

Abstract
STUDY QUESTION
Is it possible to establish a genetically engineered mouse model (GEMM) of endometriosis that mimics the natural spread of invasive endometrium?
SUMMARY ANSWER
Endometriosis occurs in an ARID1A (AT-rich interactive domain-containing protein 1A) and PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha) mutant GEMM of endometrial dysfunction following salpingectomy.
WHAT IS KNOWN ALREADY
Although mouse models of endometriosis have long been established, most models rely on intraperitoneal injection of uterine fragments, steroid hormone treatments or the use of immune-compromised mice.
STUDY DESIGN, SIZE, DURATION
Mice harboring the lactotransferrin-Cre (LtfCre0/+), Arid1afl, (Gt)R26Pik3ca*H1047R and (Gt)R26mTmG alleles were subject to unilateral salpingectomies at 6 weeks of age. Control (n = 9), LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/+ (n = 8) and LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl (n = 9) were used for the study. The (Gt)R26mTmG allele was used for the purpose of fluorescent lineage tracing of endometrial epithelium. LtfCre0/+; (Gt)R26mTmG (n = 3) and LtfCre0/+; (Gt)R26Pik3ca*H1047R/mTmG; Arid1afl/fl (n = 4) were used for this purpose. Mice were followed until the endpoint of vaginal bleeding at an average time of 17 weeks of age.
PARTICIPANTS/MATERIALS, SETTING, METHODS
At 6 weeks of age, mice were subjected to salpingectomy surgery. Mice were followed until the time point of vaginal bleeding (average 17 weeks), or aged for 1 year in the case of control mice. At time of sacrifice, endometriotic lesions, ovaries and uterus were collected for the purpose of histochemical and immunohistochemical analyses. Samples were analyzed for markers of the endometriotic tissue and other relevant biomarkers.
MAIN RESULTS AND THE ROLE OF CHANCE
Following salpingectomy, LtfCre0/+; (Gt)R26Pik3ca*H1047R/mTmG; Arid1afl/fl mice developed endometriotic lesions, including lesions on the ovary, omentum and abdominal wall. Epithelial glands within lesions were negative for ARID1A and positive for phospho-S6 staining, indicating ARID1A-PIK3CA co-mutation status, and expressed EGFP (enhanced green fluorescent protein), indicating endometrial origins.
LARGE-SCALE DATA
N/A
LIMITATIONS, REASONS FOR CAUTION
LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl mice develop vaginal bleeding as a result of endometrial dysfunction at an average age of 17 weeks and must be sacrificed. Furthermore, while this model mimics the natural spread of endometriotic tissue directly from the uterus to the peritoneum, the data presented do not reject current hypotheses on endometriosis pathogenesis.
WIDER IMPLICATIONS OF THE FINDINGS
The idea that endometriosis is the result of abnormal endometrial tissue colonizing the peritoneum via retrograde menstruation has gained widespread support over the past century. However, most models of endometriosis take for granted this possibility, relying on the surgical removal of bulk uterine tissue and subsequent transplantation into the peritoneum. Growing evidence suggests that somatic mutations in ARID1A and PIK3CA are present in the endometrial epithelium. The establishment of a GEMM which mimics the natural spread of endometrium and subsequent lesion formation supports the hypothesis that endometriosis is derived from mutant endometrial epithelium with invasive properties.
STUDY FUNDING/COMPETING INTEREST(S)
This research was supported by the American Cancer Society PF-17-163-02-DDC (M.R.W.), the Mary Kay Foundation 026-16 (R.L.C.) and the Ovarian Cancer Research Fund Alliance 457446 (R.L.C.). The authors declare no competing interests.
Keywords: endometriosis, mouse, invasion, endometrium, uterus
Introduction
Endometriosis is a disease in which cells lining the uterus (endometrium) grow outside of the uterus, causing pain and infertility in 5 to 10% of women (Alimi et al., 2018). Formation of endometriosis is thought to result from the translocation of endometrium via retrograde menstruation (Chui et al., 2017). The retrograde menstruation theory is supported by several clinical observations that obstruction of menstrual flow increases the incidence of endometriosis (Burney and Giudice, 2012). Other theories for endometriosis pathogenesis propose that lesions arise from peritoneal tissue or extra-uterine stem cells which have become transformed into ectopic endometrium, while additional theories hold that endometrium arrives into the peritoneum by lymphatic or hematogenous dissemination (Burney and Giudice, 2012). Studies have suggested that the invasive phenotype of endometriosis shares aspects with tumor metastasis, including migration and invasion (Giudice and Kao, 2004). In order for endometriosis to occur, endometrial cells may undergo epithelial-to-mesenchymal transition (EMT) in order to invade distal tissues (Bartley et al., 2014), a probable scenario given the inherent plasticity of the endometrium (Matsuzaki and Darcha, 2012).
Mutations in ARID1A (AT-rich interactive domain-containing protein 1A), a well-established tumor suppressor gene, are also found in endometriosis (Samartzis et al., 2012), and ARID1A loss may indicate malignancy in atypical endometriosis (Wiegand et al., 2011). Additionally, mutations in PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha) may be a prerequisite to ARID1A loss in endometriosis (Anglesio et al., 2017; Suda et al., 2018).
Recently, we characterized a new genetically engineered mouse model of endometrial dysfunction which results in endometrial hyperplasia, vaginal bleeding and lesion formation (Wilson et al., 2019). We utilized the lactotransferrin-Cre (LtfCre) allele, which drives Cre-recombinase expression specifically to the endometrial epithelium. The mice displayed loss of ARID1A expression and expression of the oncogenic PIK3CAH1047R mutation, resulting in endometrial hyperplasia, lesion formation and invasion into the uterine myometrium (Wilson et al., 2019). We employed genome-wide methodologies to examine changes in gene expression and chromatin accessibility of sorted endometrial epithelial cells in vivo and identified upregulation of EMT pathways and epithelial transdifferentiation in the mutant cells. We found that ARID1A and PIK3CA mutant endometrial epithelium was capable of invasion locally into the myometrium (Wilson et al., 2019). Given that endometriosis may be derived from invasive endometrial epithelium and ARID1A and PIK3CA mutations, we sought to utilize our model of endometrial dysfunction to explore the development of endometriosis.
