

Abstract
Objective
To assess support for a causal relationship between hemostatic measures and migraine susceptibility using genetic instrumental analysis.
Methods
Two-sample Mendelian randomization instrumental analyses leveraging available genome-wide association study (GWAS) summary statistics were applied to hemostatic measures as potentially causal for migraine and its subtypes, migraine with aura (MA) and migraine without aura (MO). Twelve blood-based measures of hemostasis were examined, including plasma level or activity of 8 hemostatic factors and 2 fibrinopeptides together with 2 hemostasis clinical tests.
Results
There were significant instrumental effects between increased coagulation factor VIII activity (FVIII; odds ratio [95% confidence interval] 1.05 [1.03, 1.08]/SD, p = 6.08 × 10−05), von Willebrand factor level (vWF; 1.05 [1.03, 1.08]/SD, p = 2.25 × 10−06), and phosphorylated fibrinopeptide A level (1.13 [1.07, 1.19]/SD, p = 5.44 × 10−06) with migraine susceptibility. When extended to migraine subtypes, FVIII, vWF, and phosphorylated fibrinopeptide A showed slightly stronger effects with MA than overall migraine. Fibrinogen level was inversely linked with MA (0.76 [0.64, 0.91]/SD, p = 2.32 × 10−03) but not overall migraine. None of the hemostatic factors was linked with MO. In sensitivity analysis, effects for fibrinogen and phosphorylated fibrinopeptide A were robust, whereas independent effects of FVIII and vWF could not be distinguished, and FVIII associations were potentially affected by pleiotropy at the ABO locus. Causal effects from migraine to the hemostatic measures were not supported in reverse Mendelian randomization. However, MA was not included due to lack of instruments.
Conclusions
The findings support potential causality of increased FVIII, vWF, and phosphorylated fibrinopeptide A and decreased fibrinogen in migraine susceptibility, especially for MA, potentially revealing etiologic relationships between hemostasis and migraine.
Migraine is a chronic intermittent neurologic disorder affecting up to 15% people worldwide. Migraine, especially migraine with aura (MA), is associated with increased risk of ischemic stroke and cardiovascular disease.1,2 However, the mechanisms underlying such associations are unclear. Based on prior studies, it is controversial whether enhanced atherosclerosis among individuals with MA is likely to explain the migraine association with stroke,3,4 suggesting the existence of alternative mechanisms including endothelial activation5,6 or a potential role of hypercoagulability and microemboli.5 However, findings on migraine and procoagulatory states are not consistent.7-12
In this study, we investigated potential causal relationships between hemostatic profiles and migraine by leveraging large-scale genome-wide association study (GWAS) summary statistics for migraine and migraine subtypes (MA and migraine without aura [MO]),13 and for 8 hemostatic factors (including plasma levels or activities of fibrinogen,14 D-dimer,15 coagulation factor VII [FVII],16 coagulation factor VIII [FVIII],17 coagulation factor XI [FXI],18 von Willebrand factor [vWF],17 tissue plasminogen activator [tPA],19 and plasminogen activator inhibitor-1 [PAI-1]20 among up to 120,246 individuals), 2 hemostasis clinical tests (activated partial thromboplastin time [aPTT]21 and prothrombin time/international normalized ratio [PT/INR],21 which are commonly used to screen for coagulation-factor deficiencies), as well as serum concentrations of 2 forms of fibrinopeptide A (fibrinopeptide A [ADSGEGDFXAEGGGVR*] and phosphorylated fibrinopeptide A [ADpSGEGDFXAEGGGVR*]).22
Methods
GWAS Summary Data for Migraine and Hemostatic Profiles
