

Abstract
Objective
To investigate the effects of rituximab on retinal atrophy in patients with relapsing-remitting multiple sclerosis (RRMS), we performed serial optical coherence tomography (OCT) scans among a cohort of patients with RRMS on rituximab and compared rates of ganglion cell + inner plexiform layer (GCIPL) atrophy to those observed among age- and sex-matched glatiramer acetate (GA)–and natalizumab-treated patients with RRMS and healthy controls (HCs).
Methods
In this observational study, patients with RRMS treated with a single disease-modifying therapy and HCs were followed with serial OCT for a median duration of 2.8 years. Participants with uncontrolled hypertension, diabetes mellitus, or glaucoma, and eyes with optic neuritis ≤6 months prior to baseline OCT, or during follow-up, were excluded. Statistical analyses were performed using linear mixed-effects regression.
Results
During the overall follow-up period, rates of GCIPL atrophy were −0.28 ± 0.11 µm/y among rituximab-treated patients with RRMS (n = 35). This was similar to GA-treated (n = 49; −0.33 ± 0.05 µm/y; p = 0.69) and natalizumab-treated patients (n = 88; −0.17 ± 0.10 µm/y; p = 0.13) and faster than HCs (n = 78; −0.15 ± 0.03 µm/y; p = 0.006). Rituximab-treated patients exhibited 0.55 ± 0.23 µm/y faster rates of GCIPL atrophy during the first 12 months of treatment, relative to afterwards (n = 25; p = 0.02), during which period GCIPL atrophy rates were −0.14 ± 0.13 µm/y.
Conclusions
Retinal atrophy in RRMS is modulated by rituximab. Greater attenuation of retinal atrophy may occur after 12 months of rituximab treatment, following which time GCIPL atrophy rates are similar to those observed among natalizumab-treated patients with RRMS and HCs. Our findings raise the possibility that the neuroprotective therapeutic response with rituximab in RRMS may take up to 12 months, which should be confirmed by larger studies.
Classification of Evidence
This study provides Class IV evidence on the difference in rate of change of the GCIPL thickness in patients with RRMS comparing rituximab to other disease-modifying therapies.
Multiple sclerosis (MS) is an autoimmune disorder of the CNS, in which neurodegeneration, as a sequela of neuroinflammation, is the principal substrate underlying disability progression.1 Optic neuropathy is virtually ubiquitous in MS, irrespective of optic neuritis (ON) history,2 and can be reliably assessed by optical coherence tomography (OCT), a reproducible, high-resolution imaging technique that facilitates quantification of discrete retinal layers. OCT-determined ganglion cell + inner plexiform layer (GCIPL) thickness demonstrates excellent reliability and reproducibility, with intervisit intraclass correlation coefficients as high as 0.99.3,4 Moreover, GCIPL atrophy mirrors whole brain, and in particular gray matter, atrophy over time in MS.5
Disease-modifying therapies (DMTs) for relapsing-remitting MS (RRMS) primarily suppress or modulate immune function, reducing the risk of future neuroinflammation, and related neurodegeneration.6 Accordingly, OCT has been increasingly utilized to evaluate the neuroprotective effects of DMTs in MS.7,8 Rituximab, a monoclonal antibody that depletes CD20+ B lymphocytes, similar to the Food and Drug Administration–approved therapy ocrelizumab, has been shown to be a highly effective treatment for RRMS.9,10 However, its effect on retinal atrophy in RRMS has not been investigated. The goal of this study was to evaluate the modulation of retinal atrophy by rituximab in RRMS relative to the high- and low-potency DMTs, natalizumab and glatiramer acetate (GA), respectively, and healthy controls (HCs). Our a priori hypothesis was that GCIPL atrophy rates among patients receiving rituximab would be similar to those observed among natalizumab-treated patients and HCs, and based upon observations from phase 3 clinical trials of ocrelizumab in RRMS, optimization of the neuroprotective effect of rituximab may take up to 6–12 months.11
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by the Johns Hopkins Institutional Review Board. All participants provided written informed consent.