Mice do not menstruate; therefore, the opportunity for endometrial tissue to translocate to the peritoneum via retrograde menstruation does not exist in mice. Traditional mouse models of endometriosis were developed via the surgical transplantation of autologous or syngeneic uterine tissue into the peritoneum (Cummings and Metcalf, 1995; Hirata et al., 2005; Rossi et al., 2000; Cheng et al., 2011; Greaves et al., 2014), or transplantation of human endometrial or endometriotic tissue into immune-compromised mice (Aoki et al., 1994; Grummer et al., 2001), often with estrogen supplementation (Zamah et al., 1984; Bergqvist et al., 1985). Previously developed genetically engineered mouse models of endometriosis have relied on mutation of genes in the ovarian surface epithelium rather than the endometrium (Dinulescu et al., 2005; Budiu et al., 2009). Here, we developed a model of endometriosis derived from the endometrial epithelium by surgical incision at the uterotubal junction followed by salpingectomy in mice harboring ARID1A and PIK3CA mutations in the endometrial epithelium. This procedure created an opening in the uterus at the uterotubal junction, thereby exposing the uterine cavity and providing mutant cells an opening to invade into the peritoneal cavity, resulting in endometriosis formation. In this study, we have characterized a new genetically engineered mouse model of endometriosis derived from mutant endometrial epithelium which mimics the natural spread of invasive endometrium.
Materials and Methods
Contact for reagent and resource sharing
Requests for further information, resources and reagents should be addressed to the lead contact, Ronald L. Chandler (rlc@msu.edu).
Mice
All mice were maintained on an outbred genetic background using CD-1 mice (Charles River, Wilmington, MA, USA). (Gt)R26mTmG, (Gt)R26Pik3ca*H1047R and LtfCre (Tg(Ltf-iCre)14Mmul) alleles were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and identified by PCR using published methods (Muzumdar et al., 2007; Adams et al., 2011; Daikoku et al., 2014). The Arid1afl allele was detected by PCR as previously described (Chandler et al., 2013; Chandler et al., 2015). Endpoints were vaginal bleeding, severe abdominal distension and signs of severe illness, such as dehydration, hunching, jaundice, ruffled fur, signs of infection or non-responsiveness. Sample sizes within each genotype were chosen based on the proportions of animals with vaginal bleeding between each experimental group or a Kaplan–Meyer log-rank test for survival differences. Mice were housed at the Van Andel Research Institute Animal Facility and the Michigan State University Grand Rapids Research Center in accordance with protocols approved by Michigan State University. Michigan State University is registered with the US Department of Agriculture (USDA) and has an approved Animal Welfare Assurance from the NIH Office of Laboratory Animal Welfare (OLAW). MSU is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Uterotubal incision and salpingectomy surgery
Ovarian surgical protocol was followed as previously described (Chandler et al., 2015). Mice were anaesthetized using avertin (0.3 mg/g intraperitoneal injection). Avertin was composed of sterile embryo water (Sigma, cat# W1503, St. Louis, MO, USA), 2-methyl-2-butanol (Sigma, cat# 152463) and 2,2,2-tribromoethanol (Sigma, cat# T48402). Anesthesia was confirmed by the absence of reflex withdrawal to toe pinch. Hair at the procedure site was removed with small electric shears and scrubbed with betadine solution (Povidone–Iodine Veterinary Surgical Scrub, McKesson, Pompano Beach, FL, USA). Salpingectomy surgeries were performed on 6-week-old mice. Following incision through the left flank muscle, the ovary was removed and an incision was made at the uterotubal junction, being careful to avoid the uterine artery. Following uterotubal incision, the oviduct was removed. During the procedure, the ovary was kept moistened using a gauze soaked in PBS. The ovary was gently placed back, and the muscle layer was closed using sutures. Following surgery, mice were monitored for basic biologic functions and post-procedure distress. Surgeries were performed in the afternoon. The surgeon was blinded from the genotype of the mouse. Mice were housed randomly following surgery. All surgical data has been reported to ensure the study is free of selective outcome reporting.
Histology and immunohistochemistry
Upon vaginal bleeding, mice were euthanized by carbon dioxide inhalation and uterus, ovaries and relevant tissues were collected. For indirect immunohistochemistry (IHC), 10% neutral buffered formalin (NBF)-fixed paraffin sections were processed for heat-based antigen unmasking in 10 mM sodium citrate (pH 6.0). Sections were incubated with antibodies at the following dilutions: 1:200 ARID1A (12 354, Cell Signaling, Danvers, MA); 1:400 Phospho-S6 (4585, Cell Signaling); 1:100 KRT8 (Ketatin 8) (TROMA1, DHSB); 1:100 ICAM-1 (intercellular adhesion molecule 1) (AF796-SP, R&D Systems, Minneapolis, MN, USA); 1:200 Fra2 (MABS1261, EMD Millipore, Burlington, MA, USA); 1:300 c-Jun (9165, Cell Signaling); and 1:400 Ki67 (12 202, Cell Signaling). TROMA-I antibody was deposited to the Developmental Studies Hybridoma Bank (DSHB) by Brulet, P./Kemler, R. (DSHB Hybridoma Product TROMA-I). Primary antibody dilutions and catalog numbers are compiled in Table I. The following biotin-conjugated secondary antibodies were used: donkey anti-rabbit IgG (711-065-152, Jackson ImmunoResearch Lab, West Grove, PA, USA) and donkey anti-rat IgG (#705-065-153, Jackson ImmunoResearch Lab). Secondary antibodies were detected using VECTASTAIN Elite ABC HRP Kit (Vector, Burlingame, CA, USA). Sections for IHC were lightly counter-stained with Hematoxylin QS or Methyl Green (Vector Labs). Routine hematoxylin and eosin (H&E) staining of sections was performed by the Van Andel Research Institute (VARI) Histology and Pathology Core. A VARI animal pathologist reviewed histological sections. For picrosirius red staining, the Picrosirius Red Stain Kit (Polysciences, Inc., Warrington, PA, USA, cat# 24901-250) was used according to the manufacturers’ instructions.
Table I.
Immunohistochemistry antibodies.
| Antibody | Dilution | Source | Identifier |
|---|---|---|---|
| Rabbit monoclonal anti-ARID1A/BAF250A (D2A8U) | 1:200 | Cell Signaling Technology | Cat# 12354; RRID: AB_2637010 |
| Rabbit monoclonal anti-Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) | 1:400 | Cell Signaling Technology | Cat# 4858; RRID: AB_2721245 |
| Rat monoclonal anti-TROMA-I | 1:100 | Developmental Studies Hybridoma Bank | Cat# TROMA-I; RRID: AB_531826 |
| Goat polyclonal anti-ICAM-1/CD54 | 1:100 | R&D Systems | Cat# AF796-SP; RRID: AB_2248703 |
| Rat monoclonal anti-FRA-2 (REY146C) | 1:200 | EMD Millipore | Cat# MABS1261 |
| Rabbit monoclonal anti-c-Jun (60A8) | 1:300 | Cell Signaling Technology | Cat# 9165; RRID: AB_2130165 |
| Rabbit monoclonal anti-Ki67 (D3B5) | 1:400 | Cell Signaling Technology | Cat# 12202; RRID:AB_2620142 |
RRID, Research Resource Identifier; ARID1A, AT-rich interactive domain-containing protein 1A; ICAM-1, intercellular adhesion molecule 1; Fra2, Fos-related antigen 2; TROMA-1, trophectoderm-specific marker 1.
