

Abstract
Thoracic aortic aneurysm or dissection (TAAD) is a group of life-threatening complex diseases after symptomatic onset with genetic heterogeneity accounting for approximately 20% of cases. Previously, we identified 40 rare variants in 11 TAAD-related core genes among 70 TAAD patients by next-generation sequencing. In this study, we further analyzed the variants in the disease-causing genes in 129 cases of sporadic TAAD and 22 familial cases by whole-exome sequencing. A total of 116 variants in 47 TAAD-related genes were identified, 64.7% (75/116) of which occurred in sporadic TAAD without syndromes, and among these genes, FBN1 was the most common TAAD-related gene. Of the 26.7% (31/116) that were pathogenic or likely pathogenic, almost one third were from sporadic cases without syndromes involving FBN1, SMAD3, SMAD6, MYH11, TGFBR1, MYLK, LOX and LTBP3. Interestingly, the novel VUS (variant of uncertain significance) *879Glu in MCTP2 occurred in two unrelated probands with sporadic acute aortic dissection without a bicuspid aortic valve. Furthermore, more than one variant was detected in 24 patients, and 70.8% (17/24) occurred in sporadic cases. Younger individuals were more likely to carry P/LP (pathogenic or likely pathogenic) variants and harbor more variants. P/LP carriers seem to have a larger aortic diameter, lower D-dimer levels, and a shorter ICU length of stay but longer hospitalization time. In conclusion, we expanded the candidate gene profile of TAAD, especially for sporadic cases without syndromic features. VUSs need further clarification.
Keywords: Thoracic aortic aneurysm or dissection, exome sequencing, FBN1 gene, sporadic TAAD
Introduction
Thoracic aortic aneurysm or dissection (TAAD) refers to a group of rare underrecognized life-threatening cardiovascular disorders that account for more than 17000 deaths annually in the United States [1] and are the most severe diseases of acute aortic syndrome (AAS). Medial degeneration and increased aortic wall stress are frequently involved in the formation of aortic disease [2]. Common risk factors related to TAAD include hypertension, smoking, trauma, atherosclerosis and pheochromocytoma, which may be more likely to increase aortic wall stress. Furthermore, genetic background is a specific risk factor for TAAD [3]. It has been reported that hereditary connective tissue disorders are closely associated with conditions involving medial degeneration of the aorta, such as Marfan syndrome (MFS) [OMIM: 154700] caused by the FBN1 gene encoding fibrillin-1, Loeys-Dietz syndrome (LDS) [OMIM: 609192] resulting from mutations in transforming growth factor beta receptor, the vascular form of Ehlers-Danlos syndrome (v-EDS) [OMIM: 130050] due to mutation of the COL3A1 gene, encoding type III collagen, and Shprintzen-Goldberg syndrome (SGS) [OMIM: 182212].
Genetic determinants greatly contribute to the occurrence of aortic disease, especially in TAAD with a family history [4-8]. Causative gene mutations have also been found in sporadic TAAD. In addition, certain gene mutations, such as those in TGFBR and COL genes, could also be detected in nonsyndromic hereditary TAAD [9,10]. To date, many clinicians have mainly studied syndromic and nonsyndromic hereditary TAAD cases, 20% of which were attributed to gene mutations, but the majority of patients with emergency hospitalization for acute aortic events have sporadic aneurysms or dissections without syndromic features. Lack of similar family history, abrupt asymptomatic onset, younger age at diagnosis, complex genetic heterogeneity and phenotypic diversity lead to the elaboration of the pathogenesis of sporadic TAAD remaining to be uncovered. Other than the common causative core genes FBN1, ACTA2, COL3A1, MYH11, SMAD3, TGFB2, TGFBR1, TGFBR2, MYLK and LOX, few studies have focused on other candidate genes [11-13]. With the application of next-generation sequencing (NGS) [11,14], it is possible to identify the genes causing familial or sporadic TAAD for molecular diagnosis and better prevention.
Herein, we analyzed TAAD- related genes by whole-exome sequencing (WES) in a clinically mixed cohort of sporadic and familial TAAD patients from South China, especially nonsyndromic patients without a family history.
Materials and methods
Patients
Based on the approval of the Research Ethics Committee, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, we collected the clinical information of one hundred fifty-one consecutive probands with TAAD and seventy-one relatives between 2019 and 2020 in the Department of Cardiac Surgery, Guangdong Cardiovascular Institute, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangdong Province, China. The mixed study cohort included familial and sporadic cases of TAAD-related diseases. Familial TAAD was defined as cases in which more than one patient with TAAD existed in the family pedigree. Sporadic TAAD was defined as cases in which the probands was the first to suffer from aortic disease in their own family, regardless of the presence of abnormalities in other organs. Patients with suspected MFS were assessed according to the Revised Ghent Nosology [15]. Plain and enhanced spiral CT scans of the entire aorta and Doppler echocardiographic examination were performed on patients who underwent surgery for cardiovascular system diseases, and cardiac ultrasonography was performed on the rest of the patients. The diameter of the aortic root and ascending aorta were measured by echocardiography and CT. At the same time, medical records, including age of onset, sex, tobacco use, alcohol use, hypertension history, surgical history of cardiovascular system disease, and other conditions of cardiovascular diseases, were also collected. Blood samples were obtained from probands and their relatives when possible. Written specific informed consent was obtained from all participants of this study.
Exome sequencing
DNA extraction
In our cohort, genomic DNA was extracted from peripheral blood mononuclear cells (PBMCs) from 119 patients using Magbead Blood DNA Kit (CWbiotech), and the remaining DNA was extracted with the Solpure Blood DNA kit (Magen) according to the manufacturer’s instructions.
Target capture and sequencing
Libraries were constructed according to the instructions of the manufacturer (Fast Library Prep Kit, iGeneTech) or prepared following the Illumina library preparation protocol. Briefly, 200 ng genomic DNA from 119 patients was sheared using Biorupter (Diagenode, Belgium) to obtain 150-200 bp fragments, followed by end repair, adaptor ligation and PCR amplification. Whole exons were captured from the sequencing libraries using the AIExome Enrichment Kit V1 (iGeneTech) and sequenced on the NovaSeq 6000 platform (Illumina) to obtain 150 base paired-end reads with at least 100X coverage. The other part of genomic DNA was fragmented by a Q800R Sonicator (Qsonica) to generate 300-500 bp insert fragments. Custom-designed NimbleGen SeqCap probes (Roche NimbleGen, Madison, Wis) were used for in-solution hybridization to enrich target sequences. Enriched DNA samples were indexed and sequenced on a NextSeq500 sequencer (Illumina, San Diego, Calif) with 100-150 cycles of single-end reads, according to the manufacturer’s protocols.
Variant annotation and interpretation
Primary data were obtained in the Fastq format after image analysis, and base calling was conducted using the Illumina Pipeline. Sequence alignment was performed using a Burrows-Wheeler algorithm, BWA-mem and variant calling were performed using Genome Analysis Tool Kit (GATK v4) best practices (https://software.broadinstitute.org/gatk/bestpractices/) from the Broad Institute. Sequencing reads were mapped to the reference human genome version hg19 (2009-02 release, http://genome.ucsc.edu/). Nucleotide changes observed in aligned reads were called and reviewed by using NextGENe software (SoftGenetics, State College, Pa). In addition to the detection of deleterious mutations and novel single nucleotide variants, a coverage- based algorithm developed in-house, eCNVscan, was used to detect large exonic deletions and duplications. The normalized coverage depth of each exon of a test sample was compared with the mean coverage of the same exon in the reference file, to detect copy number variants (CNVs).