Study Design and Participants
Patients with RRMS on rituximab were recruited prospectively from the Johns Hopkins MS Center between 2008 and 2018, with age- and sex-matched patients with RRMS on natalizumab or GA, and HCs, recruited retrospectively. Participant enrollment and data acquisition was performed through convenience sampling, generally during the course of routine clinical care, and was so designed to facilitate recruitment of maximum numbers of patients to each DMT subgroup, which were intended to be representative of real-world clinical cohorts. The study was observational in nature, and decisions regarding DMT selection were made by the treating neurologist, independent of the study. MS diagnosis was confirmed by the treating neurologist, according to the 2010 revised McDonald criteria, with RRMS classified according to the Lublin classification.12,13 Demographic and clinical data were recorded at baseline, including recent clinical/radiologic disease activity, and DMT use within 6 months prior to the baseline OCT scan. Prior DMT use was defined as low potency (GA/interferon β/teriflunomide), intermediate potency (dimethyl fumarate/fingolimod), or high potency (natalizumab/rituximab).
Baseline OCT assessments were performed ≤90 days prior to commencing treatment, or following treatment commencement. Participants with diabetes mellitus, uncontrolled hypertension, glaucoma, or other significant neurologic/ophthalmologic disorders were excluded, as were eyes with prior surgery/trauma, refractive errors of greater than ±6 diopters, or ON within 6 months of baseline OCT, or during follow-up. Patients on a DMT for ≥11 months and with ≥11 months of OCT follow-up were included in analyses.
Data Acquisition
Participants underwent spectral-domain OCT assessments (Cirrus HD-OCT, Carl Zeiss Meditec, Dublin, CA), as previously described.4 Briefly, macular and peripapillary scans were obtained using the Macular Cube 512 × 128 and Optic Disc Cube 200 × 200 protocols, respectively. Thickness values for the peripapillary retinal nerve fiber layer (pRNFL) were generated by conventional Cirrus HD-OCT software, as described elsewhere.4 Macular layer thicknesses including the GCIPL, inner nuclear layer (INL), and outer nuclear layer (ONL) were derived utilizing a validated, automated segmentation algorithm, as previously described.14 Average thicknesses of the GCIPL, INL, and ONL were calculated within an annulus, centered on the fovea, with an inner diameter of 1 mm, and an outer diameter of 5 mm. Qualitative review of all scans and segmentations was performed to assess for incidental pathology and to confirm the accuracy of the segmentations. Scans with signal strength <7/10, or with artifact, were excluded, in accordance with OSCAR-IB criteria.15,16 OCT methods and results are reported in accordance with the consensus APOSTEL recommendations.17
Statistical Analyses
Statistical analyses were performed using STATA version 15 (StataCorp, College Station, TX). Comparisons of demographic/clinical characteristics were performed using the Kruskal-Wallis test (age, disease duration, follow-up time, number of OCT assessments, and interval between initiation of DMT and baseline OCT assessment [gap time]), χ2 test (sex, ON history), and Fisher exact test (race, previous DMT). Rates of retinal layer thickness change were analyzed using linear mixed-effects regression models. The model used was a 3-level model, with subject- and eye-specific random intercepts (to account for nesting of longitudinal retinal thicknesses within an eye, and eyes within a participant), and random slopes in time (to account for change over time). The variance of the random effects was unstructured, and we estimated degrees of freedom using the Kenward Rogers approximation. We tested for differences between DMT groups using pairwise comparisons (1 df tests). Analyses were adjusted for demographic characteristics including age, sex, and race (which have previously been demonstrated to affect retinal atrophy rates),18–20 as well as disease duration, ON history, and gap time. In addition, a cross-product term of follow-up time with gap time was included to account for the potential cross-sectional and longitudinal effects of gap time on retinal atrophy. Gap time was dichotomized (<12 or ≥12 months) as we assumed a nonlinear relationship between gap time and time to treatment optimization.
