

Abstract
Objective: To examine the effect of autophagy on cerebral damage caused by different models and test the hypothesis that its protection mechanism acts via inhibiting expression of neuroinflammatory mediators. Methods: Autophagy was induced by rapamycin treatment. Cerebral damage was induced using models of IL-6 treatment, oxygen glucose deprivation/reoxygenation (OGD/R) in vitro, and middle cerebral artery occlusion (MCAO) in vivo. The effect and mechanism of autophagy was examined and assessed in terms of cell viability, infarction size in brain tissue, neurological score, production of inflammatory mediators IL-1β and IL-6, transcription and protein expression of autophagy markers beclin-1 and LC-3II in different experimental groups. Results: Autophagy triggered by rapamycin could protect neurons from IL-6-induced injury and astrocytes from OGD/R-induced injury in vitro and in rat brain tissue from MCAO in vivo. Autophagy significantly increased cell viability, attenuated cerebral infarction and improved neurological scores. It also inhibited production of the IL-1β and IL-6 and elevated the expression of beclin-1 and LC-3II. Conclusions: Autophagy can inhibit the inflammatory response and reduce cerebral I/R injury. There was a relationship between the extent of protection and (i) the level of the autophagic response, (ii) the stage of the cerebral I/R injury, and (iii) the time of intervention.
Keywords: Autophagy, cerebral ischemia/reperfusion, oxygen glucose deprivation, IL-1β, IL-6, neuroinflammation
Introduction
Cerebral damage caused by ischemia and reperfusion (I/R) is accompanied by a marked inflammatory response, which is initiated and aggravated by the accumulation of inflammatory cells and the expression of inflammatory mediators [1]. Thus, many researchers investigate how to block the activation of inflammatory reactions during cerebral I/R to prevent and reduce the progression of brain injury. Recently, accumulating evidence from both in vitro and in vivo study has shown that autophagy plays a critical role in inflammation by influencing and regulating inflammatory mediators [2-4]. Autophagy is a type of self-phagocytosis in eukaryotic cells which is comprised of cellular “housekeeping” processes responsible for eliminating damaged organelles, aged proteins, and unused cytoplasmic components. Under normal physiological conditions, autophagy can mobilize intracellular energy resources to meet cellular and organismal demands for metabolic substrates [5]. In the presence of pathological factors (such as nutritional and growth factor deficiencies, ischemia, hypoxia, and inflammation, damaged organelles, aged proteins, and unused cytoplasmic), components are entrapped within autophagosomes, which transport the components to lysosomes via the microtubule network to promote digestion and component recycling to restore cell function and development [6-8].
Cytokines are one of the most important inflammation mediators responsible for regulating endogenous or exogenous inflammation reactions [9]. Neuroinflammation mediated by IL-1β or IL-6 worsens brain damage caused by a cerebral I/R injury. Under ischemic and hypoxic stimuli, IL-1β and IL-6 are both expressed in neurons and glial cells [10,11]. On the contrary, inhibition of IL-1β or IL-6 was beneficial for neuroprotection. Boutin et al reported that IL-1β knockout mice resulted in notably reduced infarct volumes following experimental stroke [12]. Similarly, Sung et al found that IL-6 blockade led to a decreased brain injury in mice that experienced a traumatic and brief hypoxia event [13]. Therefore, inhibition of IL-1β and IL-6-mediated neuroinflammation can be protective in cerebral I/R-induced brain damage.
To date, diverse studies have focused widely on autophagy, including in the brain, heart, lung, kidney and liver [14-17]. However, the effect of autophagy in I/R injury is still controversial, with some researchers claiming that the process has protective effects, whereas others claim that it has disruptive effects [18-20]. Therefore, it remains elusive whether autophagy regulates the expression of inflammatory mediators such as IL-1β and IL-6 and attenuates cerebral I/R injury. Additionally, despite the various effects of autophagy which have been studied widely, the relationship among the level of autophagy, severity of inflammation, and outcome of cerebral I/R injury remains unclear. Thus, we investigated these issues in this study by using models of IL-6 treatment, oxygen glucose deprivation/reoxygenation (OGD/R), and middle cerebral artery occlusion (MCAO). An improved understanding of the association between autophagy and inflammation may be helpful in the development of new treatments for ischemic stroke.
Materials and methods
Cell culture: neurons and astrocytes
Primary cortical neuronal cells were isolated from 24-hour-old neonate Sprague Dawley (SD) rats and cultured as previous described [21,22]. Briefly, within a sterile, controllable environment, brain cortical tissue was placed in cold PBS on ice. Under a microscope, we removed the meninges, blood vessels and cut the brain into several small pieces. We transferred the cortical tissue into a new container, and 0.25% trypsin was added to digest the extracellular matrix proteins. ① The cells were plated in 24 well plates on glass coverslips and mixed with media (Neurobasal A Medium) for the neuron populations. The morphology of neurons was observed and double immunofluorescence labeling was applied to identify the expression of NeuN and MAP2 in neurons. ② The cells were plated in 24 well plates on glass coverslips and mixed with media (DMEM/F 12 Medium) for the astrocyte populations. The morphology of neurons was observed and double immunofluorescence labeling was applied to identify the expression of GAPA in astrocytes.