Sequence variants were annotated using population and literature databases including 1000 Genomes (http://www.1000genomes.org/), dbSNP, GnomAD (http://gnomad.broadinstitute.org/), Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), HGMD and Online Mendelian Inheritance in man (OMIM, http://omim.org/). Sorting Intolerant from Tolerant (SIFT, http://sift.jcvi.org/www/SIFT_BLink_submit.html) and Polymorphism Phenotyping version 2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/) software were used to analyze the structure of the protein, predict the conservation and function domains and perform multiple sequence alignment. Variant interpretation was manipulated according to the American College of Medical Genetics (ACMG) guidelines [16].
We focused on 129 TAAD-related candidate genes in the literature [17-20] and database (https://hpo.jax.org/app/). The gene list is shown in Table S1.
Sanger sequencing
Sanger sequencing was performed for 71 relatives based on the consent of some patients.
Statistical analysis
Significant differences in normally distributed continuous variables were detected by one-way ANOVA, and those in nonnormally distributed variables in multiple groups were estimated by the Kruskal-Wallis test. Fisher’s exact test was used for the assessment of categorical variables between different groups. Spearman correlation was used to compare the percentages of rank variables. P values less than 0.05 were considered statistically significant (two-sided). All calculations were performed using SPSS 24.0 software.
Results
Patient clinical data
One hundred fifty-one consecutive patients from South China were collected in our cohort, including 129 sporadic and 22 familial TAAD patients. Males composed 80.1% (121) of the cohort, with 32.2% (39) having a history of smoking and 13.2% (16) having a history of alcohol use. Eighty-eight (58.3%) patients had hypertension; of these patients, 84.1% (74) were male, and only 15.9% were female. Thoracic aortic dissection (consisting of 108 Stanford A dissections and 14 Stanford B dissections) accounted for 80.8% patients, while thoracic aortic aneurysm accounted for 15.9% (24) of patients. Of 142 TAAD patients underwent cardiovascular surgery, 9 of 30 suspected MFS did not undergo surgery because they did not meet the surgical criteria, and 5 of them did not have aortic dilation or dissection but had a distinct family history or obvious manifestations of multiple systems. In particular, one pregnant woman suffering from Stanford type A aortic dissection at the age of twenty-eight, and who underwent complex surgery for aortic repair after delivery was also included. Notably, the average age at diagnosis of sporadic TAAD was older (48.5 years, range: 2 months-76 years) than that of familial TAAD (36.0 years, range: 19-50 years). All three patients under the age of eighteen were sporadic cases. In familial cases, the mean maximum aortic dilation diameter (5.9 cm) was larger than that in cases without any family history (5.0 cm), as expected. Clinical characteristics are shown in Table 1.
Table 1.
Patient clinical data of the cohort
| Clinical characteristics of the cohort | N (%) |
|---|---|
| Number | 151 (100) |
| Male | 121 (80.1) |
| Female | 30 (19.9) |
| Age, Mean | 46.7 |
| Min | 2 M |
| Max | 76 Y |
| <40 | 43 (28.5) |
| 40-60 | 82 (54.3) |
| >60 | 26 (17.2) |
| Tobacco use | 39 (25.8) |
| Alcohol use | 16 (10.6) |
| Hypertension | 88 (58.3) |
| Family history | 22 (14.6) |
| Undergone Aortic Surgery | 142 (94.0) |
| Thoracic AD | 122 (80.8) |
| Stanford A | 108 (88.5) |
| Stanford B | 14 (11.5) |
| TAA | 24 (15.9) |
| Suspected MFS | 36 (23.8) |
| BAV | 12 (7.9) |
| Aortic Root Size | |
| Maximum Aortic Diameter (cm), Median | 4.8 |
| Min | 1.2 |
| Max | 12.3 |
| Other CV | 37 (24.5) |
AD: aortic dissection, TAA: thoracic aortic aneurysm, MFS: Marfan syndrome, BAV: Bicuspid aortic valve, CV: cardiovascular, Other CV: includes coronary artery disease, atherosclerosis of thoracic aorta, abdominal aorta, arteria cervicalis, aortic regurgitation, mitral valve prolapse or congenital heart disease or arrhythmia, M: month, Y: year.
Variants of causative genes of TAAD
In the mixed cohort, we identified 116 variants in 47 TAAD-related genes in 94 (62.3%) subjects (Figure 1). Of the 73.3% (85/116) that were variants of uncertain significance (VUS), 26.7% (31/116) were pathogenic or likely pathogenic (P/LP), including FBN1, MYH11, SMAD3, TGFBR2, TGFBR1, LOX, B3GAT3, MYLK, SMAD6, and LTBP3 (Figure 2A; Table 2), 56.0% (65/116) were novel, and three were de novo. In contrast to previous reports, the main mutations were concentrated in the following genes: FBN1 (32, 27.6%), MYH11 (5, 4.3%), MCTP2 (4, 3.4%), SMAD3 (4, 3.4%), NOTCH3 (3, 2.6%), COL5A1 (3, 2.6%), APC (3, 2.6%), COL1A2 (3, 2.6%) and FBN2 (3, 2.6%) (Figure 1). Of note, FBN1 was still the most common causative gene in this study, regardless of family history and syndrome (Figure 2B, 2C); and the only pathogenic gene in familial cases (Figure 2B). The total frequency of TAAD in our cohort with disease-causing variants (P/LP) was 22.5% (34/151), which was far greater than the prevalence of deleterious mutations (3.9%, 4/102) in 102 patients with TAAD [14]. P/LP variants of sporadic TAAD were slightly more numerous than the P/LP variants of familial TAAD (17 vs 15, Figure 2B), and Arg1125Ter in FBN1 was identified in both unrelated probands from the aforementioned two cohorts. More than one variant was identified in 24 patients, 17 with sporadic TAAD (Table S2) and 7 with familial TAAD (Table S3) (Figure 3A). Four harbored three mutations, and one harbored four mutations. Interestingly, only 37.5% (9/24) present with syndrome manifestations. (Figure 3B) Of those variants, most were occurred in sporadic TAAD, whereas FBN1, COL5A1, MYH11 occurred in both two groups (Figure 3C).
Figure 1.
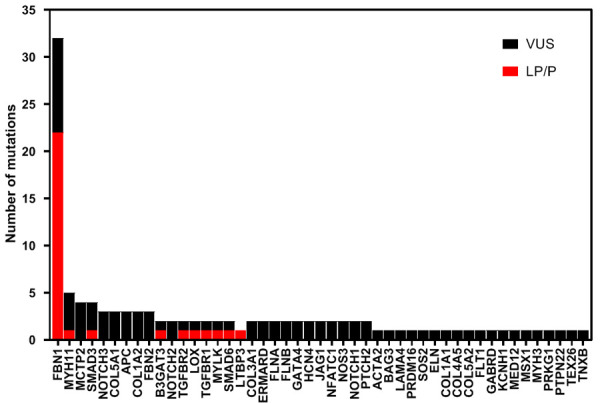
All mutations including pathogenic/likely pathogenic and variants of uncertain significance in the mixed cohort identified in 47 genes. P/LP: pathogenic or likely pathogenic, VUS: variant of uncertain significance.
Figure 2.
