

Abstract
Hepatocellular carcinoma (HCC) is the leading cause of cancer-related deaths. Previous studies have suggested that mu-opioid receptor (MOR), a member of the opioid receptor family, is involved in the pathogenesis of HCC. However, the mechanism by which MOR regulates the biological behavior of HCC is still poorly understood. To address this problem, in this study, we investigated the role of MOR in the proliferation of HCC cell lines and the underlying mechanism. First, RT-PCR, western-blot and immunohistochemistry revealed higher expression of MOR in HCC cells and tissue than in non-tumor cells or adjacent tissue, and elevated expression of MOR was associated with jeopardized survival of HCC patients, as demonstrated by bioinformatic databases. Knockdown of MOR by specific siRNA attenuated the proliferation and migration of HCC cells and this effect could be reversed by rescue experiments, confirming the essential role of MOR in the proliferation of HCC. Moreover, results of colony formation assay, CCK8 test, flow cytometry and western blot suggested that a monoclonal antibody (mAb) specifically against MOR could inhibit proliferation of HepG2 and Huh7 cells via the MOR-CD147-p53-MAPK pathway, and the interaction between MOR and CD147 was verified by immunofluorescence colocalization and co-IP analysis. The mAb against MOR also enhanced the cisplatin-induced apoptosis of HCC cells by downregulating p-ERK, Bcl-2 and upregulating Bax. Taken together, these results suggest that MOR could regulate the proliferation of HCC cells in a CD147-p53-MAPK dependent manner. MOR possesses the potential to be a therapeutic target to treat HCC.
Keywords: Hepatocellular carcinoma, mu-opioid receptor, CD147, proliferation, monoclonal antibody
Introduction
Hepatocellular carcinoma (HCC) is among the leading causes of cancer-associated mortality globally [1], and it accounts for over 80% of primary liver cancers. With rapid progression, easy metastasis, and frequent recurrence being its major characteristics [2], HCC causes substantial patient morbidity and mortality. In 2018, there were approximately 841,080 new cases of liver cancer, and the number of deaths worldwide was 781,631, which was among the top four causes of death from cancer, only after lung, colorectal and gastric cancer. Treatment strategies for HCC include surgical resection, liver transplantation, percutaneous local ablation and transarterial embolization and radiotherapy [3]. However, few effective treatment options are available for advanced and metastatic HCC, and the median survival duration is not satisfactory, for example, 33 months in North America [2]. Therefore, it is urgent to explore new therapeutic strategies and targets to improve the prognosis of HCC.
The opioid system is mainly comprised of three homologous G protein-coupled receptors, that is, the mu-opioid receptor (MOR), the delta-opioid receptor (DOR) and the kappa opioid receptor (KOR) [4,5]. The opioid system, especially the MOR, is responsible for the nociceptive effects of morphine, a well-known abused analgesic and addictive drug [6,7]. In addition to its roles in analgesics and addiction, MOR is also widely involved in the pathogenesis of a variety of tumors, such as breast cancer, lung cancer, and neuroblastoma [8-10]. The latest studies demonstrated that MOR is also involved in the pathogenesis of HCC. MOR was reported to promote the proliferation, adhesion, migration and tumorigenesis of HCC and was responsible for the regulation of the self-renewal of HCC stem cells [11]. However, the mechanism by which MOR regulates the progression of HCC is still poorly understood.
Two major characteristics of malignant tumors are invasion and metastasis. Numerous mechanisms and factors have been demonstrated to be involved in the invasion, metastasis and colonization of cancer cells to target tissue, such as changes in cell-cell junctions and key signaling pathways, including E-cadherin, EGFR, VEGF, uPA, PI3K/AKT, FAK, ZEB-1 and TGF-β, together with AP-1 and suppression of MMP activity as well as other cancer hallmarks [12-22]. In our previous work, we found that HAb18G/CD147 functions in the progression of hepatocellular carcinoma. High levels of CD147 effectively promoted the invasion and metastasis of HCC by degrading the basement membrane by MMPs (matrix metalloproteinases) [23]. In addition, HAb18 mAb, the HAb18G/CD147-specific antibody, could bind to CD147 on HCC cells with high affinity, and LICARTIN, a HAb18 mAb-based commercial drug, has been approved for clinical use in primary HCC patients since 2007 [24]. These findings indicate that CD147 has a crucial role in the progression of HCC and is an important therapeutic target for HCC. Therefore, identifying novel factors regulating CD147 is vital to understand the underlying biological processes and identify possible new therapeutic strategies for the management of HCC progression.
In this study, we explored the role of MOR in the proliferation of HCC cells and found that its expression is closely correlated with poor outcomes in HCC patients. Our preliminary results revealed that overexpressed MOR in HCC cells promoted the proliferation, migration and invasion of HCC cells. Blocking MOR with 3A5C7, a monoclonal antibody against human MOR, inhibited the proliferation of HCC cells by regulating the MOR-CD147-MAPK signaling axis in a p53-dependent manner. Moreover, MOR mAb enhanced cisplatin-induced apoptosis of HCC cells. Therefore, overexpressed MOR in HCC might be a therapeutic target for the proliferation of hepatocellular cells, and the corresponding monoclonal antibody has the potential to be a new effective treatment for HCC.
Materials and methods
Cell lines and human tissue specimens
The L02, Hep3B, SNU368, HepG2, and Huh7 cell lines were purchased from the American Type Culture Collection (ATCC, USA). All cells were cultured in RPMI 1640 media supplemented with 10% fetal bovine serum (FBS), 100 units/ml of penicillin, and 100 ug/ml of streptomycin at 37°C with 5% CO2. HCC human tissue specimens were collected from the Department of Pathology, Xijing Hospital, from 2008 to 2012 and were histologically confirmed by hematoxylin and eosin staining. This study was approved by the ethics committee of Xijing Hospital (Approval Number: KY20194064). Written informed consent was provided by all patients.
Real-time PCR
Real-time PCR was conducted as previously described [11]. Total RNA of the cell lines was extracted with an RNA isolation kit (OMEGA, USA) and were reverse transcribed into cDNA with a PrimeScript RT reagent kit (TAKARA Bio, Japan). An ABI 7200 analyzer (Applied Biosystems, USA) was used for the real-time PCR analysis of cDNA. SYBR Green I (TAKARA Bio, Japan) was used as the fluorescent probe while GAPDH as internal control. All primers were synthesized by BGI (Beijing, China) as follows: MOR, forward primer 5’-CATGCCATTCCGACCTTC-3’; reverse primer 5’-AGGCACTTTCCTAGAGAATTA-3’. GAPDH, forward primer 5’-AGAAGGCTGGGGCTCATTTG-3’; reverse primer 5’-AGGGGCCATCCACAGTCTTC-3’. p53, forward primer 5’-CATCTACAAGCAGTCACAGCACA-3’; reverse primer 5’-GGTCTCTCCCAGGACAGGC-3’. Amplification conditions were as follows: one cycle of 94°C for 4 min, 94°C 20 s, 58°C 20 s, and 72°C 20 s for additional 40 cycles on miniOpticon system (Bio-Rad, USA). The relative mRNA expression of target gene was normalized to GAPDH. Each experiment was repeated at least three times independently.
RNA interference
si-MOR (5’-GCUAUGGACUGAUGAUCUUTTtt-3’, 3’-dTd-TTCGAUACCUGACUACUAGAA-5’), si-p53 (5’-GGGUUAGUUUACAAUCAGCtt-3’, 3’-dTd-CCCAAUCAAAUGUUAGUCG-5’), and negative control siRNA, si-NC (5’-UUCUCCGAACGUGUCACGUTTtt-3’, 3’-dTd-TTAAGAGGCUUGCACUGAGCA-5’) were synthesized by Sangon Biotech, Shanghai. SiRNA transfection was performed as previously described [23]. HCC cells were transfected with siRNA using Lipofectamine 2000TM reagent according to the manufacturer’s instructions (Invitrogen, USA). Total RNA and whole cell proteins were extracted 48 hrs after transfection to determine the efficiency of interference by real-time PCR and western blot, respectively.
