

Abstract
Coenzyme A (CoA) is a ubiquitous cofactor present in all living cells and estimated to be required for up to 9% of intracellular enzymatic reactions. Mycobacterium tuberculosis (Mtb) relies on its own ability to biosynthesize CoA to meet the needs of the myriad enzymatic reactions that depend on this cofactor for activity. As such, the pathway to CoA biosynthesis is recognized as a potential source of novel tuberculosis drug targets. In prior work, we genetically validated CoaBC as a bactericidal drug target in Mtb in vitro and in vivo. Here, we describe the identification of compound 1f, a small molecule inhibitor of the 4′-phosphopantothenoyl-l-cysteine synthetase (PPCS; CoaB) domain of the bifunctional Mtb CoaBC, and show that this compound displays on-target activity in Mtb. Compound 1f was found to inhibit CoaBC uncompetitively with respect to 4′-phosphopantothenate, the substrate for the CoaB-catalyzed reaction. Furthermore, metabolomic profiling of wild-type Mtb H37Rv following exposure to compound 1f produced a signature consistent with perturbations in pantothenate and CoA biosynthesis. As the first report of a direct small molecule inhibitor of Mtb CoaBC displaying target-selective whole-cell activity, this study confirms the druggability of CoaBC and chemically validates this target.
Keywords: tuberculosis, drug discovery, coenzyme A, CoaBC
Tuberculosis (TB) remains the leading cause of death from an infectious disease worldwide, claiming an estimated 1.4 million lives in 2019.1 The high disease burden coupled with the ongoing emergence and spread of strains of Mycobacterium tuberculosis (Mtb) resistant to first- and second-line TB drugs underscores the urgent need to develop new antitubercular agents for the treatment of both drug-resistant and drug-susceptible forms of the disease.2,3 In recent years, considerable progress has been made in developing a TB drug pipeline,4 which has begun to deliver promising new drugs and drug combinations.3,5−8 However, as attrition rates are high across the entire TB drug pipeline there is an urgent need for replenishment of the pipeline, starting from the earliest stage of hit identification. This need is driving drug discovery efforts, which are aimed at delivering high-quality hit compounds with novel mechanisms of action.9−14
The coenzyme A (CoA) biosynthetic pathway has attracted attention as a source of new drug targets in a number of bacterial pathogens,15−20 including Mtb,21 owing to its ubiquity and the lack of sequence similarity between prokaryotic CoA biosynthesis enzymes and their eukaryotic counterparts.22 CoA serves as a cofactor for key metabolic enzymes involved in numerous cellular pathways, with 9% of all enzymes estimated as being dependent on this essential cofactor.18,23 Of particular significance in the case of Mtb is the critical role played by CoA in the biosynthesis of lipids,21 which comprise essential components of the cell envelope, and virulence factors,24 as well as in the catabolism of lipids, which provides the primary source of energy for the bacillus during infection.25 The availability of crystal structures of a number of prokaryotic CoA pathway enzymes has enabled structure-guided inhibitor design.26−29 However, although potent direct inhibitors of Mtb pantothenate synthetase (PanC, Rv3602c) and pantothenate kinase (PanK, CoaA, Rv1092c) have been developed using this approach, these have failed to translate into lead compounds with significant whole-cell activity against Mtb.29−32
The synthesis of CoA from its natural precursor, pantothenate (vitamin B5, Pan), occurs in five universal enzymatic reactions, the second and third of which are carried out by a bifunctional CoaBC enzyme encoded by a single gene in most bacteria, including Mtb (Rv1391).33 This enzyme comprises a C-terminal CoaB domain with 4′-phosphopantothenoyl-l-cysteine synthetase (PPCS) activity and an N-terminal CoaC domain with 4′-phosphopantothenoyl-l-cysteine decarboxylase (PPCDC) activity, which together catalyze the conversion of 4′-phosphopantothenate (PPA) into 4′-phosphopantetheine (P-PantSH)34 (Figure 1). The reaction occurs in three steps, the first two of which are carried out by CoaB to produce a 4′-phosphopantothenoyl-l-cysteine intermediate,35 which is subsequently decarboxylated at the cysteine moiety by CoaC to produce P-PantSH.36 In prior work, we genetically validated CoaBC as a drug target by demonstrating that coaBC silencing is bactericidal in Mtb in vitro as well as during both the acute and chronic stages of infection in mice.33 However, reports of potent and selective small molecule inhibitors of bacterial CoaBC enzymes are scarce, and those compounds identified have largely failed to display significant whole-cell activity.37,38 Importantly, the druggability of prokaryotic CoaBC enzymes was established by the identification of the natural product CJ-15,801, which acts as the precursor to a tight-binding CoaB (PPCS) inhibitor with potent activity against Staphylococcus aureus.19 Furthermore, the recent identification of a small molecule inhibitor of Mtb phosphopantetheinyl transferase (PptT),24 required for the transfer of the P-PantSH moiety of CoA to acyl carrier proteins (ACPs) involved in lipid production in Mtb, and the identification of PanD as the target of the bioactive form of pyrazinamide39,40 have validated the utility of key enzymes involved in CoA metabolism as antitubercular drug targets.
Figure 1.
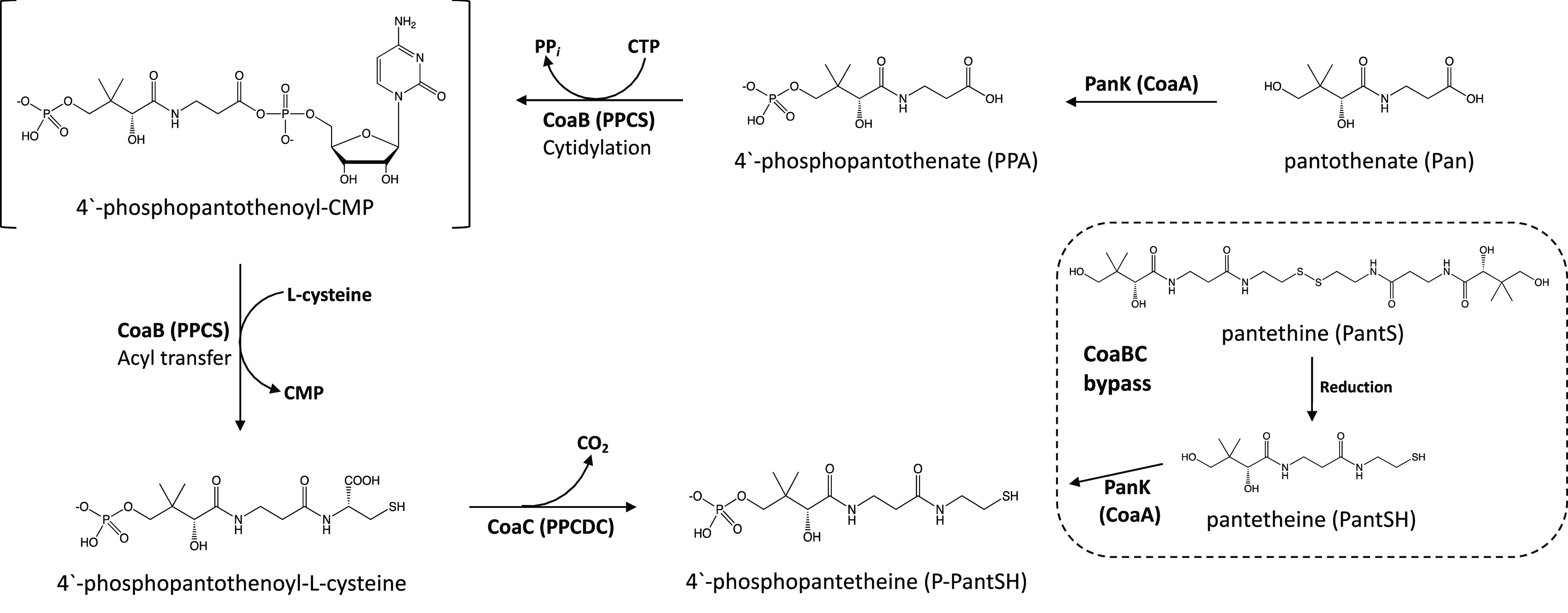
Reaction catalyzed by CoaBC in Mtb. The CoaB domain of the bifunctional protein converts 4′-phosphopantothenate (PPA) into 4′-phosphopantothenoyl-l-cysteine in two steps via the production of a 4′-phosphopantothenoyl-CMP intermediate. Decarboxylation of 4′-phosphopantothenoyl-l-cysteine by the CoaC domain results in the production of 4′-phosphopantetheine (P-PantSH). CoaBC bypass occurs via PanK-mediated phosphorylation of pantetheine (PantSH) to produce P-PantSH. PPCS, phosphopantothenoyl-l-cysteine synthetase; PPCDC, phosphopantothenoyl-l-cysteine decarboxylase.
We recently reported the first crystal structure of the mycobacterial CoaBC together with the first potent and selective inhibitors of Mtb CoaB.41 However, these potent inhibitors of CoaB activity displayed limited whole-cell activity against Mtb due to impaired permeability/efflux and intracellular metabolism.41 In this study, we confirm the druggability of Mtb CoaBC by identifying a new CoaB inhibitor that confers whole-cell activity against Mtb and providing evidence linking the growth inhibitory effects of this compound on Mtb to inhibition of CoaBC.