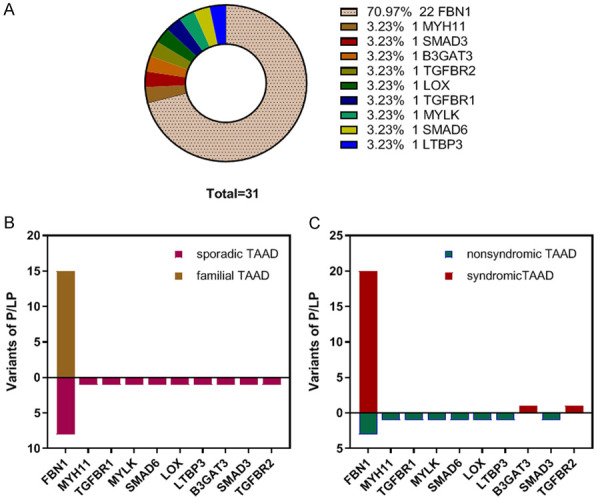
Pathogenic/likely pathogenic mutations identified in 10 genes. A: Gene distribution of pathogenic/likely pathogenic mutations; B: Pathogenic/likely pathogenic mutations in sporadic TAAD and familial TAAD. One mutation of FBN1 was identified in both unrelated probands from the aforementioned two cohorts; C: Pathogenic/likely pathogenic mutations in syndromic TAAD and non-syndromic TAAD. One mutation of FBN1 was identified in both unrelated probands from the aforementioned two cohorts. P/LP: pathogenic or likely pathogenic, VUS: variant of uncertain significance, sporadic TAAD: sporadic thoracic aortic aneurysm or dissection, familial TAAD: familial thoracic aortic aneurysm or dissection, syndromic TAAD: syndromic thoracic aortic aneurysm or dissection, nonsyndromic TAAD: nonsyndromic thoracic aortic aneurysm or dissection.
Table 2.
Clinical characteristics of patients with pathogenic/likely pathogenic mutations in the study
| ID | Sex | Age (year) | Disease | Clinical MFS | Family history | Max-aortic diameter (cm) | Surgery | Gene | Variant | Classification | Reported |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TAAD0043 | F | 37 | TAA | Y | Y | 50 | B+MVP+TVP | FBN1 | c.2023_2026del, p. Phe675ValfsX41 | P | Y |
| TAAD0027 | M | 28 | AD | Y | Y | 85 | B+TAR+DASI | FBN1 | c.2430delA, p. Glu810AspfsX36 | P | Novel |
| TAAD0038 | M | 48 | AD | Y | Y | NA | TEVER | FBN1 | c.3373C>T, p. Arg1125Ter | P | Y |
| TAAD0080 | F | 6 | / | Y | N | 18 | / | FBN1 | c.5788+5G>A | P | Y |
| TAAD0015 | M | 49 | AD | N | N | 60 | B+TAR+DASI+MVP+TVP | SMAD3 | c.1102C>T, p. Arg368Ter | P | Y |
| TAAD0046 | M | 52 | AD | Y | N | 48 | B+PAR+DASI+MVP | TGFBR2 | c.1483C>T, p. Arg495Ter | P | Y |
| TAAD0049 | F | 28 | AD | N | N | 64 | B+TAR+DASIa | FBN1 | c.3373C>T, p. Arg1125Ter | P | Y |
| TAAD0083 | M | 50 | AD | N | N | 50 | B+TAR+DASI | FBN1 | c.2860C>T, p. Arg954Cys | P | Y |
| TAAD0069 | M | 32 | AD | Y | N | 44 | AOR+TAR+DASI | FBN1 | c.6000C>A, p. Cys2000Ter | LP | Y |
| TAAD0074 | F | 23 | TAA | Y | N | 56 | B | FBN1 | c.2648G>A, p. Trp883Ter | LP | Y |
| TAAD0086 | M | 27 | AD | Y | N | 100 | B+TAR+DASI | FBN1 | C.3047delC, p. Thr1016LysfsX19 | LP | Novel |
| TAAD0091 | F | 23 | TAA | Y | N | 43 | / | FBN1 | c.6253T>G, p. Cys2085Gly | LP | Novel |
| TAAD0097 | M | 2 months | AoD | Y | N | 13 | / | B3GAT3 | C.47C>A, p. Ser16Ter | LP | Novel |
| TAAD0017 | F | 44 | AD | N | N | 44 | AOR+TAR+DASI | MYH11 | c.5838_5839del, p. Arg1946SerfsX2 | LP | Y |
| TAAD0021 | F | 50 | AD | N | N | 45 | AOR+TAR+DASI | FBN1 | c.2470delA, p. Ser824AlafsX22 | LP | Novel |
| TAAD0028 | M | 48 | AD | N | N | 63 | B+TAR+DASI+CABG | TGFBR1 | c.1459C>T, p. Arg487Trp | LP | Y |
| TAAD0031 | M | 47 | AD | N | N | 37 | AOR+TAR+DASI+AVP | MYLK | c.3665_3666del, p. Val1222GlufsX22 | LP | Novel |
| TAAD0041 | M | 58 | TAA | N | N | 43 | B | SMAD6 | c.220C>T, p. Gln74Ter | LP | Y |
| TAAD0048 | M | 38 | AD | N | N | 40 | AOR+TAR+DASI+AVP+MVR | LOX | c.433C>T, p. Gln145Ter | LP | Novel |
| TAAD0056 | M | 51 | AD | N | N | 49 | B+TAR+DASI+CABG | LTBP3 | c.169_170insAGGCGGGGGCGGGGCGC, p. Leu57GlnfsX16 | LP | Novel |
| TAAD0072 | M | 34 | TAA | Y | Y | 65 | B | FBN1 | c.4583-1G>A | LP | Y |
| TAAD0079 | M | 49 | AD | Y | Y | NA | B | FBN1 | c.7754T>C, p. Ile2585Thr | LP | Y |
| TAAD0081b | M | 31 | / | Y | Y | 30 | / | FBN1 | c.7113G>A, p. Trp2371Ter | LP | Y |
| TAAD0082b | M | 26 | TAA | Y | Y | 43 | / | FBN1 | c.7113G>A, p. Trp2371Ter | LP | Novel |
| TAAD0084 | F | 34 | TAA | Y | Y | 48 | MVP+TVP | FBN1 | c.6825_6837delins14, p. Gly2277AspfsX9 | LP | Novel |
| TAAD0085 | F | 44 | AD | Y | Y | 86 | B+TAR+DASI | FBN1 | c.2089delC, p. Gln697SerfsX21 | LP | Novel |
| TAAD0087b | F | 43 | AD | Y | Y | 72 | B+TAR+DASI+MVP+TVP | FBN1 | c.6575G>T, p. Cys2192phe | LP | Novel |
| TAAD0088b | F | 41 | TAA | Y | Y | 50 | / | FBN1 | c.6575G>T, p. Cys2192phe | LP | Novel |
| TAAD0092 | M | 49 | AD | Y | Y | 70 | B+TAR+DASI | FBN1 | c.316C>T, p. Gln106Ter | LP | Novel |
| TAAD0096 | M | 32 | TAA | Y | Y | 59 | B+TAR+DASI | FBN1 | c.1165_1166delTG, p. Cys389LeufsX62 | LP | Novel |
| TAAD0051 | M | 30 | AD | Y | Y | NA | B+TAR+DASI+MVP+TVP | FBN1 | c.2886C>G, p. Tyr962Ter | LP | Novel |
| TAAD0047 | M | 27 | AD | Y | Y | 68 | B+TAR+DASI | FBN1 | c.1838-2A>G | LP | Novel |
| TAAD0014 | M | 26 | AD | Y | Y | 74 | B+TAR+DASI | FBN1 | c.2538delA, p. Ile846MetfsX25 | LP | Novel |
| TAAD0099 | M | 27 | TAA | Y | Y | 87 | B+MVP+TVP | FBN1 | c.5296+1G>T | LP | Novel |
F: female; M: male; TAA: thoracic aortic aneurysm; AD: aortic dissection; AoD: aortic root dilatation NA: not available; B: Bentall procedure; TAR: total arch replacement; PAR: partial arch replacement; DASI: descending aorta stent implantation; MVP: mitral valve plastic; MVR: mitral valve replacement; TVP: tricuspid valve plastic; AVP: aortic valvuloplasty; AOR: ascending aorta replacement; CABG: coronary artery bypass grafting; TEVAR: thoracic endovascular aortic repair; P: pathogenic; LP: likely pathogenic; Two unrelated patients harboring the same mutation of FBN1 was Underlined.
aortic surgery was performed after Caesarean section and subtotal hysterectomy at the same stage.
two pairs of siblings.