The latest GWAS summary-level data for migraine (included 59,674 cases and 316,078 controls) and its subtypes MA (included 4,837 cases and 49,174 controls) and MO (included 4,833 cases and 106,834 controls) were obtained from the International Headache Genetics Consortium, which comprised 22 cohorts13 (appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p). GWAS summary statistics for fibrinogen levels (n = 120,246),14 D-dimer levels (n = 21,052),15 FVII activity (n = 20,014),16 FVIII activity (n = 25,897),17 FXI levels (n = 16,169),18 vWF levels (n = 42,379),17 tPA levels (n = 26,929),19 PAI-1 levels (n = 19,599),20 aPTT (n = 9,240),21 and PT/INR (n = 2,583)21 were obtained from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium Hemostasis Working Group. GWAS data for the concentrations of 2 fibrinopeptides (fibrinopeptide A or ADSGEGDFXAEGGGVR* and phosphorylated fibrinopeptide A or ADpSGEGDFXAEGGGVR*, n = 7,824) were available at mips.helmholtz-muenchen.de/proj/GWAS/gwas/ 22 (appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p). Participants in all studies had European ancestry, and the genotype data were imputed either to 1,000 Genomes Project23 or HapMap2.24
Mendelian Randomization Analysis
Bidirectional Mendelian randomization (MR) analyses were conducted between each hemostatic biomarker and migraine or migraine subtypes (MA and MO). To do this, we first extracted independent genome-wide significant SNPs for each trait using a clumping function in PLINK25 (pngu.mgh.harvard.edu/purcell/plink/), resulting in sets of independent SNPs with p value ≤5 × 10−8 and r2 < 0.05 in a 1,000-kb window size (appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p). Then, we used the selected genetic instruments for each exposure (i.e., hemostatic factors) in robust testing of whether their effects on the exposures resulted in proportional effects on the outcome (i.e., migraine) using generalized summary data–based Mendelian randomization (GSMR),26 and also in the reverse direction (i.e., from migraine to hemostatic biomarkers). Single nucleotide polymorphism (SNP) instruments with apparent pleiotropic effects were detected by heterogeneity in dependent instruments (HEIDI; pHEIDI < 0.01)26 and were excluded from the GSMR analysis to reduce potential for biased causal estimates, loss of statistical power, and potential false-positive causal relationships in the MR inference. All the instrumental estimates of hemostatic profiles with migraine and its subtypes were interpreted as the odds ratio (OR) of per SD change of the hemostatic factors, hemostasis clinical tests, and fibrinopeptides being tested. As migraine is a binary variable, we scaled the reverse causal estimates to represent the average change in hemostatic factors per doubling (2-fold increase) in the prevalence of migraine by multiplying the reverse causal estimate by 0.693 (loge2).27 All hypothesis testing was 2-sided. The threshold for nominal significance was p < 0.05. The threshold for significance among multiple hypotheses was determined by the Bonferroni standard as p < 0.05/number of tests.
Sensitivity Analysis for Mendelian Randomization
Multiple sensitivity analyses were conducted to verify the effects between hemostatic profiles and migraine. To identify and exclude potential pleiotropic instruments, we performed MR implemented with the Mendelian randomization pleiotropy residual sum and outlier test (MR-PRESSO)28 in both the forward (coagulation to migraine) and reverse (migraine to coagulation factor) directions. We further performed conventional inverse variance–weighted (IVW) MR, weighted median, simple median, MR-Egger (Egger regression), and leave-one-out analysis implemented in the R package TwoSampleMR.29 In addition, we note that the ABO locus is highly pleiotropic for many phenotypes including coagulation factors. To further test whether instruments related to ABO gene modify the instrumental effects, we conducted sensitivity analysis by excluding instruments related to the ABO gene using 3 strategies: (1) removing instruments on ABO gene, (2) removing instruments residing within 200 kb of ABO gene, and (3) removing any additional instruments on chromosome 9. We conducted cis-variants only MR for traits with a sufficient number of genome-wide significant instruments (i.e., for fibrinogen, FVII, FXI, vWF, and PAI-1) to assess whether the observed association was driven by pleiotropy effects of trans-variants.
