.18 (s, 2H), 5.12 (s, 2H); 19F NMR (376 MHz, CDCl3) δ −114.1.
The oil obtained above (67.2 mg) was hydrogenated according to General Procedure B; then, the crude residue was further purified by prepHPLC to give the desired compound 6a in 72% yield (27.0 mg) as a white fluffy solid.
1H NMR (600 MHz, DMSO-d6) δ 13.17 (br s, 1H), 8.17 (br s, 2H), 7.46–7.42 (m, 2H), 7.42 (d, J = 3.2 Hz, 1H), 7.24–7.18 (m, 2H), 6.37 (d, J = 3.2 Hz, 1H); 13C NMR (151 MHz, DMSO-d6) δ 162.5, 161.5 (d, J = 244.1 Hz), 131.7, 130.75 (d, J = 3.4 Hz), 130.69 (d, J = 8.2 Hz), 125.2, 120.8, 114.8 (d, J = 21.4 Hz), 110.7; 19F NMR (376 MHz, DMSO-d6) δ −115.8; HPLC analysis tR 11.3 min, purity >99.6%; HRMS (ESI) m/z: [M-H]− calcd for C11H8FN2O4S 283.0194, found 283.0189.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.1c00104.
Synthesis and characterization of compounds and information on enzymatic and microbiological assays and crystallography (PDF)
Author Contributions
§ A.J.M.F. and Y.E. contributed equally.
We thank our coworkers and collaborators and the Innovative Medicines Initiative (European Lead factory and ENABLE components), the Medical Research Council, the Wellcome Trust, Cancer Research UK, and the Ineos Institute for Antimicrobial Research for funding our work on antibiotics, MBL fold/metalloenzymes, and β-lactamase inhibitors. This research was funded in whole or in part by the Wellcome Trust (Grant 106244/Z/14/Z).
The authors declare no competing financial interest.
Supplementary Material
References
- Munita J. M.; Arias C. A. (2016) Mechanisms of Antibiotic Resistance. Microbiol. Spectrum 4, 1. 10.1128/microbiolspec.VMBF-0016-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demain A. L.; Sanchez S. (2009) Microbial drug discovery: 80 years of progress. J. Antibiot. 62, 5–16. 10.1038/ja.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp-Wallace K. M.; Endimiani A.; Taracila M. A.; Bonomo R. A. (2011) Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 55, 4943–4960. 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K. (2013) Proliferation and significance of clinically relevant β-lactamases. Ann. N. Y. Acad. Sci. 1277, 84–90. 10.1111/nyas.12023. [DOI] [PubMed] [Google Scholar]
- Perez F.; Endimiani A.; Hujer K. M.; Bonomo R. A. (2007) The continuing challenge of ESBLs. Curr. Opin. Pharmacol. 7, 459–469. 10.1016/j.coph.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans B. A.; Amyes S. G. B. (2014) OXA β-Lactamases. Clin. Microbiol. Rev. 27, 241–263. 10.1128/CMR.00117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K.; Bradford P. A. (2019) Interplay between β-lactamases and new β-lactamase inhibitors. Nat. Rev. Microbiol. 17, 295–306. 10.1038/s41579-019-0159-8. [DOI] [PubMed] [Google Scholar]
- Papp-Wallace K. M.; Endimiani A.; Taracila M. A.; Bonomo R. A. (2011) Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 55, 4943–4960. 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T. R.; Toleman M. A.; Poirel L.; Nordmann P. (2005) Metallo-β-Lactamases: the Quiet before the Storm?. Clin. Microbiol. Rev. 18, 306–325. 10.1128/CMR.18.2.306-325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T. R. (2005) The emergence and implications of metallo-beta-lactamases in Gram-negative bacteria. Clin. Microbiol. Infect. 11 (6), 2–9. 10.1111/j.1469-0691.2005.01264.x. [DOI] [PubMed] [Google Scholar]