Figure 3.
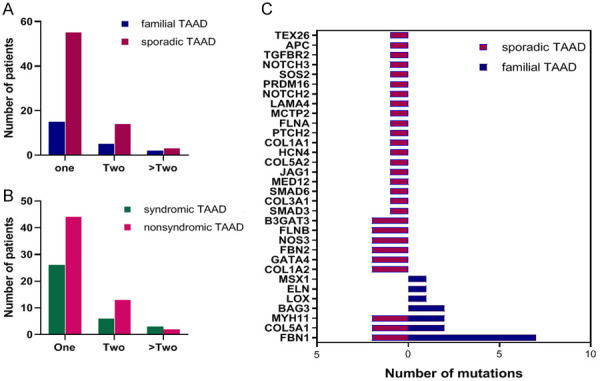
Multiple mutations identified in the study. A: The number of patients with family TAAD or sporadic TAAD in groups with different numbers of mutations; B: The number of patients with syndromic TAAD or nonsyndromic TAAD in groups with different numbers of mutations; C: Gene mutations in individuals with two or more mutations in sporadic TAAD and familial TAAD. familial TAAD: familial thoracic aortic aneurysm or dissection, sporadic TAAD: sporadic thoracic aortic aneurysm or dissection, syndromic TAAD: syndromic thoracic aortic aneurysm or dissection, nonsyndromic TAAD: nonsyndromic thoracic aortic aneurysm or dissection.
Sporadic TAAD
A total of 92 variants were identified in sporadic TAAD, the majority of which were VUSs (81.5%). Among the 17 P/LP variants, more than half (10/17, 58.8%) were in nonsyndromic subjects without a family history. Seventy-five (81.5%) variants were found to occur in 114 nonsyndromic TAAD patients without family history, and of these, 13.3% (10/75) were P/LP, which mainly involved FBN1 (3, 30.0%), SMAD3 (1, 10.0%), SMAD6 (1, 10.0%), MYH11 (1, 10.0%), TGFBR1 (1, 10.0%), MYLK (1, 10.0%), LOX (1, 10.0%) and LTBP3 (1, 10.0%) (Table 2). Among the VUSs, the proportion in FBN1 was 6.2% (4), in MCTP2 was 6.2% (4), and in each of the following genes was 4.6% (3): SMAD3, MYH11, COL1A2 and FBN2. Moreover, 44.6% of VUSs were predicted as “deleterious” by both SIFT and Polyphen2_HDIV. Interestingly, the novel VUS variant c.2635T>G (*879Glu) in MCTP2 appeared in two unrelated probands with type A aortic dissection without BAV.
Seventeen mutations were identified in 15 patients with suspected syndromic TAAD without family history, almost half of which were novel, three were de novo (two were P/LP in FBN1 and one was a VUS in TGFBR2). Of the patients displaying a phenotype highly suggestive of syndromic TAAD, ten were eventually confirmed as MFS, two were LDS with TGFBR2 mutations (P and VUS, respectively), one was B3GAT3-related syndrome (two compound heterozygous variants in B3GAT3), one harbored a VUS in COL4A5, and one patient was normal without gene mutation. The abnormal absent of α5 (type IV collagen) in the aortic media arising from the COL4A5 gene seemed to be related to the particular genotypes of early onset aortic dissection and aneurysm with Alport syndrome [21]. In addition, more than one mutation (in both APC and TEX26) was identified in patient TAAD0046 with LDS, both of which were associated with BAV [22,23], whereas the patient did not suffer aortic valve malformation and no previous evidence supports the correlation between each of the two genes and TAAD.
Unexpectedly, seven variants of the FBN1 gene were identified in seven patients (six were AD, one was thoracic aortic aneurysm [TAA] with BAV) with a mean age of 45.9 (range: 28-69) who had not been previously suspected to have MFS; three variants were P/LP, and three VUSs were predicted to be “deleterious” by both SIFT and Polyphen2_HDIV (Table 3). Indeed, after we revisited the clinical data, none of the patients presented obvious clinical manifestations according to the MFS Ghent criteria [15]. A 28-year-old pregnant woman with acute Stanford type A dissection harbored a pathogenic mutation in FBN1 (Arg1125Ter), and she gave birth to a child without congenital malformation at 36+ gestational weeks. Her mother died at the age of thirty because of an allergy as she described. Of the two patients who had a history of aortic surgery, patient TAAD0005 with a VUS (Glu616Lys) in FBN1 developed Stanford type A dissection after TEVAR (thoracic endovascular aortic repair), and patient TAAD0021 with novel LP (Ser824AlafsX22) in FBN1 underwent the Bentall procedure due to an aneurysm of the aortic sinus two years ago. Both patient TAAD0083 with the P variant Arg954Cys and patient TAAD0012 with the novel VUS Glu1449Val underwent emergency hospitalization after a sports accident. The first was tall but not thin (height of 1.83 m, weight of 85 kg) with a Z-score of 5.52, and the latter had a Z-score of 10.18 at the diagnosed age of 37. Moreover, TAAD0083 also harbored NOTCH3 (Val237Met) in addition to FBN1 variants. A variant (Val237Met) in NOTCH3 with a 0.002 frequency in East Asian was reported in a 70-year-old Japanese woman diagnosed with CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) with phenotypes of gait disturbance and dementia [24]. The same two variants were also present in the daughter of TAAD0083 without any clinical symptoms.
Table 3.
FBN1 mutation identified in nonsyndromic TAAD without family history
| ID | Sex | Age | Hypertension | Gene | Variant | Classification | SIFT | Polyphen2_HDIV | Disease | Surgery procedure | History of aortic surgery |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TAAD0005 | M | 49 | N | FBN1 | c.1846G>A p. Glu616Lys | VUS | D | D | AD | AVP+AOR+TAR+DASI | Y |
| TAAD0012 | M | 37 | N | FBN1 | c.4346A>T p. Glu1449Val | VUS | D | D | AD | B+PAR | N |
| TAAD0021 | F | 50 | N | FBN1 | c.2470delA, p. Ser824AlafsX22 | LP | / | / | AD | AOR+TAR+DASI | Y |
| TAAD0029 | M | 69 | Y | FBN1 | c.1825C>T p. Arg609Cys | VUS | D | D | AD | AVR+AOR+PAR+DASI | N |
| TAAD0037 | M | 38 | N | FBN1 | c.7559C>T p. Thr2520Met | VUS | T | D | TAA | AVR | N |
| FBN2 | c.577C>T p. Pro193Ser | VUS | T | D | |||||||
| TAAD0049 | F | 28 | Ya | FBN1 | c.3373C>T p. Arg1125Ter | P | / | / | AD | B+TAR+DASI; cesarean section + hysterectomyb | N |
| TAAD0083 | M | 50 | N | FBN1 | c.2860C>T p. Arg954Cys | P | / | / | AD | B+TAR+DASI | N |
| NOTCH3 | c.709G>A p. Val237Met | VUS | / | / |
transient gestational hypertension;
delivery before aortic repair in the same stage.
VUS: variant of uncertain significance; P: pathogenic; LP: likely pathogenic; D: deleterious; T: tolerated; B: Bentall procedure; AVP: aortic valvuloplasty; AOR: ascending aorta replacement; TAR: total arch replacement; DASI: descending aorta stent implantation; PAR: partial arch replacement; AVR: aortic valve replacement.