Multivariable MR Analysis for FVIII and vWF
Finally, as FVIII plasma levels are determined largely by vWF plasma levels owing to the carrier role of vWF for FVIII in plasma,30 it was important to investigate whether there is an independent causal role of genetically determined FVIII activities from that of genetically determined vWF levels on migraine and its subtypes or vice versa. We therefore applied a multivariable MR approach in which we adjusted for the variant effects of vWF in the estimate of causal effect between FVIII and migraine or its subtypes (or adjusted for the variant effects of FVIII when investigating the effect between vWF and migraine or its subtypes). To do this, we conducted a 2-step procedure requiring only the GWAS summary statistics. First, we performed conditional instrumental analysis using multi-trait–based conditional and joint analysis (mtCOJO) implemented in GSMR26 (i.e., SNP effects on vWF were adjusted for effects on FVIII, and vice versa), and then the adjusted instruments were used to derive the conditional instrumental estimate using the IVW method.
Data Availability
All data used in this study were restricted to the GWAS summary statistics described above and were available from cited study authors or from the public domain as indicated.
Standard Protocol Approvals, Registrations, and Patient Consents
This study uses solely GWAS summary statistics from meta-analysis, but all participants who contributed to cohorts constituting the meta-analysis provided written informed consent and each of the cohort protocols was approved by a local institutional review board.
Results
Mendelian Randomization
We observed evidence of significant effects of genetically determined increased FVIII activity (odds ratio [95% confidence interval] 1.05 [1.03, 1.08]/SD of FVIII, p = 6.08 × 10−05), vWF level (1.05 [1.03, 1.08]/SD of vWF, p = 2.25 × 10−06), and phosphorylated fibrinopeptide A level (1.13 [1.07, 1.19]/SD of phosphorylated fibrinopeptide A, p = 5.44 × 10−06) with migraine after controlling for multiple testing (p < 0.05/11) (figure 1A and appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p). At nominal significance (p < 0.05), genetically determined decreased fibrinogen levels (0.93 [0.87, 0.98]/SD of fibrinogen, p = 0.01) and short aPTT (0.98 [0.96, 1.00]/second, p = 0.01) were associated with migraine, while other hemostatic markers were not associated.
Figure 1. Instrumental Estimates of Hemostatic Profiles With Migraine, Migraine With Aura (MA), and Migraine Without Aura (MO).

Instrumental estimates of hemostatic profiles with (A) migraine, (B) MA, and (C) MO. All the analyses were conducted using generalized summary data–based Mendelian randomization (GSMR) except for D-dimer, prothrombin time/international normalized ratio (PT/INR), plasminogen activator inhibitor-1 (PAI-1), tissue plasminogen activator (tPA), fibrinopeptide A, and phosphorylated fibrinopeptide A, which were estimated using inverse variance–weighted (IVW) Mendelian randomization implemented in the R package TwoSampleMR due to limited number of instruments (less than 10). All the instrumental estimates were interpreted as the odds ratio of per SD change of the hemostatic profiles being tested. aPTT = activated partial thromboplastin time; FVII = coagulation factor VII; FVIII = coagulation factor VIII; FXI = coagulation factor XI; vWF = von Willebrand factor.
When we extended this analysis to migraine subtypes, genetically determined elevated FVIII activity (1.13 [1.05, 1.22]/SD of FVIII, p = 1.74 × 10−03), vWF level (1.12 [1.05, 1.20]/SD of vWF, p = 7.98 × 10−04), and phosphorylated fibrinopeptide A level (1.22 [1.01, 1.48]/SD of phosphorylated fibrinopeptide A, p = 0.04) showed slightly stronger effects with MA than with overall migraine in spite of considerably smaller sample size in the migraine subtype GWAS (figure 1B and appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p). Although genetically determined decreased fibrinogen level was only nominally significant for overall migraine, it was significantly related to MA (0.76 [0.64, 0.91]/unit of natural log transformed fibrinogen, p = 2.32 × 10−03) after controlling for multiple testing (figure 1B and appendix; data available from Dryad). Among the hemostasis clinical tests, aPTT (0.93 [0.87, 1.00]/second, p = 0.02) was nominally significantly linked with MA (figure 1B and appendix; data available from Dryad). None of the traits was linked with MO (figure 1C and appendix; data available from Dryad).