Results
High-Throughput Screening to Identify Inhibitors of Mycobacterial CoaB
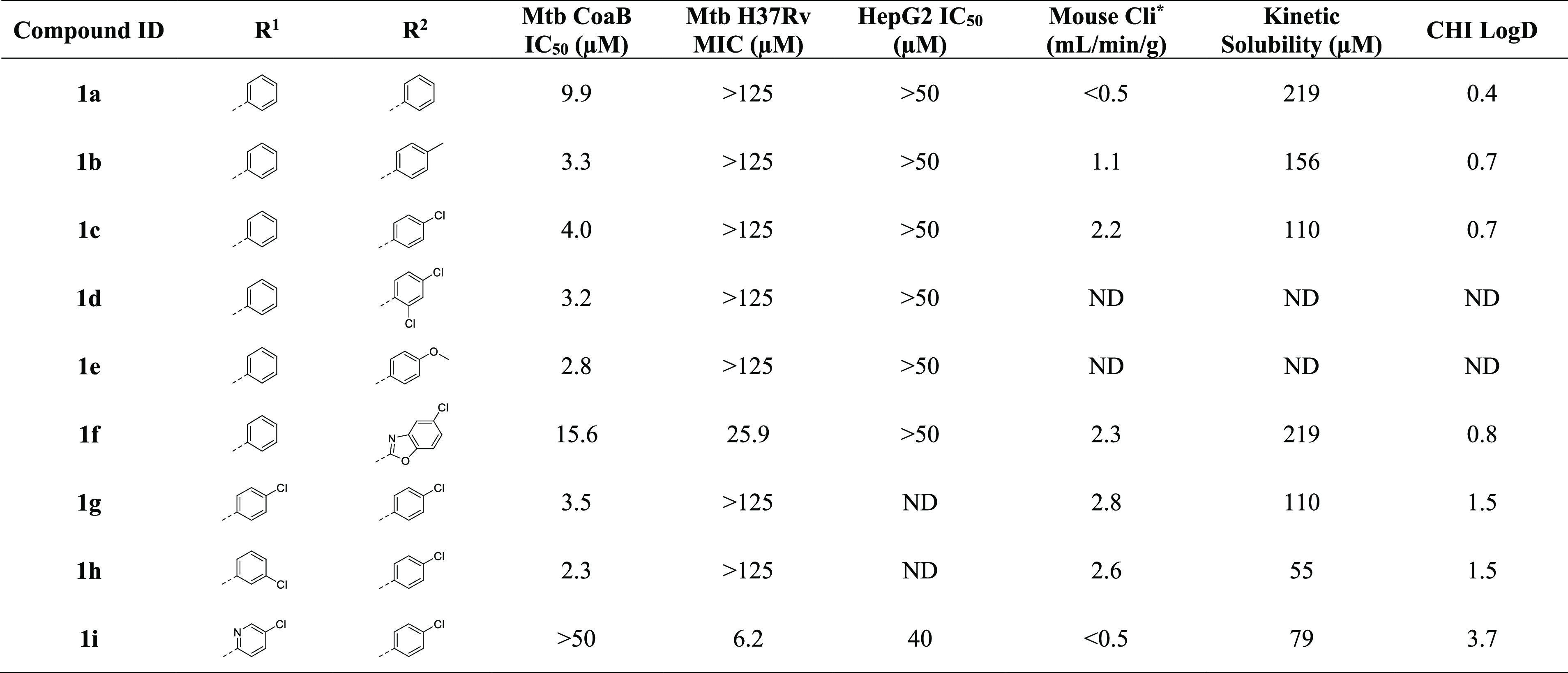
High-throughput screening of 215 000 small molecules from the DDU compound library was carried out to identify inhibitors of Mtb CoaB activity using an adaptation of the BIOLMOL Green end-point pyrophosphate quantification assay, as previously described.41 This compound library predominantly comprises commercially available molecules representing a wide range of chemical space that possessing the favorable physicochemical and molecular properties required for a potential preclinical drug candidate, with a small subset of the library (<0.5%) comprising proprietary compounds with known phenotypic activity against a number of various pathogens. The primary screen, which had an overall hit rate of 0.6%, led to the identification of compound 1a, with an IC50 of 9.9 μM against Mtb CoaB. Following confirmation of the target-specific inhibitory activity of 1a by demonstration of its inactivity in a counter-screen designed to eliminate false positive hits arising due to inactivation of the pyrophosphatase, a further 8 series analogues were synthesized, with the initial structure–activity relationship (SAR) investigation focused on probing the pharmacophore to understand the impact of the N-phenyl and S-phenyl substituents on their Mtb CoaB activity (Table 1). Replacement of the S-phenyl group of 1a with either 4-methylphenyl (1b), 4-chlorophenyl (1c), or 2,4-dichlorophenyl (1d) led to modest improvements in activity against Mtb CoaB, while introduction of 4-methoxyphenyl (1e) led to a 4-fold increase in potency. Conversely, introduction of a substituted benzoxazole (1f) led to a modest reduction in potency against Mtb CoaB (Table 1). Introduction of either a 3-chlorophenyl (1h) or 4-chlorophenyl (1g) at the N1-position was tolerated with respect to Mtb CoaB activity, while introduction of a chloropyridine at this position (1i) led to a complete loss of inhibitory activity (Table 1).
Table 1. SAR and In Vitro Profiling of Modifications to Compound 1aa.
Intrinsic clearance (Cli) using CD1 mouse liver microsomes. ND = not determined. CHI, chromatographic hydrophobicity index.
Assessment of the pharmacological properties of the compounds revealed that none were overly lipophilic, with all except 1i having CHI LogD values ≤1.5, and their kinetic solubility limits ranged between 55 and 219 μM. The compounds showed good stability in mouse liver microsomes, all having clearance values of ≤2.8 (mL/min)/g and therefore meeting our internal requirement of <5 (mL/min)/g for progression of an early hit compound. None of the compounds showed significant cytotoxicity against HepG2 cells (IC50 > 50 μM) with the exception of 1i, which showed modest activity (IC50 = 40 μM).
Whole-Cell Screening of Series Analogues against Mtb In Vitro
Minimum inhibitory concentrations (MICs) of the 9 compounds were determined for wild-type Mtb (strain H37RvMA)42 grown in Middlebrook 7H9 media supplemented with albumin-dextrose-catalase (ADC) and glycerol. With the exception of 1f and 1i, none of the compounds displayed whole-cell activity against replicating Mtb at concentrations up to 125 μM (Table 1). Although the introduction of a substituted benzoxazole did not substantially affect the activity of 1f in the biochemical assay, this modification resulted in a modest improvement in whole-cell activity against Mtb (MIC = 25.9 μM) (Table 1). The introduction of a chloropyridine at the N1-position in 1i led to complete ablation of activity in the biochemical CoaB assay; interestingly, this resulted in significantly improved activity against Mtb (MIC = 6.2 μM), thus implicating another target(s) in the mechanism of antimycobacterial action of this analogue.
Assessment of Target Selectivity in Mtb Using a coaBC Hypomorph
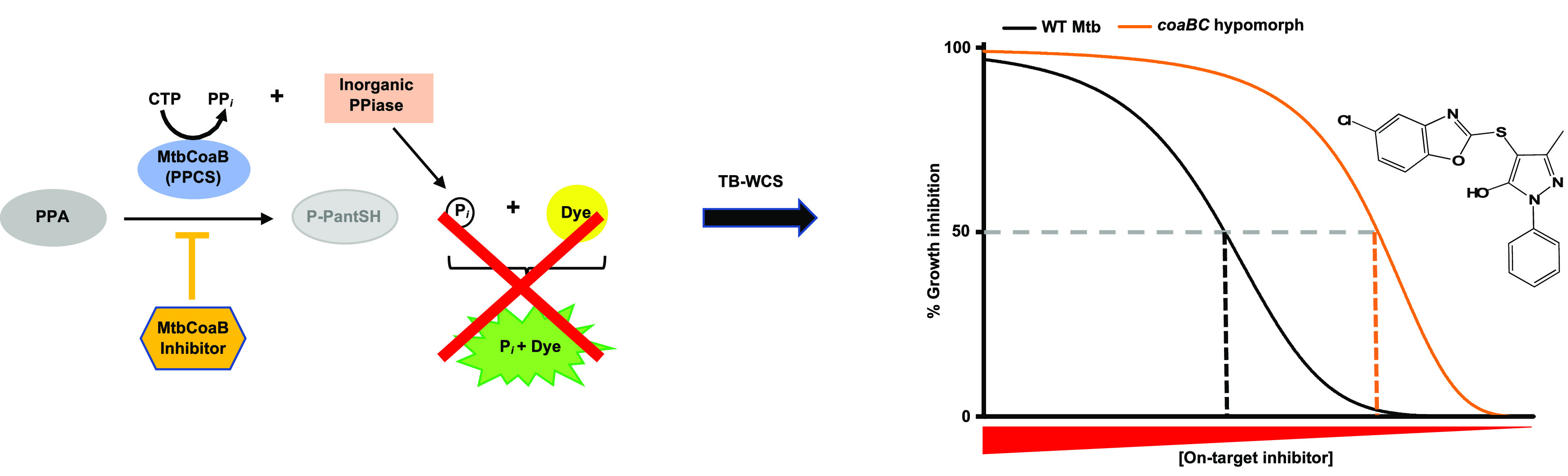
As reported previously, we and others have demonstrated the utility of conditional knockdown mutants (hypomorphs) as tools to ascertain the target selectivity of inhibitors in Mtb by means of target-based whole-cell screening.43−48 This approach entails assessing whether cellular depletion of the target of interest increases the potency of compounds with biochemical activity against that target. The parent compound and all analogues were subjected to screening in checkerboard assays using a coaBC Tet-OFF hypomorph33 in which coaBC expression is under control of a tetracycline-regulated promoter such that progressive transcriptional silencing of coaBC occurs upon addition of increasing concentrations of the inducer, anhydrotetracycline (ATc). The presence of ATc had no discernible effect on the potency of 8 of the 9 compounds against the coaBC hypomorph at inhibitor concentrations up to 125 μM. While the lack of hypersensitization of the hypomorph in the case of compound 1i (Figure 2a) is expected given its lack of biochemical activity against CoaB, the lack of hypersensitization of the hypomorph to 1a–1e, 1g, and 1h suggests that the inability of these compounds to inhibit growth of Mtb is likely attributable to compound permeation, efflux, and/or metabolism. In contrast, progressive transcriptional silencing of coaBC in the presence of increasing concentrations of ATc induced dose-dependent hypersensitization of the coaBC hypomorph to 1f, resulting in a ∼7-fold decrease in MIC in the presence of 12.5 ng/mL ATc, an observation indicative of on-target activity in Mtb (Figure 2b). Conversely, ATc had no effect on the activity of 1f against wild-type Mtb H37RvMA (Figure 2c), which had a MIC equivalent to that observed for the coaBC hypomorph at low ATc concentrations and in its absence (0–3.2 ng/mL) (Figure 2b). This result is consistent with the fact that the level of coaBC expression in the coaBC Tet-OFF hypomorph in the absence of ATc is similar to that observed in wild-type Mtb.33 Moreover, ATc did not render the coaBC hypomorph more susceptible to the first-line antitubercular drugs rifampicin and isoniazid (Figure 2d,e) whose mechanisms of action are unrelated to CoA biosynthesis or utilization. Therefore, coaBC transcriptional silencing selectively renders Mtb hypersensitive to 1f, suggesting that this compound mediates growth inhibition of Mtb, at least in part, via inhibition of CoaBC activity.
Figure 2.
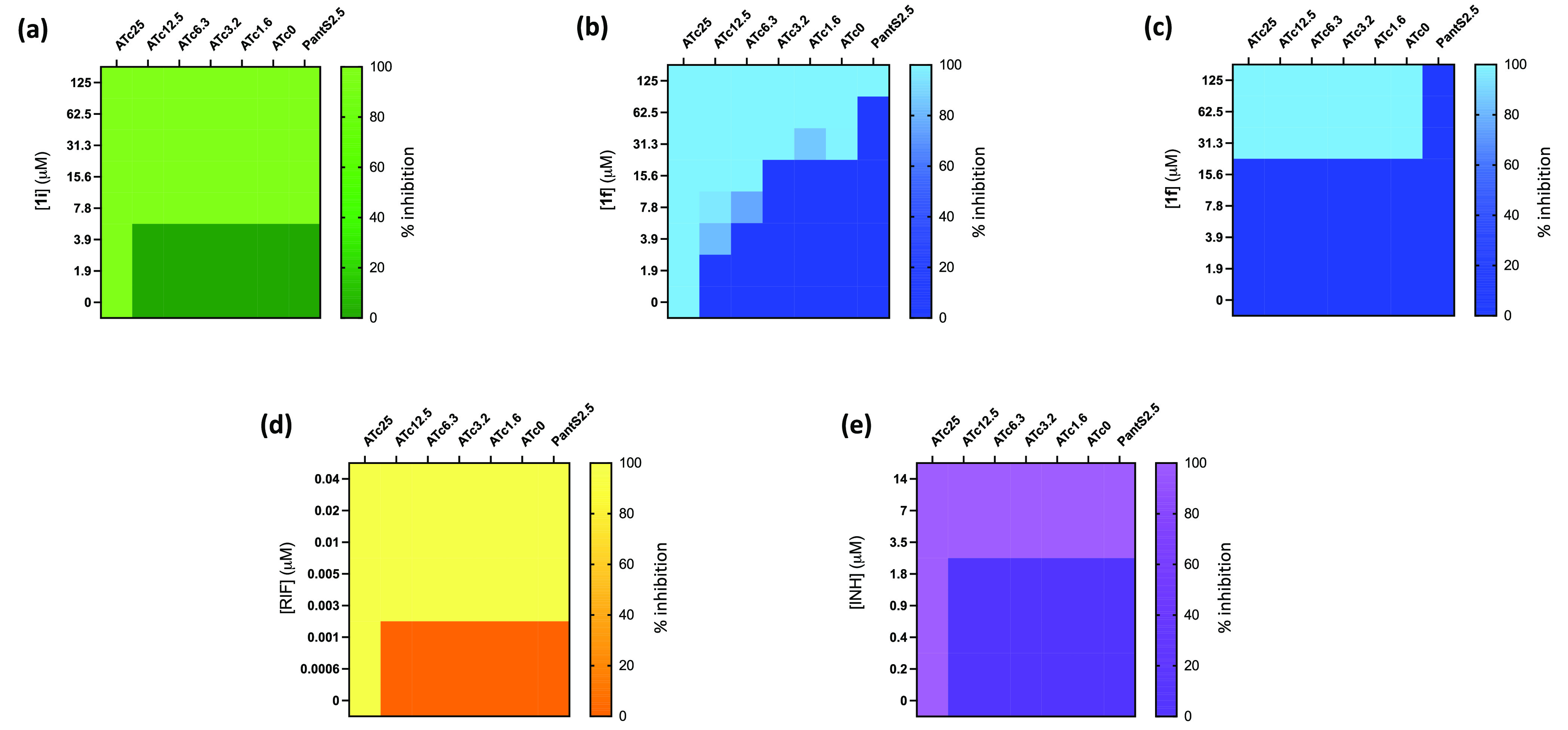
Target-based whole-cell screening of compounds 1i and 1f against a coaBC Tet-OFF hypomorph. The impact of ATc-mediated transcriptional silencing of coaBC on the relative growth of a coaBC hypomorph (a,b; d,e) and wild-type Mtb H37RvMA (c) in the presence of increasing concentrations of 1i (a), 1f (b, c), rifampicin (d), and isoniazid (e) was determined. Data are representative of the mean and SD of independent triplicates. ATc, anhydrotetracycline; Tet-OFF, mutant whose growth is inhibited in the presence of ATc; PantS2.5, pantethine (2.5 mg/mL); RIF, rifampicin; INH, isoniazid.
