

Abstract
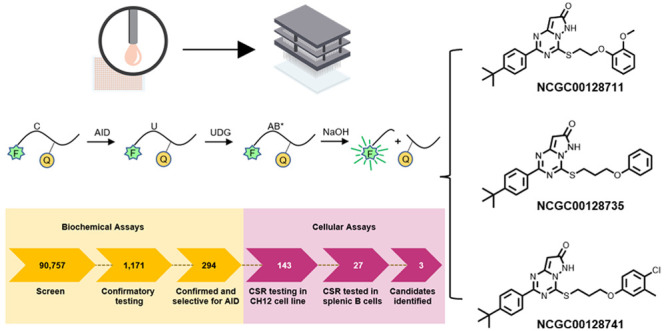
Activation-induced deaminase (AID) not only mutates DNA within the immunoglobulin loci to generate antibody diversity, but it also promotes development of B cell lymphomas. To tame this mutagen, we performed a quantitative high-throughput screen of over 90 000 compounds to see if AID activity could be mitigated. The enzymatic activity was assessed in biochemical assays to detect cytosine deamination and in cellular assays to measure class switch recombination. Three compounds showed promise via inhibition of switching in a transformed B cell line and in murine splenic B cells. These compounds have similar chemical structures, which suggests a shared mechanism of action. Importantly, the inhibitors blocked AID, but not a related cytosine DNA deaminase, APOBEC3B. We further determined that AID was continually expressed for several days after B cell activation to induce switching. This first report of small molecules that inhibit AID can be used to gain regulatory control over base editors.
Keywords: small molecule inhibitors, activation-induced deaminase, high-throughput screen, B cells, cellular assay, class switch recombination
Activation-induced deaminase (AID) is an enzyme expressed in B cells that diversifies variable genes to produce high-affinity antibodies and changes constant genes via class switch recombination (CSR) to yield different heavy chain classes. AID is targeted to subregions in three immunoglobulin loci, encompassing 2 kb around rearranged variable exons and 4–8 kb in switch regions preceding heavy chain constant genes.1,2 Little is known about how AID is recruited to these on-target sites, except that transcription is necessary and factors associated with paused RNA polymerase II are involved.3−6 Even less is known about the recruitment of AID to off-target genes, such as MYC, BCL6, and PIM1.7−10 One thread tying AID with engagement of on- and off-target sites is the stalling of RNA polymerase II,11 which is guided by promoter and enhancer activity of the genes.12−15
The concerning consequence of off-target mutagenesis by AID in B cells is the generation of cancer initiated by DNA point mutations, leading to subsequent strand breaks and translocations. Lymphomas can result after AID leaves a trail of high-frequency breaks in the heavy chain switch regions that become translocation partners with other genes sustaining low-frequency breaks.16,17 Off-target point mutations can also cause resistance to drugs in lymphomas.18−20 Furthermore, overexpression of AID in transgenic mouse models produces tumors in other cell types, such as T cells.21 A second detrimental effect of AID is the generation of autoimmunity, which arises after on-target point mutations in variable genes produce antibodies with high affinity for self-proteins.22−24 While aberrant deaminase activity can pose threats to the genome, recent biotechnological efforts have focused on harnessing and targeting deaminase activity in base editors related to AID. These dual outcomes highlight both the risk and potential associated with controlling DNA deaminase activity.
In light of AID’s pathological roles in promoting mutagenesis associated with cancer and autoimmunity, we focused on identifying small molecules that could modulate its activity. A fluorescent assay was developed to screen over 90 000 compounds that could inhibit AID. Three compounds were selected that reduced CSR and base editing in three different assays, demonstrating the potential to mitigate AID activity in biological systems.
Results
Quantitative High-Throughput Screen (qHTS) of 90 757 Compounds by Biochemical Assays for Cytosine Deamination
A FRET (fluorescence resonance energy transfer) quencher assay was devised to screen compounds for inhibition of cytosine deamination by AID. The assay employed two oligonucleotide substrates to differentiate between inhibitors of deamination and the downstream processing of AID-generated uracil. For AID activity, a 20-base oligonucleotide, DJC20, was made with a 5′-Cy3 fluorophore (green), a single C at position 6, and BHQ2 quencher at position 10. A matched uracil-containing oligonucleotide, DJC23, was constructed with a distinct 5′-Cy5 fluorophore (red) and U in lieu of C at position 6. The assay involved coincubation of DJC20 and DJC23 with purified human AID and uracil DNA glycosylase (UDG), followed by the addition of NaOH. As shown in Figure 1A, AID deaminates C to U, and UDG removes the U base; NaOH cleaves the abasic site, separating the 5′-fluorophore from the internal quencher. Cy3 fluorescence was used to track AID reactivity, while Cy5 fluorescence controls for UDG inhibition, abasic processing, or other confounding effects. To assess the assay performance, inhibition of both oligonucleotides was tested with a nonspecific inhibitor, aurintricarboxylic acid (ATA) (Figure S1A), which inhibits protein–nucleic acid interactions, in a 1536-well format. In a test for the FRET quencher assay robustness with ±150 μM ATA across a 384 well plate, Cy3 fluorescence in inhibited vs uninhibited populations was separated into clear groups (Z′-factor = 0.7) (Figure S1B), whereas the inhibited/uninhibited Cy5 populations were overlapping (Figure S1C).25 The results demonstrate that the inhibition observed in Figure S1B is specific to AID, and the screening assay was able to distinguish AID-specific inhibition. Titration curves with ATA (Figure S1D) further confirmed >10× inhibition for AID (IC50 of ∼3 μM) compared to UDG (IC50 of >50 μM).
Figure 1.

Screening compounds for inhibition of cytosine deamination by AID. (A) FRET biochemical assay to detect AID and UDG activity. Two oligonucleotides containing C (DJC20) or U (DJC23) had distinct fluorophores (Fs) linked to a quencher (Q). Fluorescence was recorded after removal of U by UDG and cleavage of the abasic site (AB) by NaOH. The dual output assay measured both activities in the same well to identify inhibition of AID or UDG. (B) Workflow for high-throughput screening of 90 757 compounds in biochemical assays for inhibiting deamination and screening of 143 compounds in cellular assays for inhibiting CSR. (C) Gel deamination assay. After incubating DJC20 substrate with AID and increasing concentrations of three inhibitors, the deaminated uracil was removed, and the abasic site was cleaved. Band intensity of the product was measured by fluorescence. (D) Inhibition values were determined by the ratio of the product (P) divided by the sum of P and substrate (S) bands. Data is shown for assays with 0.25 μM substrate. (E) Structures of the top 3 candidates.
The workflow for screening compounds by biochemical assays to cellular assays is summarized in Figure 1B. In total, 90 757 compounds were screened in qHTS (6 or 7 points, final concentration range of 3.7 nM to 114 μM) format (Tables S1–S3) on a fully integrated robotic system. Potential AID inhibitors were identified using the dual-output of Cy3 and Cy5 fluorescence quenching in high-throughput assays. The dose–response curves were then sorted into different profiles (classes 1–4) (Figure S1E–H). Because of the high enzyme concentration, we set a low bar in our hit criteria, with any compound exhibiting an inhibitory response greater than 50% at the highest concentration and/or having dose responses with curve classes ranging between −1 and −2. Using these criteria, 1552 compounds were identified as potential inhibitors (Table S2), which were then assessed for counter screen activity in the UDG channel, leading to the removal of 299 compounds. After excluding for UDG activity, 1253 compounds were identified, of which 1171 were sourced for confirmatory testing (Table S3). Of these, 294 were confirmed for inhibition against AID, with none exhibiting activity against UDG. We then removed compounds known to be frequent hitters from NCATS internal HTS projects, in addition to manually inspected compounds for chemical tractability.26−28 The remaining 143 candidates were examined in cellular assays (Table S3).
Biological Screen of 143 Compounds by Cellular Assays for CSR
Compounds were first screened in a transformed, murine CH12 cell line to test inhibition of AID as measured by CSR to IgA.29 Cell proliferation, which would also affect switching, was measured by labeling cells with eFluor 670 proliferation dye. Cells were then stimulated with anti-CD40, IL4, and TGFβ, in the absence or presence of 10 μM compound in a final concentration of 0.1% DMSO (Figure 2A). After 3 days of incubation, the cells were assessed by flow cytometry for viability with 7-AAD, for proliferation with eFluor 670, and for AID activity by switching to IgA. In the absence of compound, ∼60% of cells were viable after 3 days and between 28 and 39% of cells switched from expressing membrane IgM to IgA (Figure 2B, Figure S2), demonstrating the robust nature of the CH12 cell line for switching. Using this assay, we tested 143 compounds from the biochemical assay at a concentration of 10 μM to determine if they inhibited switching. In 2–4 independent assays, 27 compounds showed reproducible inhibition of CSR by 10–30% compared to DMSO controls, with no effect on viability or proliferation (Table S4). The remaining 116 compounds were either toxic to the cells, did not inhibit CSR, or altered cell proliferation.