- Abboud M. I.; Damblon C.; Brem J.; Smargiasso N.; Mercuri P.; Gilbert B.; Rydzik A. M.; Claridge T. D. W.; Schofield C. J.; Frère J.-M. (2016) Interaction of Avibactam with Class B Metallo-β-Lactamases. Antimicrob. Agents Chemother. 60, 5655–5662. 10.1128/AAC.00897-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queenan A. M.; Bush K. (2007) Carbapenemases: the Versatile β-Lactamases. Clin. Microbiol. Rev. 20, 440–458. 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somboro A. M.; Osei Sekyere J.; Amoako D. G.; Essack S. Y.; Bester L. A. (2018) Diversity and Proliferation of Metallo-β-Lactamases: a Clarion Call for Clinically Effective Metallo-β-Lactamase Inhibitors. Appl. Environ. Microbiol. 84, e00698–00618. 10.1128/AEM.00698-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palzkill T. (2013) Metallo-β-lactamase structure and function. In Antimicrobial Therapeutics Reviews: The Bacterial Cell Wall as an Antimicrobial Target; Bush K., Ed.; Vol. 1277, p 91–104, Blackwell, Boston, MA. [Google Scholar]
- Buynak J. D. (2013) β-Lactamase inhibitors: a review of the patent literature (2010 – 2013). Expert Opin. Ther. Pat. 23, 1469–1481. 10.1517/13543776.2013.831071. [DOI] [PubMed] [Google Scholar]
- Palacios A. R.; Rossi M.-A.; Mahler G. S.; Vila A. J. (2020) Metallo-β-Lactamase Inhibitors Inspired on Snapshots from the Catalytic Mechanism. Biomolecules 10, 854. 10.3390/biom10060854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy N.; Shungube M.; Arvidsson P. I.; Baijnath S.; Kruger H. G.; Govender T.; Naicker T. (2020) A 2018–2019 patent review of metallo beta-lactamase inhibitors. Expert Opin. Ther. Pat. 30, 541–555. 10.1080/13543776.2020.1767070. [DOI] [PubMed] [Google Scholar]
- Panduwawala T., Brandt P., Wang D., Andaloussi M., Brem J., and Schofield C. J. (2018) Inhibitors of metallo-beta-lactamases. WO2018215799A1.
- Davies D. T.; Leiris S.; Sprynski N.; Castandet J.; Lozano C.; Bousquet J.; Zalacain M.; Vasa S.; Dasari P. K.; Pattipati R.; Vempala N.; Gujjewar S.; Godi S.; Jallala R.; Sathyap R. R.; Darshanoju N. A.; Ravu V. R.; Juventhala R. R.; Pottabathini N.; Sharma S.; Pothukanuri S.; Holden K.; Warn P.; Marcoccia F.; Benvenuti M.; Pozzi C.; Mangani S.; Docquier J.-D.; Lemonnier M.; Everett M. (2020) ANT2681: SAR Studies Leading to the Identification of a Metallo-β-lactamase Inhibitor with Potential for Clinical Use in Combination with Meropenem for the Treatment of Infections Caused by NDM-Producing Enterobacteriaceae. ACS Infect. Dis. 6, 2419–2430. 10.1021/acsinfecdis.0c00207. [DOI] [PubMed] [Google Scholar]
- McGeary R. P.; Tan D. T. C.; Selleck C.; Monteiro Pedroso M.; Sidjabat H. E.; Schenk G. (2017) Structure-activity relationship study and optimization of 2-aminopyrrole-1-benzyl-4,5-diphenyl-1H-pyrrole-3-carbonitrile as a broad spectrum metallo-β-lactamase inhibitor Eur. Eur. J. Med. Chem. 137, 351–364. 10.1016/j.ejmech.2017.05.061. [DOI] [PubMed] [Google Scholar]
- Wachino J.; Jin W. C.; Kimura K.; Kurosaki H.; Sato A.; Arakawa Y. (2020) Sulfamoyl Heteroarylcarboxylic Acids as Promising Metallo-beta-Lactamase Inhibitors for Controlling Bacterial Carbapenem Resistance. mBio 11, e03144–03119. 10.1128/mBio.03144-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi N.; Lee V. E.; Chalam-Judge N.; Newman R.; Wilkinson A. J.; Cooper I. R.; Orr D.; Lee S.; Savage V. J. (2021) Restoring carbapenem efficacy: a novel carbapenem companion targeting metallo-β-lactamases in carbapenem-resistant Enterobacterales. J. Antimicrob. Chemother. 76, 460–466. 10.1093/jac/dkaa455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta F.; Supuran C. T.; Scozzafava A. (2014) Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med. Chem. 6, 1149–1165. 10.4155/fmc.14.68. [DOI] [PubMed] [Google Scholar]
- Winum J.-Y.; Scozzafava A.; Montero J.-L.; Supuran C. T. (2006) Therapeutic potential of sulfamides as enzyme inhibitors. Med. Res. Rev. 26, 767–792. 10.1002/med.20068. [DOI] [PubMed] [Google Scholar]
- Park J. D.; Kim D. H.; Kim S.-J.; Woo J.-R.; Ryu S. E. (2002) Sulfamide-Based Inhibitors for Carboxypeptidase A. Novel Type Transition State Analogue Inhibitors for Zinc Proteases. J. Med. Chem. 45, 5295–5302. 10.1021/jm020258v. [DOI] [PubMed] [Google Scholar]