Treatment of neurons and assessment of viability
Neurons were cultured at 1 × 104 cells/well in a 96-well plate in a NEUROBASAL medium and, then, we carried out the procedure depicted in Diagram 1. We treated four groups of neurons: a control group which consisted of untreated cells (blank group); a group treated with 100 ng/ml IL-6 (IL-6 group) for 2 hours to induce inflammation; a group treated with 1 mM/L rapamycin (Rap group) for 1 hour to trigger autophagy; and a group treated with 1 mM/L rapamycin for 1 hour prior to inducing inflammation with 100 ng/ml IL-6 for 2 hours (Rap+IL-6 group). The viability of untreated and treated neurons was measured with the Cell Counting Kit-8 (CCK-8 kit), following the manufacturer’s instructions (Nanjing Jiancheng Technology Co., Ltd). The CCK-8 blank well did not contain neurons, medium or CCK-8; the CCK-8 control contains medium and CCK-8, but no neurons. The absorbance was measured at 450 nm using a microplate reader. Neuronal viability (%) was calculated as follows: (OD value of experimental group - OD value of CCK-8 blank)/(OD value of CCK-8 control - OD value of CCK-8 blank).
Diagram 1.
Protocol used to treat neurons.
Treatment of astrocytes
To establish the OGD model, astrocytes were cultured in glucose-free Dulbecco’s Modified Eagle Medium F-12 (DMEM/F-12) and incubated in a humidified atmosphere of 5% CO2, 1% O2, and 94% N2 at 37°C for 2 h [23]. Five different groups were created by varying treatment and exposure, depicted in Diagram 2: an OGD-free control group which consisted of untreated cells (blank group); the first two groups were treated with OGD and washed afterwards with phosphate buffered saline and cultured in DMEM/F-12 for reoxygenation 24 h (OGD/24R group) and 48 h (OGD/48R group) to induce reperfusion injury; and the second two groups were treated first with 10 nM/L rapamycin for 48 h prior to being treated with OGD and subsequently cultured in DMEM/F-12 for reoxygenation 24 h (Rap+OGD/24R group) and 48 h (Rap+OGD/48R group).
Diagram 2.

Protocol used to treat astrocytes.
Establishment of middle cerebral artery occlusion model
Healthy male Sprague Dawley (SD) rats, SPF quality scale, weighing 280-320 g, n=36, provided by Beijing Weitong Lihua Experimental Animal Technology Co., Ltd. were randomly divided into a sham group, a ischemia-reperfusion group (I/R group), and a rapamycin treated group (Rap+I/R group). The I/R group was handled to trigger MCAO as follows. Initially, the rats were intubated with tracheal intubation, ventilator assisted respiration, 1%-2% enflurane mixed with 70% N2O and 30% O2 inhalation anesthesia. Then, a 0.26 mm diameter filament was inserted 18-20 mm from the external carotid artery to the beginning of the middle cerebral artery of the rat to create infarction in the middle cerebral artery territory. Finally, the filament was withdrawn after 2 h of ischemia to induce reperfusion. In the Rap+I/R group, the procedure was similar, but an intracerebroventricular injection was performed with 0.8 ng rapamycin, to induce autophagy, at the beginning of reperfusion.
Assessment of neurological score
The neurological score was measured after reperfusion 48 h after the reperfusion period by an assessor who was unaware of the experimental groups. The Garcia test is a composite neurological test used to evaluating the motor and sensory deficits as described below Table 1 [24].
Table 1.
Details of neurological score
| Spontaneous activity |
| Normal (3) |
| Slightly affected (2) |
| Severely affected (1) |
| No movement (0) |
| Symmetry in movement of 4 limbs assessed when rat held suspended by tail |
| Symmetric (3) |
| Asymmetric (2) |
| Hemiplegic (1) |
| Forepaw outstretching assessed by bringing rat to edge of table and making it walk on forelimbs while being held by tail and observing forelimb use |
| Symmetric forepaws (3) |
| Mild asymmetry (2) |
| Marked asymmetry (1) |
| One forelimb did not move (0) |
| Climbing determined by placing rat on the wall of a wire cage and observing climbing and strength of attachment to wall |
| Climbed easily, gripped tightly (3) |
| One side impaired (2) |
| Failed to climb or tended to circle instead of climbing (1) |
| Body proprioception |
| Equal on both sides (3) |
| Reacted slowly to stimulus on 1 side (2) |
| No response on one side (1) |
| Response to vibrissae touch determined by brushing vibrissae on each side |
| Symmetric (3) |
| Asymmetric (2) |
| No response on 1 side (1) |
Determination of cerebral infarction size
Rats in each experimental group were sacrificed by intraperitoneal injection of excessive chloral hydrate after 48 h of reperfusion. The brain was decapitated and placed in a brain slice mold, and a coronal section was made every 2 mm for a total of 6 slices. The brain slices were stained in 2% 2,3,5-triphenyltetrazolium chloride (TCC) solution prepared in 0.2 mol/L Ph7.4 PBS for 10 minutes (in the case of warm bath), and placed in a fixative solution. Photographs were taken 24 h later and the infarct volume was calculated using the Image-Pro Analysis Software 5.0 image analysis system. Cerebral infarction volume (%) = (Infarction volume/Total brain volume) × 100%.
Immunofluorescent staining
Rats in each experimental group were injected with chloral hydrate after 48 h of reperfusion and subsequently sacrificed by transcardiac perfusion with physiological saline followed by 4% paraformaldehyde dissolved in PBS (pH 7.4) to fix brain tissue. The brain sections were made by dehydration, transparency, dipping wax and embedding. The brain sections were separately incubated with antibody (GFAP 1:100 mouse anti rat; NeuN 1:100 mouse anti rat; LC-3B 1:200 rabbit anti rat; Beclin-1 1:200 rabbit anti rat; IL-1β 1:200 rabbit anti rat; IL-6 1:200 rabbit anti rat) at 4°C overnight. After washing, the sections were separately incubated with Dylight 488 labeled goat anti mouse IgG and cy3 labeled goat anti rabbit IgG for 1 hour at 37°C. The sections were given anti-fade reagent with DAPI, mounted and observed under a fluorescence microscope.