Microscopy and imaging
Fluorescent images were taken using a Nikon SMZ18 microscope, X-Cite Series 120 PC Q (Excelitas Technologies, Waltham, MA, USA) fluorescent lamp and a Nikon DS-Ri1 camera. Images were analyzed using Nikon NIS Elements Advanced Research software.
Statistics
Statistical analyses were performed using GraphPad (San Diego, CA, USA) Prism 8 software.
Results
Salpingectomy of genetically engineered mice results in a broad spectrum of endometriotic phenotypes
To determine if the invasive cellular phenotypes observed in LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/+ and LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl mice (Fig. 1A) promote the extrauterine spread of mutant endometrial epithelium, we devised a surgical protocol that mimics retrograde translocation of endometrial tissue into the peritoneal space (Fig. 1B and C). In contrast to humans, the isthmus and ampulla of mouse oviducts are tightly coiled and the infundibulum of the mouse oviduct opens into the bursal space, rather than the peritoneal cavity (Rendi et al., 2012). We reasoned that exposing the endometrial lumen and removing the anatomic barrier of the oviducts and bursal membrane via surgery would allow invasive luminal endometrial epithelial cells undergoing delamination and luminal shedding to more easily translocate into the peritoneal space. By performing unilateral salpingectomies at 6 weeks of age, we maximized the period between lactoferrin activation (beginning at 4 weeks) and expected mortality due to vaginal bleeding at 14–17 weeks (Fig. 1C).
Figure 1.
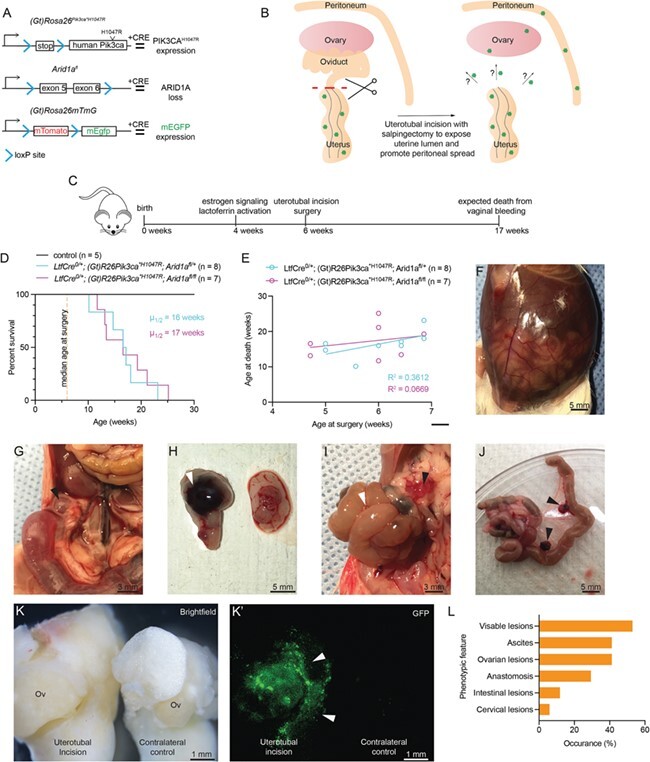
LtfCre 0/+ ; (Gt)R26Pik3ca *H1047R ; Arid1a fl/+ and LtfCre 0/+ ; (Gt)R26Pik3ca *H1047R ; Arid1a fl/fl mice develop ovarian lesions, anastomosis and ascites following salpingectomy. (A) Diagram depicting mouse alleles used in this study. ARID1A, AT-rich interactive domain-containing protein 1A; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; mEGFP, mouse enhanced green fluorescent protein. (B) Schematic representation of uterotubal incision and salpingectomy surgery. (C) Timeline for experimental mouse model. (D) Survival of mice following surgery. Survival was based on vaginal bleeding and other endpoints (see Table II). Control mice were aged for 1 year (n = 9, black line). No difference in survival was observed between LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/+ (n = 8, blue line) and LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl (n = 9, purple line) mice following surgery (P = 0.7421, unpaired t test, two-tailed). (E) Age at surgery vs. age at death. No significant relationship was observed (P = 0.1151 for LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/+, blue line; P = 0.3584 for LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl, purple line Pearson’s). (F) Example of LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl mice with ascites following surgery. (G) Ovarian cysts are observed in animals 6 weeks post-salpingectomy on the ipsilateral ovary. Arrow indicates cyst. (H) Example of animal with hemorrhagic ‘chocolate cyst’ endometrioma on ovary ipsilateral to incision. Arrow indicates lesion. (I) Intestinal obstruction in mutant mice following surgery. White arrows denote intestines, black arrows denote omental metastasis. (J) Intestinal anastomosis following salpingectomy. Black arrows denote omental lesions. (K) Bright-field image and fluorescent green image (K′). EGFP fluorescence denotes Cre + endometrial epithelium on or near the ovary. Ov: ovary. (L) Histogram compiling observational data upon necropsy.
Following surgery, mice were followed until vaginal bleeding or other endpoints (Table II). One control mouse died from complication from surgery the following day and was excluded from survival analysis (Table II). No phenotypes were observed in control mice subjected to salpingectomy and aged for approximately 1 year (Fig. 1D, Table II). LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/+ and LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl mice lived for an average of 17 weeks, 11 weeks beyond surgery (Fig. 1D). Survival differences between groups following surgery was not significant (P = 0.7421, unpaired t test, two-tailed), and age at time of surgery did not correlate with age at death (Fig. 1E) (R 2 = 0.3612 for LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/+, R 2 = 0.0669 for LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl, Pearson’s). Following salpingectomy, LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/f mice developed hemorrhagic ascites within 6 weeks following surgery (Fig. 1F). Extrauterine endometriotic lesions were observed on the ovary (Fig. 1G), with some lesions bearing resemblance to hemorrhagic ‘chocolate cyst’ endometriomas (Fig. 1H). In some cases, we observed intestinal anastomosis resulting from endometrial spread and fibrosis in the omentum (Fig. 1I and J). LtfCre contains an internal ribosomal entry site (IRES)-enhanced green fluorescent protein (EGFP) cassette. Based on EGFP expression, we detected mutant endometrial epithelial cells at sites ipsilateral to the site of incision, but not contralateral ovaries or uterine horns (Fig. 1K). Mice developed a spectrum of phenotypic conditions, including ascites, anastomosis and lesion formation (Fig. 1L). Over 50% of mice displayed visible lesion formation upon dissection (Fig. 1L). The phenotypes observed in this model reflect the broad spectrum of conditions affecting women with endometriosis.
Table II.