Confirmation of the Target Selectivity of Compound 1f in Mtb by CoaBC Bypass
We previously confirmed the functionality of CoaBC bypass in Mtb via phosphorylation of pantetheine (PantSH), the reduced form of pantethine (PantS), by the Type I PanK (CoaA) to produce P-PantSH, the product of the CoaBC-catalyzed reaction (Figure 1).33,37,49 It therefore follows that if 1f is acting by inhibition of CoaBC supplementation of the growth medium with exogenous PantS should rescue Mtb from the inhibitory effects of this compound. Indeed, supplementation with PantS rescued the wild-type strain from 1f toxicity (Figure 2c) and led to a 4-fold reduction in susceptibility of the coaBC hypomorph to 1f, although rescue of the hypomorph was not observed at the highest concentration of 1f tested (Figure 2b). To exclude the possibility that conversion of PantS to Pan via hydrolysis could result in increased production of PPA, which might in turn increase the activity of CoaBC and thereby render 1f less active against the coaBC hypomorph, we assessed the ability of Pan to rescue growth of this strain under conditions of maximal coaBC silencing (i.e., at an ATc concentration of 200 ng/mL). Although Pan fully rescued Mtb from the growth inhibitory effects of PanC deficiency in a panC Tet-OFF hypomorph,43 as expected, it was unable to restore growth of the coaBC hypomorph (Figure S1). This observation argues against the existence of pantetheinase (vanin) activity (enzymes that catalyze the hydrolysis of PantSH to Pan and are abundantly present in mammalian tissues and serum50) in Mtb and, thus, against a confounding role for PantS hydrolysis and subsequent alteration in the physiological flux through the canonical pathway in the PantS-mediated rescue of Mtb from 1f toxicity.
Given the propensity of Mtb hypomorphs to acquire suppressor mutations that abrogate their ATc responsiveness,44 especially upon prolonged propagation and/or serial passage, the coaBC hypomorph was grown to a lower cell density (OD600) than the wild-type comparator for the use in the MIC assays shown in Figure 2. We therefore hypothesized that the discrepancy in PantS rescue observed between the wild-type and coaBC hypomorph strains at the highest concentration of 1f tested (125 μM) could be due to an inoculum effect. Consistent with this hypothesis, the MICs of 1f in the presence of PantS (2.5 mg/mL) were found to be 83.3 μM and 77.2 μM for the wild-type and the coaBC hypomorph, respectively, when the cultures used to inoculate the MIC plates were at an OD600 of 0.2, but increased to >125 μM when the inoculating cultures were at a higher cell density (OD600 = 0.6). Importantly, the inhibitory activity of 1f against the wild-type strain was unaffected by the density of inoculating culture (MIC = 25.9 μM, assessed at OD600 = 0.2 or 0.6), and the presence of ATc did not confer any non-specific growth inhibitory effects at the lower cell density (Figure S2). Taken together with the ATc dose-dependent hypersensitization of the coaBC hypomorph, these results support the conclusion that the antibacterial activity of 1f is mediated through inhibition of CoaBC.
Elucidation of the Mode of Inhibition and Selectivity of Compound 1f
To assess the mode of inhibition of CoaBC by 1f, competition experiments were carried out between the compound and the three substrates of CoaB (PPA, CTP, and l-cysteine) using the EnzChek coupled enzyme assay, as previously described.41 We first confirmed that compound 1f was inactive on the coupled reporter enzymes at 100 μM. The IC50 for this compound using the EnzChek assay was determined to be 24.3 μM against the Mtb CoaBC, comparable to that observed in the high-throughput screen (Figure 3a). To evaluate the selectivity of compound 1f, the Mtb enzyme was substituted with human CoaB in the EnzChek coupled enzyme assay. A very similar dose–response profile was observed with an IC50 value of 23.4 μM, suggesting poor selectivity of this compound for the Mtb enzyme (Figure 3b).
Figure 3.

CoaBC inhibition by compound 1f. Dose response profile for MtbCoaBC (a) and HCoaB (b). Lineweaver–Burk plots showing the effect of varying concentrations of compound 1f in the presence of varying concentrations of CTP (c), PPA (d), and l-cysteine (e). Data are representative of the mean and SD of independent triplicates.
Mtb CoaB exhibited an apparent Km of 27.4 ± 2.6, 45.4 ± 5.3, and 29.8 ± 3.0 μM for CTP, PPA, and l-cysteine, respectively (Figure S3). Inhibition experiments demonstrated that 1f displays uncompetitive inhibition for PPA, with an αKi of 19.0 ± 0.6 μM, and noncompetitive inhibition for CTP and l-cysteine, with Ki values of 10.1 ± 1.4 μM and 22.7 ± 7.6 μM, respectively (Figure 3c–e), consistent with the compound binding preferentially in the presence of PPA. This is in contrast to our previous observation with allosteric inhibitors of CoaBC, which bound preferentially in the presence of all three substrates.41 We therefore decided to employ a different approach to further validate these findings. Using differential scanning fluorimetry (DSF), the melting temperature of CoaBC in the absence of natural ligands was determined to be 46 °C (Table 2; Figure S4). Evaluation of the effect of 1f on the melting temperature of CoaBC in the presence of the three substrates resulted in a substantial shift in melting temperature only when PPA was present, regardless of the presence or absence of other substrates and products (Table 2), thus further confirming that binding of compound 1f to CoaB is dependent on the presence of PPA (Table 2). The negative melting shift observed in the presence of PPA and l-cysteine is consistent with a mixed inhibition profile for l-cysteine.
Table 2. Effect of Compound 1f on the Melting Temperature of Mtb CoaBC in the Presence of the CoaB Substrates CTP, PPA, and l-Cysteine.
| Thermal
shift relative to CoaBCa (°C) |
|||
|---|---|---|---|
| Substrates (1 mM) | Substrate | Substrate + 1f (2.5 mM) | ΔTmb |
| No substrate | – | 1.5 | |
| CTP | 5.5 | 5.5 | 0 |
| CMP | 1.5 | 2.5 | 1 |
| PPA | 3.5 | 10.5 | 7 |
| l-cysteine | 0.5 | 1.5 | 1 |
| CMP + l-cysteine | 1.5 | 2.5 | 1 |
| CMP + PPA | 4.5 | 10.5 | 7 |
| CMP + l-cysteine + PPA | 4.5 | 11 | 6.5 |
| l-cysteine + PPA | 4.5 | 1.5 | –3 |
| CTP + PPA | 11.8 ± 0.8 | 18.8 ± 0.3 | 7 |
| CTP + l-cysteine | 5.2 ± 0.3 | 5.2 ± 0.3 | 0 |
CoaBC melting temperature under the assay conditions used is 46 °C. Data are representative of three independent triplicates. Where SD is not indicated, SD = 0.
ΔTm represents the difference in melting temperature in the presence and absence of compound 1f under each condition.
Although it was previously reported that binding of PPA to Enterococcus faecalis CoaB occurs after CTP binding,51 the ability of PPA alone to substantially shift the melting temperature of Mtb CoaBC suggests that PPA is able to bind to the Mtb CoaB in the absence of CTP (Table 2). In Escherichia coli, binding of PPA to CoaB causes movements in the protein that close the PPA binding site, thereby limiting access to the CTP binding site52 (Figure S5). Mechanistically, this triggers the formation of a dead-end complex in which PPA and compound 1f bind to the free enzyme, preventing CTP binding and thereby blocking catalysis. This accounts for the inhibition profile of compound 1f, which is very close to competitive inhibition in the case of CTP. A crystal structure of CoaB with 1f bound could not be obtained, but the observed mode of inhibition together with known movements that occur in the protein after PPA binding (Figure S5), with a flexible loop covering the PPA binding site upon binding of this substrate,52 suggests a different binding mode for 1f with distinct properties from the previously reported allosteric inhibitors.41
Metabolic Consequences of Exposure of Mtb to Compound 1f
The metabolic derangement resulting from exposure of Mtb to 5 μM or 50 μM of either 1f or the potent allosteric CoaB inhibitor 2b (IC50 = 0.08 μM)41 (Figure 4) for 24 h was determined, as previously described.53 Unlike 1f, which is active against Mtb (MIC = 25.9 μM), compound 2b did not display significant whole-cell activity (MIC > 250 μM).41 Interestingly, however, both compounds elicited similar alterations in the abundance of metabolites specifically associated with Pan and CoA biosynthesis as compared to untreated cells (Figure 4; Figure S6). Exposure to both compounds produced a distinct metabolomic signature involving metabolites related to the CoA pathway, which was largely consistent with that observed following transcriptional silencing of coaBC in Mtb.33 Specifically, there was substantial accumulation of dephospho-CoA, the immediate precursor of CoA, and notable depletion of the CoA thioesters propionyl-CoA, succinyl-CoA, and malonyl-CoA relative to untreated bacilli (Figure 4; Figure S6). The latter effect, which was dose-dependent in the case of 1f, is consistent with the reduced levels of malonyl- and acetyl-CoA observed upon transcriptional silencing of coaBC.33 Unlike the CoA depletion observed upon coaBC silencing,33 no detectable change in the level of CoA was observed in Mtb treated with either compound 1f or 2b under the conditions tested (Figure 4). Notably, however, while analogous alterations in the levels of all other metabolites were observed after 1.5 days of coaBC silencing, depletion of CoA was only observed in the coaBC hypomorph at day 3. This may reflect the longevity of this cofactor in Mtb, mediated by the reassembly of CoA via recycling of its P-PantSH moiety from holo-ACPs in the presence of ATP, as has been observed in E. coli.54
Figure 4.