Additional models using linear splines allowed the slope of GCIPL atrophy to change at a specific time point, as defined by the insertion of a knot or break point. Linear mixed-effects models including break points at 6 and 12 months posttreatment commencement were chosen specifically due to our a priori established research hypothesis that optimization of neuroprotective effect following commencement of B-cell–depleting therapy may take up to 6–12 months. This hypothesis was based on observations from phase 3 clinical trials showing reduced disease activity among patients receiving ocrelizumab following 48 weeks of treatment, relative to an age- and sex-matched cohort of patients with RRMS receiving interferon-β, with ocrelizumab-treated patients demonstrating improvement in disability progression from as early as 24 weeks.11 As an extension of our primary analyses, a single linear mixed-effects model with a break point at 12 months post-treatment initiation facilitated calculation of separate GCIPL atrophy rates for the period pre-12 months vs post-12 months of treatment. These analyses were adjusted for age, sex, race, disease duration, and ON history, but not gap time, as gap time was inherently accounted for in the linear spline models. Additional analyses of GCIPL atrophy pre vs post 6 months of therapy were performed, utilizing a linear spline at 6 months, and including only rituximab-treated patients. Natalizumab- and GA-treated patients were not included in these secondary analyses, as a lower proportion of these patients had undergone OCT assessments within 6 months of commencing treatment, as compared to rituximab-treated patients, which could potentially underpower or bias results.
Statistical significance was defined as p ≤ 0.05. Figures were made using GraphPad Prism version 8.2 (GraphPad Software, La Jolla, CA, graphpad.com) and R version 3.61 (Vienna, Austria).
Data Availability
Anonymized data used for this study are available from the corresponding author on reasonable request, with the proper data sharing agreements in place.
Results
Baseline Characteristics of the Study Population
A total of 172 patients with RRMS on DMTs (rituximab: n = 35; natalizumab: n = 88; GA: n = 49) and 78 HCs were eligible for inclusion. Age, sex, race, and ON history did not differ between groups (table 1). Rituximab-treated patients had a shorter median duration of follow-up (1.9 years) vs natalizumab- and GA-treated patients and HCs (3.0, 2.7, and 3.1 years, respectively; p < 0.001); a shorter median interval between OCT assessments (191.5 days) vs natalizumab and GA-treated patients and HCs (234, 364, and 385 days, respectively; p < 0.001); and a shorter median gap time (0.3 years) vs the natalizumab and GA cohorts (0.9 and 2.2 years, respectively; p < 0.001). Primarily due to differences in gap time, a higher proportion of the rituximab-treated cohort had transitioned from other DMTs (particularly intermediate- or high-potency therapies) in the 6 months preceding baseline OCT assessment (p < 0.001), due to treatment failure, intolerable side-effect profile, or patient preference. Baseline GCIPL thicknesses were lower in the RRMS cohorts relative to HCs (p < 0.001).
Table 1.
Baseline Demographic and Clinical Characteristics

Comparisons of Rates of Retinal Thinning Stratified by DMTs
In multivariable analyses, during the overall period of follow-up, rates of GCIPL thinning were −0.28 ± 0.11 µm/y among rituximab-treated patients, which were similar to those observed among GA-treated patients (−0.33 ± 0.05 µm/y; p = 0.69), and faster than HCs (−0.15 ± 0.03 µm/y; p = 0.006; figure 1). Although natalizumab-treated patients appeared to exhibit lower rates of GCIPL atrophy (−0.17 ± 0.10 µm/y) as compared to rituximab-treated patients, this difference was not statistically significant (p = 0.13). No differences in atrophy rates were observed between DMT subgroups for the INL, ONL, or pRNFL (data not shown).
Figure 1. Ganglion Cell + Inner Plexiform Layer (GCIPL) Thickness Over Time, Stratified by Disease-Modifying Therapies (DMTs).