Summary of mice.
| Genotype | Age at surgery (days) | Age at death (days) | Cause of death/reason for sacrifice | Observations |
|---|---|---|---|---|
| Control | 42 | 43 | FD | Complications from surgery |
| Control | 35 | 103 | ED | No phenotype |
| Control | 35 | 103 | ED | No phenotype |
| Control | 35 | 103 | ED | No phenotype |
| Control | 42 | 403 | ES | No phenotype |
| Control | 45 | 406 | ES | No phenotype |
| Control | 45 | 406 | ES | No phenotype |
| Control | 45 | 406 | ES | No phenotype |
| Control | 48 | 418 | ES | No phenotype |
| LtfCre0/+; Arid1afl/+; (Gt)R26Pik3ca*H1047R | 39 | 71 | VB | |
| LtfCre0/+; Arid1afl/+; (Gt)R26Pik3ca*H1047R | 35 | 103 | VB | Lesion on ipsilateral ovary |
| LtfCre0/+; Arid1afl/+; (Gt)R26Pik3ca*H1047R | 42 | 112 | AB, VB | Lesions throughout peritoneum, organ anastomosis, ascites |
| LtfCre0/+; Arid1afl/+; (Gt)R26Pik3ca*H1047R | 45 | 112 | FD | |
| LtfCre0/+; Arid1afl/+; (Gt)R26Pik3ca*H1047R | 35 | 116 | VB | Intestinal anastomosis, ascites |
| LtfCre0/+; Arid1afl/+; (Gt)R26Pik3ca*H1047R | 45 | 120 | VB, DA | Ovarian lesions, intestinal anastomosis, ascites |
| LtfCre0/+; Arid1afl/+; (Gt)R26Pik3ca*H1047R | 48 | 126 | Distressed, VB | Lesions on both ovaries and omentum, intestinal anastomosis |
| LtfCre0/+; Arid1afl/+; (Gt)R26Pik3ca*H1047R | 48 | 162 | VB, DA | Ascites |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R | 42 | 82 | Lethargic, pale | Lesion on cervix, blood in intestines |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R | 33 | 92 | VB | |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R | 45 | 94 | DA | Lesions on ovary and mesentery |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R | 48 | 98 | ED | Lesion on ovary |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R | 48 | 98 | ED | Intestinal anastomosis, ascites |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R | 33 | 116 | VB | Intestinal anastomosis, ascites |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R | 48 | 135 | Distressed, VB | Anemic intestines |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R | 42 | 148 | AB, VB | Ipsilateral ovary fused with kidney, ascites |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R | 42 | 176 | VB | Lesion on ovary |
| LtfCre0/+;(Gt)R26mTmG | 54 | 149 | ED | No phenotype |
| LtfCre0/+;(Gt)R26mTmG | 33 | 94 | ED | No phenotype |
| LtfCre0/+;(Gt)R26mTmG | 64 | 125 | ED | No phenotype |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R/mTmG | 28 | 123 | VB | Lesion on omentum, abdominal wall |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R/mTmG | 38 | 99 | VB | Lesion on ovary |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R/mTmG | 33 | 92 | VB | Lesion on abdominal wall |
| LtfCre0/+; Arid1afl/fl; (Gt)R26Pik3ca*H1047R/mTmG | 33 | 116 | VB | Lesion on omentum |
Summary of mice. VB, vaginal bleeding; AB, abdominal bulge; FD, found dead; DA, distended abdomen; ES, end of study; ED, explorative dissection.
Endometrial glands within lesions display markers of ARID1A/PIK3CA mutant endometrium
In order to determine the role of the mutant endometrium in the formation of endometriotic lesions, we used histological analysis and IHC to identify ARID1A and PIK3CA mutant endometrium within lesions. Extrauterine lesions in the ovary were composed of disorganized and dysplastic ARID1A-negative glandular epithelial cells and were observed invading abdominal smooth muscle (Fig. 2A and B). Loss of ARID1A immunoreactivity in the epithelial compartment of the lesions supports endometrial epithelial origins. The extrauterine endometriotic cells also expressed KRT8, a marker of endometrial epithelium, and phospho-S6 ribosomal protein (P-S6), a marker for the activated PI3K pathway (Fig. 2C and D). Endometriotic lesions appeared highly fibrotic, as indicated by picrosirius red staining, relative to adjacent uterine tissue (Fig. 2E–J). The identification of these markers within endometriotic lesions indicates a role for mutant endometrium in the formation of these lesions.
Figure 2.
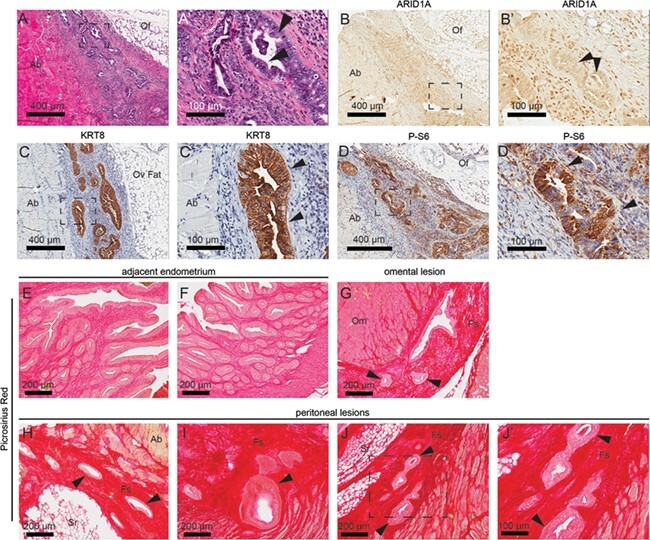
Endometrial glands formed in ovary are ARID1A-negative. (A) H&E staining in sections of ovarian lesion. (B–D) Immunohistochemistry (IHC) for ARID1A (B), KRT8 (ketatin 8) (C) and phospho-S6 (P-S6) (D) in sections of ovarian lesion. Panels A–D represent near-adjacent sections of extrauterine endometrial lesions, while panels A′–D′ represent portion of slide in panels A–D surrounded by black box. Arrows indicate epithelial gland formation. (E–J) Picrosirius red staining in sections of adjacent uterine tissue (E, F), omental lesions (G) and peritoneal lesions (H–J). Panel J′ represents portion in panel J surrounded by black box. Ab: abdominal wall; Cy: cyst; Fs: fibrotic stroma; Of: ovarian fat; Om: omentum; Oo: oocyte; Ov: ovary: Sr: serosa.