Familial TAAD
A total of 25 variants were identified among 22 familial TAAD patients; 60.0% (15) were classified as P/LP, all of which were associated only with FBN1. Of the 21 cases of clinically suspected MFS, 19 cases were ascertained to be MFS due to the FBN1 gene, one was familial TAA with an ACTA2 mutation, and one was the carrier of a VUS in APC mutation, which was predicted to be “deleterious” by SIFT and Polyphen2_HDIV. Notably, the nineteen-year-old subject with the APC mutation displayed aortic root aneurysm with BAV (Z-score of 13.28) and had a surgical history of aortic coarctation and ligation of PDA (patent ductus arteriosus) at the age of five and a family history of long slender limbs. The remaining patient suffered from TAA with BAV harboring a novel mutation in ERMARD. In addition, more than one mutation in genes other than FBN1 was detected in 7 suspected MFS patients from 4 unrelated families, including three pairs of siblings (Table S3).
Genotype-phenotype correlation
Age under 45 years at diagnosis, female sex, family history, and syndromic manifestations involving the aortic root were found to increase the likelihood of carrying a P/LP variant (Table 4). The younger patients were when they developed TAAD, the more likely they were to carry P/LP variants. The median age at diagnosis of people harboring LP/P variants was 35.5 years, which was lower than 48.5 years for VUSs (P=0.003) and then 53.0 years for no variant (P=0.000001) (Figure 4A). After ruling out syndromic TAAD, the median age at diagnosis of the P/LP group and the VUS group increased to 49.5 years and 51.0 years, respectively (Figure 4B). Moreover, the pathogenicity of the variant increased with the degree of aortic regurgitation (Ρ=0.253, P=0.002). More than one variant was identified in 25.53% (24/94) of gene-positive probands. The no variant group was significantly different from the one variant group (P=0.003). The median age decreased with the increasing number of variants in all but the group of four variants (Ρ=-0.286; P=0.000377) (Figure 4C). After excluding syndromic TAAD, the median age of the three variant group greatly increased from 39.5 years to 48 years, which was slightly higher than that of the two variants group (46 years) (Figure 4D). To compare the severity of the surgery patients with variant types, we assessed the maximum aortic diameter, length of hospital stay, length of ICU (intensive care unit) stay, and D-dimer level. P/LP variant carriers were apt to have a greater median aortic diameter (P=0.038; P=0.006) than the VUS or the N group; and a lower median D-dimer level (P=0.004) than the N group (Figure 5A, 5B). Interestingly, the median ICU duration of the P/LP group was lower than that of the N group (P=0.031), although the median hospitalization time was higher than that of the N or VUS group (differences were not significant) (Figure 5C, 5D). However, among the surgery subjects, the number of variants did not seem to correlate with length of hospital stay, length of ICU stay, maximum aortic diameter, or D-dimer level, all of which were significantly associated with disease severity.
Table 4.
Risk of carrying pathogenic /likely pathogenic mutations based on diverse phenotypes
| Total | Pathogenic/Likely pathogenic (percentage) | RR (95% CI) | P-value | |
|---|---|---|---|---|
| Family history | 22 | 17 (77.3) | 5.864 (3.565-9.644) | 2.6432E-9 |
| syndromic | 36 | 24 (66.7) | 7.667 (4.060-14.477) | 1.327E-11 |
| Age <45 | 64 | 22 (34.4) | 2.492 (1.334-4.656) | 0.003 |
| Female | 30 | 11 (36.7) | 1.929 (1.062-3.504) | 0.038 |
| Maximum aortic size>5 cm | 67 | 19 (28.4) | 1.791 (0.980-3.273) | 0.077 |
| Aortic root | 110 | 30 (27.3) | 2.795 (1.049-7.446) | 0.027 |
| D-dimmer>20000 | 21 | 3 (14.3) | 0.599 (0.147-1.928) | 0.410 |
Figure 4.
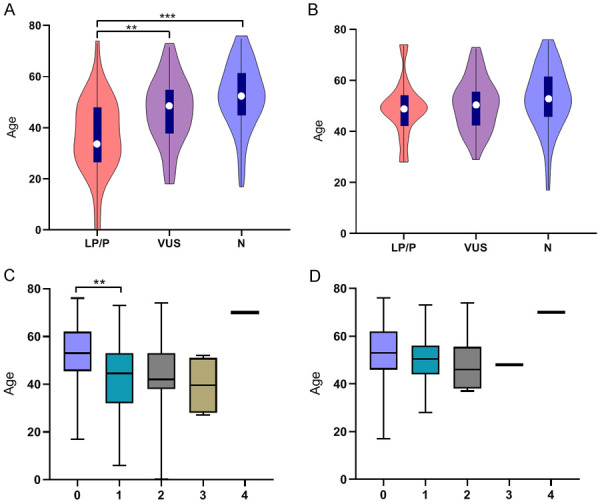
Correlations between clinical phenotype of the diagnosed age and genotypes. A: The diagnosed age of all patients with different mutation types; B: The diagnosed age of patients with different mutation types after eliminating clinically syndromic individuals; C: The diagnosed age of all patients with different numbers of mutations; D: The diagnosed age of patients with different numbers of mutations after eliminating clinically syndromic individuals. P/LP: pathogenic or likely pathogenic, VUS: variant of uncertain significance, N: no mutation. *P<0.05; **P<0.01; ***P<0.001.
Figure 5.
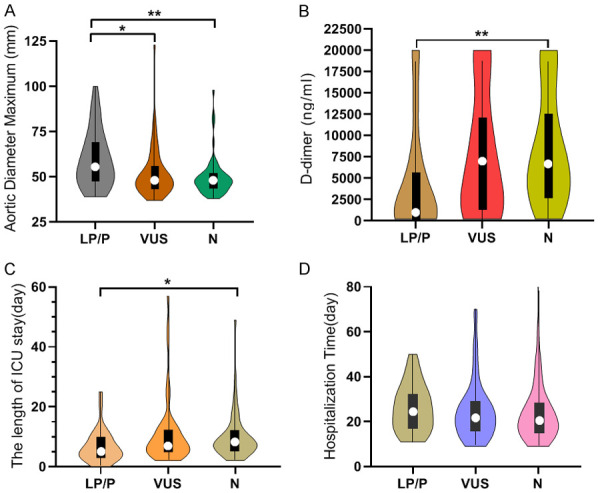
Correlations between clinical phenotypes and genotype of different mutation types in surgery subjects. A: Aortic diameter maximum of surgery patients in different mutation type groups; B: D-dimer of surgery patients in different mutation type groups; C: The length of ICU stay of surgery patients in different mutation type groups; D: The hospitalization time of surgery patients in different mutation type groups. P/LP: pathogenic or likely pathogenic, VUS: variant of uncertain significance, N: no mutation. *P<0.05; **P<0.01; ***P<0.001.
Discussion
The etiology and pathogenesis of aortic aneurysm/dissection is extremely complex and genetically heterogeneous. Causative genes could be responsible for the severe morbidity of 4.9%-18.6% TAAD [9,10,25-27]. Herein, we primarily focused on 129 sporadic TAAD patients in addition to 22 familial TAAD patients, which were categorized into syndromic and nonsyndromic groups, and spared no effort to explore the possible reason using the power of gene sequencing.
A total of 116 variants in 47 genes were identified by WES, 64.7% of which existed in sporadic TAAD without syndromes. Of the 26.7% that were pathogenic or likely pathogenic, almost one third were from sporadic patients without syndromes and involved FBN1, SMAD3, SMAD6, MYH11, TGFBR1, MYLK, LOX and LTBP3. The frequency of pathogenetic/likely pathogenetic variants was 22.5% in our cohort, which was higher than that in our previous reports [27]. The majority of variants were in FBN1 as expected, accounting for 27.6%, and 53.1% were in sporadic cases, which was in line with the suggestion of a common pathogenesis of aortic events in MFS and sporadic TAAD [28]. In contrast to other reports [27,29], the ACTA2 gene was not second to FBN1 as the most common mutated gene, as it accounted for only 0.9% of cases in this study. However, in other countries, ACTA2 mutations have been reported to be the most common cause of familial TAAD and causative of first-time sporadic and young-onset TAAD [30,31]. In agreement with a previous study, MYH11 and SMAD3 mutations were responsible for TAAD with frequencies of 4.3% and 3.4%, respectively [12]. Both altered smooth muscle cell (SMC) contraction and impaired TGF-β signaling proteins have adverse effects on the development of the aortic wall.