Figure 2.

Inhibition of CSR in B cells. (A) B cells were labeled with proliferation dye, and compounds were added in 0.1% DMSO final concentration. Cells were stimulated in culture and assayed by flow cytometry 3 days later for viability, proliferation, and CSR. (B) Inhibition of CSR in the CH12 cell line. In the absence of compound, 60% of the cells were viable by 7-AAD exclusion, and 39% of cells switched from IgM to IgA. In the presence of 143 compounds at 10 μM, 27 showed 10–30% inhibition of switching. The data is from 2 to 4 independent assays. (C) Inhibition of CSR in naïve splenic B cells. Of the 27 compounds, 3 had 100% viability, proliferation, and decreased CSR. Titration curves from 156 nM to 10 μM showed that switching to IgG1 was decreased by 20–30% at 10 μM compound (red line), without affecting viability (black line). Results are from three independent experiments; error bars signify standard deviation.
To test CSR inhibition of the 27 compounds in nontransformed B cells, naïve splenic B cells from C57BL/6 mice were isolated using negative selection with anti-CD43 and anti-CD11b. Cells were labeled with eFluor 670 proliferation dye and stimulated with lipopolysaccharide (LPS) and IL4 in the absence or presence of the compounds at 10 μM concentration in 0.1% DMSO (Table S4). Three days later, cells were evaluated by flow cytometry for viability with Zombie Aqua, for proliferation with eFluor 670, and for AID activity by switching to IgG1 (Figure S3). In nine independent assays, three compounds, NCGC00128711, NCGC00128735, and NCGC00128741, showed reproducible inhibition of CSR by 20–30%; the remaining 24 compounds had <10% inhibition of CSR or altered viability or proliferation. These three compounds were then titrated down from 10 μM to 156 nM to determine when CSR dropped relative to viability. As shown in Figure 2C, inhibition of CSR could be observed at 5–10 μM concentrations with little effect on viability. The top 3 candidates were also tested at higher concentrations, but they showed a 20% drop in viability at 20 μM. Toxicity would affect CSR, so we limited the analysis to 10 μM inhibitor concentrations.
Confirmation of Three Compounds by Gel Assay for Deamination
The three compounds were reacquired and purified in-house, and activity was confirmed in an orthogonal in vitro gel-based assay measuring deamination. AID was incubated with 0.25 mM DJC20 substrate in the presence of increasing concentrations of the inhibitors. The deaminated uracil was removed with UDG, and the abasic site was cleaved with apurinic/apyrimidinic enzyme 1 (APE1).30 The cleavage band intensity was then determined on a denaturing gel (Figure 1C). The ratio of product formation to product plus substrate of the three inhibitors was plotted versus compound concentration as shown in Figure 1D. Compound NCGC00128711 showed a lower intensity of deamination, because the gel likely had more substrate added than the other two gels; however, this did not affect the IC50 value. IC50 values were calculated using two concentrations of substrate, 0.25 and 2.5 μM, and the results were similar in range (data not shown). However, we were unable to determine if the inhibitors were binding competitively or noncompetitively, due to the requirement for a high concentration of enzyme (1 μM) in the assay relative to substrate (0.25 and 2.5 μM). Recombinant human AID has relatively low activity in vitro, and the true effective concentration is unknown.31 Thus, the assumption of behavior of competitive and noncompetitive inhibitors under non-Michaelis–Menten conditions could not be assessed.
Structures of Inhibitors and Analogues
The three compounds, shown in Figure 1E, share a common pyrazolo[1,5-a][1,3,5]triazine 7(6H)-one core and a modest inhibitory IC50 in the range of single to low double digits in the micromolar scale. However, the physiochemical and metabolic properties of these compounds are poor, with low aqueous solubility, reduced permeability, and low liver microsome stability that may contribute to their low potency in vivo. We attempted to improve the deamination inhibitory activity via modulation of physicochemical properties by designing and synthesizing six additional analogues focused around this chemotype (Figure S4A). Although the new analogues were more soluble (Figure S4B), they lacked inhibitory activity in the biochemical assays at concentrations at 100 μM and in cellular assays at 10 μM (Table S5). Nonetheless, these compounds may serve as a foundation to explore a comprehensive structure analysis of activity in the future.
Specificity for AID and APOBEC3B Deaminases
A critical test of the selected compounds is whether they specifically block deamination by AID or can also inhibit other cytosine DNA deaminases, such as APOBEC3B (A3B). A novel gain-of-function real-time fluorescent reporter32 was used to measure single base editing by AID and A3B in 293T cells. In these base editor complexes, either AID, containing amino acids 1–188, or A3B, containing the C-terminal catalytic domain, were individually fused to a Cas9 nickase-UGI polyprotein. A reporter plasmid contained both a gene for mCherry to measure transfection efficiency (red) and an inactive gene for enhanced Green Fluorescent Protein (eGFP) containing a Y93H missense mutation to measure editing by the deaminases. After C to U base editing of the Y93 mutant codon in eGFP, the reporter will fluoresce green, as demonstrated in Figure 3A and Figure S5A. The 293T cells were cotransfected with a Y93 guide RNA (gRNA) to direct each Cas9 editing complex to the eGFP reporter. Inhibitor compounds were added, and fluorescence was measured after 18 h by imaging analysis (Figure S5B).
Figure 3.
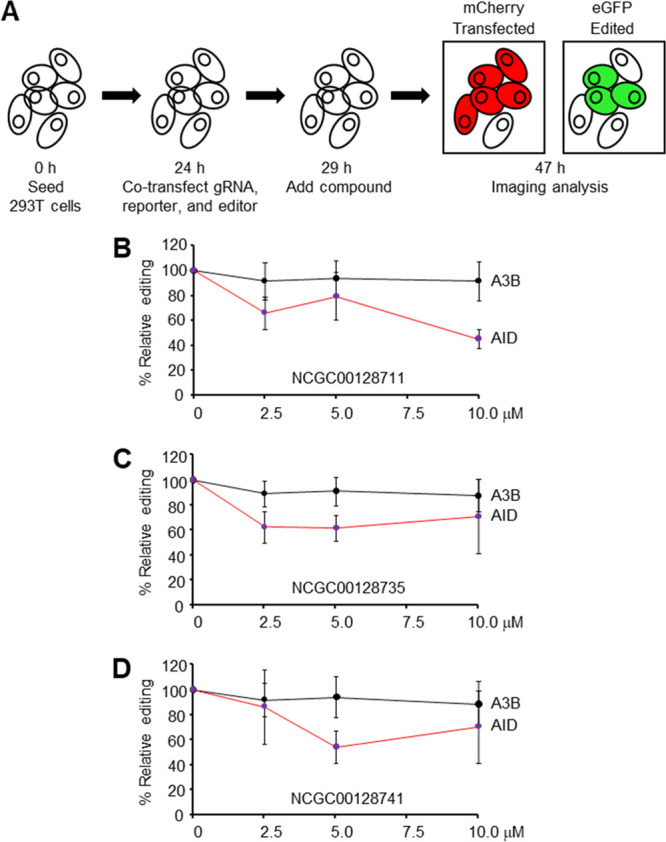
Inhibition of AID and A3B editing. (A) Fluorescent reporter assay in 293T cells. Guide RNA, dual plasmid reporter, and AID or A3B editors were added with compounds. Red and green fluorescence indicated transfection and editing, respectively. The ratio of double positive cells to mCherry positive cells was used to calculate relative editing frequencies. (B–D) Titration of compounds to measure effect on AID and A3B editing. Error bars represent the average of three independent experiments.
We first determined if our compounds inhibited AID in this assay by adding the six negative analogues described in Figure S4 and the three positive candidates at 50 μM concentration. Figure S5C shows that AID edited the reporter to 20% with DMSO only, and the six negative analogues exhibited either partial or no inhibition of base editing. Notably, the three leads identified in CSR screens showed robust inhibition with base editing down to 0–3%; however, some toxicity was evident at the 50 μM concentration. To avoid toxicity, we retested the compounds at a lower concentration of 2.5–10 μM and compared inhibition of both AID and A3B. Figure 3B shows that NCGC00128711 inhibited AID editing at increasing concentrations from 2.5 to 10 μM but had no effect on A3B editing. Similarly, NCG00128735 and NCGC00128741 inhibited AID but not A3B (Figure 3C,D). Lack of inhibition of A3B demonstrated that the compounds were not toxic to 293T cells at 10 μM. These results show that AID proficiently edited the reporter, and it was inhibited by small molecules that had no effect on A3B activity.