Real time-polymerase chain reaction
Rats in each experimental group were sacrificed by intraperitoneal injection of excessive chloral hydrate after 48 h of reperfusion. The brain tissue was removed quickly. The nucleotide sequences of primers used in this experiment were designed according to the sequence described as follows: Beclin-1 (F: AAGAGTGTAGAGAACCAGAT; R: TCGCATTGAAGACATTGG, 76 bp); LC-3B (F: AGTGATTATAGAGCGATA; R: CTAATTATCTTGATGAGTTC, 108 bp); IL1β (F: AACATAAGCCAACAAGTG; R: ACAGGACAGGTATAGATTC); IL-6 (F: GAACAACTTACAAGATAACA; R: GACTCTAACTTCTCCATTA); β-actin (F: TATGGAATCCTGTGGCATC; R: GTGTTGGCATAGAGGTCTT). Relative abundance of mRNA was calculated after normalization to β-actin RNA. The relative quantification was determined using the comparative CT method.
Western blotting analysis
① The neurons and astrocytes of each experimental group were washed by PBS twice. Then, 300 µl RIPA (containing 30 µl protease inhibitor) was added to each well which was placed on ice for splitting for 10 minutes. The supernatant was collected by cell centrifugation at 12000 rpm for 10 minutes at 4°C. ② Rats in each experimental group were sacrificed by intraperitoneal injection of excessive chloral hydrate after 48 h of reperfusion. The brain tissues were removed quickly. The brain tissue/100 mg were added 500 µl RIPA (containing 50 µl protease inhibitor) and cut by sterile ophthalmic scissors on ice. Homogenization was done by an ultrasonic cell disrupter for 20 seconds and placed on ice for 20 minutes. The supernatant was collected by cell centrifugation at 12000 rpm for 10 minutes at 4°C. Protein concentration assay were performed according to the BCA kit instructions. The protein was denatured by boiling water. Then, 80 µg of protein was loaded, electrophoresed by SDS-PAGE and transferred to film. Next, 5% BSA was blocked for 2 h, and TBS solution was used to wash 3 times, 5 min each time. Cells were incubated with antibody (Beclin-1, ab210498, abcam, 1:500, LC-3B, ab192890, abcam, 1:500) overnight at 4°C and the TBS solution washed 5 times, 5 min each time. We added HRP-labeled secondary antibody (ab7090, abcam, 1:2000), incubated at 37°C for 1 hour and washed the membrane with TBST solution 3 times, 5 min each time. The HRP substrate color development kit was used for development, and the PVDF film was scanned by a chemiluminometer. The protein band was analyzed by the targeted protein gray value/the β-actin value.
Enzyme-linked immunosorbent assays
The content from the Elisa kit was placed on a suitable work table inside a temperature controlled room or lab. We prepared the reagents and configured standard curves. Standard wells and sample wells were set separately. The standard port or a sample of 100 µl was added to each well, mixed gently by shaking and incubated for 90 minutes at 37°C. The liquid was discarded and we did not wash the wells. A total of 100 µl working solution of biotin-labeled antibody was added to each well and covered with a new plate for 1 hour at 37°C. The working solution was discarded and we washed the plate 3 times. Next, 100 µl avidin working solution of horseradish peroxidase-labeled antibody was added to each well and covered with a new plate for 30 minutes at 37°C. The avidin working solution was discarded and the plate was washed 5 times. Next, 90 µl substrate solution was added to each well in sequence and color was developed for 15 minutes at 37°C. The reaction was stopped by adding 50 µl stop solution to each well in sequence. The OD value of each well was measured at a wavelength of 450 nm using a microplate reader within 5 minutes of the termination of the reaction. We calculated IL-1β and IL-6 content in the astrocytes by standard curve.
Statistical analysis
All data were analyzed by SPSS 19.0 software and presented as mean ± SD. One-way analysis of variance was used to compare data from different groups. Post-hoc tests was performed by using Tamhane’s T2. P < 0.05 was the criterion for significance.
Results
Rapamycin-induced autophagy inhibits the inflammatory response in neurons
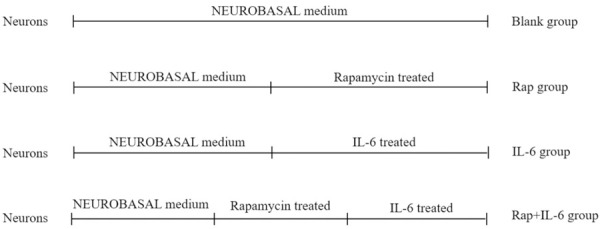
The morphology of neurons in each group was observed microscopically. IL-6-induced neurons showed an altered shape, with destroyed plasma membranes and irregular network structures. The halos around several neurons disappeared. Fewer cellular protrusions were observed; of those that were present, the morphology was defective. The pretreatment of cells with rapamycin, which induced autophagy, could reverse these morphological changes (Figure 1A). Neurons were cultured in the presence or absence of IL-6, rapamycin, and rapamycin + IL-6, and the viability of cells was measured with CCK-8. No difference in viability was observed between the blank and Rap groups (P > 0.05) (Figure 1B). However, the viability of cells cultured in the presence of IL-6 was significantly decreased, and a significant difference in viability was noticed between the blank and IL-6 groups (P < 0.01) (Figure 1B). When rapamycin was used to induce autophagy in neurons before the addition of IL-6 into the medium, the viability of cells was increased, and a significant difference in viability was noticed between IL-6 and Rap+IL-6 groups (P < 0.01) (Figure 1B). The levels of autophagy markers, beclin-1 and LC-3ll, were measured by western blotting. The results showed that the Rap+IL-6 group showed increased levels of beclin-1 and LC-3ll compared with the IL-6 group (P < 0.01) (Figure 1C-E).