(A) Spaghetti plots depict the individual trajectory in GCIPL thickness stratified by DMT group. (B) Annualized change in GCIPL thickness (adjusted β, µm/y), stratified by DMTs, for the overall period of follow-up. The error bars represent the 95% confidence interval (CI) for the annualized change in GCIPL thickness. p Values derived from linear mixed-effects regression models accounting for within-subject intereye correlations, adjusting for age at baseline, sex, and race (White vs non-White) in comparisons between rituximab and healthy controls (HCs), and additionally for optic neuritis history, disease duration, and gap time in comparisons between DMTs. (C) Annualized change in GCIPL thickness (adjusted β, µm/y), stratified by DMTs, for the period pre vs post 12 months of treatment. The error bars represent the 95% CI for the annualized change in GCIPL thickness. p Values derived from linear mixed-effects regression models accounting for within-subject intereye correlations, adjusting for age at baseline, sex, race (White vs non-White), optic neuritis history, disease duration, and gap time. Figure was made using R version 3.61 (Vienna, Austria) and GraphPad Prism version 8.2 (GraphPad Software, La Jolla, CA; graphpad.com). GA = glatiramer acetate.
Comparisons of retinal atrophy in the period pre vs post 12 months of treatment with rituximab revealed 0.55 ± 0.23 µm/y (n = 25; p = 0.02) faster rates of GCIPL thinning during the first 12 months of treatment, as compared to afterwards, when GCIPL atrophy rates were −0.14 ± 0.13 µm/y (table 2; figures 1 and 2). In order to probe whether the observed difference was influenced by disease activity or a postnatalizumab rebound phenomenon, these analyses were repeated separately, excluding patients who received natalizumab (n = 7) and patients who demonstrated clinical/radiologic disease activity (n = 17) within 6 months prior to baseline OCT assessment on rituximab. While the difference in GCIPL atrophy rates remained significant in analyses including only patients not recently on natalizumab (n = 18; difference: 0.48 ± 0.24 µm/y; p = 0.049), this difference was not significant in analyses including only patients without recent disease activity (n = 9; 0.39 ± 0.38 µm/y; p = 0.30); however, there was no difference in GCIPL atrophy rates during the first 12 months of rituximab treatment between patients with vs without recent disease activity (−0.86 ± 0.20 µm/y vs −0.74 ± 0.33 µm/y, respectively; p = 0.75). Similarly, GCIPL thinning was faster pre vs post 12 months of treatment in the natalizumab cohort (n = 46; difference: 0.32 ± 0.15 µm/y; p = 0.04), with similar post 12 months atrophy rates (−0.13 ± 0.10 µm/y) as compared to those observed among rituximab-treated patients (p = 0.94). GCIPL atrophy rates did not differ pre vs post 12 months of treatment with GA (n = 17; difference: 0.10 ± 0.35 µm/y; p = 0.78), with atrophy rates of −0.30 ± 0.04 µm/y for the period post 12 months of GA treatment (p = 0.24 vs rituximab).
Table 2.
Differences in Rates of Ganglion Cell + Inner Plexiform Layer (GCIPL) Change (µm/y) Following Disease-Modifying Therapy (DMT) Commencement

Figure 2. Difference in the Slope of Annualized Ganglion Cell + Inner Plexiform Layer (GCIPL) Thickness Over Time, Stratified by Disease-Modifying Therapies (DMTs), for the Period <12 Months vs ≥12 Months of Treatment.
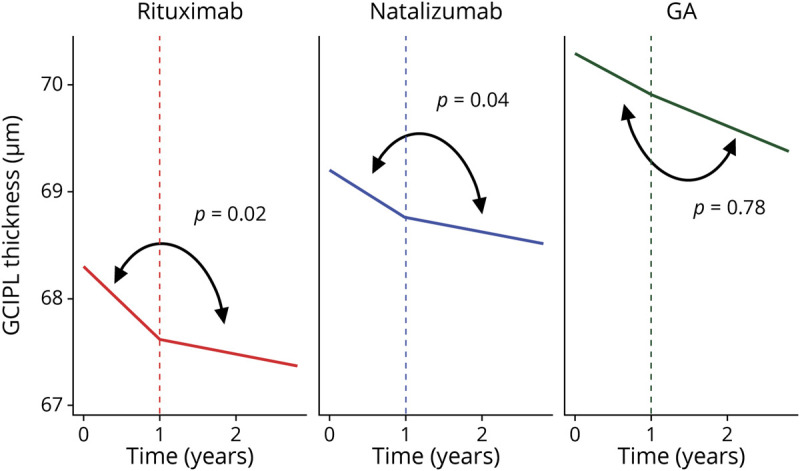
Difference, µm/y (95% confidence interval): rituximab: 0.55 (0.10–0.99); natalizumab: 0.32 (0.02–0.62); glatiramer acetate (GA) 0.10 (−0.58 to 0.78). p Values derived from linear mixed-effects regression models accounting for within-subject intereye correlations, adjusting for age at baseline, sex, race (White vs non-White), optic neuritis history, and disease duration, which assess the difference in slope <12 months vs ≥12 months of treatment for each DMT. Figure was made using R version 3.61 (Vienna, Austria) and R version 3.61.