- Krajnc A.; Lang P. A.; Panduwawala T. D.; Brem J.; Schofield C. J. (2019) Will morphing boron-based inhibitors beat the beta-lactamases?. Curr. Opin. Chem. Biol. 50, 101–110. 10.1016/j.cbpa.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radkiewicz J. L.; McAllister M. A.; Goldstein E.; Houk K. N. (1998) A Theoretical Investigation of Phosphonamidates and Sulfonamides as Protease Transition State Isosteres. J. Org. Chem. 63, 1419–1428. 10.1021/jo971425f. [DOI] [Google Scholar]
- Obreza A.; Gobec S. (2004) Recent Advances in Design, Synthesis and Biological Activity of Aminoalkylsulfonates and Sulfonamidopeptides. Curr. Med. Chem. 11, 3263–3278. 10.2174/0929867043363659. [DOI] [PubMed] [Google Scholar]
- Brem J.; van Berkel S. S.; Zollman D.; Lee S. Y.; Gileadi O.; McHugh P. J.; Walsh T. R.; McDonough M. A.; Schofield C. J. (2016) Structural Basis of Metallo-β-Lactamase Inhibition by Captopril Stereoisomers. Antimicrob. Agents Chemother. 60, 142–150. 10.1128/AAC.01335-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K.; Yanagihara K.; Kaku N.; Harada Y.; Migiyama Y.; Nagaoka K.; Morinaga Y.; Nakamura S.; Imamura Y.; Miyazaki T.; Izumikawa K.; Kakeya H.; Hasegawa H.; Yasuoka A.; Kohno S. (2013) In vivo efficacy of biapenem with ME1071, a novel metallo-β-lactamase (MBL) inhibitor, in a murine model mimicking ventilator-associated pneumonia caused by MBL-producing Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 42, 238–243. 10.1016/j.ijantimicag.2013.05.016. [DOI] [PubMed] [Google Scholar]
- Liu B.; Trout R. E. L.; Chu G.-H.; McGarry D.; Jackson R. W.; Hamrick J. C.; Daigle D. M.; Cusick S. M.; Pozzi C.; De Luca F.; Benvenuti M.; Mangani S.; Docquier J.-D.; Weiss W. J.; Pevear D. C.; Xerri L.; Burns C. J. (2020) Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- and Metallo-β-lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections. J. Med. Chem. 63, 2789–2801. 10.1021/acs.jmedchem.9b01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett F., Jiang J., Pasternak A., Dong S., Gu X., Scott J. D., Tang H., Zhao Z., Huang Y., Hunter D., Yang D., Young K., Xiao L., Zhang Z., Fu J., Bai Y., Zheng Z., and Zhang X.. Metallo-β-lactamase inhibitors. WO2016/039185, 2016.
- Brem J., Rydzik A. M., McDonough M. A., Schofield C. J., Morrison A., Hewitt J., Pannifer A., and Jones P.. Preparation of substituted indole-2-carboxylic acids as inhibitors of metallo-beta-lactamases. WO 2017093727, A1, 2017.
- Wilkinson A., Cooper I., Orr D., Finlayson J., Bunt A., Appelqvist P., Wallberg H., and Wångsell F.. Antibacterial compounds. WO 2019220125, 2019.
- Dunn G. E.; Lee G. K. J. (1971) Kinetics and Mechanism of Decarboxylation of Pyrrole-2-Carboxylic Acid in Aqueous Solution. Can. J. Chem. 49, 1032–1035. 10.1139/v71-172. [DOI] [Google Scholar]
- Mundle S. O. C.; Kluger R. (2009) Decarboxylation via Addition of Water to a Carboxyl Group: Acid Catalysis of Pyrrole-2-Carboxylic Acid. J. Am. Chem. Soc. 131, 11674–11675. 10.1021/ja905196n. [DOI] [PubMed] [Google Scholar]
- Wuts P. G. M. (2014) Protection for the Amino Group. In Greene’s Protective Groups in Organic Synthesis; Wuts P. G. M., Ed., Chapter 7, p 895–1193, Wiley, New York. [Google Scholar]
- Lu X.; Zhang H.; Tonge P. J.; Tan D. S. (2008) Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorg. Med. Chem. Lett. 18, 5963–5966. 10.1016/j.bmcl.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biftu T., Huang X., Liu W., Pan W., Park M., Pasternak A., Sun W., Tang H., and Zang Y.. Synthesis of chromane monobactam compounds for the treatment of bacterial infections. WO 2019070492, A1, 2019.