Ovarian and omental lesions are derived from lactoferrin-activated endometrium
In order to follow the lineage of cells in which LtfCre0/+ has been activated, we introduced the (Gt)R26mTmG allele into our model (Muzumdar et al., 2007). Both control LtfCre0/+; (Gt)R26mTmG and LtfCre0/+; (Gt)R26Pik3ca*H1047R/mTmG; Arid1afl/fl mice displayed prominent GFP expression in the uterus (Fig. 3). Post-salpingectomy, GFP expression in LtfCre0/+; (Gt)R26mTmG mice remained limited to the uterus (Fig. 3A and B). However, spread of metastatic cells in the LtfCre0/+; (Gt)R26Pik3ca*H1047R/mTmG; Arid1afl/fl model was observed in lesions occupying the omentum (Fig. 3C–E), the ovary (Fig. 3F and G) and throughout the peritoneum (Fig. 3H–K) by GFP expression in whole mount tissues. Endometrial epithelium and endometrial glands occupied only a small portion of visible lesions, with clear evidence of blood accumulation in afflicted tissues (Fig. 3E, F, H and J).
Figure 3.
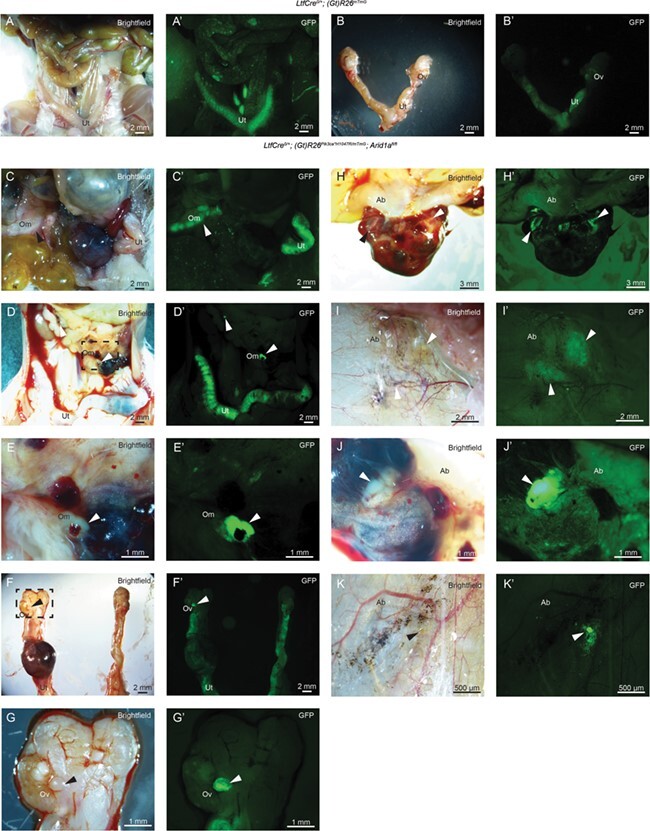
Lineage tracing of ectopic endometrium with (Gt)R26mTmG allele. (A–K) Bright-field images of mouse intestines and gynecologic tract following salpingectomy. (A′–K′) Fluorescent images of the same tissues, depicting cells derived from LtfCre0/+;(Gt)R26mTmG (n = 3) or LtfCre0/+; (Gt)R26Pik3ca*H1047R/mTmG; Arid1afl/fl (n = 4) mice. In control mice, GFP expression was confined to the uterus (A–B). In LtfCre0/+; (Gt)R26Pik3ca*H1047R/mTmG; Arid1afl/fl mice, lesions were observed on the omentum (C–E), the ovary (F–G) and the peritoneum (H–K). Ab: abdominal wall; Om: omentum; Ov: ovary; Ut: uterus.
Endometriotic lesions display novel biomarkers
Endometrial epithelium was identified within endometriotic lesions of the omentum (Fig. 4A and B). Endometriotic tissue included both endometrial epithelium as well as fibrotic stroma (Fig. 4A and B). In one case, omental tissue was fused to the kidney through endometriotic anastomosis (Fig. 4A). These endometrial glands were negative for ARID1A staining by IHC (Fig. 4C) and positive for KRT8 (Fig. 4D) and P-S6 (Fig. 4E).
Figure 4.
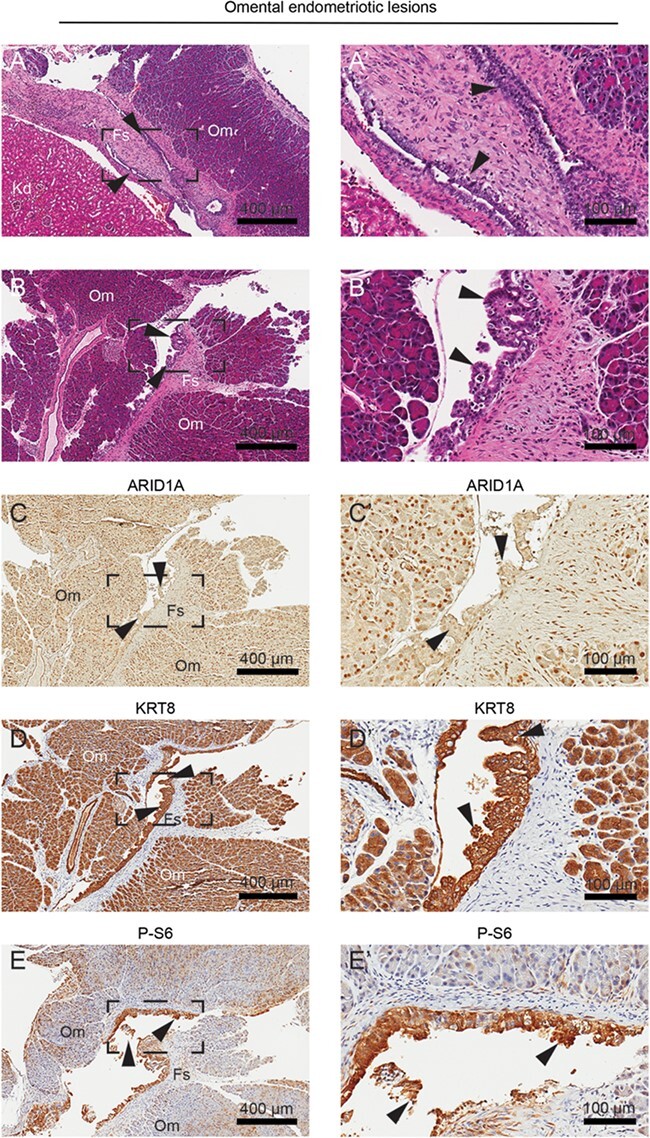
Histological characterization of omental endometriotic lesions. (A–B) H&E staining of lesions within the omentum. Endometrial glands were identified between the omentum and kidney (A) and between omental folds (B). (C–E) IHC staining for ARID1A (C), KRT8 (D) and P-S6 (E). Panels A–E represent near-adjacent sections of omental endometriotic lesions, while panels A′–E′ represent portion of slide in panels A–E surrounded by black box. Arrows indicate endometrial glands. Fs: fibrotic stroma; Om: omentum.