In secondary analyses including a linear spline at 6 months after treatment initiation, rituximab-treated patients demonstrated faster GCIPL atrophy during the initial 6 months of treatment relative to the period afterwards (n for comparison = 22; difference: 0.91 ± 0.37 µm/y; p = 0.02; GCIPL atrophy rates post 6 months of treatment: −0.18 ± 0.12 µm/y).
In addition, we performed sensitivity analyses including only OCT assessments performed at approximately yearly intervals (e.g., 0 months; 10–14 months; 22–26 months), to evaluate whether our findings may have been confounded by the varying intervals between OCT scans among the treatment cohorts. In these analyses, utilizing the same linear mixed-effects models as described previously, adjusted GCIPL atrophy rates for the overall period of follow-up did not differ markedly, as compared to atrophy rates calculated when using all OCT scans, for patients on natalizumab (−0.18 ± 0.30 µm/y vs −0.17 ± 0.10 µm/y), patients on GA (−0.36 ± 0.10 µm/y vs −0.33 ± 0.05 µm/y), and in HCs (−0.14 ± 0.05 µm/y vs −0.15 ± 0.03 µm/y). While GCIPL atrophy rates in the rituximab cohort appeared lower for the overall duration of follow-up in analyses including only yearly OCT scans, relative to those models including all scans (−0.14 ± 0.32 µm/y vs −0.28 ± 0.11 µm/y), this is likely due to the exclusion of scans performed during the initial months following rituximab commencement, when GCIPL atrophy had previously been shown to be fastest. Despite this, in analyses using yearly scans only, GCIPL atrophy remained faster during the first 12 months following rituximab commencement as compared to the period afterwards (difference: 0.91 ± 0.38 µm/y; p = 0.02). Therefore, similar trends of GCIPL atrophy were observed in analyses including yearly scans only, as compared to analyses including all scans (performed at varying intervals throughout the study).
Classification of Evidence
The primary research question was to evaluate whether, and the manner in which, rituximab modulates retinal atrophy in RRMS. This study provides Class IV evidence that for patients with RRMS, rituximab appears to be associated with an attenuation of retinal atrophy. The neuroprotective effect of rituximab was most apparent after 12 months or more of treatment, during which period rituximab-treated patients exhibited GCIPL atrophy rates of −0.14 ± 0.13 µm/y, similar to the GCIPL atrophy rate observed among a separate cohort of patients with RRMS following 12 months of treatment with natalizumab (−0.13 ± 0.10 µm/y; p = 0.94). GCIPL atrophy was significantly faster during the first 12 months of rituximab treatment, relative to afterwards (n = 25; difference: 0.55 ± 0.23 µm/y; p = 0.02).
Discussion
In this study, following 12 months of therapy, rituximab-treated patients with RRMS exhibited similar rates of GCIPL thinning as compared to natalizumab-treated patients with RRMS and HCs. Our findings provide objective support that rituximab attenuates GCIPL atrophy in RRMS.