- van Berkel S. S.; Brem J.; Rydzik A. M.; Salimraj R.; Cain R.; Verma A.; Owens R. J.; Fishwick C. W. G.; Spencer J.; Schofield C. J. (2013) Assay Platform for Clinically Relevant Metallo-beta-lactamases. J. Med. Chem. 56, 6945–6953. 10.1021/jm400769b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajnc A.; Brem J.; Hinchliffe P.; Calvopina K.; Panduwawala T. D.; Lang P. A.; Kamps J.; Tyrrell J. M.; Widlake E.; Saward B. G.; Walsh T. R.; Spencer J.; Schofield C. J. (2019) Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-beta-Lactamases. J. Med. Chem. 62, 8544–8556. 10.1021/acs.jmedchem.9b00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill S. T.; Cain R.; Wang D. Y.; Lohans C. T.; Wareham D. W.; Oswin H. P.; Mohammed J.; Spencer J.; Fishwick C. W.; McDonough M. A.; Schofield C. J.; Brem J. (2017) Cyclic Boronates Inhibit All Classes of beta-Lactamases. Antimicrob. Agents Chemother. 61, e02260–16. 10.1128/AAC.02260-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem J.; Cain R.; Cahill S.; McDonough M. A.; Clifton I. J.; Jimenez-Castellanos J. C.; Avison M. B.; Spencer J.; Fishwick C. W.; Schofield C. J. (2016) Structural basis of metallo-beta-lactamase, serine-beta-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat. Commun. 7, 12406. 10.1038/ncomms12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimraj R.; Hinchliffe P.; Kosmopoulou M.; Tyrrell J. M.; Brem J.; Berkel S. S.; Verma A.; Owens R. J.; McDonough M. A.; Walsh T. R.; Schofield C. J.; Spencer J. (2019) Crystal structures of VIM-1 complexes explain active site heterogeneity in VIM-class metallo-beta-lactamases. FEBS J. 286, 169–183. 10.1111/febs.14695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill S. T.; Tarhonskaya H.; Rydzik A. M.; Flashman E.; McDonough M. A.; Schofield C. J.; Brem J. (2016) Use of ferrous iron by metallo-β-lactamases. J. Inorg. Biochem. 163, 185–193. 10.1016/j.jinorgbio.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitha M.; Marts A. R.; Bergstrom A.; Møller A. J.; Moritz L.; Turner L.; Nix J. C.; Bonomo R. A.; Page R. C.; Tierney D. L.; Crowder M. W. (2014) Biochemical, Mechanistic, and Spectroscopic Characterization of Metallo-β-lactamase VIM-2. Biochemistry 53, 7321–7331. 10.1021/bi500916y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meini M.-R.; Llarrull L. I.; Vila A. J. (2015) Overcoming differences: The catalytic mechanism of metallo-β-lactamases. FEBS Lett. 589, 3419–3432. 10.1016/j.febslet.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Moriarty N. W.; Poon B. K.; Sobolev O. V.; Terwilliger T. C.; Adams P. D. (2017) Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol. 73, 148–157. 10.1107/S2059798316018210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinical and Laboratory Standards Institute (2018) W. P. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. M07–11th ed.
- Clinical and Laboratory Standards Institute (2018) W. P. Performance standards for antimicrobial susceptibility testing.
- Pettinati I.; Brem J.; Lee S. Y.; McHugh P. J.; Schofield C. J. (2016) The Chemical Biology of Human Metallo-β-Lactamase Fold Proteins. Trends Biochem. Sci. 41, 338–355. 10.1016/j.tibs.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z. (2007) Nucleases of the Metallo-β-lactamase Family and Their Role in DNA and RNA Metabolism. Crit. Rev. Biochem. Mol. Biol. 42, 67–93. 10.1080/10409230701279118. [DOI] [PubMed] [Google Scholar]
- Steeds H. G.; Knowles J. P.; Yu W. L.; Richardson J.; Cooper K. G.; Booker-Milburn K. I. (2020) Rapid Access to Azabicyclo[3.3.1]nonanes by a Tandem Diverted Tsuji–Trost Process. Chem. - Eur. J. 26 (63), 14330–14334. 10.1002/chem.202003762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T.; Fukuda T.; Ishibashi F.; Iwao M. (2009) Design and Synthesis of Lamellarin D Analogues Targeting Topoisomerase I. J. Org. Chem. 74 (21), 8143–8153. 10.1021/jo901589e. [DOI] [PubMed] [Google Scholar]
- Lu X.; Zhang H.; Tonge P. J.; Tan D. S. (2008) Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorg. Med. Chem. Lett. 18 (22), 5963–5966. 10.1016/j.bmcl.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.