Figure 1.
The effect of rapamycin-induced autophagy inhibits the inflammatory response in neurons. A. Representative morphological image of neurons, scale bar = X100. B. Statistics of neurons vitality by CCK-8. C. Representative Western blotting image of Beclin-1 and LC-3ll. D. Statistics of the expressional level of Beclin-1. E. Statistics of the expressional level of LC-3ll.
Rapamycin-induced autophagy inhibits the inflammatory response in astrocytes following glucose oxygen deprivation/reoxygenation
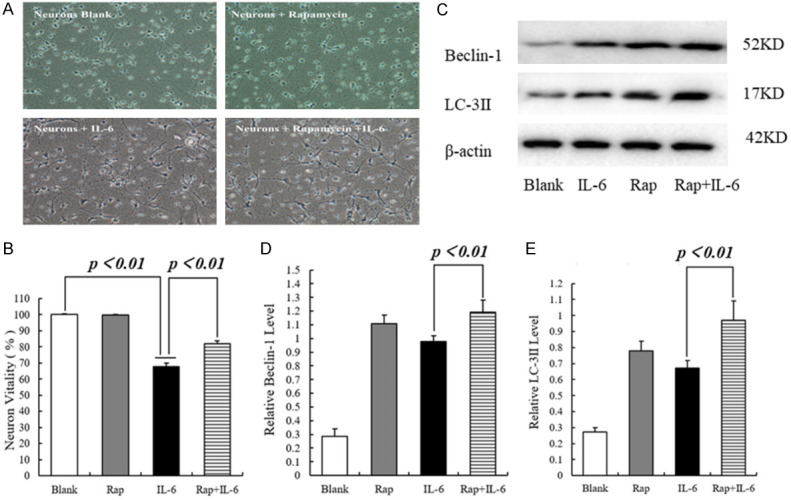
In the first two groups, astrocytes were cultured in glucose-free medium under conditions of OGD for 2 h, followed by complete medium and reoxygenation for 24 and 48 h. In the second two groups, astrocytes were pretreated with rapamycin and cultured under conditions of OGD, followed by reoxygenation. The morphology of astrocytes in each group was observed microscopically. In the OGD/R group, the astrocyte’s shape was altered, and the plasma membranes were destroyed. In addition, the synapses were shortened, and tight junctions were destroyed. There was evidence of edema and coagulative necrosis. The pretreatment of astrocytes with rapamycin could reverse these changes (Figure 2A).
Figure 2.
The effect of rapamycin-induced autophagy inhibits inflammatory responses in astrocytes. A. Representative morphological image of astrocytes, scale bar = X100. B. Statistics of the level of IL-1β. C. Statistics of the level of IL-6. D. Representative Western blotting image of Beclin-1 and LC-3ll. E. Statistics of the expressional level of Beclin-1. F. Statistics of the expressional level of LC-3ll.
Previous studies have reported that astrocytes secrete inflammatory mediators, such as IL-1β and IL-6, which can also stimulate astrocytes in an autocrine manner, thereby aggravating tissue damage during cerebral I/R injury. The levels of IL-1β and IL-6 in astrocytes of each group were detected by enzyme-linked immunosorbent assays (ELISA). The OGD/R group had higher levels of IL-1β and IL-6 than the Rap+OGD/R group (P < 0.01) (Figure 2B, 2C). The levels of autophagy markers, beclin-1 and LC-3ll, were measured by western blot. The results showed that the Rap+OGD/R group exhibited increased levels of beclin-1 and LC-3ll compared with the OGD/R group (P < 0.01) (Figure 2D-F).
Infarction size and neurological score
Brain cross-sections were stained with TTC, and the infarction size was determined. There was no evidence of cerebral infraction in rat brains of the sham group. The extent of infarction was large in rat brains of the I/R group, with tissue death noted in the cortex, subcortical region, striatum, and hippocampus. The extent of infarction was small in rat brains of the Rap+I/R group, in which autophagy was induced with rapamycin (P < 0.01) (Figure 3A, 3B).
Figure 3.

Infarction size and neurological score. A. Image of TTC staining. B. Statistics of infarction size. C. Statistics of neurological score.
Neurological scores were assigned to each group at 48 h after MCAO. No neurological deficits were observed in rats of the sham group. Poor performance in autonomic exercises, limb activities, and body sensations was noted in rats of the I/R group. Compared to the I/R group, there was a significant improvement in the neurological score in rats of the Rap+I/R group (P < 0.05) (Figure 3C).