Plasmid construction for MOR, ERK and CD147
The expression vectors pCI-neo-MOR, pcDNA3.1-ERK and pcDNA3.1-CD147 used for the overexpression of MOR, ERK and CD147 were synthesized by GenScript (Nanjing, China). The cDNA of MOR (NM_000914.4), ERK (NM_002745.5) and CD147 (NM_001728.4) was also optimized by OptimumGene TM Codon Analysis of GenScript to insert into EcoR I/Not I of the backbone vector pCI-neo and pcDNA3.1 accordingly. The expression of MOR, ERK and CD147 was analyzed by western blot after plasmids were transfected into cells for 48 hrs.
Transfection
HCC cells were seeded in 6-well plates and grown about 85% confluence before transfection. All plasmids or siRNAs were transfected with a Lipofectamine 2000TM transfection kit (Invitrogen, USA) according to the manufacture’s instructions.
Western blot and antibodies
Western blot analysis was performed according to previous report [10]. Total cell proteins were extracted with RIPA buffer (Sigma-Aldrich, USA) supplemented with protease inhibitor and phosphorylation inhibitor cocktail. Each sample of 30 μg protein was run on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Total proteins were transferred to polyvinylidene fluoride membrane (PVDF, Hybond, UK) and membranes were incubated with primary antibodies after being blocked with 5% non-fat milk in TBS with 0.1% Tween 20 for 1 hr at room temperature. Primary antibodies include anti-MOR (Abcam, ab17934), anti-p53 (Proteintech, 60283-2-Ig), anti-p21 (Proteintech, 10355-1-AP), anti-Cyclin D1 (Proteintech, 60186-1-Ig), anti-H-Ras (Proteintech, 18295-1-Ig), anti-ERK (HUABio, ET1061-29), anti-pERK (CST, 4370s), anti-Bax (Poteintech, 60267-1), anti-Bcl-2 (HUABio, ER0602), anti-CD147 (prepared by National Translational Science Center for Molecular Medicine, Xi’an, China), anti-GAPDH (Proteintech, 60004-1-Ig), and anti-α-tubulin (made by our laboratory according to manufacturer’s instructions). Horseradish peroxidase-conjugated (HRP) secondary antibodies (Invitrogen, A28177SMPLE, USA) and enhanced chemiluminescence reagents (Pierce Biotechnology, USA) were applied to visualize interested proteins. GAPDH or α-tubulin was used as an internal control.
Cell proliferation assay
Cell Counting Kit-8 (CCK-8) assays were used to determine the proliferation capacity of HCC cells. After incubation with antibody or IgG control for 24 hrs, HCC cells were re-seeded into 96-well plates with 4×103 cells/well and cultured for 24, 48 and 72 hrs, respectively. Cell viability was determined using a CCK-8 kit (ImmunoWay Biotechnology Co., TX, USA) with optical OD450 nm. We also used colony formation assays to assess the tumorigenicity of HCC cells. The treated cells were seeded into 6-well plates with 2×103 cells/well and incubated at 37°C with 5% CO2 for two weeks. When turning into visible, the colonies were fixed in methanol and calculated after staining with 0.2% crystal violet. Each experiment was repeated three times independently.
Transwell assay
To investigate the invasion and migration capacities of HCC cells, transwell assays were performed using transwell chambers (Costar, USA) as previously described [23]. Chambers were pre-coated with Matrigel to assess invasion ability, while chambers without Matrigel were used to examine migration ability. 4×105 of HCC cells suspended in serum-free media were added to the upper transwell chambers (8 μm, Corning Inc., USA). RPMI 1640 with 20% FBS was applied to the bottom chambers. After incubation for 48 hrs at 37°C with 5% CO2, the HCC cells that invaded or migrated into the lower membrane surface were fixed in methanol for about half an hour. The cells were counted after staining with 0.2% crystal violet staining solution in three randomly selected fields.
Wound healing assay
Wound healing assays were performed as previously described [25]. Briefly, 8×105 cells/well were seeded into a 6-well plate and were incubated up to 90% confluence. The monolayer cells were scratched with a tip and cultured in serum-free media, then wound area images were taken at 0 hr and 48 hrs after scratching. The migration area was calculated as (A0 - An)/A0 ×100%. A0 represents the initial wound area, An represents the remaining area at the time of photographing.
Cell cycle analysis
Propidium iodide (PI) staining was adopted to analyze cell cycle as previously described [25]. 1×106 cells were collected from 6-well plate and washed twice with 0.01 M, pH 7.2 PBS after being fixed with 70% (w/w) ethanol for 12 hrs. Then the cells were incubated with 50 μM PI containing 5 μg/ml RNase A at 4°C for 30 min. The stained cells were immediately analyzed by flow cytometry (FACs) (Calibur, Beckman Coulter, USA) and Modfit Software (Flow J 7.6).
Cell apoptosis analysis
Cell apoptosis was detected using the fluorescein isothiocyanate annexin V-FITC and propidium iodide (PI) Apoptosis Detection Kit I (Biolegend, USA) according to previous report [25]. Briefly, 1×106 of 3A5C7-treated, mouse IgG-treated and non-treated cells were collected and washed in ice-cold 0.01 M, pH 7.2 PBS for two times, and resuspended in 100 μL of binding buffer. After incubation with 5 μL of annexin V-FITC and 3 μL of PI for 30 min at room temperature in a dark room, the cells were analyzed by Flow cytometric (Calibur, Beckman Coulter, USA).
Immunohistochemical staining (IHC)
HCC tissue specimens were subjected to immunohistochemical staining for MOR as previously described [25]. Briefly, sections were dewaxed and achieved antigen retrieval with 10 μM boiling citrate buffer at pH 6.0 for five minutes. Sections were incubated with anti-MOR antibody (ab17934, Abcam, Cambridge, UK) for 60 min at room temperature. The percentages of staining-positive cells were categorized into four classifications: 3 (67%-100%), 2 (34%-66%), 1 (1%-33%), and 0 (0%). The staining intensities were evaluated into four grades: 3 (strong), 2 (moderate), 1 (weak), and 0 (none). The staining score was defined as the product of the percentage classifications and intensity grades.
Immunofluorescence
Immunofluorescence test was performed as previously described [24]. 1×105 of Huh7 or HepG2 cells were seeded into dishes and cultured overnight. Cells were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature and washed with PBS for three times. After being blocked with 5% goat serum for 30 min at room temperature, cells were incubated with anti-MOR (Invitrogen, 44-308G) and HAb18 mAb (prepared by our laboratory) overnight at 4°C. FITC-conjugated goat anti-mouse IgG (Invitrogen, A21202) and Alexa 555-conjugated goat anti-rabbit IgG (Invitrogen, A31572) were used as secondary antibodies. 4’,6-diamidino-2-phenylindole (DAPI) was used to counterstain nucleus. Each experiment was performed three times independently. Images were obtained with an FV1000 laser scanning confocal microscope (Nikon, Japan).
Coimmunoprecipitation (Co-IP)
Coimmunoprecipitation was performed as previously described [25]. Protein extracts were incubated for 2 hrs at 4°C with IgG and protein A+G-agarose to eliminate nonspecific binding. Primary antibodies against MOR were added to cell extract tubes for incubation at 4°C overnight. The protein A/G-agarose was collected by centrifugation. Immunoprecipitated proteins were analyzed by 10% SDS-PAGE at 100 V for 1.5 hrs. Membranes were blocked with 5% nonfat milk. Primary antibodies of anti-MOR (Invitrogen, PA5-26138) and HAb18 mAb (prepared by our laboratory) diluted at 1:200 were incubated at 4°C overnight, and secondary antibodies were incubated for 1 hr at room temperature before detection with enhanced chemiluminescence (PerkinElmer Life Sciences, USA).