Use of Inhibitors to Measure AID Function in Activated B Cells
To examine if these inhibitors could be used to address an open biologic question of when AID is active after stimulation, we added them to splenic B cells at various time points. We hypothesized that the enzyme would be sensitive to inhibitors over time and prevent CSR. Splenic B cells were stimulated with LPS and IL4 ex vivo, and compounds in DMSO, or DMSO only as a control, were added to the cultures at either 0, 12, 24, 36, 48, or 60 h (Figure 4A). Flow cytometry was performed at 72 h to measure switching to IgG1. Viability dropped off markedly in these cultures by 96 h, so the 72 h time point was chosen for analysis of CSR. The percent of IgG1 (% IgG1) in DMSO was set as 100%, and the % IgG1 with compound was compared to that value as percent inhibition. When calculating the percent of CSR (% CSR), DMSO controls ranged from 22 to 31% IgG1, and inhibitors ranged from 12 to 26% IgG1. As shown in Figure 4B, CSR at 72 h was inhibited ∼10% when compounds were added at 0 h. Because AID protein is detectable as early as 24 h by immunoblots,33,34 these results suggest that the inhibitors remained stable for at least 24 h. Maximum inhibition of ∼20% was seen when the inhibitors were added at either 12, 24, 36, or 48 h after stimulation, when AID would be abundant and functional. Surprisingly, AID could be inhibited, albeit less efficiently, when inhibitors were added as late as 60 h. The results indicate that AID is continually promoting CSR events 60 h after receiving the initial stimulus.
Figure 4.

Time-dependent inhibition of CSR after B cell activation. (A) Splenic B cells were stimulated with LPS and IL4. Compounds in DMSO, or DMSO only, were added 0, 12, 24, 36, 48, and 60 h later, and flow cytometry was performed at 72 h. (B) Percent inhibition of IgG1 by compounds relative to the DMSO only control, which was set at 100% IgG1. Error bars depict the standard deviation from 4 to 9 assays. Significance measured by paired two-tailed t test; * p < 0.05.
Discussion
DNA cytosine deaminases are potent enzymes that instigate genomic diversity for both beneficial and harmful outcomes in humans. To gain small molecule control over these enzymes, research initially focused on members of the APOBEC3 family, which mutate both viral and cellular genomes. Inhibitors of A3G were identified by high-throughput screens, with the potential of modulating catalytic activity during HIV infection.35,36 A3B also promotes mutagenesis in a variety of cancer cells and is an appropriate target for manipulation using small molecules.37−39 However, controlling the master deaminase of the adaptive immune system, AID, has proved elusive, in part due to the difficulty in purifying the enzyme for high-throughput analysis. Previous results have suggested that AID can be modulated by chemicals that inhibit heat shock protein 90, which stabilizes AID, in order to promote its degradation and thereby decrease CSR or cancer progression.40,41 Conversely, some agents increase AID retention in the nucleus and elevate its activity for CSR and chromosome translocations.42 Other pharmacologic approaches to modulating AID activity have included siRNA to decrease AID expression in tumor cells,43 and inhibition of repair of AID-initiated breaks by RAD51 to induce apoptosis.44,45
Here, we took a more direct approach by targeting AID itself with small molecules. Our biochemical assay leveraged a soluble and more active version of human AID,46 along with a dual substrate pair that can be used to enhance specificity in a high-throughput amenable screen of over 90 000 compounds. Filtering of hits, followed by examination of 27 candidates in cellular assays of CSR, led to the identification of three inhibitors that reduced switching by 30% without affecting cell viability and proliferation. Importantly, the three inhibitors were selective for AID and did not inhibit A3B in a GFP-reporter assay in 293T cells. AID and A3B target cytosine in distinct DNA motifs, and their overall structures are different.47 The differential inhibition of the proteins indicates that the inhibitors bind to a unique site on AID that is not present in A3B.
The fact that the lead inhibitors all have a similar core chemical structure suggests a shared mechanism of action. Related analogues were explored to improve solubility and permeability, but they showed no inhibitory activity. Nonetheless, more systematic efforts to explore structure–activity relationships could help improve potency and reveal the binding mode of the compounds. DNA-bound structures of a related family member APOBEC3A have highlighted the nucleobases and backbone which differentiate DNA deaminases from their similar nucleoside/nucleotide deaminase relatives.39 For AID, the mechanism of action of the leads is not known, although their structures resemble purines in AID’s trinucleotide WRC hotspot motif (W = A/T, R = purines).46,48−51 The leads could thus function by competitively binding via the −1 position upstream of cytosine in the substrate to block the catalytic site. To assess whether the compounds could affect AID expression in B cells, we examined AID expression during CSR by Western blot with the different compounds and did not observe any decrease in protein levels in preliminary experiments. However, this analysis needs further experimentation with more sensitive techniques to fully understand the mechanism of AID inhibition. Future efforts to understand the mechanism or improve these leads would benefit from structural modeling approaches, particularly because the crystal structure of AID has recently been resolved.52,53
We then used the three inhibitors to address a biological question of when AID is functional in B cells after stimulation. Previous reports have demonstrated that AID is first expressed at day 1 in ex-vivo-stimulated cells by PCR of cDNA or Western blots of protein. AID then increases by days 2–3,33,34 suggesting that its expression is linked to cell division during CSR.54,55 Our small molecule approach allowed us to ask if AID is required continuously to increase CSR or if previously switched cells just divide more. The inhibitors were most effective at preventing CSR by 25% when added 12–48 h after stimulation, but CSR was still prevented at later times, albeit at a lower frequency of 10%. This suggests that AID promotes CSR continuously by introducing productive breaks up to 60 h after stimulation. Thus, the increased CSR at later time points is due to both the introduction of new switching events by AID and increased division of class-switched cells.
In addition to a role as probe of AID structure and biology, the long-term goal of finding small molecule inhibitors is to regulate AID therapeutically in an attempt to reduce off-target effects on cancer and lower mutational loads in autoimmune antibodies. Our studies have uniquely defined the first generation of AID inhibitors, which can be used as a starting point for the discovery of more efficacious compounds to gain regulatory control over this base editor.
Materials and Methods
CH12 Cell Stimulation
The murine CH12 cell line was obtained from F.W. Alt (Harvard University), and cells were cultured in 1X RPMI (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 50 μM β-mercaptoethanol (Sigma-Aldrich), 1X GlutaMAX (Thermo Fisher Scientific), 1X Penicillin plus Streptomycin (Thermo Fisher Scientific), and 5% Medium NCTC-109 (Thermo Fisher Scientific). To measure proliferation, 10 million cells were incubated with 5 μM Cell Proliferation Dye eFluor 670 (Thermo Fisher Scientific) in 2 mL of PBS for 10 min at 37 °C in an incubator containing 5% CO2. Staining was then quenched by diluting the reaction 1:5 with prechilled culture media and left on ice for a minimum of 5 min. Cells were spun for 5 min at 1300 rpm; the supernatant was aspirated, and the cells were washed three times in 1 mL of culture media prior to being counted. To induce optimal CSR from IgM to IgA in a 96-well format, 250 μL of cultures at 62 500 cells/mL were stimulated for 72 h in medium containing 1 μg/mL antimouse CD40 antibody (Clone no. FGK45, Enzo Life Sciences), 10 ng/mL recombinant murine IL4 (R&D Systems), and 2 ng/mL recombinant human TGF-β (R&D Systems). Inhibitor compounds were dissolved in 100% DMSO and diluted 1:10 in media for a 10% DMSO solution. This solution was subsequently diluted 1:100 by adding 2.5 μL into the 250 μL of culture containing cells, for a final DMSO concentration of 0.1%. As a negative and positive control, some culture wells were supplemented with and without media containing 0.1% DMSO, respectively.
Splenic B Cell Stimulation
Naive splenic B cells were isolated from C57BL/6 mice by negative selection with anti-CD43 and anti-CD11b magnetic beads (Miltenyi Biotec). For proliferation, cells were labeled with eFluor 670 dye as described above and cultured in the same media as CH12 cells minus the NCTC-109 supplement. To induce CSR to IgG1 in a 96-well format, the cells were stimulated for 72 h in medium containing 5 μg/mL lipopolysaccharide (LPS) (E. coli serotype 0111:B4; Sigma-Aldrich), and 10 ng/mL recombinant murine IL4. Inhibitors were added in a final concentration of 0.1% DMSO. Viability was assessed with Zombie Aqua (BioLegend). Mice were provided by the NIA breeding colony, and both sexes were used at 2–4 months of age. All animal protocols were reviewed and approved by the Animal Care and Use Committee of the National Institute on Aging.