There are few reports about the LTBP3 gene (latent-transforming growth factor beta-binding protein 3) in correlation with thoracic aneurysm or dissection. Recently, three rare variants from two unrelated families were found to be causative of heritable TAAD, and heterozygous mutations in LTBP3 might increase the risk for later-onset thoracic aortic disease [7]. Moreover, the novel LP variant Leu57GlnfsX16 in LTBP3 was identified in a 51-year-old male who suffered acute Stanford type A aortic dissection without either family or syndromic history in our cohort. In fact, LTBP-3 mRNA is expressed at high levels in some tissues, including the human heart and skeletal muscle, which participate in the formation of a large latent complex (LLX) with LAP (latency-associated peptide) and TGFβ and the regulation of TGFβ activity [32-34]. Interestingly, LTBP3 has a similar structure of 8-Cys domains specific to FBN1; they colocalize in tissues, and loss of fibrillin 1 in the ECM prevents matrix incorporation of LTBP-3 [33,35]. Zilberberg et al proved that improper localization of LLX composed of LTBP-3 and TGFβ contributes to aortic disease progression in MFS [36].
To the best of our knowledge, four variants of the MCTP2 (multiple C2 domain and transmembrane region protein) gene were first reported to be responsible for nonsyndromic TAAD without a family history, accounting for 3.4% of TAAD cases (Figure 6A, 6B). The VUS *879Glu was present in two unrelated TAAD probands without a history of familial or syndromic TAAD. Both were Stanford Type A aortic dissections without BAV, which contrasts with what has been reported in a study identifying the MCTP2 gene in BAV patients [20,23]. However, the patient with Arg542* underwent surgery for aortic valve replacement due to the BAV phenotype. The MCTP2 gene located at chromosomal 15q26.2 is composed of three significant C2 domains, two transmembrane domains, a variable N-terminal region and a C-terminal sequence, which are involved in the biological processes of calcium-mediated signaling and multicellular organism development. In contrast to the other three families of trafficking proteins containing multiple C2 domains and one single transmembrane region, such as synaptotagmins, ferlins and E-Syts, the MCTP2 protein binds Ca2+ via the C2 domains in the absence of phospholipids with a high apparent affinity [37]. It was proven that Y235C located in the first C2 domain was attributed to the altered calcium-binding affinity of MCTP2 protein [38]. In this study, Lys232Serfs*3, Arg542* and Tyr496* were also located in the significant C2 domain (the first and the third, respectively), which includes perfect Ca2+-binding modules and was highly conserved.
Figure 6.
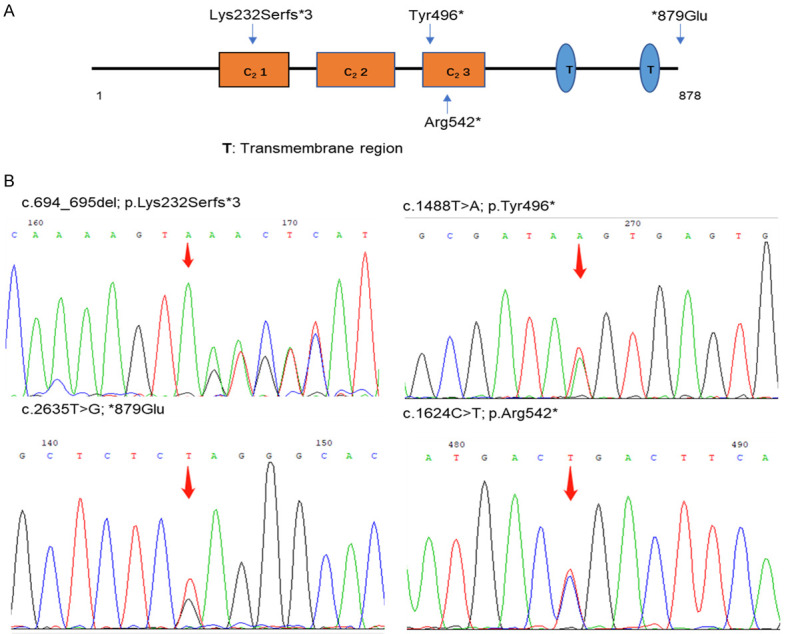
Four novel mutations identified in the MCTP2 gene. A. The structure of MCTP2 gene. B. Sequencing chromatographs of four mutations in MCTP2 gene.
It has been proposed that rare MCTP2 mutations might be attributed to cardiac malformations in the failure of endocardial cells to undergo EMT (epithelial-mesenchymal transformation) [38]. There is emerging evidence that the MCTP2 gene is primarily distributed in rat heart muscle except the testis, and functional knockdown could result in abnormal cardiac development such as coarctation of the aorta (CoA) [37,38]. EMT of endocardial cells and the morphogenesis of the endocardial cushion are fundamental to the remodeling of the outflow tract from the aorta, which is absent in MCTP2 morphants. Further mechanistic studies are needed to explain the inconsistent phenotypes of CoA and aortic dilation/dissection with the MCTP2 mutant.
Causative mutations tend to occur in young patients under the age of 45 years at diagnosis. Likewise, it seems that de novo variants were easily found in severely syndromic TAAD patients with young age at diagnosis. Furthermore, female sex, family history, and syndromic manifestations, involving the aortic root were found to increase the likelihood of carrying a P/LP variant. P/LP carriers seem to have low D-dimer levels, short ICU stays but increased maximum aortic diameter, and long hospitalization times, which could be because most young patients with early apparent symptoms were hospitalized in a state of nonstress and a nonemergency state before surgery. Consistent with a previous study, people with one or more mutations appeared to be younger [39]. However, the number of mutations did not correlate with these aforementioned indicators of disease severity.
Exome sequencing made it easier to obtain a definitive diagnosis despite complicated overlapping phenotypes between multiple syndromes or low gene penetrance among mutation carriers. In sporadic cases, we diagnosed a two-month-old boy, under the suspicion of MFS due to arachnodactyly and mild aortic root dilation as linkeropathies owing to novel rare B3GAT3 variants, which is an extremely rare hereditary connective tissue disorder affected by various glycosyltransferases in the biosynthesis of proteoglycans [40]. In addition, seven individuals with a mean age of 45.9 years who did not fulfill the revised Ghent criteria (prior to genetic testing), were found to harbor FBN1 variants, three of which were P/LP. Unexpectedly, none of the first-degree-relatives presented any suspected clinical features of MFS. As expert consensus suggests, screening immediate relatives, especially asymptomatic relatives, is particularly important to avoid the risk of sudden rupture of aortic aneurysms or dissection [41]. However, even among siblings with TAAD in each family, the identified variants were not entirely identical (Table S3). The FBN1 gene might be responsible for the remarkable phenotypes, but we cannot ignore the potential mutual effect for similar phenotypes between multiple genes. Instead of Sanger sequencing, performing WES among siblings when available will be more beneficial for disease diagnosis and discovery of new candidate genes.