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 8.0 (San Diego, California, USA). The expression levels of MOR in tumor tissues and adjacent nontumor tissues were compared using unpaired t-test. Differences between gene expression were assessed with one-way ANOVA. Data are presented as mean ± SEM. The results were considered statistically significant with P < 0.05.
Results
MOR expression is upregulated in HCC and is correlated with poor outcomes
To explore the expression of MOR in human HCC tissue and cell lines, real-time PCR and western blot were performed accordingly. Real-time PCR showed that MOR mRNA exists in different HCC cell lines and is significantly higher in HepG2, Huh7, Bel7404, and Hep3B cells but not SNU368 cells or in L02 cells, a human fetal hepatocyte cell line (Figure 1A). Consistently, TCGA datasets also revealed higher mRNA expression in HCC tissue than in adjacent HCC tissue (Figure 1B). Western blot analysis showed that MOR was highly expressed in HepG2, Huh7 and Hep3B cells but expressed at low levels in the SNU368, Bel7404 and L02 cell lines (Figure 1C and 1D). In tissue specimens from HCC patients, immunochemistry (IHC) showed that MOR expression was higher in HCC tissue than in adjacent peritumor tissue (Figure 1E and 1F). These results suggest that compared with the nontumor control, MOR is upregulated in human HCC tissue and HCC cell lines of HepG2 and Huh7. Thus, the following experiments were conducted in HepG2 and Huh7 cells.
Figure 1.
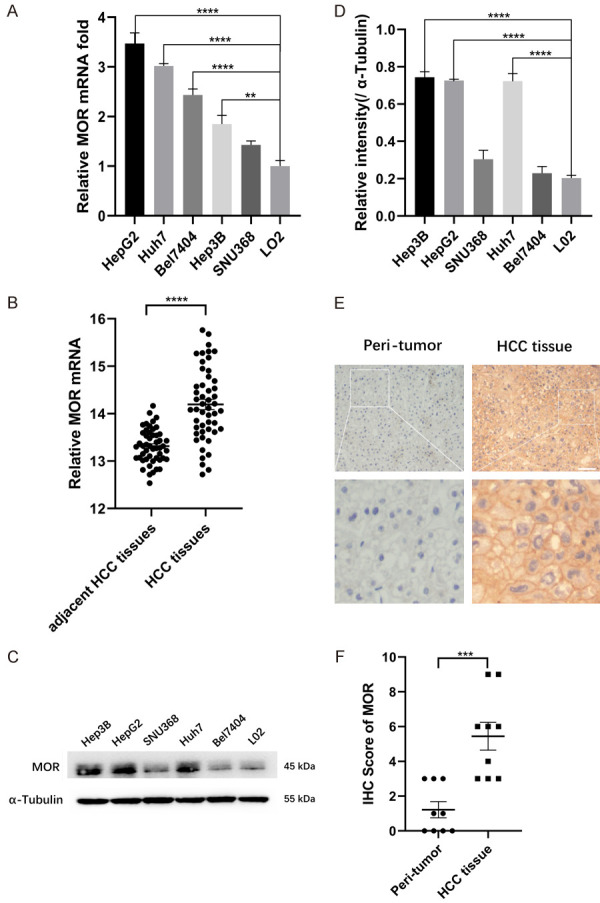
MOR is highly expressed in HCC cell lines and human tumor tissue. (A) Total RNA was isolated and subjected to real-time PCR analysis for MOR expression among the liver cell line LO2 and various HCC cancer cell lines (**P < 0.01, ****P < 0.0001). (B) MOR mRNA levels in 50 paired HCC and adjacent nontumor tissues were determined in data from TCGA datasets by t-test (****P < 0.0001). (C) Western blot analysis of MOR protein expression among different human hepatocellular carcinoma cell lines and L02 cells. See Supplementary File Raw Blot Images (Figure 1C) for original blot images. (D) Scanning densitometry analysis of western blot data in (C) (****P < 0.0001). (E) Representative immunohistochemistry images of MOR expression in 9 paired HCC tissues and adjacent nontumor tissues. The length of the scale bars was 50 μm. (F) IHC scores of each case in (E) (***P < 0.001). Data were presented as the mean ± SEM. Student’s t-test and one-way ANOVA were used for statistical analysis.
To further investigate the clinical significance of MOR in HCC, Kaplan-Meier plotter analysis was performed on the data from TCGA dataset mining. Log-rank tests of overall survival (OS) and disease-free survival (DFS) curves revealed that overexpression of MOR was significantly associated with shorter OS and DFS in HCC patients (OS, P = 0.025. DFS, P = 0.0031, Figure 2A and 2B). Moreover, the progression-free survival (PFS) curve also showed that MOR overexpression was significantly correlated with faster progression in HCC patients (P = 0.0054, Figure 2C). These results suggested that MOR had prognostic value for the overall survival of HCC patients. Hence, we determined that the upregulated MOR in HCC might contribute to the pathogenesis of HCC.
Figure 2.
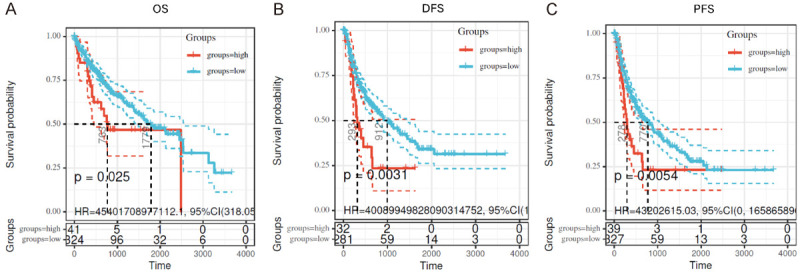
High MOR expression is related to poor survival and faster progression of HCC. (A-C) The correlation between MOR expression, overall survival (A), disease-free survival (B), and progression-free survival (C) analysis was determined in data from TCGA datasets by Kaplan-Meier survival analysis.
Silencing MOR attenuates the proliferation and migration of HCC cells
To unveil the biological role of MOR in HCC, RNA interference was adopted to knock down the expression of MOR in HepG2 and Huh7 cells. After treatment with MOR-specific small interfering RNA (si-MOR), real-time quantitative PCR showed that si-MOR could effectively decrease the mRNA expression of MOR compared with that of silencer negative control siRNA (si-NC) (Figure 3A). Consistently, western blot analysis showed that the expression of MOR decreased significantly in si-MOR-treated cells compared with that in si-NC-treated cells (Figure 3B).
Figure 3.