Flow Cytometry
After 72 h, 96-well plates containing cells were spun for 10 min at 1300 rpm. After removal of the supernatant, CH12 cells were incubated for 20 min on ice with 200 μL of 1:200 antimouse IgA (Southern Biotech), while splenic B cells were incubated with 1:1500 antimouse IgG1 (Clone no. RMG1-1, Biolegend) using Sort Media (2.5 mM HEPES, 1 mM EDTA, 1% FBS, 1X PBS) as a diluent. Cells were then centrifuged and resuspended in Sort Media with 7-aminoactinomycin D (7-AAD) (Sigma-Aldrich) for CH12 cells or Zombie Aqua (BioLegend) for splenic B cells to measure viability.
Plasmids for Single Base Editing Assay
A3Bctd-Cas9n-UGI has been reported.32 The human AID cDNA encoding amino acids 1–188 was amplified from a pcDNA 4/TO expression construct56 using primers 5′-AGATCCGCGGCCGCGCCGCCACCATGGACAGCCTCTTGATGAACC and 5′-TGAGGTCCCGGGAGTCTCGCTGCCGCTGTCATCAACCTCATACAGGGGC, restricted with NotI and XmaI and cloned into similarly digested BE3.57 The MLM3636 gRNA vector (43860) was optimized for RNA pol III using primers 5′-CGATTAATGCGTCTCGTTCTAGAGCTAGAAATAGCAAGTTAGAATAAGGCTAGTC and 5′-GACTAGCCTTATTCTAACTTGCTATTTCTAGCTCTAGAACGAGACGCATTAATCG and then modified for interchangeable protospacer use with a lentiCRISPR plasmid58 using primers 5′-GGACGAAACACCGGAGACGATTAATGC, 5′-GCATTAATCGTCTCCGGTGTTTCGTCC, 5′-GACGATTAATGCGTCTCTGTTCTAGAGCTAG, and 5′-CTAGCTCTAGAACAGAGACGCATTAATCGTC using site-directed mutagenesis (SDM).59 The gRNA targeting eGFP codon 93 was constructed by annealing complementary oligonucleotides 5′-CACCGCCGAAGGTCACGTCCAGGAG and 5′-GAACCTCCTGGACGTGACCTTCGGC and Golden Gate cloning60 into the PolIII-optimized MLM3636.58 The CMV mCherry-T2A-eGFP expression cassette has been reported.61 This cassette was cloned into a modified HIV-1 NL4-3 backbone that contains a multiple cloning site-IRES-Hygromycin cassette excised from a pQXCIH retroviral vector using NotI and SphI restriction sites. The CMV mCherry-T2A-eGFP cassette was amplified using primers 5′-GGTGGCGCGGCCGCTTAATAG and 5′-ATTAAGCGTACGTTATTTATATAATTCATCCATACCGAGAGTG, restricted with NotI and BsiWI and cloned into similarly digested NL4-3-IRES-hygromycin. A tyrosine to histidine mutation in codon 93 as well as silent mutation to add a PAM site in codon 97 of eGFP was generated using SDM with primers 5′-GCCATGCCCGAAGGTCACGTCCAGGAGCGGACCATCTTCTTCAA and 5′-TTGAAGAAGATGGTCCGCTCCTGGACGTGACCTTCGGGCATGGC.
Episomal Editing Experiments
The 293T cells were maintained in RPMI media supplemented with 10% FBS and 1% penicillin/streptomycin. Semiconfluent 10 cm plates were transfected with 2200 ng of gRNA, 4400 ng of reporter, and 6600 ng of editor incubated with 39.5 μL of TransIT 2020 (Mirus Bio) and 1500 μL of serum-free RPMI for 25 min. Cells were harvested 5 h after transfection via trypsinization. Next, 50 000 cells diluted in 150 μL of media were plated into the center 24 wells of a prewarmed Nunclon flat bottom 48-well plate (Thermo Fisher) containing 150 μL of compound dilutions (initial concentrations were 100, 20, and 5 μM) for final concentrations of 50, 10, 5, and 2.5 μM. The outer wells contained 500 μL of supplemented RPMI to reduce the edge effect.
Live Cell Imaging and Analysis
Fluorescent images were taken using the Cytation1Multi-Mode Reader (Bio-Tek) configured with Texas Red (586/647) and GFP (469/525) light cubes. Live-cell images were captured using a 4x objective 18 h after the initial transfection. Temperature was maintained at 37 °C for the duration of imaging. Four images were taken in each well in a fixed 2 × 2 grid. After background subtraction, a dual mask analysis was used to quantify edited versus nonedited cells. All mCherry positive cells were first identified as the primary mask using the fluorescence threshold as well as size exclusion. Next, a secondary mask was created within the primary mask to identify eGFP positive cells using a GFP fluorescence threshold. The ratio of double positive cells to mCherry positive cells was then used to calculate relative editing frequencies.
Reagents for Biochemical Assays
Chemicals used for the initial biochemical screening were dimethyl sulfoxide (DMSO; ACS grade, Thermo Fisher Scientific), NaOH (Thermo Fisher Scientific), Tris–HCl pH 8 (Thermo Fisher Scientific), EDTA pH 8 (Quality Biological), dithiothreitol (DTT; MP Biomedicals), and Tween20 (Sigma-Aldrich).
Enzymes and Fluorescent Substrates
Human AID was prepared as a maltose binding protein fused to a C-terminally truncated protein (AID-1-181)46 and stored in a solution of 50 mM Tris 7.5, 150 mM NaCl, 10 μM ZnCl2, 10% glycerol, and 10 mM maltose. Uracil-DNA glycosylase (UDG) was obtained from New England Biolabs. Two substrates with fluorophores (Cy3 or Cy5) and quenchers (Black Hole Quencher-2, BHQ2) were synthesized and purified in house using phosphoamidite building blocks (Glen Research): DJC 20 (5′-Cy3-ATT AGC TTT (T-BHQ2)AT TTA TGG TGT TTG GTG TGG TT-3′) and DJC 23 (5′-Cy5-ATT AGU TTT (T-BHQ2)AT TTA TGG TGT TTG GTG TGG TT-3′). Both substrates were stored in water.
Compound Libraries
Aurintricarboxylic acid (ATA) was purchased from Sigma-Aldrich. Three compound libraries were used: Chemical Biology Consortium (CBC) containing 46 018 compounds (https://next.cancer.gov/discoveryResources/resources_ndl.htm), Sytravon collection (NCATS) comprising 43 984 druglike compounds (a retired Pharma screening collection that contains a diversity of novel small molecules, with an emphasis on medicinal chemistry-tractable scaffolds) (https://pubchem.ncbi.nlm.nih.gov/bioassay/1347069 and https://pubchem.ncbi.nlm.nih.gov/bioassay/1347077), and LOPAC1280, a 1280-member library of pharmacological-active compounds, for a total of 90 757 nonoverlapping compounds. All compounds were initially sourced from the National Center for Advancing Translational Sciences (NCATS)/National Institutes of Health (NIH). Compounds for cellular assays were subjected to quality control and exhibited >90% purity.
384-Well Assay Optimization
The final concentrations of enzyme and substrate were determined by initial tests in 384-well format (solid-bottom black plate; Greiner Bio One). We first chose substrate concentrations of 250 nM each for DJC20 and DJC23, based on Km of ∼100 nM (10 μL assay volume), which exhibited S:B values of ∼2 and ∼10, respectively, vs buffer (20 mM Tris–HCl pH 8, 1 mM EDTA pH 8, 1 mM DTT prepared fresh, with 0.05% Tween20) based on RFUs. Next, we dispensed 10 μL of (±) AID and (±) UDG, at 2.2 μM and 50 U/mL (final concentrations, respectively; n = 3/solution), onto a 384-well plate, followed by the addition of 10 μL of a substrate mixture containing 250 nM DJC20 and 250 nM DJC23 (final concentrations; Km = 5–10 μM, data not shown). Separate wells containing only buffer were also added. Solutions were incubated at room temperature (RT) for 4 h, followed by the addition of 20 μL of quenching solution (250 mM NaOH), for an additional 1 h of incubation (RT); then, they were read for fluorescence on an Envision multimodal detector at (Ex/Em) 525(45)/590(20) and 595(10)/665(7.5) for AID and UDG, respectively, and calculated for fluorescence (RFU). For plus/minus AID, a Z′ of 0.8 was observed along with an S:B of ∼3.5, while for UDG, a Z′ of 0.9 and S:B of ∼76 was observed. We left UDG in excess to be used as a counterscreen.