Our study expands upon prior work by Button et al,7 in which natalizumab-treated patients with RRMS demonstrated slower rates of GCIPL atrophy, as compared to patients treated with interferon-β and GA, and similar rates to HCs. Despite being an off-label treatment for RRMS, based on phase 2 clinical trial data, rituximab is considered to be a highly effective treatment for RRMS.9,10 It is unsurprising, therefore, that rituximab may exhibit a similar effect to natalizumab, another high-potency DMT (albeit with a very different mechanism of action), in slowing GCIPL atrophy in RRMS. Phase 3 clinical trials of ocrelizumab, a compound closely related to rituximab, in RRMS have demonstrated a greater reduction in radiologic activity and disability progression following 48 weeks of treatment, with reduction in disability progression evident from 24 weeks onwards, as compared to patients with RRMS treated with interferon-β.11 These findings suggest that a delayed optimization of therapeutic response may occur among patients with RRMS commencing B-cell therapies. This concept is potentially supported by our study's findings of faster GCIPL atrophy in the period up to 12 months following commencement of rituximab therapy relative to afterwards.
We performed additional analyses to probe whether ancillary factors may have contributed to the faster GCIPL atrophy rates observed during the first 12 months of rituximab treatment, separately excluding patients who demonstrated new clinical/radiologic disease activity, or who discontinued natalizumab, within 6 months of baseline OCT assessment on rituximab. While GCIPL atrophy remained faster during the first 12 months vs afterwards when excluding patients recently on natalizumab (n = 7), this difference was no longer significant following exclusion of patients with recent disease activity (n = 17). However, the similar atrophy rates observed during the first 12 months of treatment between patients with vs without recent disease activity suggests that these exploratory analyses were likely underpowered to detect a significant difference. Therefore, while we determined that a postnatalizumab discontinuation rebound phenomenon was unlikely to have contributed to the observed differences in GCIPL atrophy rates pre vs post 12 months of rituximab treatment, we could not rule out recent disease activity as a potential contributing factor to the faster GCIPL atrophy rates observed during the first 12 months following treatment initiation.
While the difference in GCIPL atrophy rates appeared to be greatest in comparisons between the first 6 months following commencement of rituximab treatment and the interval afterwards, these findings should be interpreted with caution due to the relatively low number of patients with OCT data pre and post 6 months of rituximab treatment (n = 22 patients). However, the observed trend of GCIPL atrophy rates for the overall period of follow-up (−0.28 ± 0.11 µm/y), as compared to the periods following 6 months of treatment (−0.18 ± 0.12 µm/y), and following 12 months of treatment (−0.14 ± 0.13 µm/y), may suggest partial optimization of therapeutic effect commencing within the first 6 months following treatment commencement, with additional optimization occurring over months 7–12.
Interestingly, natalizumab-treated patients also demonstrated faster GCIPL atrophy during the initial 12 months of treatment vs afterwards. However, a significant limitation of this study is that natalizumab- and GA-treated patients were recruited retrospectively; subsequently, a lower proportion of these patients underwent serial OCT assessments within 12 months following treatment commencement, relative to rituximab-treated patients. Therefore, analyses were underpowered to determine whether the temporal difference in GCIPL atrophy rates was, in fact, primarily driven by accelerated atrophy at a relatively early stage during the initial 12 months following natalizumab commencement. This theory is supported by several studies demonstrating sustained reductions in MS disease activity as early as 1–3 months following first natalizumab infusion.21,22 Further longitudinal studies, with OCT scans performed at early, more regular intervals, are needed to evaluate temporal changes in GCIPL atrophy rates during the initial months following natalizumab commencement. In addition, while GA-treated patients appeared to exhibit numerically faster GCIPL atrophy rates than rituximab-treated patients for the period post 12 months of treatment, this comparison did not reach statistical significance. This was likely due to statistical underpowering, as comparatively fewer rituximab-treated patients underwent serial OCT assessments after completing 12 months of treatment. Our study has additional limitations that warrant discussion. OCT scans were performed at variable intervals, with differences in median gap times among participants in different cohorts. As detailed previously, we performed sensitivity analyses to address these limitations, such as including only scans performed at approximately yearly intervals in analyses, but did not find marked differences in GCIPL atrophy rates, as compared to our primary analyses, in which all scans performed at varying intervals were included. In addition, our study did not include assessment of MRI scans to evaluate the presence of optic radiation lesions, which have been associated with GCIPL thickness reductions in a hemi-macular pattern in previous studies.23,24 This pattern of GCIPL thinning is generally hypothesized to occur secondary to transsynaptic degeneration (TSD); however, the proportion of optic radiation lesions that lead to TSD is currently unknown, as are the factors that may contribute to TSD occurring in individual patients. Future longitudinal studies could evaluate whether, and the extent to which, optic radiation lesions affect rates of GCIPL atrophy in MS, and whether the use of DMTs may attenuate this process. In addition, future larger, prospective, longitudinal studies of sufficient follow-up are required to assess the clinical significance of our study findings and elucidate more specifically how the modulation of retinal atrophy by DMTs is correlated with clinical outcome measures, such as visual acuity and Expanded Disability Status Scale scores.