Reverse MR results showed no significant effects of migraine or MO on any of the hemostatic factors (all p > 0.05, figure 2 and appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p). However, MA was not included in the reverse analysis due to the lack of genome-wide significant genetic instruments for this class of migraine. The results of these causal inference analyses support a direct role of hemostatic factors in migraine etiology.
Figure 2. Instrumental Estimates of Migraine and Migraine Without Aura (MO) With Hemostatic Profiles.

Instrumental estimates of hemostatic profiles with (A) migraine and (B) MO. All the analyses for migraine without aura were estimated using inverse variance–weighted (IVW) Mendelian randomization implemented in the R package TwoSampleMR due to limited number of instruments (less than 10). As migraine is a binary variable, the reverse causal estimates were scaled to reflect the average change in hemostatic factors (in native units specific to each factor) per doubling (2-fold increase) in prevalence of migraine (Methods). aPTT = activated partial thromboplastin time; FVII = coagulation factor VII; FVIII = coagulation factor VIII; FXI = coagulation factor XI; PAI-1 = plasminogen activator inhibitor-1; PT/INR = prothrombin time/international normalized ratio; tPA = tissue plasminogen activator; vWF = von Willebrand factor.
Sensitivity Analysis
Results for a series of MR sensitivity analyses, including MR-PRESSO, IVW, weighted median, simple median, and MR-Egger (appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p) were consistent with results from GSMR. For example, using MR-PRESSO, FVIII (OR 1.05, p = 1.81 × 10−04) and vWF (OR 1.05, p = 1.51 × 10−05) were significantly related to migraine after accounting for multiple testing. When stratified by migraine subtypes, there were stronger effects with MA (OR 1.13, p = 4.52 × 10−05 for FVIII; OR 1.12, p = 3.11 × 10−04 for vWF) but no significant effect with MO (p = 0.16 and 0.95 for FVIII and vWF, respectively). Phosphorylated fibrinopeptide A was not included in the sensitivity analysis because only 2 instruments for phosphorylated fibrinopeptide A were available.
The ABO gene is highly pleiotropic and may therefore bias the instrumental estimates of hemostatic factors on migraine. Although GSMR and MR-PRESSO address pleiotropy, we conducted sensitivity analysis by explicitly removing instruments potentially exerting effects through the ABO gene. After removing instruments proximal to the ABO gene, the association of genetically determined elevated FVIII activity (1.05/SD of FVIII, p = 6.48 × 10−03), vWF level (1.05/SD of vWF, p = 8.10 × 10−04), and phosphorylated fibrinopeptide A level (1.13/SD of phosphorylated fibrinopeptide A, p = 5.44 × 10−06) with migraine remained the same (appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p). However, when we further excluded instruments within 200 kb around the ABO gene, the effect of genetically determined elevated FVIII activity with 20 instruments retained was attenuated and nonsignificant (OR 1.02, p = 0.45), while the effects of genetically determined vWF level with 43 instruments and phosphorylated fibrinopeptide A level with 1 instrument remained similar (appendix; data available from Dryad). Results were again similar when we excluded all instruments on chromosome 9 (appendix; data available from Dryad). For migraine subtypes, we observed similar patterns with overall migraine except the effects of fibrinogen level on MA were not affected (appendix; data available from Dryad). The robust effects with vWF, phosphorylated fibrinopeptide A, and fibrinogen were further supported by leave-one-out sensitivity analysis that yielded instrumental effects with migraine and its subtypes similar to those in the primary analysis (appendix; data available from Dryad).
A final MR sensitivity analysis including only cis-variants available for 5 of the protein measures (fibrinogen, FVII, FXI, vWF, and PAI-1) showed similar patterns and magnitudes of association as the primary MR results, with fibrinogen and vWF being marginally significant (p = 0.06, appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p).