Metabolic impact of exposure of wild-type Mtb H37RvMA to 1f and 2b at 5 μM and 50 μM. Relative metabolite abundances (based on ion intensities) are indicated in heat map format, where relative abundance refers to the change in the abundance of a given metabolite in the presence of varying concentrations of 1f and 2b as compared to an untreated (DMSO) control. Data are log2 transformed, with columns representing individual treatments, as indicated, and vertical clustering by rows revealing groups of metabolites exhibiting similar responses to drug exposure. Data are representative of independent triplicates and were parsed by uncentered Pearson’s correlation with centroid linkage clustering and rendered using the image generation program Java TreeView (http://jtreeview.sourceforge.net/).
Finally, the fate of the compounds upon exposure to Mtb was followed by monitoring their intracellular accumulation. Both 1f and 2b were found to accumulate intracellularly in Mtb in a dose-dependent manner, but the intracellular abundance of 1f was substantially greater than 2b at both concentrations tested (Figure S7). To exclude the possibility that this could be due to inherent or bacterial-induced differences in the ionization efficiencies of these compounds, standard curves of 1f and 2b spiked into bacterial extracts were generated. The comparable ionization efficiencies observed for both compounds (Figure S8) further support our observation that the intracellular abundances of these compounds vary substantially, corroborating our previous observation that the phenotypic divergence between these compounds is likely due to the rapid biotransformation of 2b.41,55,56 It is therefore reasonable to conclude that the comparatively low intracellular level to which compound 2b accumulates in Mtb over 24 h is sufficient to induce metabolomic perturbations but does not reach the threshold level required to manifest in a growth phenotype through inhibition of CoaBC.
Discussion
CoA is a ubiquitous cofactor across all domains of life. The sequence divergence between prokaryotic and eukaryotic components of the CoA biosynthesis pathway22 makes it a potentially attractive source of novel drug targets in pathogens that include Mtb.21 This notion has been reinforced by the identification of PanD as the target of pyrazinoic acid, the bioactive form of the first-line antitubercular drug pyrazinamide,39,40 and the discovery of an inhibitor of Mtb PanK requiring activation by the monooxygenase, EthA.57 However, efforts to identify inhibitors of enzymes in the CoA pathway of Mtb by target-based approaches have largely stalled at the level of whole-cell activity. Using hypomorphs of Mtb as tools to assess the target selectivity of small-molecule inhibitors of PanC and PanK, we43 and others31 concluded that these failures might be explained, at least in part, by the relative invulnerability of the corresponding targets.33 The identification of CoaBC as a bactericidal target in the CoA biosynthesis pathway in Mtb33 therefore prompted us to focus our efforts on this bifunctional enzyme. This led to the first reported crystal structure of the bifunctional CoaBC from M. smegmatis and identification of two distinct chemical scaffolds that potently inhibit the CoaB domain of Mtb CoaBC.41 Although neither inhibitor showed significant whole-cell activity against Mtb, these promising biochemical and structural data reinforced the potential of CoaBC as a target and encouraged us to seek alternative inhibitory scaffolds.
A high-throughput screen for inhibitors of the CoaB activity of Mtb CoaBC yielded a primary screening hit, 1a, with activity against the enzyme in the low micromolar range (IC50 = 9.9 μM); however, this compound was inactive against replicating Mtb (MIC > 125 μM). Limited exploration around compound 1a led to the identification of an analogue, 1f, which showed similar activity against the enzyme (IC50 = 15.6 μM) but displayed modest whole-cell inhibitory activity against Mtb (MIC = 25.9 μM). Importantly, the observed ATc-dependent hypersensitization of a coaBC hypomorph33 to this compound suggested that it engages CoaBC in whole Mtb cells. This conclusion is supported by two further lines of evidence: first, supplementation of the culture medium with PantS, which specifically enables CoaBC bypass, rescued Mtb from 1f toxicity, thus establishing a direct link between CoaBC engagement and growth inhibition. Second, 1f elicited a metabolomic profile with distinctive features consistent with CoA pathway disruption and reminiscent of that observed upon transcriptional silencing of coaBC, as evidenced by accumulation of dephospho-CoA and dose-dependent depletion of the thioesters, propionyl-, succinyl-, and malonyl-CoA, which are involved in multiple cellular processes. However, unlike transcriptional silencing of coaBC for 3 days, exposure of Mtb to 1f for 24 h had no discernible impact on the level of CoA. While this may reflect the longevity of this cofactor,54 it is worth noting that genetic inhibition of a target (via its depletion within the cell) is an imperfect surrogate for chemical inhibition of that target,44,47,58 which complicates direct comparison of the metabolic consequences of transcriptional silencing vs. drug treatment.
As noted above, the substantial accumulation of dephospho-CoA and lack of accumulation of PPA following exposure of Mtb to compounds 1f and 2b as well as in response to coaBC silencing were not readily predicted, consistent with our assertion that the impact of CoaBC inhibition is more complex than alteration of its specific substrate and product levels alone.33 PanK, which catalyzes the first committed step in the conversion of Pan to CoA, is considered the rate-limiting enzyme in the CoA biosynthesis pathway59 and is regulated by feedback inhibition by CoA and its thioesters.60 However, Mtb CoaD is also regulated by feedback inhibition by CoA, as well as by dephospho-CoA, the product of its own reaction,27,61−63 and we recently reported that CoA and several acyl-CoAs are additionally capable of inhibiting CoaB activity in Mtb.41 Together, these observations suggest that the regulatory mechanisms mediating CoA biosynthesis in Mtb are complex and incompletely understood. As such, it is interesting to speculate that the depletion of ATP observed following exposure of Mtb to compounds 1f and 2b (Figure 4) may contribute to the observed accumulation of dephospho-CoA by reducing the catalytic efficiency of the ATP-dependent Mtb CoaE,64 which catalyzes the final step in the pathway by conversion of dephospho-CoA to CoA. Additionally, Mtb CoaE is competitively inhibited by CTP, for which it has an affinity similar to that of its substrate, dephospho-CoA.64 Under physiological conditions, CoaE is predominantly found in an inactive trimeric form, which is converted to an active monomeric form in the presence of dephospho-CoA.65 However, due to overlap of the dephospho-CoA and CTP binding sites, binding of CTP to CoaE completely abolishes its enzymatic activity by preventing conversion to the active form despite the presence of dephospho-CoA, with 100 μM CTP resulting in a greater than 60% decrease in CoaE activity.65 Given that 1f displays noncompetitive (but very close to competitive) inhibition for CTP (Ki = 10.1 ± 1.4 μM) and that Mtb CoaB exhibits an apparent Km of 27.4 ± 2.6 μM for CTP, the inhibitory activity of 1f may cause a transient increase in intracellular CTP pools, thus limiting the catalytic efficiency of CoaE and contributing to the observed accumulation of dephospho-CoA. This hypothesis is further supported by the observation that Mtb CoaBC can bind PPA in the absence of CTP, triggering the formation of a dead-end complex in which PPA and 1f are bound to the free enzyme, thereby preventing the binding of CTP. Notably, this mode of binding, with both 1f and 2b showing uncompetitive inhibition for PPA, could also account for the lack of accumulation of this CoaBC substrate observed following exposure of Mtb to both of these compounds for 24 h. Importantly, however, despite the induction of these unpredictable metabolomic alterations, the comparable signatures observed following exposure of Mtb to 1f or 2b and upon transcriptional silencing of coaBC provide further evidence in support of the target selectivity of these compounds.
The lack of correlation between CoaBC inhibition and whole-cell activity observed for the other compounds investigated in this study provides yet another example of the profound challenges of target-based TB drug discovery.3 In this regard, the chemical validation of CoaBC resulting from the identification of 1f as a compound that can penetrate Mtb, evade metabolism, and engage its cellular target, thereby inhibiting Mtb growth, represents a significant advance and provides a platform for further drug discovery efforts. Although 1f lacks selectivity with respect to the human enzyme, ongoing efforts to exploit additional chemical properties of these molecules are underway, with the aim of identifying compounds that not only show selectivity for Mtb CoaB but also demonstrate more potent inhibition of biochemical and phenotypic activity. The finding that 1f uncompetitively inhibits the mycobacterial CoaB, binding preferentially in the presence of PPA, presents a mode of inhibition distinct from those previously described41 and highlights the versatility of Mtb CoaB as a target that is potentially amenable to chemical inhibition via multiple approaches and by a variety of diverse chemical scaffolds.
Methods
Compound Synthesis
Solvents and reagents were purchased from commercial suppliers and used without further purification. Dry solvents were purchased in sure sealed bottles stored over molecular sieves. Reactions using microwave irradiation were carried out in a Biotage Initiator microwave. Normal phase thin layer chromatography (TLC) was performed using precoated silica plates (Kieselgel 60 F254, BDH) with visualization via UV light (UV254/365 nm) or ninhydrin solution. Flash chromatography was performed using Combiflash Companion Rf (Teledyne ISCO) and prepacked silica gel columns (Teledyne ISCO). Mass-directed preparative high performance liquid chromatography (HPLC) separations were performed using an Agilent 1260 Infinity system equipped with diode array detector (DAD) and mass detector, using Waters Sunfire C18 OBD Prep Column, 100 Å, 5 μm, 19 mm × 100 mm with SunFire C18 Prep Guard Cartridge, 100 Å, 10 μm, 19 mm × 10 mm; using water (solvent A) and methanol (solvent B). 1H NMR spectra were recorded on a Bruker Avance DPX 500 spectrometer (1H at 500.1 MHz, 13C at 125 MHz, 19F at 470.5 MHz), or a Bruker Avance DPX 400 (1H at 400 MHz). Chemical shifts (δ) are expressed in ppm, using the residual solvent as the internal reference in all cases. Signal splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), broad (br), or a combination thereof. Coupling constants (J) are quoted to the nearest 0.1 Hz.
High resolution mass spectrometry (HRMS) was carried out using a Bruker Daltonics MicrOTOF mass spectrometer run in positive mode. LC-MS analysis and chromatographic separation were conducted using (i) Bruker Daltonics MicrOTOF mass spectrometer or an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole LC/MS, where both instruments were connected to an Agilent diode array detector, (ii) Bruker MicrOTOF II focus ESI mass spectrometer connected in parallel to a Thermo Dionex Ultimate 3000 RSLC system with diode array detector, (iii) Advion Expression mass spectrometer connected to a Thermo Dionex Ultimate 3000 HPLC with diode array detector, using either a Waters XBridge column (50 mm × 2.1 mm, 3.5 μm particle size) or a Waters X-select column (30 mm × 2.1 mm, 2.5 μm particle size) with a gradient of 5–90% acetonitrile/water + 0.1% formic acid, or (iv) Shimadzu LCMS-2020 single quadrupole liquid chromatograph mass spectrometer (LC/MS) system equipped with a Hypersil gold column 50 mm × 2.1 mm 1.9 μm pore size. All assay compounds had a measured purity of ≥95% as determined using these analytical LC-MS systems.
Compound 1a, 4-Phenylsulfanyl-5-methyl-2-phenyl-pyrazol-3-ol
To a stirred solution of 5-methyl-2-phenyl-pyrazol-3-ol (50 mg, 0.29 mmol) in acetonitrile (3 mL) were added benzenethiol (47.4 mg, 0.43 mmol) and sodium hydroxide (13.8 mg, 0.34 mmol), and the reaction was heated at 80 °C for 18 h. The reaction mixture was concentrated in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate:hexane) to give the title compound (58 mg, 0.20 mmol, 68.0% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 12.19 (s, 1H), 7.75 (d, J = 7.6 Hz, 2H), 7.48 (dd, J = 7.9, 7.9 Hz, 2H), 7.32–7.26 (m, 3H), 7.15–7.07 (m, 4H), 2.13 (s, 3H). HRMS ES+ 283.0895, C16H15N2OS requires 283.0900.