Our study demonstrates that retinal atrophy in RRMS is modulated by rituximab, as evidenced by similar rates of GCIPL atrophy during the period following 12 months of rituximab treatment, as compared to those observed among natalizumab-treated patients with RRMS and HCs. Our findings suggest that optimization of neuroprotective response with rituximab in RRMS may potentially take up to 12 months.
Glossary
- DMT
disease-modifying therapy
- GA
glatiramer acetate
- GCIPL
ganglion cell + inner plexiform layer
- HC
healthy control
- INL
inner nuclear layer
- MS
multiple sclerosis
- OCT
optical coherence tomography
- ON
optic neuritis
- ONL
outer nuclear layer
- pRNFL
peripapillary retinal nerve fiber layer
- RRMS
relapsing-remitting multiple sclerosis
- TSD
transsynaptic degeneration
Appendix. Authors
Footnotes
Editorial, page 927
Class of Evidence: NPub.org/coe
Study Funding
This study was funded by the National MS Society (RG-1606-08,768 to S.S.), Race to Erase MS (to S.S.), and NIH/NINDS (R01NS082347 to P.A.C.). K.C.F. is supported by NIH/NIMH K01-MH121582 and a Career Transition Fellowship from the National MS Society (TA-1805-31,136).
Disclosure
Dr. Lambe, H. Risher, Dr. Filippatou, Dr. Murphy, Dr. Ehrhardt, E. Ogbuokiri, N. Pellegrini, B. Toliver, N.J. Luciano, S. Davis, N. Fioravante, and O. Kwakyi report no disclosures. Dr. Sotirchos has served on a scientific advisory boards for Viela Bio and Genentech and is funded by a Sylvia Lawry physician fellowship award from NMSS. Dr. Prince is a founder of Sonovex, Inc. and serves on its Board of Directors, has received consulting fees from JuneBrain LLC, and is PI on research grants to Johns Hopkins from 12Sigma Technologies and Biogen. Dr. Calabresi has received consulting fees from Disarm and Biogen and is PI on grants to JHU from Biogen and Annexon. Dr. Fitzgerald is funded by NIH/NIMH K01-MH121582 and a Career Transition Fellowship from the National MS Society (NMSS). Dr. Saidha has received consulting fees from Medical Logix for the development of CME programs in neurology; has served on scientific advisory boards for Biogen, Genzyme, Genentech Corporation, EMD Serono, and Celgene; is the PI of investigator-initiated studies funded by Genentech Corporation and Biogen Idec; received support from the Race to Erase MS foundation; has received equity compensation for consulting from JuneBrain LLC, a retinal imaging device developer; and is the site investigator of a trial sponsored by MedDay Pharmaceuticals. Go to Neurology.org/N for full disclosures.