Multivariable MR Results for FVIII and vWF
Twenty-four instruments were shared (same instruments or instruments in high linkage disequilibrium) by FVIII and vWF while 7 and 31 instruments were unique for FVIII and vWF, respectively (appendix; data available from Dryad at doi.org/10.5061/dryad.zkh18938p). We observed that the instrumental effects of FVIII and vWF on migraine and its subtypes were not independent by adjusting each on the other in multivariable MR analysis (figure 3). Specifically, the instrumental association of FVIII on migraine with conditioning on vWF (0.96 [0.81–1.15], p = 0.70) and the effect of vWF on migraine with conditioning on FVIII (1.03 [0.90–1.18], p = 0.68) were both attenuated and no longer significant (figure 3).
Figure 3. Multivariable Mendelian Randomization Analysis of Coagulation Factor VIII (FVIII) and von Willebrand Factor (vWF) With Migraine, Migraine With Aura, and Migraine Without Aura.
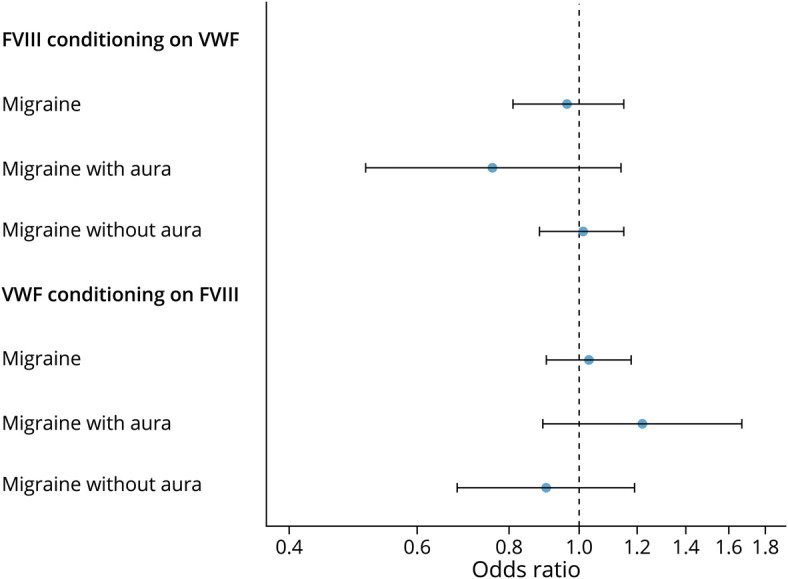
Conditional instrumental analyses were first performed using multi-trait–based conditional and joint analysis (mtCOJO) implemented in generalized summary data–based Mendelian randomization and then the instrumental estimates were calculated using inverse variance–weighted Mendelian randomization implemented in the R package TwoSampleMR based on the conditioned instruments. All the instrumental estimates were interpreted as the odds ratio of per SD change of FVIII and vWF.
Discussion
The current study provides insights that support coagulation and thrombosis as a potential etiology of migraine susceptibility. We find that genetically increased FVIII, vWF, and phosphorylated fibrinopeptide A and genetically decreased fibrinogen have potential causal effects on increased migraine susceptibility. This same pattern was slightly stronger for MA but not observed for MO. However, the effects of FVIII and vWF on migraine were not independent from each other. There was little evidence of reverse causation from reverse MR analysis (i.e., potential causation from migraine to hemostatic factor measures). However, MA was not included in the reverse MR due to lack of instruments.