Compound 1b, 4-(4-Methylphenyl)sulfanyl-5-methyl-2-phenyl-pyrazol-3-ol
To a stirred solution of 5-methyl-2-phenyl-pyrazol-3-ol (50 mg, 0.29 mmol) in acetonitrile (3 mL) were added sodium 4-methylbenzenethiolate (42 mg, 0.29 mmol) and sodium hydroxide (13.8 mg, 0.34 mmol), and the reaction was heated at 80 °C for 18 h. The reaction mixture was concentrated in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate/hexane) to give the title compound (41 mg, 0.13 mmol, 46.7% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 12.13 (s, 1H), 7.74 (d, J = 7.6 Hz, 2H), 7.48 (dd, J = 8.0, 8.0 Hz, 2H), 7.29 (t, J = 7.3 Hz, 1H), 7.11 (d, J = 8.1 Hz, 2H), 7.00 (d, J = 7.8 Hz, 2H), 2.24 (s, 3H), 2.09 (s, 3H). HRMS ES+ 297.1043, C17H14N2OS requires 297.1056.
Compound 1c, 4-(4-Chlorophenyl)sulfanyl-5-methyl-2-phenyl-pyrazol-3-ol
To a stirred solution of 5-methyl-2-phenyl-pyrazol-3-ol (55 mg, 0.32 mmol) in acetonitrile (3 mL) were added 4-chlorobenzenethiol (68.5 mg, 0.48 mmol) and sodium hydroxide (15.1 mg, 0.38 mmol), and the reaction was heated at 80 °C for 18 h. The reaction mixture was concentrated in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate/hexane) to give the title compound (25 mg, 0.07 mmol, 23.5% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 12.30 (s, 1H), 7.75 (d, J = 7.6 Hz, 2H), 7.50–7.46 (m, 2H), 7.35 (d, J = 8.5 Hz, 2H), 7.29 (t, J = 7.4 Hz, 1H), 7.10 (d, J = 8.5 Hz, 2H), 2.12 (s, 3H). HRMS ES+ 317.0488, C16H14N2OSCl requires 317.0509.
Compound 1d, 4-(3,4-Dichlorophenyl)sulfanyl-5-methyl-2-phenyl-pyrazol-3-ol
To a stirred solution of 5-methyl-2-phenyl-pyrazol-3-ol (55 mg, 0.32 mmol) in acetonitrile (3 mL) were added 3,4-dichlorobenzenethiol (77.1 mg, 0.43 mmol) and sodium hydroxide (15.1 mg, 0.38 mmol), and the reaction was heated at 80 °C for 18 h. The reaction mixture was concentrated in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate/hexane) to give the title compound (58 mg, 0.16 mmol, 54.6% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 12.38 (s, 1H), 7.77–7.75 (m, 2H), 7.64 (d, J = 2.3 Hz, 1H), 7.51–7.48 (m, 2H), 7.37–7.29 (m, 2H), 6.78 (d, J = 7.0 Hz, 1H), 2.13 (s, 3H). HRMS ES+ 351.0122, C16H13N2OSCl2 requires 351.0120.
Compound 1e, 4-(4-Methoxyphenyl)sulfanyl-5-methyl-2-phenyl-pyrazol-3-ol
To a stirred solution of 5-methyl-2-phenyl-pyrazol-3-ol (55 mg, 0.32 mmol) in acetonitrile (3 mL) were added 4-methoxybenzenethiol (66.4 mg, 0.47 mmol) and sodium hydroxide (15.1 mg, 0.38 mmol), and the reaction was heated at 80 °C for 18 h. The reaction mixture was concentrated in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate/hexane) to give the title compound (25 mg, 0.07 mmol, 24% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 12.10 (m, 1H), 7.73 (d, J = 7.6 Hz, 2H), 7.49–7.45 (m, 2H), 7.27 (t, J = 7.3 Hz, 1H), 7.09 (d, J = 8.3 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 3.71 (s, 3H), 2.11 (s, 3H). HRMS ES+ 313.0983, C17H17N2O2S requires 313.1005.
Compound 1f, 4-(5-Chloro-1,3-benzoxazol-2-yl)sulfanyl-5-methyl-2-phenyl-pyrazol-3-ol
To a stirred solution of 5-methyl-2-phenyl-pyrazol-3-ol (75 mg, 0.43 mmol) in acetonitrile (3 mL) were added 5-chloro-1,3-benzoxazole-2-thiol (119.8 mg, 0.65 mmol) and sodium hydroxide (20.7 mg, 0.52 mmol), and the reaction was heated at 80 °C for 18 h. The reaction mixture was concentrated in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate:hexane) to give the title compound (15 mg, 0.04 mmol, 9.3% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 12.10 (s, 1H), 7.81–7.79 (m, 2H), 7.68–7.65 (m, 1H), 7.53–7.39 (m, 3H), 7.33–7.26 (m, 2H), 2.07 (s, 3H). HRMS ES+ 358.0378, C17H13N2O2SCl requires 358.0411.
Compound 1g, 2-(4-Chlorophenyl)-4-(4-chlorophenyl)sulfanyl-5-methyl-pyrazol-3-ol
Step 1, 2-(4-Chlorophenyl)-5-methyl-pyrazol-3-ol
To a stirred solution of 4-chlorophenylhydrazine hydrochloride (275 mg, 1.54 mmol) in acetic acid (3 mL) was added ethyl-3-oxobutanoate (200 mg, 1.54 mmol), and the reaction was heated at reflux for 3 h. The solvent was removed in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate/hexane) to give the title compound (115 mg, 0.5 mmol, 32.2% yield), which was used without further purification. LRMS (ESI) m/z [MH+] = 209.1.
Step 2, 2-(4-Chlorophenyl)-4-(4-chlorophenyl)sulfanyl-5-methyl-pyrazol-3-ol
To a stirred solution of 2-(4-chlorophenyl)-5-methyl-pyrazol-3-ol (93 mg, 0.43 mmol) in acetonitrile (3 mL) were added 4-chlorothiophenol (96.7 mg, 0.67 mmol) and sodium hydroxide (21.4 mg, 0.54 mmol), and the reaction was heated at 80 °C for 18 h. The reaction mixture was concentrated in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate/hexane) to give the title compound (70.3 mg, 0.19 mmol, 42.7% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 12.43 (s, 1H), 7.82–7.78 (m, 2H), 7.56–7.53 (m, 2H), 7.36–7.33 (m, 2H), 7.12–7.08 (m, 2H), 2.13 (s, 3H). HRMS ES+ 351.0120, C16H13N2O2SCl2 requires 351.0121.
Compound 1h, 2-(3-Chlorophenyl)-4-(4-chlorophenyl)sulfanyl-5-methyl-pyrazol-3-ol
To a stirred solution of ethyl 2-chloro-3-oxobutanoate (45.9 mg, 0.28 mmol) in DMF (1 mL) were added 4-chlorobenzenethiol (40.3 mg, 0.28 mmol) and DIPEA (58.3 μL, 0.34 mmol); the reaction was stirred at room temperature for 1 h and then heated at 100 °C for 9 h. The solvent was removed in vacuo to give a crude residue, which was dissolved in acetic acid (0.7 mL). To this (3-chlorophenyl)hydrazine hydrochloride (50 mg, 0.28 mmol) and sodium acetate (25 mg, 0.31 mmol) were added, and the reaction mixture stirred at room temperature for 1 h followed by heating at 100 °C for 12 h.
The solvent was removed in vacuo to give a crude residue, which was dissolved in DMSO and filtered, and the filtrate was subjected to HPLC purification to give the title compound (26.7 mg, 0.076 mmol, 27.2% yield) as a pink solid.
1H NMR (400 MHz, DMSO-d6) δ 12.54 (s, 1H), 7.85 (s, 1H), 7.79–7.76 (m, 1H), 7.51 (t, J = 8.2 Hz, 1H), 7.36–7.32 (m, 3H), 7.13–7.09 (m, 2H), 2.12 (s, 3H). LRMS (ESI) m/z [MH+] = 351.0.
Compound 1i, 2-(4-Chlorophenyl)-4-(4-chlorophenyl)sulfanyl-5-methyl-pyrazol-3-ol
Step 1, 2-(5-Chloro-2-pyridyl)-5-methyl-pyrazol-3-ol
To a stirred solution of (5-chloro-2-pyridyl)hydrazine (220.6 mg, 1.54 mmol) in acetic acid (3 mL) was added ethyl-3-oxobutanoate (200 mg, 1.54 mmol), and the reaction was heated at reflux for 3 h. The solvent was removed in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate/hexane) to give the title compound (242 mg, 1.1 mmol, 71.3% yield) as an off-white solid.
1H NMR (500 MHz, DMSO-d6) δ 12.03 (s, 1H), 8.46 (d, J = 2.6 Hz, 1H), 8.40 (s, 1H), 8.02 (dd, J = 2.6, 9.0 Hz, 1H), 5.16 (s, 1H), 2.17 (s, 3H). LRMS (ESI) m/z [MH+] = 210.1
Step 2, 2-(4-Chlorophenyl)-4-(4-chlorophenyl)sulfanyl-5-methyl-pyrazol-3-ol
To a stirred solution of 2-(5-chloro-2-pyridyl)-5-methyl-pyrazol-3-ol (96 mg, 0.46 mmol) in acetonitrile (3 mL) were added 4-chlorothiophenol (99.3 mg, 0.69 mmol) and sodium hydroxide (22 mg, 0.55 mmol), and the reaction was heated at 80 °C for 18 h. The reaction mixture was concentrated in vacuo to give a crude residue, which was purified by column chromatography (SiO2, 0–100% ethyl acetate/hexane) to give the title compound (96 mg, 0.26 mmol, 55.9% yield) as a yellow solid.
1H NMR (500 MHz, DMSO-d6) δ 12.43 (s, 1H), 7.82–7.78 (m, 2H), 7.56–7.53 (m, 2H), 7.36–7.33 (m, 2H), 7.12–7.08 (m, 2H), 2.13 (s, 3H). LRMS (ESI) m/z [MH+] = 352.1.
High-Throughput Screening of Small Molecule Libraries
A library of 215 000 small molecules from the DDU compound collection was screened to identify potential inhibitors of Mtb CoaB at 30 μM using an adaptation of the BIOMOL Green (Enzo Life Sciences) pyrophosphate quantification assay, as previously described.41 Briefly, decreased production of phosphate following addition of a pyrophosphatase was indicative of reduced pyrophosphate production by CoaB and, hence, inhibition.
Counter-screening of Primary Screen Hits
Counter-screens carried out to exclude false positive compounds were performed in 50 μL reactions containing 0.5 U/mL pyrophosphatase and 2 μM pyrophosphate in 100 mM Tris-HCl, pH 7.6, buffer containing 1 mM MgCl2 and 1 mM TCEP for 2 h at room temperature in 384-well microplates (Greiner Bio-One). Control reactions were performed in the absence of the pyrophosphatase. The reactions were terminated by addition of 50 μL BIOMOL Green (Enzo Life Sciences), and product formation was measured by absorbance (650 nm) using a PHERAstar microplate reader (BMG Labtech) following incubation for 20 min. Liquid dispensing was performed using a Thermo Scientific Matrix Wellmate, and data were processed and analyzed using ActivityBase (IDBS).