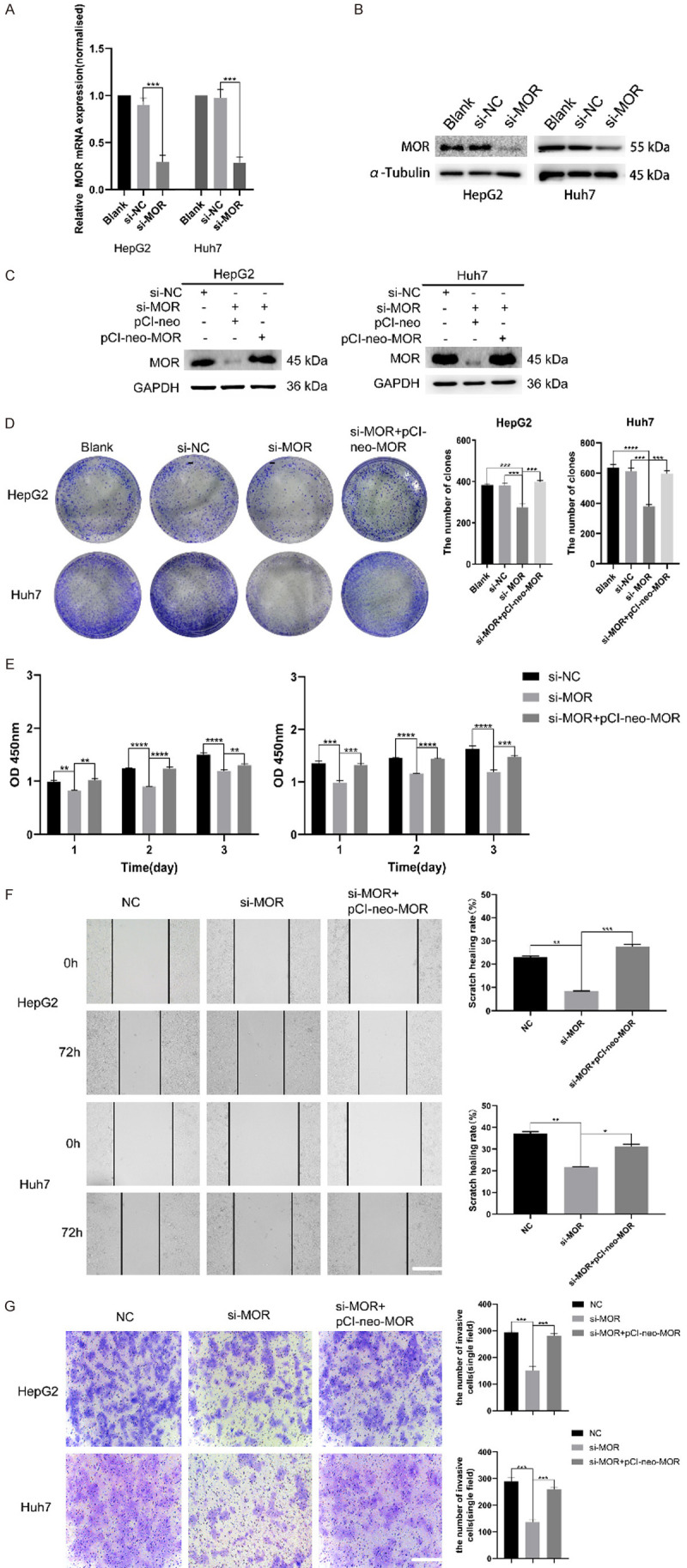
Knockdown of MOR attenuates the proliferation and migration of HCC cells. (A and B) HepG2 and Huh7 cells were transfected with control or MOR siRNA, and forty-eight hours later, total RNA and whole cell lysates were extracted and subjected to real-time PCR and western blot analysis for MOR expression, respectively. MOR was effectively silenced, as shown by the mRNA and protein levels of the two cell lines (***P < 0.001). (C) Recovered MOR expression in HepG2 and Huh7 cells after transfection with the pCI-neo-MOR plasmid was analyzed by western blot. (D) The colony formation ability of MOR-silenced HepG2 and Huh7 cells was inhibited, while overexpressing MOR using the pCI-neo-MOR plasmid reversed the inhibition effects (***P < 0.001; ****P < 0.0001). (E) The proliferation of MOR-silenced HepG2 and Huh7 cells was suppressed, and regaining MOR rescued proliferation (**P < 0.01; ***P < 0.001; ****P < 0.0001). (F) Wound healing assays showed that the migration ability of HepG2 and Huh7 cells was attenuated by MOR knockdown, and transfection with pCI-neo-MOR rescued migration (scale bars, 100 μm; *P < 0.05, **P < 0.01; ***P < 0.001). (G) Transwell assays showed that the migration ability of HepG2 and Huh7 cells was attenuated by MOR knockdown, and transfection with pCI-neo-MOR also rescued migration (scale bars, 100 μm; *P <0.05, **P < 0.01; ***P < 0.001). All experiments were repeated three times independently. See Supplementary File Raw Blot Images (Figure 3B and 3C) for original blot images. Data were presented as the mean ± SEM. One-way ANOVA methods were used for statistical analysis.
To examine the effects of MOR on HCC cells, we first performed colony formation assays to determine the proliferation of HCC cells. The results showed that the proliferation of MOR-silenced HepG2 and Huh7 cells was significantly decreased compared with that of si-NC-cells (P < 0.001) (Figure 3D). CCK8 assays yielded consistent results with those of colony formation assays (Figure 3E). Next, rescue experiments were conducted by transfecting two cell lines with the pCI-neo-MOR plasmid to overexpress MOR (Figure 3C). As expected, the decrease in cell growth induced by MOR silencing was rescued in response to MOR overexpression (Figure 3D and 3E). In vitro wound healing assays showed that the healing ability of scratch in si-MOR-transfected HepG2 and Huh7 cells was significantly decreased compared with that of si-NC-treated cells. Similarly, the decrease in scratch healing ability induced by MOR silencing was also rescued in response to MOR overexpression (Figure 3F). Transwell assay also demonstrated the decreased migration ability of si-MOR-transfected HepG2 and Huh7 cells compared with si-NC-treated cell, and it was rescued by MOR overexpression (Figure 3G). Taken together, these data suggest that MOR expression is closely involved in the proliferation and migration of HCC cells.
mAb against MOR inhibits the growth of HCC cells
To further validate the effect of MOR on the progression of HCC, a specific mouse monoclonal antibody against human MOR, named 3A5C7, was generated through the traditional hybridoma technique in our lab. After treating HepG2 and Huh7 cells with the mAb 3A5C7, colony formation assays showed that compared with the negative control, 3A5C7 mAb treatment significantly inhibited cell growth in a concentration-dependent manner, while the IgG control did not influence cell growth (Figure 4A). Consistently, CCK-8 assays showed that compared with the negative control, the 3A5C7 mAb treatment significantly inhibited cell growth in a concentration-dependent manner at 24, 48 and 72 hrs after the administration of mAb, while the IgG control did not influence cell growth (Figure 4B). The inhibitory effect of the 3A5C7 mAb on cell growth was further confirmed by cell cycle analysis. Flow cytometry analysis indicated that HepG2 and Huh7 cells were arrested at G1/S phase in response to 3A5C7 mAb treatment (Figure 4C). Moreover, wound healing and transwell assays showed that mAb 3A5C7 could also inhibit the migration and invasion of HCC cells (Supplementary Figure 1A and 1B). These results verify that anti-MOR mAb could inhibit the proliferation, migration and invasion of HCC cells.
Figure 4.

Antibody against MOR (3A5C7) inhibit the proliferation of hepatocellular carcinoma cells. (A) Colony formation assay of HCC cells subjected to normal saline, mouse IgG and different concentrations of 3A5C7 mAb (**P < 0.01; ***P < 0.001; ****P < 0.0001). (B) Proliferation ability test of HepG2 (left panel) and Huh7 (right panel) cells exposed to various concentrations of the 3A5C7 mAb (*P < 0.05; **P < 0.01; ***P < 0.001). (C) Flow cytometric assays determined the percentage of cells in different phases of the cell cycle (*P < 0.05; **P < 0.01). All experiments were done in triplicate. Data were presented as the mean ± SEM. One-way ANOVA methods were used for statistical analysis.
MOR mAb activates the p53 signaling pathway in HCC
The tumor suppressor gene p53 is a crucial regulator of DNA repair and cell cycle transition. Hence, we speculated that p53 may be involved in the function of MOR mAb in HCC. Western blot analysis showed that the MOR mAb 3A5C7 activated the p53-p21-Cyclin D1 pathway, indicating that the cell cycle was arrested in HepG2 and Huh7 cells (Figure 5A). Consistently, in p53-knockdown HepG2 and Huh7 cells, the cell cycle was not altered with 3A5C7 treatment (Figure 5C). These data suggested that the 3A5C7 mAb functioned as an HCC suppressor in a p53-dependent manner.
Figure 5.