Rapamycin-induced autophagy inhibits the expression of pro-inflammatory cytokines IL-1β and IL-6
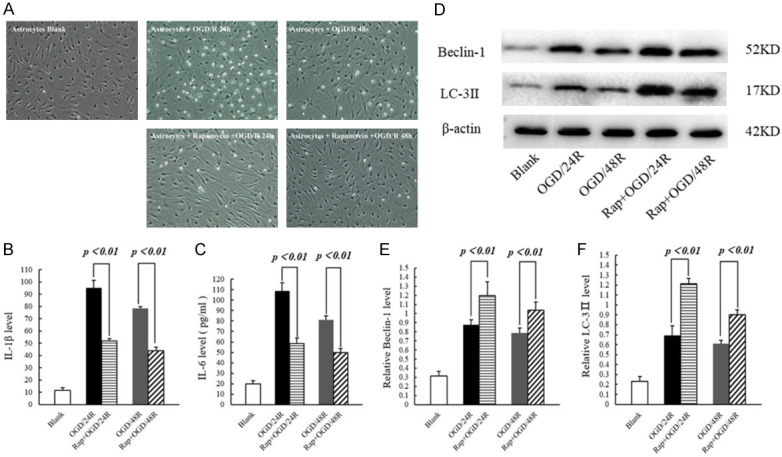
As inflammatory mediators, IL-1β and IL-6 are secreted in large amounts during cerebral I/R injury, thereby inducing tissue damage. Compared to rat brains of the I/R group, the mRNA expression of IL-1β (P < 0.01) and IL-6 (P < 0.01) was significantly decreased in brains of the Rap+I/R group (Figure 4A, 4D). Similarly, the protein expression of IL-1β (P < 0.05) and IL-6 (P < 0.01) was significantly decreased in brains of the Rap+I/R group (Figure 4B, 4C, 4E, 4F). To examine the activation of autophagy markers, we used real-time PCR and western blotting to examine the mRNA and protein expression of beclin-1 and LC-3ll. Compared to the I/R group, the mRNA expression of beclin-1 (P < 0.01) and LC-3ll (P < 0.01) was significantly increased in the Rap+I/R group (Figure 5A, 5D). Similarly, brains of the Rap+I/R group showed significantly decreased protein expression of beclin-1 (P < 0.01) and LC-3ll (P < 0.05) (Figure 5B, 5C, 5E, 5F). Finally, we used dual immunofluorescent staining to observe the expression of IL-1β and IL-6, as well as beclin-1 and LC-3ll, in the rat brain. The results showed IL-1β, IL-6, beclin-1, and LC-3ll expression in neurons and astrocytes (Supplementary Figures 1, 2, 3 and 4).
Figure 4.
The transcription and expression of IL-1β and IL-6 in rat brain tissue. A. Statistics of the transcription level of IL-1β. B. Statistics of the protein expression level of IL-1β. C. Representative Western blotting image of IL-1β. D. Statistics of the transcription level of IL-6. E. Statistics of the protein expression level of IL-6. F. Representative Western blotting image of IL-6.
Figure 5.
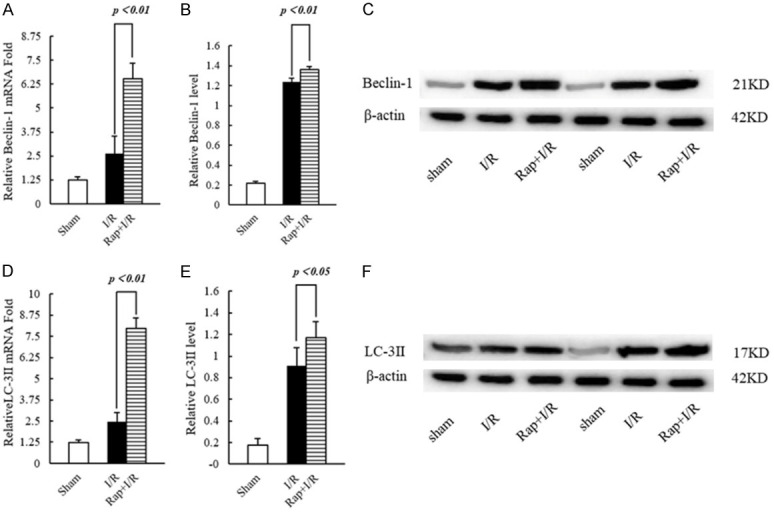
The transcription and expression of beclin-1 and LC-3ll in rat brain tissue. A. Statistics of the transcription level of beclin-1. B. Statistics of the protein expression level of beclin-1. C. Representative Western blotting image of beclin-1. D. Statistics of the transcription level of LC-3ll. E. Statistics of the protein expression level of LC-3ll. F. Representative Western blotting image of LC-3ll.
Discussion
Numerous studies have reported that cerebral I/R injury involves various pathophysiological mechanisms, including free radical generation, lipid peroxidation, excitatory amino acid toxicity, mitochondrial dysfunction, and intracellular Ca2+ overload [25-30]. The inflammatory response caused by cerebral I/R injury, characterized by the rapid activation of microglia and astrocytes, is followed by the production of inflammatory mediators and inflammatory cells, such as neutrophils, different subtypes of T cells, and mononuclear macrophages.
These inflammatory mediators penetrate the ischemic brain tissue and induce damage [31]. Damaged nerve cells can release large amounts of IL-1β and IL-6 [32,33], which mediate the inflammatory response in cerebral I/R injury [34]. The levels of IL-6 and IL-1β are low in normal brain tissues, but high under ischemic and hypoxic conditions. Furthermore, IL-1β can not only promote the production of free radicals and proteolytic enzymes, which affect the metabolic activities of nerve cells and the permeability of blood vessels (causing brain edema and aggravating brain damage), but also induce the expression of IL-6, which stimulates macrophages to produce more IL-1β [35]. IL-6, a multifunctional cytokine that participates in the inflammatory response, is especially important in the acute phase of cerebral I/R injury [36]. Our experimental results of dual immunofluorescent staining confirmed that IL-1β and IL-6 are expressed in damaged brain tissue after I/R injury. Additionally, real-time PCR, western blotting, and ELISA showed that the levels of IL-1β and IL-6 increased in neurons and astrocytes too. During inflammation, neurons and astrocytes showed morphological changes and decreased neuron viability in vitro. Significant infarction and a decline in neurological function were also observed in vivo.