Within endometriotic lesions bound to the abdominal muscle in the peritoneum, we observed endometrial gland formation between the muscle and serosa layers (Fig. 5A). Glands were negative for ARID1A (Fig. 5B) and positive for KRT8 (Fig. 5C), P-S6 (Fig. 5D) and Ki67, a marker of proliferation (Fig. 5E). Previously, we identified ICAM-1 as a marker of mutated endometrium in the LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl model, which we again observed in the peritoneal endometriotic epithelium (Fig. 5F). Furthermore, we identified expression of AP-1 (activator protein 1) subunits c-Jun and Fra2 (FOSL2) among endometriotic epithelia. Previous reports have indicated a role for AP-1 subunits in the regulation of gene transcription changes upon ARID1A loss (Kelso et al., 2017; Vierbuchen et al., 2017). AP-1 subunit expression has been observed in endometriotic lesions (Hastings et al., 2006). Here, we identified expression of c-Jun and Fra2 in the glandular epithelium of endometriotic lesions (Fig. 5G and H).
Figure 5.
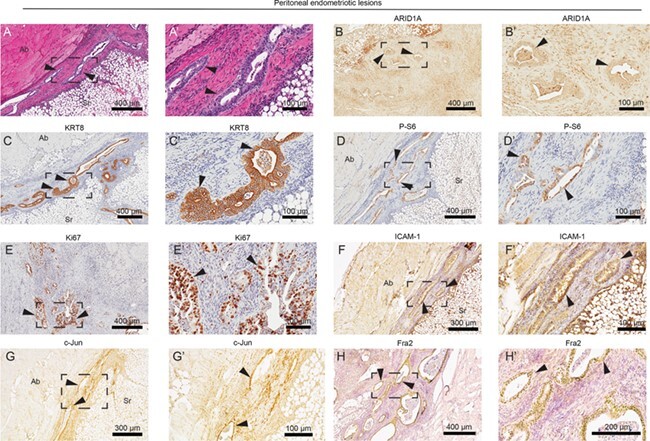
Biomarker identification in peritoneal endometriotic lesions. (A) H&E staining of endometrial glands identified in the peritoneum. Endometriotic lesions were fused to the abdominal wall. (B–H) Immunohistochemical staining of peritoneal lesions for ARID1A (B), KRT8 (C), P-S6 (D), Ki67 (E), ICAM-1 (intercellular adhesion molecule 1) (F) and AP-1 (activator protein 1) subunits c-Jun (G) and Fra2 (Fos-related antigen 2) (H). Panels A–H represent near-adjacent sections of peritoneal endometriotic lesions, while panels A′–H′ represent portion of slide in panels A–H surrounded by black box. Arrows indicate endometrial glands. Ab: abdominal wall; Sr: serosa.
Among lesions on the ovary, we observed endometrial glands and associated fibrotic stroma within the ovarian fat (Fig. 6A and B), often with hemorrhagic blood cells (Fig. 6B). Within ovarian lesions, we identified the similar biomarkers within the endometrial glands (Fig. 6). Epithelia were positively stained for KRT8 (Fig. 6C), P-S6 (Fig. 6D), Ki67 (Fig. 6E) and ICAM-1 (Fig. 6F). AP-1 subunit expression was detected in these epithelial glands, including c-Jun (Fig. 6G) and Fra2 (Fig. 6H). These data suggest that LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl endometrial cells are capable of metastasis to the omentum, peritoneum and ovaries and subsequent endometriotic lesion formation following salpingectomy.
Figure 6.
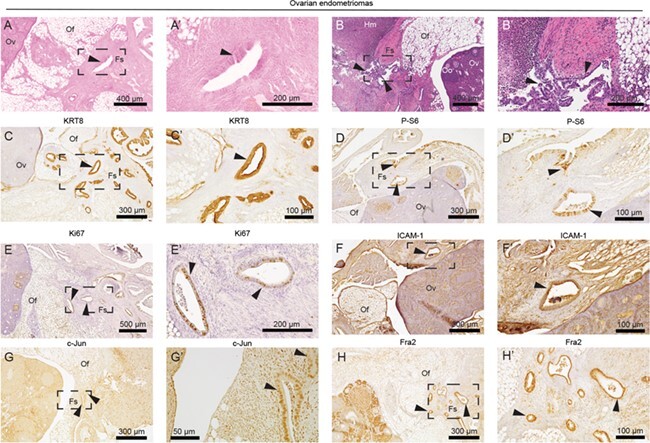
Endometrial glands formed on ovary display ICAM-1 and AP-1 subunit expression. (A–B) H&E staining of endometrial glands identified in the peritoneum. Endometriotic lesions were fused to the abdominal wall. (C–H) Immunohistochemical staining of peritoneal lesions for KRT8 (C), P-S6 (D), Ki67 (E), ICAM-1 (F) and AP-1 subunits c-Jun (G) and Fra2 (H). Panels A–H represent near-adjacent sections of ovarian endometriomas, while panels A′–H′ represent portion of slide in panels A–H surrounded by black box. Arrows indicate endometrial glands. Fs: fibrotic stroma; Hm: hemorrhage; Of: ovarian fat; Oo: oocyte; Ov: ovary.
Discussion
A long-standing hypothesis suggests that retrograde menstruation and the regurgitation of endometrium tissue into the peritoneal cavity may play a role in the development of endometriosis (Sampson, 1927). Previously, we characterized the invasive phenotype of LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl endometrium, which resulted in myometrial invasion and adenomyosis locally within the uterus. Here, we detail a new model of endometriotic spread based on our GEMM of endometrial dysfunction. ARID1A has previously been implicated as a repressor of invasion (Yan et al., 2014; Lakshminarasimhan et al., 2017; Li et al., 2017; Sun et al., 2017; Wilson et al., 2019). The present study provides in vivo mechanistic evidence suggesting that metastatic spread of endometriotic tissue to the ovaries, omentum and peritoneum occurs as a result of invasive endometrial phenotypes. ARID1A and PIK3CA are commonly mutated in the endometrium, and mutation of the endometrium can result in a number of invasive phenotypes, including endometriosis, adenomyosis, endometrial cancer and ovarian cancer. Adenomyosis is the result of direct invasion into the myometrium, while endometriosis forms following invasion distally into the peritoneum or ovary. The collective migration of mutant endometrial cells undergoing partial EMT may enhance the invasive properties of ectopic endometriotic tissue, permitting invasion of the surrounding tissue.