The MOR mAb 3A5C7 regulates the proliferation of HCC by affecting p53. (A) Western blot analysis showed expression changes in MOR-, CD147- and p53/MAPK pathway-related molecules in two HepG2 and Huh7 cell lines exposed to 3A5C7. (B) Western blotting and real-time PCR were conducted to verify the knockdown of p53 (****P < 0.0001). (C) Flow cytometry analysis of the cell cycle in cells that were silenced in p53. (D) Tumor formation capability was rescued by downregulating p53 in HepG2 and Huh7 cells (*P < 0.05, **P < 0.01). (E) HepG2 and Huh7 cells transfected with si-NC or p53 siRNA were assessed for proliferation ability at 24, 48 and 72 hrs after 3A5C7 was applied by CCK8 analysis (*P < 0.05, ****P < 0.0001). Supplementary File Raw Blot Images (Figure 5A and 5B) for original blot images. Data were presented as the mean ± SEM. Student’s t-test and one-way ANOVA were used for statistical analysis.
Next, to validate whether 3A5C7 exerted anti-HCC effects via p53, rescue experiments were also performed. The p53 gene was knocked down in HepG2 and Huh7 cells by using siRNA, and the efficiency of knockdown was confirmed by real-time quantitative PCR and western blot (Figure 5B). Colony formation and CCK-8 assays showed that p53 knockdown rescued the inhibitory effect of the 3A5C7 mAb on the proliferation of HepG2 and Huh7 cells (Figure 5D and 5E).
MOR mAb represses the MAPK pathway in the presence of p53 in HCC
To further unveil the mechanisms of the inhibitory effect of the 3A5C7 mAb on HCC, the mRNA levels of several classical pathway molecules correlated with proliferation were compared between HCC cells treated with control IgG and the 3A5C7 mAb. The results from real-time PCR demonstrated that the expression of NFAT5 and its downstream target gene MAPK was distinctly suppressed in the 3A5C7-treated group compared with the IgG-treated group (Figure 6A). Western blot analysis drew an anastomotic conclusion that the MAPK pathway was significantly suppressed after the two cell lines were subjected to the 3A5C7 mAb, indicating that the MAPK pathway is essential in the function of 3A5C7 (Figure 5A). Next, rescue experiments were conducted by overexpressing ERK using the pcDNA3.1-ERK plasmid. As expected, inhibition of cell growth induced by MOR mAb was rescued in response to ERK overexpression (Figure 6B, 6C and Supplementary Figure 2). Furthermore, regulation of p-ERK by MOR required the presence of p53 (Figure 6D). These observations show that the MAPK signaling pathway played a critical role in the function of MOR in HCC.
Figure 6.
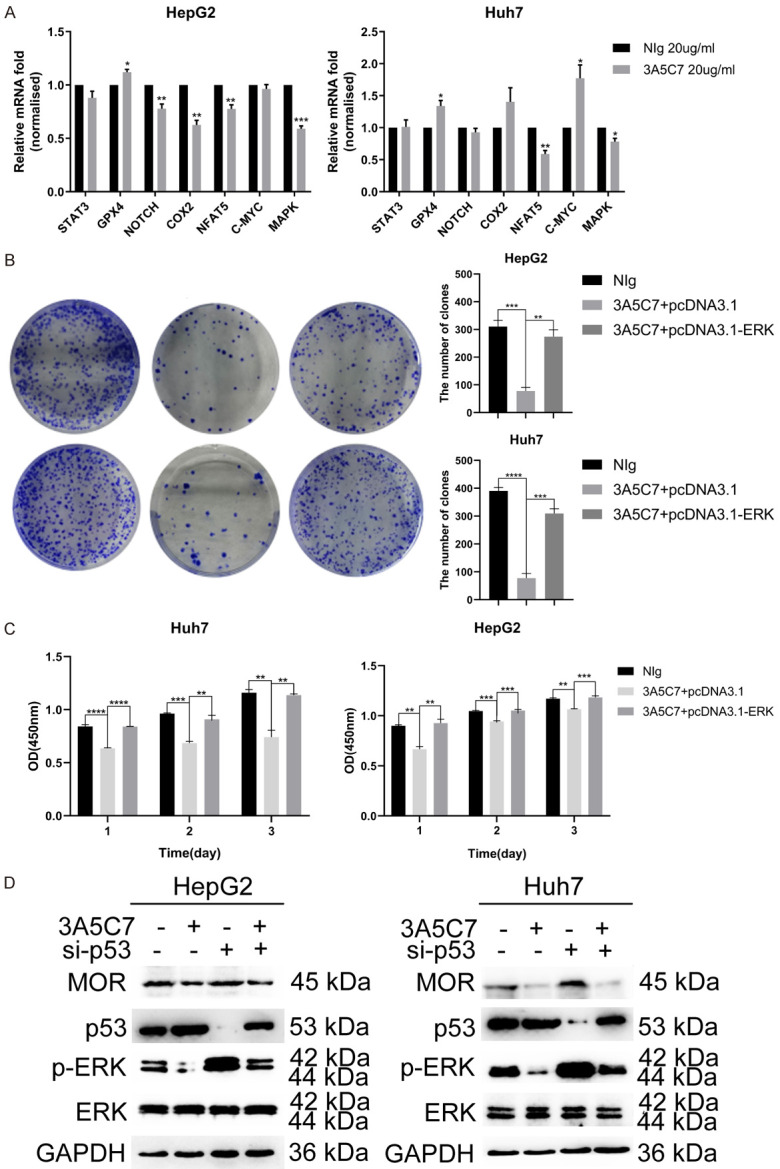
MOR mAb inhibits the proliferation of HCC cells by downregulating the MAPK pathway in the presence of p53. (A) Pathway array analysis of gene expression from HCC cells subjected to 3A5C7 antibody or mouse IgG control (*P < 0.05; **P < 0.01, ***P < 0.001). (B) Colony formation capability was rescued by overexpressing ERK in HCC cells (**P < 0.01, ***P < 0.001, ****P < 0.0001). (C) HCC cells transfected with pcDNA3.1-ERK or the control vector plasmid were subjected to CCK-8 assays (**P < 0.01, ***P < 0.001, ****P < 0.0001). (D) Western blot analysis indicated that the 3A5C7 antibody regulated the expression of p-ERK in the presence of p53. See Supplementary File Raw Blot Images (Figure 6D) for original blot images. Data were presented as the mean ± SEM. Student’s t-test and one-way ANOVA were used for statistical analysis.
MOR mAb 3A5C7 inhibits the proliferation of HCC by downregulating the CD147-p53-ERK pathway
CD147 is a tumor-associated antigen that plays a key regulatory role in HCC progression. We previously reported that CD147 inhibited p53 expression at the posttranscriptional level in HCC [24]. Hence, we speculated that CD147 might be involved in the inhibitory function of MOR mAb in HCC.
To investigate the role of CD147 in 3A5C7-related functions in HCC, we first observed the distribution of MOR and CD147 in HCC cells. In HepG2 and Huh7 cells, immunofluorescence staining showed that MOR was mainly localized on the plasma membrane, and some of them were located in the cytoplasm, but CD147 was uniformly localized on the plasma membrane. Colocalization of MOR and CD147 was detected on the plasma membrane (Figure 7A). Next, co-IP assays were performed to validate the correlation between these two proteins. As expected, the protein-protein interaction between MOR and CD147 was verified in HepG2 and Huh7 cells by co-IP assays (Figure 7B).
Figure 7.

MOR mAb suppresses the proliferation of HCC cells by downregulating CD147. (A) Immunofluorescence staining indicated the colocalization of MOR (red) and CD147 (green) together with DAPI (blue) in HepG2 and Huh7 cells. The length of scale bars was 10 μm. (B) Co-IP assays were performed to determine the interaction between MOR and CD147. (C) Western blot assay to reveal the relationship between MOR and CD147 in the presence of 3A5C7. (D) CD147 regulates the expression of p53 and p-ERK in HepG2 and Huh7 cells subjected to the 3A5C7 mAb. See Supplementary File Raw Blot Images (Figure 7B-D) for original blot images.
To identify whether CD147 plays a role in the effects of the 3A5C7 mAb on HCC, the expression of CD147 was examined after HCC cells were exposed to 3A5C7. Western blot analysis showed that the expression of CD147 was significantly decreased in 3A5C7-treated cells (Figure 5A). Moreover, to further determine whether the decrease in CD147 induced by 3A5C7 mAb was specifically dependent on MOR, rescue experiments were performed by overexpressing MOR using a pCI-neo-MOR plasmid. Restoring the expression of MOR almost reversed the reduction in CD147 (Figure 7C), indicating that the 3A5C7 mAb regulates the expression of CD147 by MOR.