Pharmacological Profiling of Compounds
Compounds were profiled for HepG2 cytotoxicity, mouse liver microsomal clearance, and aqueous solubility, as previously described.66
Bacterial Strains and Growth Conditions
The coaBC Tet-OFF hypomorph used in this study was constructed as previously described,33 and derived from the virulent, PDIM-producing parental strain, Mtb H37RvMA.42 All strains were routinely grown in Difco Middlebrook 7H9 broth (BD) supplemented with Middlebrook albumin-dextrose-catalase (ADC) enrichment (BD), 0.2% glycerol (Sigma-Aldrich), and 0.05% Tween-80, unless otherwise indicated. Hygromycin and kanamycin were used at concentrations of 50 and 25 μg/mL, and pantethine (Sigma-Aldrich) was included at 2.5 mg/mL where required. In order to repress the expression of target genes in cells expressing revTetR, cultures were routinely grown in the absence of supplementation to OD600 ≈ 0.2 prior to dilution in Middlebrook 7H9 broth containing the ATc inducer at concentrations up to 200 ng/mL in order to transcriptionally silence coaBC. To avoid inactivation of the inducer, all cultures containing ATc were incubated in the dark, and exposure of the cultures to light was minimized.
MICs and Checkerboard Assays for Assessing Target Selectivity
The minimum inhibitory concentrations (MICs) of the compounds were determined using the microbroth dilution assay and growth inhibition was quantitatively determined using Alamar Blue as a measure of fluorescence output, as previously described.46 Briefly, 2-fold serial dilutions of each compound were inoculated with a suspension of Mtb H37RvMA42 or the coaBC hypomorph33 at cell densities of ∼105 CFU/mL (OD600 = 0.6) and ∼104 CFU/mL (OD600 = 0.2), respectively, to a final volume of 100 μL in a U-bottom 96-well microtiter plate and incubated at 37 °C for 10 days. A volume of 10 μL of Alamar Blue solution was then added to each well, and the plates were incubated at 37 °C for a further 24 h. Fluorescence as an indication of growth was measured using a SpectraMax i3x multimode microplate reader (Molecular Devices) in bottom-reading mode with excitation at 544 nm and emission at 590 nm. All data are representative of independent triplicates.
Protein Purification and Enzymatic Assays
E. coli BL21(DE3) containing a pET28aSUMO-CoaBC construct or a human CoaB construct with a cleavable N-terminal 6×His tag was grown to mid-exponential growth phase (OD600 = 0.6–0.8) in 2×YT media supplemented with kanamycin (30 μg/mL) at 37 °C. Overexpression of the enzymes was induced by the addition of 0.5 mM IPTG and incubation at 18 °C for 18–20 h. Both enzymes were purified, and enzymatic assays were performed using the EnzChek pyrophosphate assay kit (E-6645) (Life Technologies) except in the case of l-cysteine competition, where a CMP quantification assay was used, as previously described.41 Briefly, the IC50 for compound 1f was determined in 100 mM Tris, pH 7.5, 1 mM TCEP, 2% DMSO, 1 mM MgCl2, 200 μM MSEG, 0.03 U/mL inorganic pyrophosphatase, 1 U/mL purine nucleoside phosphorylase, with 32 nM Mtb CoaBC, 125 μM CTP, 125 μM PPA, 500 μM l-cysteine, and variable concentrations of 1f (2–256 μM). In the assays with human CoaB, an enzyme concentration of 1 μM was used. The inhibition of the coupling enzymes by 1f was assessed, and the compound was inactive against these enzymes at the tested range of concentrations. Kinetic parameters and competition assays for CTP and PPA were determined under the same conditions but with variable concentrations of CTP (31.25 μM, 62.5 μM, 125 μM, 250 μM, and 500 μM) with 125 μM PPA and 500 μM l-cysteine or variable concentrations of PPA (31.25 μM, 62.5 μM, 125 μM, 250 μM, and 500 μM) with 125 μM CTP and 500 μM l-cysteine.
Kinetic parameters and competition assays for l-cysteine were determined in 1 mM MgCl2, 100 mM Tris, pH 7.5, 1 mM TCEP, 2% DMSO, 32 nM Mtb CoaBC, 125 μM CTP, 125 μM PPA, and variable concentrations of l-cysteine (31.25 μM, 62.5 μM, 93.75 μM, 125 μM, and 250 μM). All reactions were carried out in at least triplicate. IC50, Michaelis–Menten, and inhibition constants were calculated using GraphPad Prism.
Differential Scanning Fluorimetry
Differential scanning fluorimetry (DSF) was used to assess inhibitor binding to the CoaB domain of CoaBC. The assay was performed in a 96 well plate format using a CFX Connect (Bio-Rad). Each well contained a solution of 5 μM M. tuberculosis CoaBC, 25 mM Tris, pH 8.0, 150 mM NaCl, 1 mM MgCl2, 2.5% DMSO, 5× Sypro Orange. Substrates or products were added individually to each well at a final concentration of 1 mM in the presence and absence of 2.5 mM 1f. Combinations of substrates and products (CMP and l-cysteine; CTP and l-cysteine; CMP and PPA; CTP and PPA; CMP, PPA, and l-cysteine; PPA and l-cysteine) each at a final concentration of 1 mM were also tested in the presence or absence of 2.5 mM 1f. Unfolding of CoaBC in the absence of any substrate or product was measured in parallel. All data are representative of independent triplicates.
Metabolite Extraction and Metabolomic Profiling
Mtb-laden filters used for metabolomic analyses were generated as previously described53 and incubated at 37 °C for 4 days to expand biomass. Mtb-laden filters were then transferred onto a fresh 7H10 agar plate with or without 1f and 2b at 5 and 50 μM for a further 24 h. Metabolite extraction and LC-MS-based metabolomic profiling were carried out as previously described.33 All data obtained by metabolomics were the average of independent triplicates.
Mycobacterium tuberculosis Compound Uptake
Compound uptake was determined using a filter culture system, as previously described.41,53 Briefly, Mtb was grown on nylon Durapore 0.22 μm membrane filters placed on top of Middlebrook 7H11 agar plates supplemented with 0.2% glycerol and 10% OADC for 1 week at 37 °C to expand the biomass. The membranes were then placed atop a reservoir containing Middlebrook 7H9 broth with or without compound, such that the underside of the bacteria-laden filters was in direct contact with the media. Following 24 h of incubation at 37 °C, which we previously determined to be pre-lethal for most frontline drugs,11 the filters were plunged into 40:40:20 methanol:acetonitrile:water precooled to −20 °C, and cells were lysed using a bead beater. Cell lysates were mixed with equal volumes of 50% acetonitrile and 0.2% formic acid, and drug accumulation was measured using mass spectrometry in positive and negative ion mode, as previously described.53 Relative drug levels were quantified by comparison with standard curves generated from bacterial lysates spiked with compound, using the method of standard addition.67 Internal standards were routinely included with each sample run, and data were additionally normalized to sample protein biomass.
Acknowledgments
This work was supported by HIT-TB (OPP1024021) and SHORTEN-TB (OPP1158806) grants from the TB Drug Accelerator program of the Bill and Melinda Gates Foundation (to C. E. Barry III, K. Y. Rhee, and V. Mizrahi), the South African Medical Research Council, the National Research Foundation of South Africa, and an International Research Scholar’s grant from the HHMI (to V. Mizrahi) and, in part, by the Intramural Research Program of NIAID (H. I. M. Boshoff and C. E. Barry III). J. Hess was financially supported by the Swiss National Science Foundation (SNSF Early PostDoc Mobility Fellowship, P2ZHP2_164947) and the Marie Curie Research Grants Scheme, EU H2020 Framework Programme (H2020-MSCA-IF-2017, ID: 789607). C. Spry was funded in part by a NHMRC Overseas Biomedical Fellowship (1016357) and in part by the Bill and Melinda Gates Foundation HIT-TB (OPP1024021). T. L. Blundell is funded by the Wellcome Trust (Wellcome Trust Investigator Award 200814_Z_16_Z: RG83114). CoaBC screening was funded by a MRC-CinC (Grant No. MC_PC_14099). We thank Alex Cookson, Kirsty Cookson, Emma Gutcher, and Desiree Zeller for assistance with compound logistics.
Glossary
Abbreviations
- ATc
anhydrotetracycline
- CMP
cytidine-5′-monophosphate
- CoaB
4′-phosphopantothenoyl-l-cysteine synthetase (PPCS)
- CoaC
4′-phosphopantothenoyl-cysteine decarboxylase (PPCDC)
- CTP
cytidine-5′-triphosphate
- DSF
differential scanning fluorimetry
- EthA
monooxygenase
- Mtb
Mycobacterium tuberculosis
- Pan
pantothenate (vitamin B5)
- PanC
pantothenate synthetase
- PanD
aspartate decarboxylase
- PanK (CoaA)
type I pantothenate kinase
- PantS
pantethine
- PantSH
pantetheine
- PPA
4′-phosphopantothenate
- P-PantSH
4′-phosphopantetheine
- PptT
phosphopantetheinyl transferase
- TCEP
tris(2-carboxyethyl)phosphine
- PDIM
phthiocerol dimycocerosate
- OADC
oleic-albumin-dextrose-catalase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00904.
Effect of exogenous supplementation with pantothenate on the growth of CoaBC- and PanC-deficient Mtb, lack of effect ATc on the susceptibility of Mtb H37RvMA to compound 1f when assessed at low cell density, Mtb CoaBC saturation curve fit for Michaelis–Menten model, DSF melting profile of CoaBC, movement of a flexible loop covering PPA binding site, impact of exposure of Mtb H37Rv to 1f and 2b on acyl-CoA species and CoA pathway metabolites, total intracellular accumulation of compound 1f and 2b, and standard curves showing comparable ionization efficiencies of compounds 1f and 2b spiked into bacterial extracts (PDF)
Author Present Address
¶ Center for Discovery and Innovation, Hackensack Meridian Health, Nutley, NJ 07110, USA.
Author Present Address
∇ Shanghai Key Laboratory of Veterinary Biotechnology, School of Agriculture and Biology, Shanghai Jiao Tong University, Shanghai, 200240, China.
Author Present Address
$ Research School of Biology, The Australian National University, Canberra, ACT 2601, Australia.
Author Contributions
J. C. Evans and S. L. Lynch designed and performed the microbiology experiments. D. Murugesan, J. M. Post, S. Thompson, S. R. Green, P. C. Ray, and C. Spry developed and performed the high-throughput screening. V. Mendes and J. Hess designed and performed the kinetic experiments. J. Hess and C. Spry synthesized 4′-phosphopantothenate. Z. Wang and N. Nahiyaan performed the metabolomic and compound uptake experiments. J. C. Evans, V. Mendes, S. Thompson, S. R. Green, P. C. Ray, A. G. Coyne, C. Abell, H. I. M. Boshoff, P. G. Wyatt, K. Y. Rhee, T. L. Blundell, C. E. Barry III, and V. Mizrahi managed the project. J. C. Evans and V. Mizrahi wrote the manuscript. All authors approved the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Global Tuberculosis Report, WHO. 2020.