FVIII functions as a cofactor for the proteolytic activation of factor X by factor IXa in the blood coagulation cascade, while vWF acts as a carrier for FVIII and the deficiency of vWF has a profound effect on the stability of FVIII.30 vWF has been previously found to be increased during a migraine attack.10,31,32 However, to our knowledge, no study has investigated effects of increased FVIII level or activity (which can contribute to the development of small thrombi33) and migraine in adults. Functions of vWF may be more diverse than for FVIII, and separate studies have demonstrated that elevated vWF is related to increased platelet aggregation, decreased fibrinolysis, increased inflammation, increased oxidative stress, and decreased cerebrovascular reactivity.5,10 All are relevant to endothelial dysfunction that is thought to be important for migraine pathophysiology.34 However, the multivariable MR showed that the instrumental effects of both FVIII and vWF are not independent from each other, and therefore, the current data are not able to prioritize the effects of FVIII and vWF on migraine and its subtypes. Similarly, our instrumental analysis cannot address whether other vWF concentration related features, such as its multimeric state, may also be relevant to potential causal effects.
Fibrinogen plays critical roles in the last stages of the blood coagulation cascade. Fibrinopeptide A, products of the cleavage of the N-termini of the fibrinogen Aα and Bβ chains by thrombin, has both phosphorylated and nonphosphorylated forms.35 Fibrinogen phosphorylation increases sharply under acute phase conditions, such that up to 60% of the segment corresponding to fibrinopeptide A may be phosphorylated,35 while phosphorylated fibrinopeptide A is reliable marker of coagulation activity.36,37 The association between fibrinopeptides and migraine has not been previously explored but studies of fibrinogen and migraine have shown inconsistent associations.7,8,11,12 Our observations of opposing effects of fibrinogen and the phosphorylated fibrinopeptide A may reflect increased degradation from fibrinogen to fibrin and release of phosphorylated fibrinopeptides A in susceptibility to migraine. Reduced concentration of fibrinogen due to increased degradation may also increase overall coagulation activity.
We note that the MR study design implies that the findings are more readily interpreted as associations between levels of hemostatic factors and migraine susceptibility rather than altered levels of hemostatic factors as a consequence of a migraine attack. The genetic instruments were identified primarily for their association with normal variation in the hemostatic factors among individuals who were neither enriched as migraineurs nor likely to be experiencing an attack during the blood draw (since an attack is highly debilitating). These loci are not of genome-wide significance for migraine even though the sample size for the migraine GWAS was larger than for the GWASs hemostatic factors.13 In addition, MR is less likely to be affected by reverse causality in MR than nongenetic observational analysis due to the nature of genetic inheritance.38 Therefore, the findings suggest that the normal variation precedes, or at worst shares pathophysiology with, migraine susceptibility.
Limitations of MR as a tool to infer potential causal roles of hemostatic factors in migraine susceptibility merit consideration. First, we only used summary statistics for participants of European ancestry, possibly limiting generalizability to other populations. Second, since an individual's genetic profile is established at conception, our findings pertain to lifelong exposure to the influences of inherited alleles on the coagulation factors, rather than at a discrete moment, for example, in adulthood, when migraine was ascertained for the GWAS. Finally, like other MR studies, we are not able to directly evaluate the potential of unmeasured confounding, for example, due to pleiotropy, even as our sensitivity analyses suggested limited evidence of bias due to such effects.
Our findings suggest potential causal roles of genetically determined elevated FVIII, vWF, phosphorylated fibrinopeptide A, and decreased fibrinogen in migraine susceptibility, especially for MA, but the effects of FVIII and vWF on migraine are not independent from each other. These findings motivate further research into roles of hypercoagulation and endothelial dysfunction—and their targeting for therapeutic strategies—in migraine susceptibility.
Acknowledgment
The authors thank the participants and researchers from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Hemostasis Working Group, 23andMe, Inc., and the International Headache Genetics Consortium (IHGC), who contributed to or collected data.