Moreover, to further explore whether CD147 is a mediator of the regulatory effects of 3A5C7 on p53 and ERK in HCC cells, we overexpressed CD147 in HCC cells by using the pcDNA3.1-CD147 plasmid. Regaining of CD147 nearly reversed the upregulation of p53 and downregulation of p-ERK induced by 3A5C7 (Figure 7D). Taken together, these results suggest that the inhibitory effects of the 3A5C7 mAb on HCC were mediated by the MOR-CD147-p53-ERK pathway.
MOR mAb enhances cisplatin-induced apoptosis of HCC cells
Cisplatin, a widely used chemotherapeutic agent against HCC, faces severe problems related to drug resistance. Our data indicate that the 3A5C7 mAb can inhibit the proliferation of HCC cells, opening the possibility that the 3A5C7 mAb could be a potential therapeutic strategy to treat HCC and to enhance the efficacy of cisplatin. To test this possibility, we tested the effects of the mAb 3A5C7 combined with cisplatin on the apoptosis of HCC cells. Annexin V-FITC/PI staining showed that the apoptotic cell proportion (early apoptosis) for cisplatin plus 3A5C7 mAb was 13.6%, which was higher than that of cisplatin plus mouse IgG (5.93%) or cisplatin alone (3.11%) in HepG2 cells. Similarly, the apoptotic cell proportion (late apoptosis) for cisplatin plus 3A5C7 mAb was 25%, which was higher than that of cisplatin plus mouse IgG (14%) or cisplatin alone (12.7%) in Huh7 cells (Figure 8A). This enhancement effect was confirmed by western blot analysis, which showed greater intensity of pro-apoptotic protein (Bax) and a lower concentration of anti-apoptotic protein bands (Bcl-2) in the cisplatin plus 3A5C7 mAb group in a concentration-dependent manner (Figure 8B). Moreover, to further verify the existence of such an enhancement effect, the expression levels of drug resistance genes in HepG2 and Huh7 cells were examined by real-time PCR. The results showed that the expression of the ABCC1, ABCC2, ABCB1, and ABCG2 genes was lower in HCC cells subjected to cisplatin plus 3A5C7 mAb than in HCC cells treated with cisplatin plus control IgG (Figure 8C).
Figure 8.

MOR mAb (3A5C7) enhances cisplatin-induced apoptosis of hepatocellular carcinoma cells. (A) Cell apoptosis was evaluated by Annexin V-FITC/PI staining in HCC cells treated with 3A5C7 plus cisplatin or cisplatin alone at 48 hrs (***P < 0.001; ****P < 0.0001). (B) The levels of p-ERK, Bax, and Bcl-2 were determined by western blot in HCC cells exposed to 3A5C7 mAb plus cisplatin, control IgG plus cisplatin and cisplatin alone at 48 hrs. (C) Q-PCR was performed to evaluate changes in drug resistance genes in HepG2 and Huh7 cells exposed to cisplatin plus 3A5C7 mAb (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). (D and E) Flow cytometry assay and western blot to assess the apoptosis of HepG2 and Huh7 cells with or without MOR silencing (****P < 0.0001). (F and G) Flow cytometry assay and Western blot assay to detect the apoptosis of two cell lines with or without ERK overexpression (***P < 0.001; ****P < 0.0001). See Supplementary File Raw Blot Images (Figure 8B, 8E and 8G) for original blot images. Data were presented as the mean ± SEM. Student’s t-test and one-way ANOVA were used for statistical analysis.
Next, to ensure that mAb 3A5C7 specifically binds to MOR to cause this enhancement effect, the MORs of HepG2 and Huh7 cells were knocked down by siRNA. Flow cytometry showed that knocking down MOR by siRNA alone had no significant effects on the apoptotic rate of either HepG2 or Huh7 cells (Supplementary Figure 3). Then both MOR-silenced HCC cells and control cells were treated with cisplatin plus 3A5C7 for 24 hrs. As a consequence, MOR-silenced cells showed no enhancement of the apoptotic effect of cisplatin compared to the control cells, indicating that the enhancement of cisplatin induced by the 3A5C7 mAb was achieved specifically through MOR (Figure 8D). Conforming to this phenomenon, western blot demonstrated that there were no obvious differences in Bax or Bcl-2 levels between two groups after MOR was silenced (Figure 8E), that is to say, MOR is essential for the 3A5C7 mAb to exert its enhancement effect on cisplatin.
To investigate whether the enhancement effects of the 3A5C7 mAb on cisplatin were caused by inhibition of p-ERK (Figure 8B and 8E), HepG2 and Huh7 cells were transfected with pcDNA3.1-ERK or control plasmid for 48 hrs to overexpress ERK and cultured with 1 μg/ml cisplatin plus 3A5C7 mAb at 20 μg/ml for another 24 hrs. Flow cytometry assays showed that the number of apoptotic cells was significantly decreased in the ERK-overexpression group compared with the control plasmid-transfected group, and western blot assays also showed the upregulation of Bcl-2 protein and the downregulation of Bax protein in the pcDNA3.1-ERK-transfected group (Figure 8F and 8G). These results suggest that the mAb 3A5C7 enhanced the antitumor effect of cisplatin on HCC by regulating the expression levels of p-ERK, Bax and Bcl-2.
Discussion
In this study, we confirmed that MOR contributed to the proliferation of HCC cells and that elevated expression of MOR was associated with decreased survival of patients with HCC. In addition, by using a monoclonal antibody against MOR, we elucidated that MOR mediated the proliferation of HCC cells in a CD147-p53-MAPK-dependent manner (Figure 9). MOR could be a potential therapeutic target for HCC, in that mAb combined with cisplatin, the conventional antitumor agent, could enhance the capability of cisplatin to induce apoptosis in HCC cells. These results suggest that MOR plays important regulatory roles in HCC proliferation and could be a new target for HCC treatment.
Figure 9.
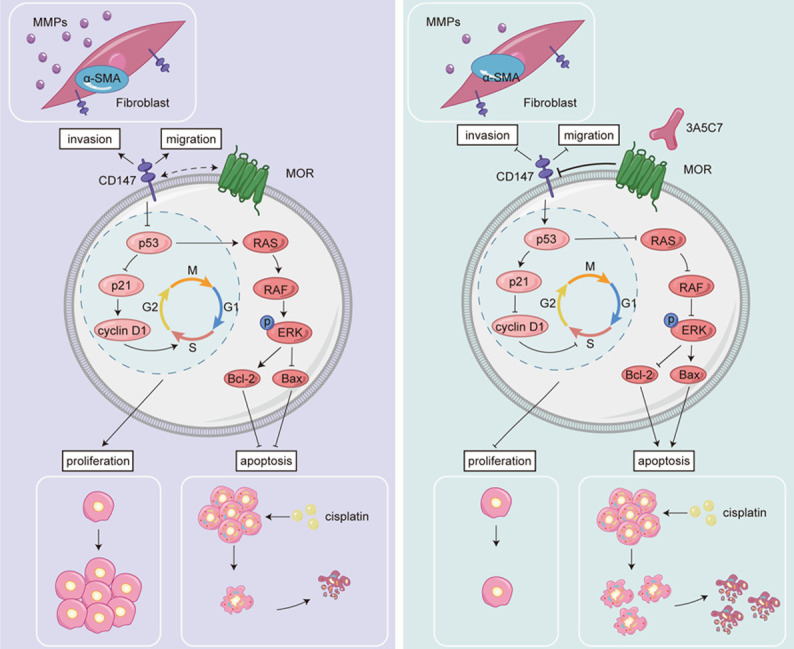
A model chart describes the mechanism by which MOR mAb inhibits the progression of HCC cells.