- Zumla A.; Nahid P.; Cole S. T. (2013) Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discovery 12 (5), 388–404. 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- Evans J. C.; Mizrahi V. (2018) Priming the tuberculosis drug pipeline: new antimycobacterial targets and agents. Curr. Opin. Microbiol. 45, 39–46. 10.1016/j.mib.2018.02.006. [DOI] [PubMed] [Google Scholar]
- J Libardo M D.; Boshoff H. I.; Barry C. E (2018) The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol. 42, 81–94. 10.1016/j.coph.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanico A.; Moreira R.; Lopes F. (2018) Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. Eur. J. Med. Chem. 150, 525–545. 10.1016/j.ejmech.2018.03.020. [DOI] [PubMed] [Google Scholar]
- Conradie F.; Everitt D.; Crook A. M. (2020) Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. Reply. N Engl J. Med. 382 (24), 2377. 10.1056/NEJMc2009939. [DOI] [PubMed] [Google Scholar]
- Tiberi S.; Munoz-Torrico M.; Duarte R.; Dalcolmo M.; D’Ambrosio L.; Migliori G. B. (2018) New drugs and perspectives for new anti-tuberculosis regimens. Pulmonology 24 (2), 86–98. 10.1016/j.rppnen.2017.10.009. [DOI] [PubMed] [Google Scholar]
- Wellington S.; Hung D. T. (2018) The Expanding Diversity of Mycobacterium tuberculosis Drug Targets. ACS Infect. Dis. 4 (5), 696–714. 10.1021/acsinfecdis.7b00255. [DOI] [PubMed] [Google Scholar]
- Andries K.; Verhasselt P.; Guillemont J.; Gohlmann H. W.; Neefs J. M.; Winkler H.; Van Gestel J.; Timmerman P.; Zhu M.; Lee E.; Williams P.; de Chaffoy D.; Huitric E.; Hoffner S.; Cambau E.; Truffot-Pernot C.; Lounis N.; Jarlier V. (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307 (5707), 223–7. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Matsumoto M.; Hashizume H.; Tomishige T.; Kawasaki M.; Tsubouchi H.; Sasaki H.; Shimokawa Y.; Komatsu M. (2006) OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3 (11), e466. 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar M.; Nathan C.; Rhee K. Y. (2014) Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat. Commun. 5, 4306. 10.1038/ncomms5306. [DOI] [PubMed] [Google Scholar]
- Pethe K.; Bifani P.; Jang J.; Kang S.; Park S.; Ahn S.; Jiricek J.; Jung J.; Jeon H. K.; Cechetto J.; Christophe T.; Lee H.; Kempf M.; Jackson M.; Lenaerts A. J.; Pham H.; Jones V.; Seo M. J.; Kim Y. M.; Seo M.; Seo J. J.; Park D.; Ko Y.; Choi I.; Kim R.; Kim S. Y.; Lim S.; Yim S. A.; Nam J.; Kang H.; Kwon H.; Oh C. T.; Cho Y.; Jang Y.; Kim J.; Chua A.; Tan B. H.; Nanjundappa M. B.; Rao S. P.; Barnes W. S.; Wintjens R.; Walker J. R.; Alonso S.; Lee S.; Kim J.; Oh S.; Oh T.; Nehrbass U.; Han S. J.; No Z.; Lee J.; Brodin P.; Cho S. N.; Nam K.; Kim J. (2013) Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 19 (9), 1157–60. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Stover C. K.; Warrener P.; VanDevanter D. R.; Sherman D. R.; Arain T. M.; Langhorne M. H.; Anderson S. W.; Towell J. A.; Yuan Y.; McMurray D. N.; Kreiswirth B. N.; Barry C. E.; Baker W. R. (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405 (6789), 962–6. 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- Wellington S.; Nag P. P.; Michalska K.; Johnston S. E.; Jedrzejczak R. P.; Kaushik V. K.; Clatworthy A. E.; Siddiqi N.; McCarren P.; Bajrami B.; Maltseva N. I.; Combs S.; Fisher S. L.; Joachimiak A.; Schreiber S. L.; Hung D. T. (2017) A small-molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat. Chem. Biol. 13 (9), 943–950. 10.1038/nchembio.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambady A.; Awasthy D.; Yadav R.; Basuthkar S.; Seshadri K.; Sharma U. (2012) Evaluation of CoA biosynthesis proteins of Mycobacterium tuberculosis as potential drug targets. Tuberculosis (Oxford, U. K.) 92 (6), 521–8. 10.1016/j.tube.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Cheng C. S.; Jia K. F.; Chen T.; Chang S. Y.; Lin M. S.; Yin H. S. (2013) Experimentally validated novel inhibitors of Helicobacter pylori phosphopantetheine adenylyltransferase discovered by virtual high-throughput screening. PLoS One 8 (9), e74271. 10.1371/journal.pone.0074271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Tempel W.; Smil D.; Bolshan Y.; Schapira M.; Park H. W. (2013) Crystal structures of Klebsiella pneumoniae pantothenate kinase in complex with N-substituted pantothenamides. Proteins: Struct., Funct., Genet. 81 (8), 1466–72. 10.1002/prot.24290. [DOI] [PubMed] [Google Scholar]
- Spry C.; Kirk K.; Saliba K. J. (2008) Coenzyme A biosynthesis: an antimicrobial drug target. FEMS Microbiol Rev. 32 (1), 56–106. 10.1111/j.1574-6976.2007.00093.x. [DOI] [PubMed] [Google Scholar]
- van der Westhuyzen R.; Hammons J. C.; Meier J. L.; Dahesh S.; Moolman W. J.; Pelly S. C.; Nizet V.; Burkart M. D.; Strauss E. (2012) The antibiotic CJ-15,801 is an antimetabolite that hijacks and then inhibits CoA biosynthesis. Chem. Biol. 19 (5), 559–71. 10.1016/j.chembiol.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Allanson N. M.; Thomson S. P.; Maclean J. K.; Barker J. J.; Primrose W. U.; Tyler P. D.; Lewendon A. (2003) Inhibitors of phosphopantetheine adenylyltransferase. Eur. J. Med. Chem. 38 (4), 345–9. 10.1016/S0223-5234(03)00047-3. [DOI] [PubMed] [Google Scholar]
- Butman H. S.; Kotze T. J.; Dowd C. S.; Strauss E. (2020) Vitamin in the Crosshairs: Targeting Pantothenate and Coenzyme A Biosynthesis for New Antituberculosis Agents. Front. Cell. Infect. Microbiol. 10, 721. 10.3389/fcimb.2020.605662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genschel U. (2004) Coenzyme A biosynthesis: reconstruction of the pathway in archaea and an evolutionary scenario based on comparative genomics. Mol. Biol. Evol. 21 (7), 1242–51. 10.1093/molbev/msh119. [DOI] [PubMed] [Google Scholar]
- Begley T. P.; Kinsland C.; Strauss E. (2001) The biosynthesis of coenzyme A in bacteria. Vitam. Horm. 61, 157–71. 10.1016/S0083-6729(01)61005-7. [DOI] [PubMed] [Google Scholar]
- Ballinger E.; Mosior J.; Hartman T.; Burns-Huang K.; Gold B.; Morris R.; Goullieux L.; Blanc I.; Vaubourgeix J.; Lagrange S.; Fraisse L.; Sans S.; Couturier C.; Bacque E.; Rhee K.; Scarry S. M.; Aube J.; Yang G.; Ouerfelli O.; Schnappinger D.; Ioerger T. R.; Engelhart C. A.; McConnell J. A.; McAulay K.; Fay A.; Roubert C.; Sacchettini J.; Nathan C. (2019) Opposing reactions in coenzyme A metabolism sensitize Mycobacterium tuberculosis to enzyme inhibition. Science 363, eaau8959. 10.1126/science.aau8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.; VanderVen B. C.; Fahey R. J.; Russell D. G. (2013) Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J. Biol. Chem. 288 (10), 6788–800. 10.1074/jbc.M112.445056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciulli A.; Scott D. E.; Ando M.; Reyes F.; Saldanha S. A.; Tuck K. L.; Chirgadze D. Y.; Blundell T. L.; Abell C. (2008) Inhibition of Mycobacterium tuberculosis pantothenate synthetase by analogues of the reaction intermediate. ChemBioChem 9 (16), 2606–11. 10.1002/cbic.200800437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev V.; Smirnova E.; Chupova L.; Esipov R.; Kuranova I. (2012) X-ray study of the conformational changes in the molecule of phosphopantetheine adenylyltransferase from Mycobacterium tuberculosis during the catalyzed reaction. Acta Crystallogr., Sect. D: Biol. Crystallogr. 68, 1660–70. 10.1107/S0907444912040206. [DOI] [PubMed] [Google Scholar]
- Venkatraman J.; Bhat J.; Solapure S. M.; Sandesh J.; Sarkar D.; Aishwarya S.; Mukherjee K.; Datta S.; Malolanarasimhan K.; Bandodkar B.; Das K. S. (2012) Screening, identification, and characterization of mechanistically diverse inhibitors of the Mycobacterium tuberculosis enzyme, pantothenate kinase (CoaA). J. Biomol. Screening 17 (3), 293–302. 10.1177/1087057111423069. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Yin W.; Martinelli L. K.; Evans J.; Chen J.; Yu Y.; Wilson D. J.; Mizrahi V.; Qiao C.; Aldrich C. C. (2014) Reaction intermediate analogues as bisubstrate inhibitors of pantothenate synthetase. Bioorg. Med. Chem. 22 (5), 1726–35. 10.1016/j.bmc.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkelid C.; Bergfors T.; Raichurkar A. K.; Mukherjee K.; Malolanarasimhan K.; Bandodkar B.; Jones T. A. (2013) Structural and biochemical characterization of compounds inhibiting Mycobacterium tuberculosis pantothenate kinase. J. Biol. Chem. 288 (25), 18260–70. 10.1074/jbc.M113.476473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy B. K.; Landge S.; Ravishankar S.; Patil V.; Shinde V.; Tantry S.; Kale M.; Raichurkar A.; Menasinakai S.; Mudugal N. V.; Ambady A.; Ghosh A.; Tunduguru R.; Kaur P.; Singh R.; Kumar N.; Bharath S.; Sundaram A.; Bhat J.; Sambandamurthy V. K.; Bjorkelid C.; Jones T. A.; Das K.; Bandodkar B.; Malolanarasimhan K.; Mukherjee K.; Ramachandran V. (2014) Assessment of Mycobacterium tuberculosis pantothenate kinase vulnerability through target knockdown and mechanistically diverse inhibitors. Antimicrob. Agents Chemother. 58 (6), 3312–26. 10.1128/AAC.00140-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Casey A.; Odingo J.; Kesicki E. A.; Abrahams G.; Vieth M.; Masquelin T.; Mizrahi V.; Hipskind P. A.; Sherman D. R.; Parish T. (2013) A high-throughput screen against pantothenate synthetase (PanC) identifies 3-biphenyl-4-cyanopyrrole-2-carboxylic acids as a new class of inhibitor with activity against Mycobacterium tuberculosis. PLoS One 8 (11), e72786. 10.1371/journal.pone.0072786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. C.; Trujillo C.; Wang Z.; Eoh H.; Ehrt S.; Schnappinger D.; Boshoff H. I.; Rhee K. Y.; Barry C. E. 3rd; Mizrahi V. (2016) Validation of CoaBC as a Bactericidal Target in the Coenzyme A Pathway of Mycobacterium tuberculosis. ACS Infect. Dis. 2 (12), 958–968. 10.1021/acsinfecdis.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss E.; Kinsland C.; Ge Y.; McLafferty F. W.; Begley T. P. (2001) Phosphopantothenoylcysteine synthetase from Escherichia coli. Identification and characterization of the last unidentified coenzyme A biosynthetic enzyme in bacteria. J. Biol. Chem. 276 (17), 13513–6. 10.1074/jbc.C100033200. [DOI] [PubMed] [Google Scholar]