The model presented herein mimics the natural spread of endometriotic tissue via retrograde translocation. While mice do not menstruate, our data support the idea that anatomic barriers obstructing the retrograde spread of invasive endometrial tissue prevent the disease. In our mouse model, removal of the oviducts and bursal membrane promoted the spread of invasive endometrial cells harboring ARID1A and PIK3CA mutations from the ipsilateral uterine horn. Established mouse models of endometriosis have been limited by the requirement of tissue transplantation, steroid hormone treatment or genetic perturbation in tissues other than the endometrial epithelium (King et al., 2016). The present model recapitulates the retrograde traversal of tissue, while mimicking the heterogeneous phenotypes presented in endometriosis patients. This model may provide utility in studying the mechanisms by which endometriosis spreads to particular tissues. However, the present model has its own limits. Due to uterine endometrial dysfunction and subsequent vaginal bleeding (Wilson et al., 2019), LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl mice have a short lifespan, and disease progression cannot be tracked in the long term. Furthermore, LtfCre0/+; (Gt)R26Pik3ca*H1047R; Arid1afl/fl display a phenotype which is 100% penetrant throughout the endometrium and, as such, all endometrial epithelia harbor ARID1A and PIK3CA mutations, which is not the case in patients with endometriosis. Even still, many endometriotic lesions harbor mutations other than ARID1A or PIK3CA, suggesting that other genetic mutations may suffice for the invasive phenotypes observed in endometriosis.
Given the widespread acceptance of the implantation theory, it has been suggested that an inappropriate differentiation of the endometrium may be the direct cause of endometriosis (Grummer et al., 2001). Loss of ARID1A in the mouse endometrium results in infertility (Kim et al., 2015) and EMT (Wilson et al., 2019), and the present study provides evidence for endometriotic lesion formation as a result of invasive ARID1A-PIK3CA mutant endometrium. While endometrial cancer and endometriosis have several gene mutations in common, including ARID1A, PIK3CA and PTEN, epidemiologic studies of endometrial cancer risk among women with endometriosis have yielded mixed results (Mogensen et al., 2016; Poole et al., 2017). PIK3CA mutations exist in normal endometrial epithelial cells without transformation, and PIK3CA mutation is thought to be an early event in both endometriosis and atypical endometrial hyperplasia (Berg et al., 2015; Suda et al., 2018; Lac et al., 2019). LtfCre0/+; (Gt)R26Pik3ca*H1047R mice display no phenotype, while LtfCre0/+; Arid1afl/fl epithelium undergo apoptosis (Wilson et al., 2019). ARID1A mutations are found in the epithelial compartment of ectopic endometriosis, but not in normal endometrium (Suda et al., 2018; Lac et al., 2019).
Relative to the present model, mutations which contribute to endometriosis are likely acquired more gradually, such that endometrium may harbor a single mutation in PIK3CA, while endometriotic lesions gain additional mutations acquired during or subsequent to its spreading to the peritoneum. Based on our findings, we predict that ARID1A loss occurs subsequent to PIK3CA mutation. The addition of ARID1A mutation in a cell harboring PIK3CA mutation results in an invasive phenotype capable of endometriotic spread. While retrograde menstruation occurs in most women (Halme et al., 1984), most women do not develop endometriosis. The acquisition of invasive phenotypes in the endometrial epithelium is necessary but not sufficient for the development of endometriosis, as retrograde menstruation, hematogenous spread, lymphatic spread or other factors are required for the transplantation of tissue into the peritoneal cavity.
Acknowledgements
We thank Drs John Risinger, Jose Teixeira and Kathy Cho for the helpful discussions. We thank the Van Andel Research Institute Histology and Pathology Core for the histology services.
Authors’ roles
Conceptualization, M.R.W. and R.L.C.; investigation, M.R.W. and J.H.; methodology, M.R.W. and R.L.C.; formal analysis, M.R.W.; writing—original draft, M.R.W.; writing—review & editing, M.R.W. and R.L.C.; funding acquisition, M.R.W. and R.L.C.; supervision, R.L.C.
Funding
American Cancer Society Postdoctoral Fellowship (PF-17-163-02-DDC to M.R.W.); Innovative Translational Grant from the Mary Kay Foundation (026-16 to R.L.C.); Liz Tilberis Early Career Award from the Ovarian Cancer Research Fund Alliance (OCRFA) (457446 to R.L.C.).
Conflict of interest
The authors declare no competing interests.
References
- Adams JR, Xu K, Liu JC, Agamez NM, Loch AJ, Wong RG, Wang W, Wright KL, Lane TF, Zacksenhaus E et al. Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res 2011;71:2706–2717. [DOI] [PubMed] [Google Scholar]
- Alimi Y, Iwanaga J, Loukas M, Tubbs RS. The clinical anatomy of endometriosis: a review. Cureus 2018;10:e3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noe M, Horlings HM, Lum A, Jones S, Senz J, Seckin T et al. Cancer-associated mutations in endometriosis without cancer. N Engl J Med 2017;376:1835–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki D, Katsuki Y, Shimizu A, Kakinuma C, Nozawa S. Successful heterotransplantation of human endometrium in SCID mice. Obstet Gynecol 1994;83:220–228. [PubMed] [Google Scholar]
- Bartley J, Julicher A, Hotz B, Mechsner S, Hotz H. Epithelial to mesenchymal transition (EMT) seems to be regulated differently in endometriosis and the endometrium. Arch Gynecol Obstet 2014;289:871–881. [DOI] [PubMed] [Google Scholar]
- Berg A, Hoivik EA, Mjos S, Holst F, Werner HM, Tangen IL, Taylor-Weiner A, Gibson WJ, Kusonmano K, Wik E et al. Molecular profiling of endometrial carcinoma precursor, primary and metastatic lesions suggests different targets for treatment in obese compared to non-obese patients. Oncotarget 2015;6:1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergqvist A, Jeppsson S, Kullander S, Ljungberg O. Human uterine endometrium and endometriotic tissue transplanted into nude mice. Morphologic effects of various steroid hormones. Am J Pathol 1985;121:337–341. [PMC free article] [PubMed] [Google Scholar]
- Budiu RA, Diaconu I, Chrissluis R, Dricu A, Edwards RP, Vlad AM. A conditional mouse model for human MUC1-positive endometriosis shows the presence of anti-MUC1 antibodies and Foxp3+ regulatory T cells. Dis Model Mech 2009;2:593–603. [DOI] [PubMed] [Google Scholar]