1536-Well Assay Optimization
We then moved the above reaction conditions to 1536-well format (Greiner black solid-bottom medium-binding assay plate) and prepared an intraplate titration (10 mM, 1:2 dilution, 8-point, n = 2) of aurintricarboxylic acid (ATA), a tool compound that inhibits protein–nucleic acid interactions, to assist in assessing assay performance. Briefly, 3 μL of enzymes (2.2 μM AID and 50 U/mL UDG, final concentration; neutral control) or enzyme (50 U/mL UDG; AID positive control) in assay buffer (20 mM Tris–HCl pH 8, 1 mM EDTA pH 8, 1 mM DTT prepared fresh, with 0.05% Tween20) was dispensed into a 1536-well Greiner black solid-bottom assay plate. Controls (DMSO; positive and neutral) and intraplate control (ATA; final concentration range from 888 nM to 114 μM) were transferred (23 or 46 nL) via Wako PinTool equipped with 1536-pin array. The plate was covered and incubated for 15 min at room temperature, followed by addition of a 1 μL aliquot of a substrate solution (250 nM DJC20 and 250 nM DJC23) to start the reaction, for a final assay volume of 4 μL. The reaction was stopped after 4 h by the addition of an equal volume (4 μL) of NaOH solution (final concentrations of 250 mM). After a short spin (1000 rpm for 15 s), the assay plate was incubated at room temperature, and the fluorescence (Ex/Em) at 525(45)/590(20) and 595(10)/665(7.5), for DJC20 and DJC23, respectively, were recorded using an EnVision multimodal detector (PerkinElmer) using standard protocol settings, at 15, 30, 45, and 60 min, to determine the quench solution’s optimal incubation time. We chose 30 min, with a signal window of ∼578 000 RFUs and Z′ of 0.77, and we observed an IC50 for ATA of ∼2 μM. We then performed an enzyme titration, where we chose 0, 0.5, 1, and 2 μM AID (all containing 50 U/mL UDG) to test (n = 64/condition) using the above protocol, modifying the incubation times to 1, 2, 4, and 6 h after a 1 μL addition of 250 nM substrate mixture. Balancing assay performance, reagent consumption, and percent conversion (∼25%), we selected 0.5 μM enzyme and an incubation time of 6 h (Z′ = 0.6; Table S1).
Quantitative High-Throughput Screening (qHTS) for Pilot Screen
With these conditions in place, we performed a pilot screen against the LOPAC1280 compound library, consisting of 6 plates in qHTS format (10 mM, 1:5 dilution) resulting in a final assay concentration of 91.5 nM to 114 μM. Our protocol was similar to the above with minor modifications to the control solutions. Briefly, 3 μL of enzymes (0.5 μM AID and 50 U/mL UDG, final concentration; neutral control) or enzyme (50 U/mL UDG; AID positive control) or enzyme (0.5 μM AID; UDG positive control) in assay buffer (20 mM Tris–HCl pH 8, 1 mM EDTA pH 8, 1 mM DTT prepared fresh, with 0.05% Tween20) was dispensed into a 1536-well Greiner black solid-bottom assay plates. The remainder of the assay was performed as previously described. Reagents were kept on ice. Average Z′-factors of 0.5 ± 0.3 and 0.8 ± 0.0, signal windows of ∼345 000 and ∼788 000, S:B values of ∼1.4 and 2.1, for both AID and UDG, respectively, were observed along with an intraplate control IC50 of 0.57 ± 0.05 μM (MSR = 1.3) for ATA. All screening operations were performed on a fully integrated robotic system (Wako Automation) for potential AID inhibitors using a dual-output Cy3 and Cy5 fluorescence quenching for the qHTS assay. During dispensing, enzyme and substrate bottles were kept on ice to minimize degradation. Plates containing DMSO only (instead of compound solutions) were randomly inserted throughout the screen to monitor any systematic trend in the assay signal associated with reagent dispenser variation or a decrease in enzyme specific activity.
Due to owning a limited number of filter sets, we modified the above by changing the DJC20 excitation fluorescence filter from 525(45) to 540(8). We decreased UDG from 50 to 2 U/mL, while still keeping it in excess so as not to let it be the limiting reagent. Separately, we also introduced Mitoxantrone as intraplate control (identified from LOPAC1280 screen), but we switched back to ATA. Finally, we increased the enzyme concentration from 500 nM up to 1000 nM. These modifications were done to balance reagent quantities and signal strength, while also allowing for the best chance to identify inhibitors based on reaction progress and substrate concentration. In addition to the LOPAC1280 compound library, we screened 90 757 compounds from CBC and Sytravon in qHTS format dose–response (4- or 5-points, final concentration range from 114 nM to 114 μM). Average Z′ values of 0.7 ± 0.2 and 0.8 ± 0.1, for both AID and UDG, respectively, were observed across 285 plates. We used intraplate control IC50 values of 3.2 ± 3.7 μM (MSR = 4.7) and 7.5 ± 5.6 μM (MSR = 5.2), for ATA and Mitoxantrone, respectively.
qHTS Protocol for Compound Confirmation
Reactions were prepared in a 1536-well plate format using 3 μL of enzymes (1 μM AID and 2 U/mL UDG final concentration, or UDG alone) in assay buffer. Compounds (qHTS format dose–response: 6 or 7 points, final concentration range of 3.7 nM to 114 μM) and controls in DMSO (final concentration of 0.6% for positive and neutral controls) and an intraplate control (ATA; final concentration range from 888 nM to 114 μM) were transferred (23 or 46 nL) via Wako PinTool equipped with 1536-pin array. Plates were covered and incubated for 15 min at room temperature, followed by addition of 1 μL of substrate mixture (250 nM DJC20 and 250 nM DJC23) to start the reaction, and incubated for 4 h, followed by the addition of an equal volume (4 μL) of NaOH solution (final concentration of 250 mM). After a short spin (1000 rpm for 15 s), the assay plate was incubated at room temperature for 1 h and read for fluorescence by Ex/Em (bandpass) at 525(45)/590(20) and 595(10)/665(7.5) for DJC20 and DJC23, respectively, on an EnVision detector using standard protocol settings (Table S1). The fluorescence intensity was normalized against no-inhibitor and no-enzyme controls.
In Vitro Deamination Gel Assay
Briefly, 15 μL of enzyme (final concentration 1 μM) or buffer was dispensed into a 384-well polypropylene compound storage plate (Greiner catalog no. 784201). Compounds (final concentration range from 6 to 196 μM) and controls (DMSO; positive and neutral, 2% final) were transferred (200 nL) via acoustic droplet ejection (ADE; Labcyte Echo). The plate was covered and incubated for 15 min at room temperature, followed by addition of a 5 μL aliquot of a substrate solution (DJC20 and UDG, final concentrations of 0.25 μM and 2 U/mL, respectively) to start the reaction, for a final assay volume of 20 μL. Samples were incubated for 4 h (∼90% conversion; data not shown), followed by cleavage of the abasic sites with APE1 (2 nM; Trevigen), incubated at room temperature for 30 min, and the cleavage reaction was terminated by heating to 95 °C for 10 min. Samples were mixed with an equal volume of formamide/sucrose loading solution (5% formamide, 25% sucrose in 2X TBE buffer), run on 15% TBE polyacrylamide (1.5 mm, 15-lane) native gel made using the ready-to-use solution of 30% (w/v) acrylamide:bis(acrylamide) (29:1; AccuGel, National Diagnosticsfor 50 min (120 V)). The gel was scanned at 532 nm using a Typhoon FLA-9500 laser scanner for band intensity quantification using ImageQuant TL v8.1. The activity was determined by the ratio of the signal from the product, divided by the sum of the product and substrate bands.