References
- 1.Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med 2018;378:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petzold A, Balcer LJ, Calabresi PA, et al. . Retinal layer segmentation in multiple sclerosis: a systematic review and meta-analysis. Lancet Neurol 2017;16:797–812. [DOI] [PubMed] [Google Scholar]
- 3.Saidha S, Syc SB, Durbin MK, et al. . Visual dysfunction in multiple sclerosis correlates better with optical coherence tomography derived estimates of macular ganglion cell layer thickness than peripapillary retinal nerve fiber layer thickness. Mult Scler 2011;17:1449–1463. [DOI] [PubMed] [Google Scholar]
- 4.Syc SB, Warner CV, Hiremath GS, et al. . Reproducibility of high-resolution optical coherence tomography in multiple sclerosis. Mult Scler 2010;16:829–839. [DOI] [PubMed] [Google Scholar]
- 5.Saidha S, Al-Louzi O, Ratchford JN, et al. . Optical coherence tomography reflects brain atrophy in multiple sclerosis: a four-year study: retinal atrophy reflects brain atrophy in MS. Ann Neurol 2015;78:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.National Multiple Sclerosis Society. The Use of Disease-Modifying Therapies in Multiple Sclerosis: Principles and Current Evidence. Multiple Sclerosis Coalition; 2014. [Google Scholar]
- 7.Button J, Al-Louzi O, Lang A, et al. . Disease-modifying therapies modulate retinal atrophy in multiple sclerosis: a retrospective study. Neurology 2017;88:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zivadinov R, Tavazzi E, Hagemeier J, et al. . The effect of glatiramer acetate on retinal nerve fiber layer thickness in patients with relapsing–remitting multiple sclerosis: a longitudinal optical coherence tomography study. CNS Drugs 2018;32:763–770. [DOI] [PubMed] [Google Scholar]
- 9.Hauser SL, Waubant E, Arnold DL, et al. . B-Cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008;358:676–688. [DOI] [PubMed] [Google Scholar]
- 10.Naismith RT, Piccio L, Lyons JA, et al. . Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: a 52-week phase II trial. Neurology 2010;74:1860–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauser SL, Bar-Or A, Comi G, et al. . Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017;376:221–234. [DOI] [PubMed] [Google Scholar]
- 12.Polman CH, Reingold SC, Banwell B, et al. . Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald Criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lublin FD, Reingold SC, Cohen JA, et al. . Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang A, Carass A, Hauser M, et al. . Retinal layer segmentation of macular OCT images using boundary classification. Biomed Opt Express 2013;4:1133–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tewarie P, Balk L, Costello F, et al. . The OSCAR-IB consensus criteria for retinal OCT quality assessment. PLoS One 2012;7:e34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schippling S, Balk L, Costello F, et al. . Quality control for retinal OCT in multiple sclerosis: validation of the OSCAR-IB criteria. Mult Scler 2015;21:163–170. [DOI] [PubMed] [Google Scholar]
- 17.Cruz-Herranz A, Balk LJ, Oberwahrenbrock T, et al. . The APOSTEL recommendations for reporting quantitative optical coherence tomography studies. Neurology 2016;86:2303–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sotirchos ES, Gonzalez Caldito N, Filippatou A, et al. . Progressive multiple sclerosis is associated with faster and specific retinal layer atrophy. Ann Neurol 2020;87:885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filippatou AG, Lambe J, Sotirchos ES, et al. . Association of body mass index with longitudinal rates of retinal atrophy in multiple sclerosis. Mult Scler 2020;26:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caldito NG, Saidha S, Sotirchos ES, et al. . Brain and retinal atrophy in African-Americans versus Caucasian-Americans with multiple sclerosis: a longitudinal study. Brain 2018;141:3115–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kappos L, O'Connor PW, Polman CH, et al. . Clinical effects of natalizumab on multiple sclerosis appear early in treatment course. Neurology 2013;260:1388–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller DH, Khan OA, Sheremata WA, et al. . A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2003;348:15–23. [DOI] [PubMed] [Google Scholar]
- 23.Huang-Link Y-M, Al-Hawasi A, Eveman I. Retrograde degeneration of visual pathway: hemimacular thinning of retinal ganglion cell layer in progressive and active multiple sclerosis. Neurology 2014;261:2453–2456. [DOI] [PubMed] [Google Scholar]
- 24.Al-Louzi O, Button J, Newsome SD, et al. . Retrograde trans-synaptic visual pathway degeneration in multiple sclerosis: a case series. Mult Scler 2017;23:1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data used for this study are available from the corresponding author on reasonable request, with the proper data sharing agreements in place.