One of the limitations of our study is that we did not eliminate other risk factors, such as hypertension, which was responsible for a considerable proportion of aortic dissection. Second, it would be better to identify the association between genotype and phenotype if TAA and dissection were grouped separately. Third, our study had a relatively limited sample size, so more data are needed to verify and confirm the results. We focused on exome sequencing of 129 TAAD-related genes, which could account for most of the phenotypes of TAAD, but might miss other potential regions or new genes. Whole-genome sequencing could make all the difference with the development of cost-effective gene analysis.
In summary, we found that the frequency of gene positivity (including VUS and LP/P) was 62.3% in the mixed cohort of sporadic and familial TAAD, and 80.6% of cases were nonsyndromic TAAD without a family history. Moreover, 26.7% of mutations were pathogenic or likely pathogenic, and almost one third of these were from sporadic cases without syndromes. In addition to focusing on core pathogenic genes involved in hereditary TAAD, paying more attention to de novo variants in sporadic cases and candidate genes in nonsyndromic cases without family history is more conducive to further mechanistic research about silent but life-threatening aortic events.
Acknowledgements
The study was supported by the Department of Cardiovascular Surgery, Guangdong Cardiovascular Institute, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences and grants from the National Key Research and Development Program of China (2017YFC1308003) and Guangzhou Science and Technology Program key projects (202002020037). Thanks for the participation and support of those families in the study.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldfinger JZ, Halperin JL, Marin ML, Stewart AS, Eagle KA, Fuster V. Thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2014;64:1725–1739. doi: 10.1016/j.jacc.2014.08.025. [DOI] [PubMed] [Google Scholar]
- 3.Isselbacher EM, Lino Cardenas CL, Lindsay ME. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation. 2016;133:2516–2528. doi: 10.1161/CIRCULATIONAHA.116.009762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbier M, Gross MS, Aubart M, Hanna N, Kessler K, Guo DC, Tosolini L, Ho-Tin-Noe B, Regalado E, Varret M, Abifadel M, Milleron O, Odent S, Dupuis-Girod S, Faivre L, Edouard T, Dulac Y, Busa T, Gouya L, Milewicz DM, Jondeau G, Boileau C. MFAP5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet. 2014;95:736–743. doi: 10.1016/j.ajhg.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, Varret M, Prakash SK, Li AH, d’Indy H, Braverman AC, Grandchamp B, Kwartler CS, Gouya L, Santos-Cortez RL, Abifadel M, Leal SM, Muti C, Shendure J, Gross MS, Rieder MJ, Vahanian A, Nickerson DA, Michel JB, Jondeau G, Milewicz DM. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milewicz DM, Regalado ES, Shendure J, Nickerson DA, Guo DC. Successes and challenges of using whole exome sequencing to identify novel genes underlying an inherited predisposition for thoracic aortic aneurysms and acute aortic dissections. Trends Cardiovasc Med. 2014;24:53–60. doi: 10.1016/j.tcm.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo DC, Regalado ES, Pinard A, Chen J, Lee K, Rigelsky C, Zilberberg L, Hostetler EM, Aldred M, Wallace SE, Prakash SK, Leal SM, Bamshad MJ, Nickerson DA, Natowicz M, Rifkin DB, Milewicz DM. LTBP3 pathogenic variants predispose individuals to thoracic aortic aneurysms and dissections. Am J Hum Genet. 2018;102:706–712. doi: 10.1016/j.ajhg.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gould RA, Aziz H, Woods CE, Seman-Senderos MA, Sparks E, Preuss C, Wünnemann F, Bedja D, Moats CR, McClymont SA, Rose R, Sobreira N, Ling H, MacCarrick G, Kumar AA, Luyckx I, Cannaerts E, Verstraeten A, Björk HM, Lehsau AC, Jaskula-Ranga V, Lauridsen H, Shah AA, Bennett CL, Ellinor PT, Lin H, Isselbacher EM, Lino Cardenas CL, Butcher JT, Hughes GC, Lindsay ME, Mertens L, Franco-Cereceda A, Verhagen JMA, Wessels M, Mohamed SA, Eriksson P, Mital S, Van Laer L, Loeys BL, Andelfinger G, McCallion AS, Dietz HC. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat Genet. 2019;51:42–50. doi: 10.1038/s41588-018-0265-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnaud P, Hanna N, Benarroch L, Aubart M, Bal L, Bouvagnet P, Busa T, Dulac Y, Dupuis-Girod S, Edouard T, Faivre L, Gouya L, Lacombe D, Langeois M, Leheup B, Milleron O, Naudion S, Odent S, Tchitchinadze M, Ropers J, Jondeau G, Boileau C. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD) Genet Med. 2019;21:2015–2024. doi: 10.1038/s41436-019-0444-y. [DOI] [PubMed] [Google Scholar]
- 10.Weerakkody R, Ross D, Parry DA, Ziganshin B, Vandrovcova J, Gampawar P, Abdullah A, Biggs J, Dumfarth J, Ibrahim Y, Bicknell C, Field M, Elefteriades J, Cheshire N, Aitman TJ. Targeted genetic analysis in a large cohort of familial and sporadic cases of aneurysm or dissection of the thoracic aorta. Genet Med. 2018;20:1414–1422. doi: 10.1038/gim.2018.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Proost D, Vandeweyer G, Meester JA, Salemink S, Kempers M, Ingram C, Peeters N, Saenen J, Vrints C, Lacro RV, Roden D, Wuyts W, Dietz HC, Mortier G, Loeys BL, Van Laer L. Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Hum Mutat. 2015;36:808–814. doi: 10.1002/humu.22802. [DOI] [PubMed] [Google Scholar]
- 12.Poninska JK, Bilinska ZT, Franaszczyk M, Michalak E, Rydzanicz M, Szpakowski E, Pollak A, Milanowska B, Truszkowska G, Chmielewski P, Sioma A, Janaszek-Sitkowska H, Klisiewicz A, Michalowska I, Makowiecka-Ciesla M, Kolsut P, Stawinski P, Foss-Nieradko B, Szperl M, Grzybowski J, Hoffman P, Januszewicz A, Kusmierczyk M, Ploski R. Next-generation sequencing for diagnosis of thoracic aortic aneurysms and dissections: diagnostic yield, novel mutations and genotype phenotype correlations. J Transl Med. 2016;14:115. doi: 10.1186/s12967-016-0870-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Lu C, Wu W, Liu Y, Wang R, Si N, Meng X, Zhang S, Zhang X. Application of next-generation sequencing to screen for pathogenic mutations in 123 unrelated Chinese patients with Marfan syndrome or a related disease. Sci China Life Sci. 2019;62:1630–1637. doi: 10.1007/s11427-018-9491-8. [DOI] [PubMed] [Google Scholar]
- 14.Ziganshin BA, Bailey AE, Coons C, Dykas D, Charilaou P, Tanriverdi LH, Liu L, Tranquilli M, Bale AE, Elefteriades JA. Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Ann Thorac Surg. 2015;100:1604–1611. doi: 10.1016/j.athoracsur.2015.04.106. [DOI] [PubMed] [Google Scholar]