Kinetic Solubility Assay
pIon’s patented μSOL assay was used for kinetic solubility determination.62,63 In this assay, the classical saturation shake-flask solubility method was adapted as previously described.64 Test compounds were prepared in 10 mM DMSO stock and diluted to a final drug concentration of 150 μM in the aqueous solution (pH 7.4, 100 mM phosphate buffer). Samples were incubated at room temperature for 6 h and vacuum-filtered using Tecan Te-Vac to remove any precipitates. The concentration of the compound in the filtrate was measured via UV absorbance (λ: 250–498 nm). The unknown drug concentration was determined by comparing the fully solubilized reference plate which contained 17 μM compound dissolved in spectroscopically pure n-propanol. All compounds were tested in duplicates. The kinetic solubility (μg/mL) of compounds was calculated using the μSOL Evolution software. The three controls used were albendazole (low solubility), phenazolpyridine (moderate solubility), and furosemide (high solubility). This method has been previously published.65
Parallel Artificial Membrane Permeability Assay (PAMPA)
The stirring double-sink PAMPA method (patented by pION Inc.) was employed to determine the permeability of compounds via PAMPA. The PAMPA lipid membrane consisted of an artificial membrane of a proprietary lipid mixture and dodecane (pIon Inc.), optimized to predict gastrointestinal tract (GIT) passive permeability. The lipid was immobilized on a plastic matrix of a 96-well “donor” filter plate placed above a 96-well “acceptor” plate. The pH 7.4 solution was used in both donor and acceptor wells. The test articles, stocked in 10 mM DMSO solutions, were diluted to 0.05 mM in aqueous buffer (pH 7.4), and the concentration of DMSO was 0.5% in the final solution. During the 30 min permeation period at room temperature, the test samples in the donor compartment were stirred using the Gutbox technology (pIon Inc.) to reduce the aqueous boundary layer. The test article concentrations in the donor and acceptor compartments were measured using a UV plate reader (Nano Quant, Infinite 200 PRO, Tecan Inc.). Permeability calculations were performed using Pion Inc. software and were expressed in units of 10–6 cm/s. Compounds with low or weak UV signal were analyzed using high-resolution LC/MS (Thermo QExactive). The three controls used were ranitidine (low permeability), dexamethasone (moderate permeability), and verapamil (high permeability). This method has been previously published.66
Rat Liver Microsome Stability Assay
Single time point microsomal stability was determined in a 96-well HTS format. Sample preparation was automated using a Tecan EVO 200 robot. A High Resolution LC/MS (Thermo QExactive) instrument was used to measure the percentage of compound remaining after incubation using a previously described method.67 Six standard controls were tested in each run: buspirone and propranolol (for short half-life), loperamide and diclofenac (for short to medium half-life), and carbamazepine and antipyrine (for long half-life). Briefly, the incubation consisted of 0.5 mg/mL microsomal protein, 1.0 μM drug concentration, and NADPH regeneration system (containing 0.650 mM NADP+, 1.65 mM glucose 6-phosphate, 1.65 mM MgCl2, and 0.2 unit/mL G6PDH) in 100 mM phosphate buffer at pH 7.4. The incubation was carried out at 37 °C for 15 min. The reaction was quenched by adding 555 μL of acetonitrile (∼1:2 ratio) containing 0.28 μM albendazole (internal standard). Sample acquisition and data analysis was done using a previously described method.67
General Methods for Chemistry
All air or moisture sensitive reactions were performed under positive pressure of nitrogen or argon with oven-dried glassware. Anhydrous solvents and bases such as dichloromethane, N,N-dimethylforamide (DMF), acetonitrile, ethanol, DMSO, dioxane, DIPEA (diisopropylethylamine), and triethylamine were purchased from Sigma-Aldrich. Palladium catalysts were purchased from Strem chemicals and used as such. All other reagents were purchased from Sigma-Aldrich or Combi-Blocks in the highest available purity and used as received. Preparative purification was performed on a Waters semipreparative HPLC system using a Phenomenex Luna C18 column (5 μm, 30 mm × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10–50% acetonitrile over 8 min was used during the purification. Fraction collection was triggered by UV detection (220 nm). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies).
Method 1
A 3 min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 min run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 μm, 3 mm × 100 mm) was used at a temperature of 50 °C.
Method 2
A 7 min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 min run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 μm, 3 mm × 75 mm) was used at a temperature of 50 °C.
Purity determination was performed using an Agilent Diode Array Detector for both Method 1 and Method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. Chemical shifts for final compounds are reported in parts per million (ppm) with undeuterated solvent (DMSO-d6 at 2.50 ppm) as internal standard for DMSO-d6 solutions. Chemical shifts for intermediate compounds are reported in parts per million (ppm) with undeuterated solvent (CDCl3 at 7.26 ppm) as internal standard for CDCl3 solutions. All the analogues tested in the biological assays have purity greater than 95%, based on both analytical methods. High-resolution mass spectrometry was recorded on an Agilent 6210 Time-of-Flight LC/MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
Phenyl-4-thioxopyrazolo[1,5-a][1,3,5]triazine-7(6H)-one was prepared as described in https://www.mdpi.com/1422-8599/2017/4/M970. To a mixture of phenyl-4-thioxopyrazolo[1,5-a][1,3,5]triazine-7(6H)-one (0.12 g, 0.5 mmol) and potassium carbonate (0.138 g, 1 mmol) in acetone (6 mL), 3-(1H-imidazol-1-yl)propyl methanesulfonate (0.12g, 0.6 mmol) was added. The reaction was heated to 65 °C for 18 h. The mixture was acidified with TFA (4 equiv), concentrated, and dissolved in DMSO. The mixture was purified by preparative HPLC. LC-MS t1 = 3.54 min, M + H = 353. 1H NMR (400 MHz, DMSO-d6): δ 11.73 (s, 1H), 9.09 (s, 1H), 8.35–8.28 (m, 2H), 7.85–7.80 (m, 1H), 7.71–7.66 (m, 1H), 7.62–7.50 (m, 3H), 5.99 (s, 1H), 4.40 (t, J = 6.9 Hz, 2H), 3.48 (t, J = 7.2 Hz, 2H), 2.40 (q, J = 7.1 Hz, 2H).
qHTS Data Analysis and Statistics
Data from each assay were normalized plate-wise to corresponding intraplate controls (neutral control was DMSO and positive control was as noted). The same controls were also used for the calculation of the Z-factor, which is a measure of assay quality control.25 Percent activity was derived using in-house software (http://tripod.nih.gov/curvefit/). All concentration response curves (CRCs) were fitted using in-house developed software (http:/ncgc.nih.gov/pub/openhts/). Curves were categorized into four classes as shown in Figure S1: complete response curves (class 1), partial curves (class 2), single point actives (class 3), and inactives (class 4). CRCs were fitted and classified, and IC50 was determined (add ref Inglese and Wang). Otherwise, CRCs were fitted and IC50 values were calculated using Prism software (version 7.04, GraphPad Software, Inc.) sigmoidal dose–response (variable slope). The chemical structures were standardized using the Layered Chemical Identifier program (LyChi version 20141028).68 We used the Palantir data integration platform, which is configured to ingest all HTS results generated at NCATS, and harmonized this data with other sources, such as ChEMBL and OrthoMCL. All qHTS results are publicly available at PubChem (htttps://pubchem.ncbi.nlm.nih.gov/source/NCGC; AID 1347431, 1347432, 1347433, 1347434).
Acknowledgments
We acknowledge initial contributions by Steve Finckbeiner, NHGRI, NIH. This work was partially supported through the Intramural Research Program at the National Institutes of Health, National Institute on Aging (AG000732), National Center for Advancing Translational Sciences (NCATS), and extramural support (R01-HG010646 to R.M.K. and PO1-CA234228 to R.S.H.). We would like to thank the NCATS Compound Management, Automation, ADME, and Analytical groups for their support. We thank Kyle Brimacombe at NCATS for graphic design assistance; Ajit Jadhav, Richard Eastman, and David Maloney for thoughtful discussion; and Kelli Wilson for curation of molecules and data integration. D.J.S. received salary support from the University of Minnesota Craniofacial Research Training (MinnCResT) program (NIH T90DE022732) and from an NIAID K99/R00 transition award (K99-AI147811).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00064.
Author Present Address
○ D.J.C.: Intellia Therapeutics, Cambridge, MA 02139, USA.
Author Present Address
◆ D.J.S.: Renaissance School of Medicine, Department of Microbiology and Immunology, Stony Brook University, Stony Brook, NY 11794, USA.
Author Contributions
J.A.-G. and A.Y. contributed equally. The concept was conceived by A.S., R.S.H., R.M.K., and P.J.G. The overall study design was developed by J.A.G., A.Y., R.W.M., A.E.R., D.J.C., and J.M. Additional experiments were performed by D.J.S., D.D., A.V.Z., D.J., and G.R. The manuscript was written by J.A.G., A.Y., R.W.M., A.E.R., R.S.H., R.M.K., and P.J.G.
The authors declare the following competing financial interest(s): R.S.H. is a co-founder, shareholder, and consultant of ApoGen Biotechnologies Inc.