- Kupke T. (2002) Molecular characterization of the 4′-phosphopantothenoylcysteine synthetase domain of bacterial dfp flavoproteins. J. Biol. Chem. 277 (39), 36137–45. 10.1074/jbc.M206188200. [DOI] [PubMed] [Google Scholar]
- Strauss E.; Begley T. P. (2001) Mechanistic studies on phosphopantothenoylcysteine decarboxylase. J. Am. Chem. Soc. 123 (26), 6449–50. 10.1021/ja016020y. [DOI] [PubMed] [Google Scholar]
- Moolman W. J.; de Villiers M.; Strauss E. (2014) Recent advances in targeting coenzyme A biosynthesis and utilization for antimicrobial drug development. Biochem. Soc. Trans. 42 (4), 1080–6. 10.1042/BST20140131. [DOI] [PubMed] [Google Scholar]
- Patrone J. D.; Yao J.; Scott N. E.; Dotson G. D. (2009) Selective inhibitors of bacterial phosphopantothenoylcysteine synthetase. J. Am. Chem. Soc. 131 (45), 16340–1. 10.1021/ja906537f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal P.; Sarathy J. P.; Yee M.; Ragunathan P.; Shin J.; Bhushan S.; Zhu J.; Akopian T.; Kandror O.; Lim T. K.; Gengenbacher M.; Lin Q.; Rubin E. J.; Gruber G.; Dick T. (2020) Pyrazinamide triggers degradation of its target aspartate decarboxylase. Nat. Commun. 11, 1661. 10.1038/s41467-020-15516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal P.; Nartey W.; Ragunathan P.; Sarathy J.; Kaya F.; Yee M.; Setzer C.; Manimekalai M. S. S.; Dartois V.; Gruber G.; Dick T. (2017) Pyrazinoic Acid Inhibits Mycobacterial Coenzyme A Biosynthesis by Binding to Aspartate Decarboxylase PanD. ACS Infect. Dis. 3 (11), 807–819. 10.1021/acsinfecdis.7b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes V. G. S. R.; Evans J. C.; Hess J.; Blaszczyk M.; Spry C.; Bryant O.; Cory-Wright J.; Chan D. S.-H.; Torres P. H. M.; Wang Z.; Nahiyaan N.; O’Neill S.; Damerow S.; Post J.; Bayliss T.; Lynch S. L.; Coyne A. G.; Ray P. C.; Abell C.; Rhee K. Y.; Boshoff H. I. M.; Barry C. E.; Mizrahi V.; Wyatt P. G.; Blundell T. L.; Green S. R. (2021) Inhibiting Mycobacterium tuberculosis CoaBC by targeting an allosteric site. Nat. Commun. 12 (1), 143. 10.1038/s41467-020-20224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger T. R.; Feng Y.; Ganesula K.; Chen X.; Dobos K. M.; Fortune S.; Jacobs W. R. Jr.; Mizrahi V.; Parish T.; Rubin E.; Sassetti C.; Sacchettini J. C. (2010) Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J. Bacteriol. 192 (14), 3645–53. 10.1128/JB.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams G. L.; Kumar A.; Savvi S.; Hung A. W.; Wen S.; Abell C.; Barry C. E. 3rd; Sherman D. R.; Boshoff H. I.; Mizrahi V. (2012) Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 19 (7), 844–54. 10.1016/j.chembiol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. C.; Mizrahi V. (2015) The application of tetracyclineregulated gene expression systems in the validation of novel drug targets in Mycobacterium tuberculosis. Front. Microbiol. 6, 812. 10.3389/fmicb.2015.00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Donini S.; Pacitto A.; Sala C.; Hartkoorn R. C.; Dhar N.; Keri G.; Ascher D. B.; Mondesert G.; Vocat A.; Lupien A.; Sommer R.; Vermet H.; Lagrange S.; Buechler J.; Warner D. F.; McKinney J. D.; Pato J.; Cole S. T.; Blundell T. L.; Rizzi M.; Mizrahi V. (2017) The Inosine Monophosphate Dehydrogenase, GuaB2, Is a Vulnerable New Bactericidal Drug Target for Tuberculosis. ACS Infect. Dis. 3 (1), 5–17. 10.1021/acsinfecdis.6b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Brecik M.; Mukherjee R.; Evans J. C.; Svetlikova Z.; Blasko J.; Surade S.; Blackburn J.; Warner D. F.; Mikusova K.; Mizrahi V. (2015) The complex mechanism of antimycobacterial action of 5-fluorouracil. Chem. Biol. 22 (1), 63–75. 10.1016/j.chembiol.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Wei J. R.; Krishnamoorthy V.; Murphy K.; Kim J. H.; Schnappinger D.; Alber T.; Sassetti C. M.; Rhee K. Y.; Rubin E. J. (2011) Depletion of antibiotic targets has widely varying effects on growth. Proc. Natl. Acad. Sci. U. S. A. 108 (10), 4176–81. 10.1073/pnas.1018301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bakali J. B. M., Evans J. C., Boland J. A., McCarthy W., Dias M. V. B., Coyne A. G., Mizrahi V., Blundell T. L., Abell C., and Spry C. (2020) Fragment Linking Applied to the Discovery of Mycobacterium tuberculosis Phosphopantetheine Adenylyltransferase Inhibitors. bioRxiv, 10.1101/2020.09.04.280388v1. [DOI]
- Balibar C. J.; Hollis-Symynkywicz M. F.; Tao J. (2011) Pantethine rescues phosphopantothenoylcysteine synthetase and phosphopantothenoylcysteine decarboxylase deficiency in Escherichia coli but not in Pseudomonas aeruginosa. J. Bacteriol. 193 (13), 3304–12. 10.1128/JB.00334-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaskow B. J.; Proffitt J. M.; Blangero J.; Moses E. K.; Abraham L. J. (2012) Diverse biological activities of the vascular non-inflammatory molecules - the Vanin pantetheinases. Biochem. Biophys. Res. Commun. 417 (2), 653–8. 10.1016/j.bbrc.2011.11.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J.; Patrone J. D.; Dotson G. D. (2009) Characterization and kinetics of phosphopantothenoylcysteine synthetase from Enterococcus faecalis. Biochemistry 48 (12), 2799–806. 10.1021/bi802240w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanitzek S.; Augustin M. A.; Huber R.; Kupke T.; Steinbacher S. (2004) Structural basis of CTP-dependent peptide bond formation in coenzyme A biosynthesis catalyzed by Escherichia coli PPC synthetase. Structure 12 (11), 1977–88. 10.1016/j.str.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Nandakumar M.; Prosser G. A.; de Carvalho L. P.; Rhee K. (2015) Metabolomics of Mycobacterium tuberculosis. Methods Mol. Biol. 1285, 105–15. 10.1007/978-1-4939-2450-9_6. [DOI] [PubMed] [Google Scholar]
- Hartl J.; Kiefer P.; Meyer F.; Vorholt J. A. (2017) Longevity of major coenzymes allows minimal de novo synthesis in microorganisms. Nat. Microbiol 2, 17073. 10.1038/nmicrobiol.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi D.; Freundlich J. S. (2017) Antimycobacterial Metabolism: Illuminating Mycobacterium tuberculosis Biology and Drug Discovery. Trends Microbiol. 25 (9), 756–767. 10.1016/j.tim.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes V. G. S. R., Evans J. C., Hess J., Blaszczyk M., Spry C., Bryant O., Cory-Wright J., D.S-H C., Torres P. H. M., Wang Z., O’Neill S., Damerow S., Post J., Bayliss T., Lynch S. L., Coyne A. G., Ray P. C., Abell C., Rhee Y. K., Boshoff H. I. M., Barry C. E. III, Mizrahi M., Wyatt P. G., and Blundell T. L (2019) Inhibiting Mycobacterium tuberculosis CoaBC by targeting a new allosteric site. bioRxiv, www.biorxiv.org/content/10.1101/870154v3. [DOI] [PMC free article] [PubMed]
- Chiarelli L. R.; Mori G.; Orena B. S.; Esposito M.; Lane T.; de Jesus Lopes Ribeiro A. L.; Degiacomi G.; Zemanova J.; Szadocka S.; Huszar S.; Palcekova Z.; Manfredi M.; Gosetti F.; Lelievre J.; Ballell L.; Kazakova E.; Makarov V.; Marengo E.; Mikusova K.; Cole S. T.; Riccardi G.; Ekins S.; Pasca M. R. (2018) A multitarget approach to drug discovery inhibiting Mycobacterium tuberculosis PyrG and PanK. Sci. Rep. 8 (1), 3187. 10.1038/s41598-018-21614-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D.; Ehrt S. (2014) Regulated Expression Systems for Mycobacteria and Their Applications. Microbiol. Spectrum 10.1128/microbiolspec.MGM2-0018-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackowski S.; Rock C. O. (1981) Regulation of coenzyme A biosynthesis. J. Bacteriol. 148 (3), 926–32. 10.1128/JB.148.3.926-932.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallari D. S.; Jackowski S.; Rock C. O. (1987) Regulation of pantothenate kinase by coenzyme A and its thioesters. J. Biol. Chem. 262 (6), 2468–71. 10.1016/S0021-9258(18)61527-3. [DOI] [PubMed] [Google Scholar]
- Wubben T. J.; Mesecar A. D. (2010) Kinetic, thermodynamic, and structural insight into the mechanism of phosphopantetheine adenylyltransferase from Mycobacterium tuberculosis. J. Mol. Biol. 404 (2), 202–19. 10.1016/j.jmb.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Miller J. R.; Ohren J.; Sarver R. W.; Mueller W. T.; de Dreu P.; Case H.; Thanabal V. (2007) Phosphopantetheine adenylyltransferase from Escherichia coli: investigation of the kinetic mechanism and role in regulation of coenzyme A biosynthesis. J. Bacteriol. 189 (22), 8196–205. 10.1128/JB.00732-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerlof A.; Lewendon A.; Shaw W. V. (1999) Purification and characterization of phosphopantetheine adenylyltransferase from Escherichia coli. J. Biol. Chem. 274 (38), 27105–11. 10.1074/jbc.274.38.27105. [DOI] [PubMed] [Google Scholar]
- Walia G.; Kumar P.; Surolia A. (2009) The role of UPF0157 in the folding of M. tuberculosis dephosphocoenzyme A kinase and the regulation of the latter by CTP. PLoS One 4 (10), e7645. 10.1371/journal.pone.0007645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia G.; Surolia A. (2011) Insights into the regulatory characteristics of the mycobacterial dephosphocoenzyme A kinase: implications for the universal CoA biosynthesis pathway. PLoS One 6 (6), e21390. 10.1371/journal.pone.0021390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P. C.; Huggett M.; Turner P. A.; Taylor M.; Cleghorn L. A. T.; Early J.; Kumar A.; Bonnett S. A.; Flint L.; Joerss D.; Johnson J.; Korkegian A.; Mullen S.; Moure A. L.; Davis S. H.; Murugesan D.; Mathieson M.; Caldwell N.; Engelhart C. A.; Schnappinger D.; Epemolu O.; Zuccotto F.; Riley J.; Scullion P.; Stojanovski L.; Massoudi L.; Robertson G. T.; Lenaerts A. J.; Freiberg G.; Kempf D. J.; Masquelin T.; Hipskind P. A.; Odingo J.; Read K. D.; Green S. R.; Wyatt P. G.; Parish T. (2021) Spirocycle MmpL3 Inhibitors with Improved hERG and Cytotoxicity Profiles as Inhibitors of Mycobacterium tuberculosis Growth. ACS Omega 6 (3), 2284–2311. 10.1021/acsomega.0c05589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D. C. (2003) Quantitative Chemical Analysis, 6th ed., W.H. Freeman, New York. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.