- Burney RO, Giudice LC. Pathogenesis and pathophysiology of endometriosis. Fertil Steril 2012;98:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RL, Brennan J, Schisler JC, Serber D, Patterson C, Magnuson T. ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol Cell Biol 2013;33:265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RL, Damrauer JS, Raab JR, Schisler JC, Wilkerson MD, Didion JP, Starmer J, Serber D, Yee D, Xiong J et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat Commun 2015;6:6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CW, Licence D, Cook E, Luo F, Arends MJ, Smith SK, Print CG, Charnock-Jones DS. Activation of mutated K-ras in donor endometrial epithelium and stroma promotes lesion growth in an intact immunocompetent murine model of endometriosis. J Pathol 2011;224:261–269. [DOI] [PubMed] [Google Scholar]
- Chui MH, Wang TL, Shih IM. Endometriosis: benign, malignant, or something in between? Oncotarget 2017;8:78263–78264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings AM, Metcalf JL. Induction of endometriosis in mice: a new model sensitive to estrogen. Reprod Toxicol 1995;9:233–238. [DOI] [PubMed] [Google Scholar]
- Daikoku T, Ogawa Y, Terakawa J, Ogawa A, DeFalco T, Dey SK. Lactoferrin-iCre: a new mouse line to study uterine epithelial gene function. Endocrinology 2014;155:2718–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med 2005;11:63–70. [DOI] [PubMed] [Google Scholar]
- Giudice LC, Kao LC. Endometriosis. Lancet 2004;364:1789–1799. [DOI] [PubMed] [Google Scholar]
- Greaves E, Cousins FL, Murray A, Esnal-Zufiaurre A, Fassbender A, Horne AW, Saunders PT. A novel mouse model of endometriosis mimics human phenotype and reveals insights into the inflammatory contribution of shed endometrium. Am J Pathol 2014;184:1930–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grummer R, Schwarzer F, Bainczyk K, Hess-Stumpp H, Regidor PA, Schindler AE, Winterhager E. Peritoneal endometriosis: validation of an in-vivo model. Hum Reprod 2001;16:1736–1743. [DOI] [PubMed] [Google Scholar]
- Halme J, Hammond MG, Hulka JF, Raj SG, Talbert LM. Retrograde menstruation in healthy women and in patients with endometriosis. Obstet Gynecol 1984;64:151–154. [PubMed] [Google Scholar]
- Hastings JM, Jackson KS, Mavrogianis PA, Fazleabas AT. The estrogen early response gene FOS is altered in a baboon model of endometriosis. Biol Reprod 2006;75:176–182. [DOI] [PubMed] [Google Scholar]
- Hirata T, Osuga Y, Yoshino O, Hirota Y, Harada M, Takemura Y, Morimoto C, Koga K, Yano T, Tsutsumi O et al. Development of an experimental model of endometriosis using mice that ubiquitously express green fluorescent protein. Hum Reprod 2005;20:2092–2096. [DOI] [PubMed] [Google Scholar]
- Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, Hargreaves DC. Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Yoo JY, Wang Z, Lydon JP, Khatri S, Hawkins SM, Leach RE, Fazleabas AT, Young SL, Lessey BA et al. ARID1A is essential for endometrial function during early pregnancy. PLoS Genet 2015;11:e1005537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CM, Barbara C, Prentice A, Brenton JD, Charnock-Jones DS. Models of endometriosis and their utility in studying progression to ovarian clear cell carcinoma. J Pathol 2016;238:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lac V, Nazeran TM, Tessier-Cloutier B, Aguirre-Hernandez R, Albert A, Lum A, Khattra J, Praetorius T, Mason M, Chiu D et al. Oncogenic mutations in histologically normal endometrium: the new normal? J Pathol 2019. [DOI] [PubMed] [Google Scholar]
- Lakshminarasimhan R, Andreu-Vieyra C, Lawrenson K, Duymich CE, Gayther SA, Liang G, Jones PA. Down-regulation of ARID1A is sufficient to initiate neoplastic transformation along with epigenetic reprogramming in non-tumorigenic endometriotic cells. Cancer Lett 2017;401:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Xu ZL, Zhao Z, An Q, Wang L, Yu Y, Piao DX. ARID1A gene knockdown promotes neuroblastoma migration and invasion. Neoplasma 2017;64:367–376. [DOI] [PubMed] [Google Scholar]
- Matsuzaki S, Darcha C. Epithelial to mesenchymal transition-like and mesenchymal to epithelial transition-like processes might be involved in the pathogenesis of pelvic endometriosis. Hum Reprod 2012;27:712–721. [DOI] [PubMed] [Google Scholar]
- Mogensen JB, Kjaer SK, Mellemkjaer L, Jensen A. Endometriosis and risks for ovarian, endometrial and breast cancers: a nationwide cohort study. Gynecol Oncol 2016;143:87–92. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- Poole EM, Lin WT, Kvaskoff M, De Vivo I, Terry KL, Missmer SA. Endometriosis and risk of ovarian and endometrial cancers in a large prospective cohort of U.S. nurses. Cancer Causes Control 2017;28:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendi MH, Muehlenbachs A, Garcia RL, Boyd KL. Female Reproductive System. In: Comparative Anatomy and Histology. Elsevier, 2012,253–284 [Google Scholar]
- Rossi G, Somigliana E, Moschetta M, Santorsola R, Cozzolino S, Filardo P, Salmaso A, Zingrillo B. Dynamic aspects of endometriosis in a mouse model through analysis of implantation and progression. Arch Gynecol Obstet 2000;263:102–107. [DOI] [PubMed] [Google Scholar]
- Samartzis EP, Samartzis N, Noske A, Fedier A, Caduff R, Dedes KJ, Fink D, Imesch P. Loss of ARID1A/BAF250a-expression in endometriosis: a biomarker for risk of carcinogenic transformation? Mod Pathol 2012;25:885–892. [DOI] [PubMed] [Google Scholar]
- Sampson JA. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol 1927;14:422–469. [Google Scholar]
- Suda K, Nakaoka H, Yoshihara K, Ishiguro T, Tamura R, Mori Y, Yamawaki K, Adachi S, Takahashi T, Kase H et al. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Rep 2018;24:1777–1789. [DOI] [PubMed] [Google Scholar]
- Sun X, Wang SC, Wei Y, Luo X, Jia Y, Li L, Gopal P, Zhu M, Nassour I, Chuang JC et al. Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell 2017;32:574–589 e576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, Greenberg ME. AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol Cell 2017;68:1067–1082 e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand KC, Lee AF, Al-Agha OM, Chow C, Kalloger SE, Scott DW, Steidl C, Wiseman SM, Gascoyne RD, Gilks B et al. Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J Pathol 2011;224:328–333. [DOI] [PubMed] [Google Scholar]
- Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, Adams M, Johnson B, Su RW, Joshi NR et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat Commun 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang YH, Fan HZ, Sun YH, Yang PY, Liu F. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis 2014;35:867–876. [DOI] [PubMed] [Google Scholar]
- Zamah NM, Dodson MG, Stephens LC, Buttram VCJ, Besch PK, Kaufman RH. Transplantation of normal and ectopic human endometrial tissue into athymic nude mice. Am J Obstet Gynecol 1984;149:591–597. [DOI] [PubMed] [Google Scholar]