Supplementary Material
References
- Heltzel J. M. H.; Gearhart P. J. (2020) What Targets Somatic Hypermutation to the Immunoglobulin Loci?. Viral Immunol. 33, 277–281. 10.1089/vim.2019.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudinet C.; Braikia F. Z.; Dauba A.; Khamlichi A. A. (2020) Mechanism and regulation of class switch recombination by IgH transcriptional control elements. Adv. Immunol. 147, 89–137. 10.1016/bs.ai.2020.06.003. [DOI] [PubMed] [Google Scholar]
- Rajagopal D.; Maul R. W.; Ghosh A.; Chakraborty T.; Khamlichi A. A.; Sen R.; Gearhart P. J. (2009) Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J. Exp. Med. 206, 1237–1244. 10.1084/jem.20082514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Wuerffel R.; Feldman S.; Khamlichi A. A.; Kenter A. L. (2009) S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J. Exp. Med. 206, 1817–1830. 10.1084/jem.20081678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R.; Gazumyan A.; Jankovic M.; Di Virgilio M.; Klein I.; Ansarah-Sobrinho C.; Resch W.; Yamane A.; Reina San-Martin B.; Barreto V.; Nieland T. J.; Root D. E.; Casellas R.; Nussenzweig M. C. (2010) Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143, 122–133. S0092-8674(10)01065-2 [pii] 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul R. W.; Cao Z.; Venkataraman L.; Giorgetti C. A.; Press J. L.; Denizot Y.; Du H.; Sen R.; Gearhart P. J. (2014) Spt5 accumulation at variable genes distinguishes somatic hypermutation in germinal center B cells from ex vivo-activated cells. J. Exp. Med. 211, 2297–2306. 10.1084/jem.20131512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H. M.; Peters A.; Baron B.; Zhu X.; Storb U. (1998) Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science 280, 1750–1752. 10.1126/science.280.5370.1750. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L.; Neumeister P.; Goossens T.; Nanjangud G.; Chaganti R. S.; Kuppers R.; Dalla-Favera R. (2001) Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 412, 341–346. 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- Liu M.; Duke J. L.; Richter D. J.; Vinuesa C. G.; Goodnow C. C.; Kleinstein S. H.; Schatz D. G. (2008) Two levels of protection for the B cell genome during somatic hypermutation. Nature 451, 841–845. 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- Yamane A.; Resch W.; Kuo N.; Kuchen S.; Li Z.; Sun H. W.; Robbiani D. F.; McBride K.; Nussenzweig M. C.; Casellas R. (2011) Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat. Immunol. 12, 62–69. 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Prado A. F.; Perez-Duran P.; Perez-Garcia A.; Benguria A.; Torroja C.; de Yebenes V. G.; Ramiro A. R. (2018) A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J. Exp. Med. 215, 761–771. 10.1084/jem.20171738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J.; Wang Q.; Dose M.; Pruett N.; Kieffer-Kwon K. R.; Resch W.; Liang G.; Tang Z.; Mathe E.; Benner C.; Dubois W.; Nelson S.; Vian L.; Oliveira T. Y.; Jankovic M.; Hakim O.; Gazumyan A.; Pavri R.; Awasthi P.; Song B.; Liu G.; Chen L.; Zhu S.; Feigenbaum L.; Staudt L.; Murre C.; Ruan Y.; Robbiani D. F.; Pan-Hammarstrom Q.; Nussenzweig M. C.; Casellas R. (2014) B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 159, 1524–1537. 10.1016/j.cell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F. L.; Du Z.; Federation A.; Hu J.; Wang Q.; Kieffer-Kwon K. R.; Meyers R. M.; Amor C.; Wasserman C. R.; Neuberg D.; Casellas R.; Nussenzweig M. C.; Bradner J. E.; Liu X. S.; Alt F. W. (2014) Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell 159, 1538–1548. 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot S. P.; Litzler L. C.; Subramani P. G.; Eranki A. K.; Fifield H.; Patenaude A. M.; Gilmore J. C.; Santiago G. E.; Bagci H.; Cote J. F.; Larijani M.; Verdun R. E.; Di Noia J. M. (2018) A licensing step links AID to transcription elongation for mutagenesis in B cells. Nat. Commun. 9, 1248. 10.1038/s41467-018-03387-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senigl F.; Maman Y.; Dinesh R. K.; Alinikula J.; Seth R. B.; Pecnova L.; Omer A. D.; Rao S. S. P.; Weisz D.; Buerstedde J. M.; Aiden E. L.; Casellas R.; Hejnar J.; Schatz D. G. (2019) Topologically Associated Domains Delineate Susceptibility to Somatic Hypermutation. Cell Rep. 29, 3902–3915. e3908. 10.1016/j.celrep.2019.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani D. F.; Nussenzweig M. C. (2013) Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu. Rev. Pathol.: Mech. Dis. 8, 79–103. 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- Alt F. W.; Zhang Y.; Meng F. L.; Guo C.; Schwer B. (2013) Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 152, 417–429. 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldhahn N.; Henke N.; Melchior K.; Duy C.; Soh B. N.; Klein F.; von Levetzow G.; Giebel B.; Li A.; Hofmann W. K.; Jumaa H.; Muschen M. (2007) Activation-induced cytidine deaminase acts as a mutator in BCR-ABL1-transformed acute lymphoblastic leukemia cells. J. Exp. Med. 204, 1157–1166. 10.1084/jem.20062662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm L.; Duy C.; Iacobucci I.; Kuchen S.; von Levetzow G.; Feldhahn N.; Henke N.; Li Z.; Hoffmann T. K.; Kim Y. M.; Hofmann W. K.; Jumaa H.; Groffen J.; Heisterkamp N.; Martinelli G.; Lieber M. R.; Casellas R.; Muschen M. (2009) The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell 16, 232–245. 10.1016/j.ccr.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber T. A.; Chang M. S.; Sposto R.; Muschen M. (2010) Activation-induced cytidine deaminase accelerates clonal evolution in BCR-ABL1-driven B-cell lineage acute lymphoblastic leukemia. Cancer Res. 70, 7411–7420. 10.1158/0008-5472.CAN-10-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki I. M.; Hiai H.; Kakazu N.; Yamada S.; Muramatsu M.; Kinoshita K.; Honjo T. (2003) Constitutive expression of AID leads to tumorigenesis. J. Exp. Med. 197, 1173–1181. 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H. C.; Wu Y.; Yang P.; Wu Q.; Job G.; Chen J.; Wang J.; Accavitti-Loper M. A.; Grizzle W. E.; Carter R. H.; Mountz J. D. (2007) Overexpression of activation-induced cytidine deaminase in B cells is associated with production of highly pathogenic autoantibodies. J. Immunol. 178, 5357–5365. 10.4049/jimmunol.178.8.5357. [DOI] [PubMed] [Google Scholar]
- Umiker B. R.; McDonald G.; Larbi A.; Medina C. O.; Hobeika E.; Reth M.; Imanishi-Kari T. (2014) Production of IgG autoantibody requires expression of activation-induced deaminase in early-developing B cells in a mouse model of SLE. Eur. J. Immunol. 44, 3093–3108. 10.1002/eji.201344282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C.; Foley J.; Clayton N.; Kissling G.; Jokinen M.; Herbert R.; Diaz M. (2007) Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J. Immunol. 178, 7422–7431. 10.4049/jimmunol.178.11.7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. H.; Chung T. D.; Oldenburg K. R. (1999) A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screening 4, 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Bruns R. F.; Watson I. A. (2012) Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 55, 9763–9772. 10.1021/jm301008n. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Thorne N.; Auld D. S.; Inglese J. (2010) Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 14, 315–324. 10.1016/j.cbpa.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M.; Kondo S.; Sugai M.; Nazarea M.; Imamura S.; Honjo T. (1996) High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int. Immunol. 8, 193–201. 10.1093/intimm/8.2.193. [DOI] [PubMed] [Google Scholar]
- McNeill D. R.; Whitaker A. M.; Stark W. J.; Illuzzi J. L.; McKinnon P. J.; Freudenthal B. D.; Wilson D. M. 3rd. (2020) Functions of the major abasic endonuclease (APE1) in cell viability and genotoxin resistance. Mutagenesis 35, 27–38. 10.1093/mutage/gez046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larijani M.; Petrov A. P.; Kolenchenko O.; Berru M.; Krylov S. N.; Martin A. (2007) AID associates with single-stranded DNA with high affinity and a long complex half-life in a sequence-independent manner. Mol. Cell. Biol. 27, 20–30. 10.1128/MCB.00824-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A. S.; Salamango D. J.; Serebrenik A. A.; Shaban N. M.; Brown W. L.; Harris R. S. (2019) A panel of eGFP reporters for single base editing by APOBEC-Cas9 editosome complexes. Sci. Rep. 9, 497. 10.1038/s41598-018-36739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader C. E.; Linehan E. K.; Mochegova S. N.; Woodland R. T.; Stavnezer J. (2005) Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J. Exp. Med. 202, 561–568. 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul R. W.; Saribasak H.; Martomo S. A.; McClure R. L.; Yang W.; Vaisman A.; Gramlich H. S.; Schatz D. G.; Woodgate R.; Wilson D. M. 3rd; Gearhart P. J. (2011) Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat. Immunol. 12, 70–76. 10.1038/ni.