MOR is a member of the opioid receptors that belong to the G protein-coupled receptor superfamily [5]. MOR is located not only in the central nervous system but also in peripheral tissues, such as the colon, breast, prostate, lung and liver [4]. The importance of MOR in the pathogenesis and progression of HCC has drawn increasing attention in recent years. Jin et al. found that RNA interference or a specific inhibitor of MOR resulted in inhibited proliferation and an increased apoptotic rate of HepG2 and L02 cells, and the downregulation of MOR could increase the phosphorylation of MKK7 and lead to JNK activation [26]. Similarly, D.T. Chen et al. found that depletion of MOR could suppress the migration, invasion, and proliferation of human MHCC-97H and Huh7 cells, while overexpression of MOR promoted their growth and metastasis [27]. Consistently, in our study, we also found that siRNA-induced knockdown of MOR could inhibit the proliferation and migration of HCC cell lines, confirming the essential regulatory role of MOR in the pathogenesis of HCC.
Despite the established contribution of MOR to the proliferation of HCC, the mechanism by which MOR exerts its regulatory effects remains unclear. In our study, by using a mAb targeting MOR, we found that targeting MOR with a mAb could suppress the expression of CD147 and thus activate p53, which in turn inhibited the MAPK pathway and exerted an antiproliferative effect in HCC cells. This result provides us with the potential mechanism by which MOR regulates the proliferation of HCC, the CD147-p53-MAPK pathway. CD147, also known as basin or EMMPRIN, belongs to the immunoglobulin superfamily and is a transmembrane glycoprotein [28]. CD147 is overexpressed in a variety of cancers and is associated with the progression, invasiveness, and chemoresistance of cancer, and this overexpression also indicates adverse tumor outcomes [29-31]. Notably, CD147 is a key regulator of the pathogenesis of HCC, promoting its invasion, metastasis, and resistance [23,25,32-36]. In this study, we found that mAb targeting MOR could effectively downregulate the expression of CD147. We also verified the interaction between MOR and CD147 by co-IP and confocal colocalization. This could be the mechanism by which MOR is involved in the regulation of HCC. However, we still do not know how MOR interacts with CD147. The activation of MOR could recruit two different pathways, i.e., the G-protein-mediated pathway and the β-arrestin pathway, depending on the bound ligands [37,38]. Thus, we surmised that the downstream molecules of MOR might be involved in the transcription of BSG, the gene that encodes CD147 [28], or the translation, trafficking and endocytosis of CD147. Further studies are needed to fully illustrate the exact mechanism of the interaction between MOR and CD147.
The treatment strategy for HCC is a multidisciplinary approach, including orthotopic liver transplantation, liver resection, interventional treatment, and systemic treatment [39]. Despite these developed strategies, the survival of HCC after treatment is still not satisfactory, ranging from 31 months to 60 months, depending on the region [40]. Moreover, the mortality of HCC is still astonishingly high, with 782,000 cases diagnosed and 746,000 deaths in 2012 [1]. In our study, we found that the expression level of MOR was negatively correlated with the survival of HCC patients in terms of the overall survival rate, disease-free survival rate, and progression-free survival rate. These results suggest that MOR could be a prognostic marker for HCC patients. In other words, we might be able to predict the outcome of HCC patients by evaluating the expression level of MOR. In addition, we found that in HCC cell lines where MOR is highly expressed, mAb combined with cisplatin, a widely studied and used chemotherapy agent [41-43], could significantly enhance the efficacy of cisplatin to induce apoptosis in HepG2 and Huh-7 cell lines. This result again opens the possibility that MOR could be a therapeutic target for HCC and emphasizes the importance of treatment strategies with multiple targets.
Several limitations in our study should be noted. First, the roles of MOR in HCC and the effects of mAb 3A5C7 were only examined in vitro. Future studies harnessing a nude mouse model are urgently needed to illustrate the in vivo effects of mAb 3A5C7. Second, the mechanism of how mAb regulates the expression of MOR and how MOR interacts with CD147 is still unexplained in our study. It is of great importance to determine these mechanisms in further studies.
In conclusion, our study confirmed that MOR is overexpressed in both HCC tissue and cells, and the increased MOR was associated with the adverse prognosis of HCC patients. MOR is involved in the proliferation, metastasis and invasion of HCC. We further demonstrated that the MOR mAb 3A5C7 could suppress the proliferation of HCC via the MOR-CD147-p53-MAPK pathway. Targeting MOR with the mAb 3A5C7 could significantly enhance the ability of cisplatin to induce apoptosis of HCC cells. Our study reemphasized the involvement of MOR in the pathogenesis of HCC, and we hypothesized that MOR could be a new therapeutic target for HCC.
Acknowledgements
This study was supported by the National Science and Technology Major Project (2019ZX09732001) and the National Basic Research Program of China (2015CB553701).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 2.Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16:589–604. doi: 10.1038/s41575-019-0186-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sim HW, Knox J. Hepatocellular carcinoma in the era of immunotherapy. Curr Probl Cancer. 2018;42:40–48. doi: 10.1016/j.currproblcancer.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Stein C. Opioid receptors. Annu Rev Med. 2016;67:433–451. doi: 10.1146/annurev-med-062613-093100. [DOI] [PubMed] [Google Scholar]
- 5.Pasternak GW, Pan YX. Mu opioids and their receptors: evolution of a concept. Pharmacol Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 7.Darcq E, Kieffer BL. Opioid receptors: drivers to addiction? Nat Rev Neurosci. 2018;19:499–514. doi: 10.1038/s41583-018-0028-x. [DOI] [PubMed] [Google Scholar]
- 8.Bortsov AV, Millikan RC, Belfer I, Boortz-Marx RL, Arora H, McLean SA. μ-Opioid receptor gene A118G polymorphism predicts survival in patients with breast cancer. Anesthesiology. 2012;116:896–902. doi: 10.1097/ALN.0b013e31824b96a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lennon FE, Mirzapoiazova T, Mambetsariev B, Salgia R, Moss J, Singleton PA. Overexpression of the μ-opioid receptor in human non-small cell lung cancer promotes Akt and mTOR activation, tumor growth, and metastasis. Anesthesiology. 2012;116:857–867. doi: 10.1097/ALN.0b013e31824babe2. [DOI] [PubMed] [Google Scholar]
- 10.Yuen JW, So IY, Kam AY, Wong YH. Regulation of STAT3 by mu-opioid receptors in human neuroblastoma SH-SY5Y cells. Neuroreport. 2004;15:1431–1435. doi: 10.1097/01.wnr.0000130433.90962.6e. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Li G, Tao T, Kang X, Liu C, Zhang X, Wang C, Li C, Guo X. The μ-opioid receptor (MOR) promotes tumor initiation in hepatocellular carcinoma. Cancer Lett. 2019;453:1–9. doi: 10.1016/j.canlet.2019.03.038. [DOI] [PubMed] [Google Scholar]