We used rapamycin to induce autophagy in both neurons, astrocytes and rat brain tissue to inhibit inflammation, Beclin-1 and LC3II were examined as key markers for autophagy. Our results showed that autophagy triggered by rapamycin could protect neurons from IL-6-induced injury and astrocytes from OGD/R-induced injury in vitro and the rat brain tissue from I/R injury in vivo had a reduced inflammatory response, characterized by increased viability of neurons, decreased infarction size in the brain, recovery of neurological function, decreased expression of IL-1β and IL-6, and increased expression of beclin-1 and LC-3ll. The protective mechanism of autophagy on neurons, astrocytes, and the brain tissue indicates that rapamycin could effectively inhibit the inflammatory response caused by cerebral I/R injury.
Under pathological conditions, autophagy’s effect is critical for neuronal function [37]. However, the current literature in this matter is contradictory [38]. Shi and colleagues reported that activity of autophagy had a protective role in ischemic-hypoxic injury [39]. On the other hand, Zhou and colleagues indicated that autophagy could contribute to the apoptosis process [40]. We believe that these contradictory outcomes must be understood taking into consideration the level of autophagy per se: considerably high or low autophagy extent might be harmful. For instance, the work of Sun and colleagues [41] showed that an excessive level of autophagy promoted cell death and, hence, a decrease in autophagy proved beneficial for brain tissue. In our study, we found out that autophagy markers beclin-1 and LC3ll in neurons can be detected and indicated the activity of autophagy in the IL-6 group, but neuronal viability decreased. It has been considered that an insufficient level of autophagy in the IL-6 group may be also harmful, but neurons treated with rapamycin prior to IL-6 treatment in the Rap+IL-6 group had a higher beclin-1 and LC3ll expression, increased neuron viability and improved neuronal shape, as compared to the IL-6 group, even though rapamycin increases autophagy levels. Thus, we believe that both extremes, either an insufficient or an excessive degree of autophagy, can be harmful for the neurons; therefore, moderate autophagy may protect neurons from death.
We also observed that the effects of autophagy on astrocytes were linked with the extent of ischemic and hypoxic events. Our results showed that the levels of beclin-1 and LC-3II were higher in the Rap+OGD/24R group than those in the Rap+OGD/48R group. However, the levels of IL-1β and IL-6 were lower in the Rap+OGD/48R group than those in the Rap+OGD/24R group, contrary to our expectations. The inflammatory response caused by a cerebral I/R injury is a gradually escalating pathological process, which reaches its peak of IL-1β and IL-6 production at 24 hours after starting reperfusion [42,43]; hence, this suggests that the protective effects of autophagy may be counteracted by the peak of inflammatory response. Therefore, our results showed the ability of autophagy to suppress the inflammatory response was limited and was influenced by the severity of inflammation on cerebral I/R injury.
Cerebral I/R injury is an acute and long-lasting process that proceeds from ischemia to reperfusion, the protective effects of autophagy related to the rapamycin administration time are also relevant. Wu and colleagues reported that rapamycin treatment at 30 minutes and 12 hours prior to cerebral ischemia exerts a protective effects on cerebral I/R injury in rats [44]. Similarly, Lv and colleagues reported that rapamycin treatment 2 days prior to cerebral ischemia also downregulated I/R injury related inflammatory response [45]. However, in clinical practice, it is difficult to predict when a stroke will occur and treatments can only take place once ischemia or reperfusion has occurred. We think that the value of autophagy could be obtained at the beginning of the reperfusion. In our study, we induced autophagy in rats through an intracerebroventricular injection of rapamycin at the beginning of the reperfusion period and observed a reduction in the infarction size, a recovery of the neuronal function, and a decreased expression of IL-1β and IL-6, which essentially maintained neuronal function and protected brain tissue. Our results also suggest that the induced autophagy at the beginning of reperfusion could also effectively inhibit the inflammatory response, thereby protecting the brain from cerebral I/R injury.
Conclusion
Autophagy can inhibit the inflammatory response caused by IL-6 treated, OGD and cerebral I/R injury, as well as protect neurons, astrocytes and rat brain tissue. There is a relationship between the extent of protection and the level of the autophagic response, the stage of the cerebral ischemic event, and the time of intervention. The relationship between autophagy and the inflammatory response is complex. An effective understanding of this relationship is essential for reducing cerebral I/R injury. Further studies are needed to address (i) how autophagy can be adjusted to moderate levels in various models, and (ii) how the balance between autophagy and the inflammatory response can be maintained. Meanwhile, we plan to study the best time to induce the autophagy during other time points after the cerebral I/R episode.