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Shandilya S. M.; Carpenter M. A.; Rathore A.; Brown W. L.; Perkins A. L.; Harki D. A.; Solberg J.; Hook D. J.; Pandey K. K.; Parniak M. A.; Johnson J. R.; Krogan N. J.; Somasundaran M.; Ali A.; Schiffer C. A.; Harris R. S. (2012) First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 7, 506–517. 10.1021/cb200440y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M. E.; Li M.; Harris R. S.; Harki D. A. (2013) Small-molecule APOBEC3G DNA cytosine deaminase inhibitors based on a 4-amino-1,2,4-triazole-3-thiol scaffold. ChemMedChem 8, 112–117. 10.1002/cmdc.201200411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. B.; Lackey L.; Carpenter M. A.; Rathore A.; Land A. M.; Leonard B.; Refsland E. W.; Kotandeniya D.; Tretyakova N.; Nikas J. B.; Yee D.; Temiz N. A.; Donohue D. E.; McDougle R. M.; Brown W. L.; Law E. K.; Harris R. S. (2013) APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370. 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. B.; Temiz N. A.; Harris R. S. (2013) Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 45, 977–983. 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; Carpenter M. A.; Banerjee S.; Shaban N. M.; Kurahashi K.; Salamango D. J.; McCann J. L.; Starrett G. J.; Duffy J. V.; Demir O.; Amaro R. E.; Harki D. A.; Harris R. S.; Aihara H. (2017) Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 24, 131–139. 10.1038/nsmb.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A.; Patenaude A. M.; Affar el B.; Lamarre A.; Young J. C.; Di Noia J. M. (2010) Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J. Exp. Med. 207, 2751–2765. 10.1084/jem.20101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montamat-Sicotte D.; Litzler L. C.; Abreu C.; Safavi S.; Zahn A.; Orthwein A.; Muschen M.; Oppezzo P.; Munoz D. P.; Di Noia J. M. (2015) HSP90 inhibitors decrease AID levels and activity in mice and in human cells. Eur. J. Immunol. 45, 2365–2376. 10.1002/eji.201545462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot S. P.; Litzler L. C.; Trajtenberg F.; Zahn A.; Robert F.; Pelletier J.; Buschiazzo A.; Magor B. G.; Di Noia J. M. (2015) Consecutive interactions with HSP90 and eEF1A underlie a functional maturation and storage pathway of AID in the cytoplasm. J. Exp. Med. 212, 581–596. 10.1084/jem.20141157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. Q.; Joshi P. S.; Wang C.; El-Omrani H. Y.; Xiao Y.; Liu X.; Hagan J. P.; Liu C. G.; Wu L. C.; Bai X. F. (2010) Targeting activation-induced cytidine deaminase overcomes tumor evasion of immunotherapy by CTLs. J. Immunol. 184, 5435–5443. 10.4049/jimmunol.0903322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont K. R.; Hasham M. G.; Donghia N. M.; Branca J.; Chavaree M.; Chase B.; Breggia A.; Hedlund J.; Emery I.; Cavallo F.; Jasin M.; Ruter J.; Mills K. D. (2013) Attenuating homologous recombination stimulates an AID-induced antileukemic effect. J. Exp. Med. 210, 1021–1033. 10.1084/jem.20121258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratiu J. J.; Racine J. J.; Hasham M. G.; Wang Q.; Branca J. A.; Chapman H. D.; Zhu J.; Donghia N.; Philip V.; Schott W. H.; Wasserfall C.; Atkinson M. A.; Mills K. D.; Leeth C. M.; Serreze D. V. (2017) Genetic and Small Molecule Disruption of the AID/RAD51 Axis Similarly Protects Nonobese Diabetic Mice from Type 1 Diabetes through Expansion of Regulatory B Lymphocytes. J. Immunol. 198, 4255–4267. 10.4049/jimmunol.1700024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R. M.; Abrams S. R.; Gajula K. S.; Maul R. W.; Gearhart P. J.; Stivers J. T. (2009) A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J. Biol. Chem. 284, 22898–22904. 10.1074/jbc.M109.025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M. E.; Harris R. S.; Harki D. A. (2018) APOBEC Enzymes as Targets for Virus and Cancer Therapy. Cell Chem. Biol. 25, 36–49. 10.1016/j.chembiol.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham P.; Bransteitter R.; Petruska J.; Goodman M. F. (2003) Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 424, 103–107. 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- Yu K.; Huang F. T.; Lieber M. R. (2004) DNA substrate length and surrounding sequence affect the activation-induced deaminase activity at cytidine. J. Biol. Chem. 279, 6496–6500. 10.1074/jbc.M311616200. [DOI] [PubMed] [Google Scholar]
- Larijani M.; Frieder D.; Basit W.; Martin A. (2005) The mutation spectrum of purified AID is similar to the mutability index in Ramos cells and in ung(−/−)msh2(−/−) mice. Immunogenetics 56, 840–845. 10.1007/s00251-004-0748-0. [DOI] [PubMed] [Google Scholar]
- Wang M.; Rada C.; Neuberger M. S. (2010) Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID. J. Exp. Med. 207, 141–153. S141–146. 10.1084/jem.20092238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham P.; Afif S. A.; Shimoda M.; Maeda K.; Sakaguchi N.; Pedersen L. C.; Goodman M. F. (2016) Structural analysis of the activation-induced deoxycytidine deaminase required in immunoglobulin diversification. DNA Repair 43, 48–56. 10.1016/j.dnarep.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Q.; Wang L.; Meng F. L.; Hwang J. K.; Alt F. W.; Wu H. (2017) AID Recognizes Structured DNA for Class Switch Recombination. Mol. Cell 67, 361–373. e364. 10.1016/j.molcel.2017.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin P. D.; Lee J. H.; Lyons A. B. (1996) B cell differentiation and isotype switching is related to division cycle number. J. Exp. Med. 184, 277–281. 10.1084/jem.184.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush J.; Liu M.; Odegard V.; Unniraman S.; Schatz D. (2005) Expression of activation-induced cytidine deaminase is regulated by cell division, providing a mechanistic basis for division-linked class switch recombination. Proc. Natl. Acad. Sci. U. S. A. 102, 13242–13247. 10.1073/pnas.0502779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamango D. J.; Becker J. T.; McCann J. L.; Cheng A. Z.; Demir O.; Amaro R. E.; Brown W. L.; Shaban N. M.; Harris R. S. (2018) APOBEC3H Subcellular Localization Determinants Define Zipcode for Targeting HIV-1 for Restriction. Mol. Cell. Biol. 38, 1. 10.1128/MCB.00356-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor A. C.; Kim Y. B.; Packer M. S.; Zuris J. A.; Liu D. R. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter M. A.; Law E. K.; Serebrenik A.; Brown W. L.; Harris R. S. (2019) A lentivirus-based system for Cas9/gRNA expression and subsequent removal by Cre-mediated recombination. Methods 156, 79–84. 10.1016/j.ymeth.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Naismith J. H. (2008) An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91. 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C.; Kandzia R.; Marillonnet S. (2008) A one pot, one step, precision cloning method with high throughput capability. PLoS One 3, e3647 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Martin A.; Salamango D.; Serebrenik A.; Shaban N.; Brown W. L.; Donati F.; Munagala U.; Conticello S. G.; Harris R. S. (2018) A fluorescent reporter for quantification and enrichment of DNA editing by APOBEC-Cas9 or cleavage by Cas9 in living cells. Nucleic Acids Res. 46, e84 10.1093/nar/gky332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdeef A.; Bendels S.; Tsinman O.; Tsinman K.; Kansy M. (2007) Solubility-excipient classification gradient maps. Pharm. Res. 24, 530–545. 10.1007/s11095-006-9169-0. [DOI] [PubMed] [Google Scholar]
- Kerns E. H.; Di L.; Carter G. T. (2008) In vitro solubility assays in drug discovery. Curr. Drug Metab. 9, 879–885. 10.2174/138920008786485100. [DOI] [PubMed] [Google Scholar]
- Avdeef A.; Strafford M.; Block E.; Balogh M. P.; Chambliss W.; Khan I. (2001) Drug absorption in vitro model: filter-immobilized artificial membranes. 2. Studies of the permeability properties of lactones in Piper methysticum Forst. Eur. J. Pharm. Sci. 14, 271–280. 10.1016/S0928-0987(01)00191-9. [DOI] [PubMed] [Google Scholar]
- Sun H.; Shah P.; Nguyen K.; Yu K. R.; Kerns E.; Kabir M.; Wang Y.; Xu X. (2019) Predictive models of aqueous solubility of organic compounds built on A large dataset of high integrity. Bioorg. Med. Chem. 27, 3110–3114. 10.1016/j.bmc.2019.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Nguyen K.; Kerns E.; Yan Z.; Yu K. R.; Shah P.; Jadhav A.; Xu X. (2017) Highly predictive and interpretable models for PAMPA permeability. Bioorg. Med. Chem. 25, 1266–1276. 10.1016/j.bmc.2016.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P.; Kerns E.; Nguyen D. T.; Obach R. S.; Wang A. Q.; Zakharov A.; McKew J.; Simeonov A.; Hop C. E.; Xu X. (2016) An Automated High-Throughput Metabolic Stability Assay Using an Integrated High-Resolution Accurate Mass Method and Automated Data Analysis Software. Drug Metab. Dispos. 44, 1653–1661. 10.1124/dmd.116.072017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefaniak F.Prediction of Compounds Activity in Nuclear Receptor Signaling and Stress Pathway Assays Using Machine Learning Algorithms and Low-Dimensional Molecular Descriptors. Front. Environ. Sci. 2015, 3, 10.3389/fenvs.2015.00077. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.