Glossary
- aPTT
activated partial thromboplastin time
- FVII
coagulation factor VII
- FVIII
coagulation factor VIII
- FXI
coagulation factor XI
- GSMR
generalized summary data–based Mendelian randomization
- GWAS
genome-wide association study
- HEIDI
heterogeneity in dependent instruments
- IVW
inverse variance–weighted
- MA
migraine with aura
- MO
migraine without aura
- MR
Mendelian randomization
- MR-PRESSO
Mendelian randomization pleiotropy residual sum and outlier test
- OR
odds ratio
- PAI-1
plasminogen activator inhibitor-1
- PT/INR
prothrombin time/international normalized ratio
- SNP
single nucleotide polymorphism
- tPA
tissue plasminogen activator
- vWF
von Willebrand factor
Appendix. Authors
Footnotes
CME Course: NPub.org/cmelist
Study Funding
Daniel I. Chasman is funded by the US NIH and US National Institute of Neurologic Disorders and Stroke (R21NS09296 and R21NS104398). Pamela M. Rist is funded by K01 HL128791. Maria Sabater-Lleal is supported by a Miguel Servet contract from the ISCIII Spanish Health Institute (CP17/00142) and co-financed the European Social Fund. Nicholas L. Smith and Paul S. de Vries were supported by NHLBI grants R01HL134894, R01HL139553, and R01HL141291. Paul S. de Vries was additionally supported by American Heart Association grant 18CDA34110116.
Disclosure
T.K. provided methodologic expertise to Amgen and CoLucid, for which the Charité–Universitätsmedizin Berlin has received financial compensation, and received honoraria from Novartis and Daiichi Sankyo for a scientific presentation and from Lilly, Newsenselab, and Total for methodological advice. The remaining authors have nothing to disclose. Go to Neurology.org/N for full disclosures.
References
- 1.Mahmoud AN, Mentias A, Elgendy AY, et al. Migraine and the risk of cardiovascular and cerebrovascular events: a meta-analysis of 16 cohort studies including 1 152 407 subjects. BMJ Open. 2018;8(3):e020498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurth T, Rist PM, Ridker PM, Kotler G, Bubes V, Buring JE. Association of migraine with aura and other risk factors with incident cardiovascular disease in women. JAMA. 2020;323(22):2281-2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stam AH, Weller CM, Janssens AC, et al. Migraine is not associated with enhanced atherosclerosis. Cephalalgia. 2013;33(4):228-235. [DOI] [PubMed] [Google Scholar]
- 4.Tietjen GE, Maly EF. Migraine and ischemic stroke in women: a narrative review. Headache. 2020;60(5):843-863. [DOI] [PubMed] [Google Scholar]
- 5.Tietjen GE, Collins SA. Hypercoagulability and migraine. Headache. 2018;58(1):173-183. [DOI] [PubMed] [Google Scholar]
- 6.Tietjen GE, Khubchandani J. Vascular biomarkers in migraine. Cephalalgia. 2015;35(2):95-117. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi A, Pitari G, Amenta V, et al. Endothelial, haemostatic and haemorheological modifications in migraineurs. Artery. 1996;22(2):93-100. [PubMed] [Google Scholar]
- 8.Kurth T, Ridker PM, Buring JE. Migraine and biomarkers of cardiovascular disease in women. Cephalalgia. 2008;28(1):49-56. [DOI] [PubMed] [Google Scholar]
- 9.Nossel HL, Ti M, Kaplan KL, Spanondis K, Soland T, Butler VP Jr. The generation of fibrinopeptide A in clinical blood samples: evidence for thrombin activity. J Clin Invest. 1976;58(5):1136-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tietjen GE, Herial NA, White L, Utley C, Kosmyna JM, Khuder SA. Migraine and biomarkers of endothelial activation in young women. Stroke. 2009;40(9):2977-2982. [DOI] [PubMed] [Google Scholar]
- 11.Tietjen GE, Khubchandani J, Herial N, et al. Migraine and vascular disease biomarkers: a population-based case-control study. Cephalalgia. 2018;38(3):511-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yucel Y, Tanriverdi H, Arikanoglu A, et al. Increased fibrinogen, D-dimer and galectin-3 levels in patients with migraine. Neurol Sci. 2014;35(4):545-549. [DOI] [PubMed] [Google Scholar]