Acknowledgements
This work was supported by Yunnan Provincial Basic Research Project (2017FE468-153).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Shukla V, Shakya AK, Perez-Pinzon MA, Dave KR. Cerebral ischemic damage in diabetes: an inflammatory perspective. J Neuroinflammation. 2017;14:21. doi: 10.1186/s12974-016-0774-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6:24. doi: 10.1186/s40169-017-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bussi C, Peralta Ramos JM, Arroyo DS, Gaviglio EA, Gallea JI, Wang JM, Celej MS, Iribarren P. Autophagy down regulates pro-inflammatory mediators in BV2 microglial cells and rescues both LPS and alpha-synuclein induced neuronal cell death. Sci Rep. 2017;7:43153. doi: 10.1038/srep43153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng Y, Gao J, Cui Y, Li M, Li R, Cui C, Cui J. Neuroprotective effects of resatorvid against traumatic brain injury in rat: involvement of neuronal autophagy and TLR4 signaling pathway. Cell Mol Neurobiol. 2017;37:155–168. doi: 10.1007/s10571-016-0356-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 8.Hamasaki M, Yoshimori T. Where do they come from? Insights into autophagosome formation. FEBS Lett. 2010;584:1296–1301. doi: 10.1016/j.febslet.2010.02.061. [DOI] [PubMed] [Google Scholar]
- 9.Kawabori M, Yenari MA. Inflammatory responses in brain ischemia. Curr Med Chem. 2015;22:1258–1277. doi: 10.2174/0929867322666150209154036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denes A, Pinteaux E, Rothwell NJ, Allan SM. Interleukin-1 and stroke: biomarker, harbinger of damage, and therapeutic target. Cerebrovasc Dis. 2011;32:517–527. doi: 10.1159/000332205. [DOI] [PubMed] [Google Scholar]
- 11.Cojocaru IM, Cojocaru M, Tănăsescu R, Iliescu I, Dumitrescu L, Silosi I. Expression of IL-6 activity in patients with acute ischemic stroke. Rom J Intern Med. 2009;47:393–396. [PubMed] [Google Scholar]
- 12.Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci. 2001;21:5528–5534. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang SH, Gangidine M, Pritts TA, Goodman MD, Lentsch AB. Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock. 2013;40:471–475. doi: 10.1097/SHK.0000000000000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tong G, Wang Y, Xu C, Xu Y, Ye X, Zhou L, Zhu G, Zhou Z, Huang J. Long non-coding RNA FOXD3-AS1 aggravates ischemia/reperfusion injury of cardiomyocytes through promoting autophagy. Am J Transl Res. 2019;11:5634–5644. [PMC free article] [PubMed] [Google Scholar]
- 15.Ruan Z, Liang M, Deng X, Lai M, Shang L, Su X. Exogenous hydrogen sulfide protects fatty liver against ischemia-reperfusion injury by regulating endoplasmic reticulum stress-induced autophagy in macrophage through mediating the class A scavenger receptor pathway in rats. Cell Biol Int. 2019 doi: 10.1002/cbin.11234. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Cao H, Li J, Wang B, Zhang P, Dong Zhang X, Liu Z, Yuan H, Zhan Z. Autophagy induced by DAMPs facilitates the inflammation response in lungs undergoing ischemia-reperfusion injury through promoting TRAF6 ubiquitination. Cell Death Differ. 2017;24:683–693. doi: 10.1038/cdd.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ling H, Chen H, Wei M, Meng X, Yu Y, Xie K. The effect of autophagy on inflammation cytokines in renal ischemia/reperfusion injury. Inflammation. 2016;39:347–356. doi: 10.1007/s10753-015-0255-5. [DOI] [PubMed] [Google Scholar]
- 18.Jin J, Sun H, Liu D, Wang H, Liu Q, Chen H, Zhong D, Li G. LRG1 promotes apoptosis and autophagy through the TGFβ-smad1/5 signaling pathway to exacerbate ischemia/reperfusion injury. Neuroscience. 2019;413:123–134. doi: 10.1016/j.neuroscience.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Xie C, Ginet V, Sun Y, Koike M, Zhou K, Li T, Li H, Li Q, Wang X, Uchiyama Y, Truttmann AC, Kroemer G, Puyal J, Blomgren K, Zhu C. Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury. Autophagy. 2016;12:410–423. doi: 10.1080/15548627.2015.1132134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ni Y, Gu WW, Liu ZH, Zhu YM, Rong JG, Kent TA, Li M, Qiao SG, An JZ, Zhang HL. RIP1K contributes to neuronal and astrocytic cell death in ischemic stroke via activating autophagic-lysosomal pathway. Neuroscience. 2018;371:60–74. doi: 10.1016/j.neuroscience.2017.10.038. [DOI] [PubMed] [Google Scholar]
- 21.Muramatsu R, Yamashita T. Primary culture of cortical neurons. BIO-PROTOCOL. 2013:3. [Google Scholar]
- 22.Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. J Vis Exp. 2013:50079. doi: 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hua R, Han S, Zhang N, Dai Q, Liu T, Li J. cPKCγ-modulated sequential reactivation of mtor inhibited autophagic flux in neurons exposed to oxygen glucose deprivation/reperfusion. Int J Mol Sci. 2018;19:1380. doi: 10.3390/ijms19051380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- 25.Xiong XY, Liu L, Yang QW. Refocusing neuroprotection in cerebral reperfusion era: new challenges and strategies. Front Neurol. 2018;9:249. doi: 10.3389/fneur.2018.00249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin Z, Zhu D, Yan Y, Yu B, Wang Q, Shen P, Ruan K. An antioxidant phytotherapy to rescue neuronal oxidative stress. Evid Based Complement Alternat Med. 2011;2011:519517. doi: 10.1093/ecam/nen053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caliskan M, Mogulkoc R, Baltaci AK, Menevse E. The effect of 3’,4’-dihydroxyflavonol on lipid peroxidation in rats with cerebral ischemia reperfusion injury. Neurochem Res. 2016;41:1732–1740. doi: 10.1007/s11064-016-1889-x. [DOI] [PubMed] [Google Scholar]