- 12.Chen B, Chen B, Zhu Z, Ye W, Zeng J, Liu G, Wang S, Gao J, Xu G, Huang Z. Prognostic value of ZEB-1 in solid tumors: a meta-analysis. BMC Cancer. 2019;19:635. doi: 10.1186/s12885-019-5830-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Cozzi PJ. Targeting uPA/uPAR in prostate cancer. Cancer Treat Rev. 2007;33:521–527. doi: 10.1016/j.ctrv.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Frezzetti D, Gallo M, Maiello MR, D’Alessio A, Esposito C, Chicchinelli N, Normanno N, De Luca A. VEGF as a potential target in lung cancer. Expert Opin Ther Targets. 2017;21:959–966. doi: 10.1080/14728222.2017.1371137. [DOI] [PubMed] [Google Scholar]
- 15.Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol Cancer Ther. 2018;17:1147–1155. doi: 10.1158/1535-7163.MCT-17-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12:3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218–1227. doi: 10.1038/ncb3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mendonsa AM, Na TY, Gumbiner BM. E-cadherin in contact inhibition and cancer. Oncogene. 2018;37:4769–4780. doi: 10.1038/s41388-018-0304-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bazzoun D, Lelièvre S, Talhouk R. Polarity proteins as regulators of cell junction complexes: implications for breast cancer. Pharmacol Ther. 2013;138:418–427. doi: 10.1016/j.pharmthera.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. 2018;48:91–103. doi: 10.1016/j.semcancer.2017.04.015. [DOI] [PubMed] [Google Scholar]
- 22.Colak S, Ten Dijke P. Targeting TGF-β signaling in cancer. Trends Cancer. 2017;3:56–71. doi: 10.1016/j.trecan.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Xu J, Xu HY, Zhang Q, Song F, Jiang JL, Yang XM, Mi L, Wen N, Tian R, Wang L, Yao H, Feng Q, Zhang Y, Xing JL, Zhu P, Chen ZN. HAb18G/CD147 functions in invasion and metastasis of hepatocellular carcinoma. Mol Cancer Res. 2007;5:605–614. doi: 10.1158/1541-7786.MCR-06-0286. [DOI] [PubMed] [Google Scholar]
- 24.Huang Q, Li J, Xing J, Li W, Li H, Ke X, Zhang J, Ren T, Shang Y, Yang H, Jiang J, Chen Z. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J Hepatol. 2014;61:859–866. doi: 10.1016/j.jhep.2014.04.035. [DOI] [PubMed] [Google Scholar]
- 25.Jin J, Wang SJ, Cui J, Li L, Li JY, Liu FL, Sun XX, Jiang JL, Cui HY, Chen ZN. Hypo-phosphorylated CD147 promotes migration and invasion of hepatocellular carcinoma cells and predicts a poor prognosis. Cell Oncol (Dordr) 2019;42:537–554. doi: 10.1007/s13402-019-00444-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu JIN, Liu Z, Zhao L, Tian H, Liu X, Yuan C. In vivo and in vitro inhibition of human liver cancer progress by downregulation of the μ-opioid receptor and relevant mechanisms. Oncol Rep. 2013;30:1731–1738. doi: 10.3892/or.2013.2640. [DOI] [PubMed] [Google Scholar]
- 27.Chen DT, Pan JH, Chen YH, Xing W, Yan Y, Yuan YF, Zeng WA. The mu-opioid receptor is a molecular marker for poor prognosis in hepatocellular carcinoma and represents a potential therapeutic target. Br J Anaesth. 2019;122:e157–e167. doi: 10.1016/j.bja.2018.09.030. [DOI] [PubMed] [Google Scholar]
- 28.Muramatsu T. Basigin (CD147), a multifunctional transmembrane glycoprotein with various binding partners. J Biochem. 2016;159:481–490. doi: 10.1093/jb/mvv127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grass GD, Dai L, Qin Z, Parsons C, Toole BP. CD147: regulator of hyaluronan signaling in invasiveness and chemoresistance. Hyaluronan Signaling and Turnover. 2014. pp. 351–373. [DOI] [PubMed] [Google Scholar]
- 30.Kanekura T, Chen X. CD147/basigin promotes progression of malignant melanoma and other cancers. J Dermatol Sci. 2010;57:149–154. doi: 10.1016/j.jdermsci.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Xin X, Zeng X, Gu H, Li M, Tan H, Jin Z, Hua T, Shi R, Wang H. CD147/EMMPRIN overexpression and prognosis in cancer: a systematic review and meta-analysis. Sci Rep. 2016;6:32804. doi: 10.1038/srep32804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Huang Q, Long X, Zhang J, Huang X, Aa J, Yang H, Chen Z, Xing J. CD147 reprograms fatty acid metabolism in hepatocellular carcinoma cells through Akt/mTOR/SREBP1c and P38/PPARα pathways. J Hepatol. 2015;63:1378–1389. doi: 10.1016/j.jhep.2015.07.039. [DOI] [PubMed] [Google Scholar]
- 33.Lu M, Wu J, Hao ZW, Shang YK, Xu J, Nan G, Li X, Chen ZN, Bian H. Basolateral CD147 induces hepatocyte polarity loss by E-cadherin ubiquitination and degradation in hepatocellular carcinoma progress. Hepatology. 2018;68:317–332. doi: 10.1002/hep.29798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang J, Guo YS, Zhang Y, Yu XL, Li L, Huang W, Li Y, Chen B, Jiang JL, Chen ZN. CD147 induces UPR to inhibit apoptosis and chemosensitivity by increasing the transcription of Bip in hepatocellular carcinoma. Cell Death Differ. 2012;19:1779–1790. doi: 10.1038/cdd.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang SJ, Chao D, Wei W, Nan G, Li JY, Liu FL, Li L, Jiang JL, Cui HY, Chen ZN. CD147 promotes collective invasion through cathepsin B in hepatocellular carcinoma. J Exp Clin Cancer Res. 2020;39:145. doi: 10.1186/s13046-020-01647-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu J, Li Y, Dang YZ, Gao HX, Jiang JL, Chen ZN. HAb18G/CD147 promotes radioresistance in hepatocellular carcinoma cells: a potential role for integrin β1 signaling. Mol Cancer Ther. 2015;14:553–563. doi: 10.1158/1535-7163.MCT-14-0618. [DOI] [PubMed] [Google Scholar]
- 37.Chan HCS, McCarthy D, Li J, Palczewski K, Yuan S. Designing safer analgesics via mu-opioid receptor pathways. Trends Pharmacol Sci. 2017;38:1016–1037. doi: 10.1016/j.tips.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madariaga-Mazon A, Marmolejo-Valencia AF, Li Y, Toll L, Houghten RA, Martinez-Mayorga K. Mu-Opioid receptor biased ligands: a safer and painless discovery of analgesics? Drug Discov Today. 2017;22:1719–1729. doi: 10.1016/j.drudis.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lurje I, Czigany Z, Bednarsch J, Roderburg C, Isfort P, Neumann UP, Lurje G. Treatment strategies for hepatocellular carcinoma (-) a multidisciplinary approach. Int J Mol Sci. 2019;20:1465. doi: 10.3390/ijms20061465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park JW, Chen M, Colombo M, Roberts LR, Schwartz M, Chen PJ, Kudo M, Johnson P, Wagner S, Orsini LS, Sherman M. Global patterns of hepatocellular carcinoma management from diagnosis to death: the BRIDGE study. Liver Int. 2015;35:2155–2166. doi: 10.1111/liv.12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kondo M, Morimoto M, Kobayashi S, Ohkawa S, Hidaka H, Nakazawa T, Aikata H, Hatanaka T, Takizawa D, Matsunaga K, Okuse C, Suzuki M, Taguri M, Ishibashi T, Numata K, Maeda S, Tanaka K. Randomized, phase II trial of sequential hepatic arterial infusion chemotherapy and sorafenib versus sorafenib alone as initial therapy for advanced hepatocellular carcinoma: SCOOP-2 trial. BMC Cancer. 2019;19:954. doi: 10.1186/s12885-019-6198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kudo M, Ueshima K, Yokosuka O, Ogasawara S, Obi S, Izumi N, Aikata H, Nagano H, Hatano E, Sasaki Y, Hino K, Kumada T, Yamamoto K, Imai Y, Iwadou S, Ogawa C, Okusaka T, Kanai F, Akazawa K, Yoshimura KI, Johnson P, Arai Y. Sorafenib plus low-dose cisplatin and fluorouracil hepatic arterial infusion chemotherapy versus sorafenib alone in patients with advanced hepatocellular carcinoma (SILIUS): a randomised, open label, phase 3 trial. Lancet Gastroenterol Hepatol. 2018;3:424–432. doi: 10.1016/S2468-1253(18)30078-5. [DOI] [PubMed] [Google Scholar]
- 43.Brower V. Sorafenib plus cisplatin for hepatocellular carcinoma. Lancet Oncol. 2016;17:e424. doi: 10.1016/S1470-2045(16)30447-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.