- 13.Gormley P, Anttila V, Winsvold BS, et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet. 2016;48(8):856-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Vries PS, Chasman DI, Sabater-Lleal M, et al. A meta-analysis of 120 246 individuals identifies 18 new loci for fibrinogen concentration. Hum Mol Genet. 2016;25(2):358-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith NL, Huffman JE, Strachan DP, et al. Genetic predictors of fibrin D-dimer levels in healthy adults. Circulation. 2011;123(17):1864-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Vries PS, Sabater-Lleal M, Huffman JE, et al. A genome-wide association study identifies new loci for factor VII and implicates factor VII in ischemic stroke etiology. Blood. 2019;133(9):967-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabater-Lleal M, Huffman JE, de Vries PS, et al. Genome-wide association transethnic meta-analyses identifies novel associations regulating coagulation factor VIII and von Willebrand factor plasma levels. Circulation. 2019;139(5):620-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sennblad B, Basu S, Mazur J, et al. Genome-wide association study with additional genetic and post-transcriptional analyses reveals novel regulators of plasma factor XI levels. Hum Mol Genet. 2017;26(3):637-649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang J, Huffman JE, Yamakuchi M, et al. Genome-wide association study for circulating tissue plasminogen activator levels and functional follow-up implicates endothelial STXBP5 and STX2. Arterioscler Thromb Vasc Biol. 2014;34(5):1093-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang J, Sabater-Lleal M, Asselbergs FW, et al. Genome-wide association study for circulating levels of PAI-1 provides novel insights into its regulation. Blood. 2012;120(24):4873-4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang W, Schwienbacher C, Lopez LM, et al. Genetic associations for activated partial thromboplastin time and prothrombin time, their gene expression profiles, and risk of coronary artery disease. Am J Hum Genet. 2012;91(1):152-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin SY, Fauman EB, Petersen AK, et al. An atlas of genetic influences on human blood metabolites. Nat Genet. 2014;46(6):543-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Genomes Project Consortium, Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437(7063):1299-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Z, Zheng Z, Zhang F, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun. 2018;9(1):224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess S, Labrecque JA. Mendelian randomization with a binary exposure variable: interpretation and presentation of causal estimates. Eur J Epidemiol. 2018;33(10):947-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palta S, Saroa R, Palta A. Overview of the coagulation system. Indian J Anaesth. 2014;58(5):515-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cesar JM, Garcia-Avello A, Vecino AM, Sastre JL, Alvarez-Cermeno JC. Increased levels of plasma von Willebrand factor in migraine crisis. Acta Neurol Scand. 1995;91(5):412-413. [DOI] [PubMed] [Google Scholar]
- 32.Tietjen GE, Al-Qasmi MM, Athanas K, Dafer RM, Khuder SA. Increased von Willebrand factor in migraine. Neurology. 2001;57(2):334-336. [DOI] [PubMed] [Google Scholar]
- 33.Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol. 2001;21(5):731-738. [DOI] [PubMed] [Google Scholar]
- 34.Dalkara T, Nozari A, Moskowitz MA. Migraine aura pathophysiology: the role of blood vessels and microembolisation. Lancet Neurol. 2010;9(3):309-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seydewitz HH, Witt I. Increased phosphorylation of human fibrinopeptide A under acute phase conditions. Thromb Res. 1985;40(1):29-39. [DOI] [PubMed] [Google Scholar]
- 36.Wilensky RL, Zeller JA, Wish M, Tulchinsky M. Urinary fibrinopeptide A levels in ischemic heart disease. J Am Coll Cardiol. 1989;14(3):597-603. [DOI] [PubMed] [Google Scholar]
- 37.Stegnar M, Vene N, Bozic M. Do haemostasis activation markers that predict cardiovascular disease exist? Pathophysiol Haemost Thromb. 2003;33(5-6):302-308. [DOI] [PubMed] [Google Scholar]
- 38.Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this study were restricted to the GWAS summary statistics described above and were available from cited study authors or from the public domain as indicated.