- 28.Wei J, Wu X, Luo P, Yue K, Yu Y, Pu J, Zhang L, Dai S, Han D, Fei Z. Homer1a attenuates endoplasmic reticulum stress-induced mitochondrial stress after ischemic reperfusion injury by inhibiting the perk pathway. Front Cell Neurosci. 2019;13:101. doi: 10.3389/fncel.2019.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu YR, Lei RY, Wang CE, Zhang BA, Lu H, Zhu HC, Zhang GB. Effects of catalpol on ATPase and amino acids in gerbils with cerebral ischemia/reperfusion injury. Neurol Sci. 2014;35:1229–1233. doi: 10.1007/s10072-014-1687-7. [DOI] [PubMed] [Google Scholar]
- 30.Cui Y, Wang JQ, Shi XH, Wang YY, Liu HY, Li Z, Dong Y, Mang J, Xu ZX. Nodal mitigates cerebral ischemia-reperfusion injury via inhibiting oxidative stress and inflammation. Eur Rev Med Pharmacol Sci. 2019;23:5923–5933. doi: 10.26355/eurrev_201907_18337. [DOI] [PubMed] [Google Scholar]
- 31.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ge JB, Lu HJ, Song XJ, Li M, Chen DD, Wu F. Protective effects of LBP on cerebral ischemia reperfusion injury in mice and mechanism of inhibiting NF-κB, TNF-α, IL-6 and IL-1β. Zhongguo Zhong Yao Za Zhi. 2017;42:326–331. doi: 10.19540/j.cnki.cjcmm.20161222.016. [DOI] [PubMed] [Google Scholar]
- 33.Lv Z, Liu C, Zhai M, Zhang Q, Li J, Zheng F, Peng M. LPS pretreatment attenuates cerebral ischaemia/reperfusion injury by inhibiting inflammation and apoptosis. Cell Physiol Biochem. 2018;45:2246–2256. doi: 10.1159/000488170. [DOI] [PubMed] [Google Scholar]
- 34.Chen S, Yin ZJ, Jiang C, Ma ZQ, Fu Q, Qu R, Ma SP. Asiaticoside attenuates memory impairment induced by transient cerebral ischemia-reperfusion in mice through anti-inflammatory mechanism. Pharmacol Biochem Behav. 2014;122:7–15. doi: 10.1016/j.pbb.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Zhu S, Zou Y, Wang T, Fu X. Knockdown of IL-1β improves hypoxia-ischemia brain associated with IL-6 up-regulation in cell and animal models. Mol Neurobiol. 2015;51:743–752. doi: 10.1007/s12035-014-8764-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang L, Chen C, Zhang X, Li X, Chen Z, Yang C, Liang X, Zhu G, Xu Z. Neuroprotective effect of curcumin against cerebral ischemia-reperfusion via mediating autophagy and inflammation. J Mol Neurosci. 2018;64:129–139. doi: 10.1007/s12031-017-1006-x. [DOI] [PubMed] [Google Scholar]
- 37.Lim Y, Cho H, Kim EK. Brain metabolism as a modulator of autophagy in neurodegeneration. Brain Res. 2016;1649:158–165. doi: 10.1016/j.brainres.2016.02.049. [DOI] [PubMed] [Google Scholar]
- 38.Puyal J, Ginet V, Vaslin A, Truttmann AC, Clarke PG. The two faces of autophagy in the nervous system. Med Sci (Paris) 2009;25:383–390. doi: 10.1051/medsci/2009254383. [DOI] [PubMed] [Google Scholar]
- 39.Shi ZY, Deng JX, Fu S, Wang L, Wang Q, Liu B, Li YQ, Deng JB. Protective effect of autophagy in neural ischemia and hypoxia: negative regulation of the Wnt/β-catenin pathway. Int J Mol Med. 2017;40:1699–1708. doi: 10.3892/ijmm.2017.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou XY, Luo Y, Zhu YM, Liu ZH, Kent TA, Rong JG, Li W, Qiao SG, Li M, Ni Y, Ishidoh K, Zhang HL. Inhibition of autophagy blocks cathepsins-tBid-mitochondrial apoptotic signaling pathway via stabilization of lysosomal membrane in ischemic astrocytes. Cell Death Dis. 2017;8:e2618. doi: 10.1038/cddis.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun D, Wang W, Wang X, Wang Y, Xu X, Ping F, Du Y, Jiang W, Cui D. bFGF plays a neuroprotective role by suppressing excessive autophagy and apoptosis after transient global cerebral ischemia in rats. Cell Death Dis. 2018;9:172. doi: 10.1038/s41419-017-0229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamashita T, Sawamoto K, Suzuki S, Suzuki N, Adachi K, Kawase T, Mihara M, Ohsugi Y, Abe K, Okano H. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: possible involvement of Stat3 activation in the protection of neurons. J Neurochem. 2005;94:459–468. doi: 10.1111/j.1471-4159.2005.03227.x. [DOI] [PubMed] [Google Scholar]
- 43.Caso JR, Moro MA, Lorenzo P, Lizasoain I, Leza JC. Involvement of IL-1beta in acute stress-induced worsening of cerebral ischaemia in rats. Eur Neuropsychopharmacol. 2007;17:600–607. doi: 10.1016/j.euroneuro.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 44.Wang L, Dengyan WU, Anchun HU, Yang C, Jing Y. Study on the protective effects of rapamycin on cerebral ischemia-reperfusion injury in rats. Journal of Chongqing Medical University. 2012 [Google Scholar]
- 45.Lv P, Lu N, Xu CR. The improvement of Rapamycin on the cerebral ischemia reperfusion injury related to downregulated TLR4/MyD88/NF-κB signaling pathway in rats. Progress of Anatomical Sciences. 2019;25:200–203. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.