- 15.Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arslan-Kirchner M, Arbustini E, Boileau C, Charron P, Child AH, Collod-Beroud G, De Backer J, De Paepe A, Dierking A, Faivre L, Hoffjan S, Jondeau G, Keyser B, Loeys B, Mayer K, Robinson PN, Schmidtke J. Clinical utility gene card for: hereditary thoracic aortic aneurysm and dissection including next-generation sequencing-based approaches. Eur J Hum Genet. 2016;24:e1–5. doi: 10.1038/ejhg.2015.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Renard M, Francis C, Ghosh R, Scott AF, Witmer PD, Ades LC, Andelfinger GU, Arnaud P, Boileau C, Callewaert BL, Guo D, Hanna N, Lindsay ME, Morisaki H, Morisaki T, Pachter N, Robert L, Van Laer L, Dietz HC, Loeys BL, Milewicz DM, De Backer J. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2018;72:605–615. doi: 10.1016/j.jacc.2018.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faggion Vinholo T, Brownstein AJ, Ziganshin BA, Zafar MA, Kuivaniemi H, Body SC, Bale AE, Elefteriades JA. Genes associated with thoracic aortic aneurysm and dissection: 2019 update and clinical implications. Aorta (Stamford) 2019;7:99–107. doi: 10.1055/s-0039-3400233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu B, Wang Y, Xiao F, Butcher JT, Yutzey KE, Zhou B. Developmental mechanisms of aortic valve malformation and disease. Annu Rev Physiol. 2017;79:21–41. doi: 10.1146/annurev-physiol-022516-034001. [DOI] [PubMed] [Google Scholar]
- 21.Kashtan CE, Segal Y, Flinter F, Makanjuola D, Gan JS, Watnick T. Aortic abnormalities in males with Alport syndrome. Nephrol Dial Transplant. 2010;25:3554–3560. doi: 10.1093/ndt/gfq271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dargis N, Lamontagne M, Gaudreault N, Sbarra L, Henry C, Pibarot P, Mathieu P, Bossé Y. Identification of gender-specific genetic variants in patients with bicuspid aortic valve. Am J Cardiol. 2016;117:420–426. doi: 10.1016/j.amjcard.2015.10.058. [DOI] [PubMed] [Google Scholar]
- 23.Bonachea EM, Zender G, White P, Corsmeier D, Newsom D, Fitzgerald-Butt S, Garg V, McBride KL. Use of a targeted, combinatorial next-generation sequencing approach for the study of bicuspid aortic valve. BMC Med Genomics. 2014;7:56. doi: 10.1186/1755-8794-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uchino M, Hirano T, Uyama E, Hashimoto Y. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) and CADASIL-like disorders in Japan. Ann N Y Acad Sci. 2002;977:273–278. doi: 10.1111/j.1749-6632.2002.tb04826.x. [DOI] [PubMed] [Google Scholar]
- 25.Overwater E, Marsili L, Baars MJH, Baas AF, van de Beek I, Dulfer E, van Hagen JM, Hilhorst-Hofstee Y, Kempers M, Krapels IP, Menke LA, Verhagen JMA, Yeung KK, Zwijnenburg PJG, Groenink M, van Rijn P, Weiss MM, Voorhoeve E, van Tintelen JP, Houweling AC, Maugeri A. Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Hum Mutat. 2018;39:1173–1192. doi: 10.1002/humu.23565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campens L, Callewaert B, Muiño Mosquera L, Renard M, Symoens S, De Paepe A, Coucke P, De Backer J. Gene panel sequencing in heritable thoracic aortic disorders and related entities - results of comprehensive testing in a cohort of 264 patients. Orphanet J Rare Dis. 2015;10:9. doi: 10.1186/s13023-014-0221-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang M, Yu C, Chen S, Xiong W, Li X, Zeng R, Zhuang J, Fan R. Identification of novel clinically relevant variants in 70 Southern Chinese patients with thoracic aortic aneurysm and dissection by next-generation sequencing. Sci Rep. 2017;7:10035. doi: 10.1038/s41598-017-09785-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LeMaire SA, McDonald ML, Guo DC, Russell L, Miller CC, Johnson RJ, Bekheirnia MR, Franco LM, Nguyen M, Pyeritz RE, Bavaria JE, Devereux R, Maslen C, Holmes KW, Eagle K, Body SC, Seidman C, Seidman JG, Isselbacher EM, Bray M, Coselli JS, Estrera AL, Safi HJ, Belmont JW, Leal SM, Milewicz DM. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43:996–1000. doi: 10.1038/ng.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng J, Guo J, Huang L, Wu Q, Yin K, Wang L, Zhang T, Quan L, Zhao Q, Cheng J. Genetic diagnosis of acute aortic dissection in South China Han population using next-generation sequencing. Int J Legal Med. 2018;132:1273–1280. doi: 10.1007/s00414-018-1890-9. [DOI] [PubMed] [Google Scholar]
- 30.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 31.Morisaki H, Akutsu K, Ogino H, Kondo N, Yamanaka I, Tsutsumi Y, Yoshimuta T, Okajima T, Matsuda H, Minatoya K, Sasaki H, Tanaka H, Ishibashi-Ueda H, Morisaki T. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD) Hum Mutat. 2009;30:1406–1411. doi: 10.1002/humu.21081. [DOI] [PubMed] [Google Scholar]
- 32.Penttinen C, Saharinen J, Weikkolainen K, Hyytiäinen M, Keski-Oja J. Secretion of human latent TGF-beta-binding protein-3 (LTBP-3) is dependent on co-expression of TGF-beta. J Cell Sci. 2002;115:3457–3468. doi: 10.1242/jcs.115.17.3457. [DOI] [PubMed] [Google Scholar]
- 33.Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-β-binding proteins. Matrix Biol. 2015;47:44–53. doi: 10.1016/j.matbio.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Dabovic B, Annes JP, Rifkin DB. Latent TGF-beta binding protein-3 (LTBP-3) requires binding to TGF-beta for secretion. FEBS Lett. 2002;517:277–280. doi: 10.1016/s0014-5793(02)02648-0. [DOI] [PubMed] [Google Scholar]
- 35.Robertson I, Jensen S, Handford P. TB domain proteins: evolutionary insights into the multifaceted roles of fibrillins and LTBPs. Biochem J. 2011;433:263–276. doi: 10.1042/BJ20101320. [DOI] [PubMed] [Google Scholar]
- 36.Zilberberg L, Phoon CK, Robertson I, Dabovic B, Ramirez F, Rifkin DB. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc Natl Acad Sci U S A. 2015;112:14012–14017. doi: 10.1073/pnas.1507652112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin OH, Han W, Wang Y, Südhof TC. Evolutionarily conserved multiple C2 domain proteins with two transmembrane regions (MCTPs) and unusual Ca2+ binding properties. J Biol Chem. 2005;280:1641–1651. doi: 10.1074/jbc.M407305200. [DOI] [PubMed] [Google Scholar]
- 38.Lalani SR, Ware SM, Wang X, Zapata G, Tian Q, Franco LM, Jiang Z, Bucasas K, Scott DA, Campeau PM, Hanchard N, Umaña L, Cast A, Patel A, Cheung SW, McBride KL, Bray M, Craig Chinault A, Boggs BA, Huang M, Baker MR, Hamilton S, Towbin J, Jefferies JL, Fernbach SD, Potocki L, Belmont JW. MCTP2 is a dosage-sensitive gene required for cardiac outflow tract development. Hum Mol Genet. 2013;22:4339–4348. doi: 10.1093/hmg/ddt283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Z, Zhou C, Tan L, Chen P, Cao Y, Li X, Yan J, Zeng H, Wang DW, Wang DW. A targeted sequencing approach to find novel pathogenic genes associated with sporadic aortic dissection. Sci China Life Sci. 2018;61:1545–1553. doi: 10.1007/s11427-018-9382-0. [DOI] [PubMed] [Google Scholar]
- 40.Baasanjav S, Al-Gazali L, Hashiguchi T, Mizumoto S, Fischer B, Horn D, Seelow D, Ali BR, Aziz SA, Langer R, Saleh AA, Becker C, Nurnberg G, Cantagrel V, Gleeson JG, Gomez D, Michel JB, Stricker S, Lindner TH, Nurnberg P, Sugahara K, Mundlos S, Hoffmann K. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am J Hum Genet. 2011;89:15–27. doi: 10.1016/j.ajhg.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verhagen JMA, Kempers M, Cozijnsen L, Bouma BJ, Duijnhouwer AL, Post JG, Hilhorst-Hofstee Y, Bekkers SCAM, Kerstjens-Frederikse WS, van Brakel TJ, Lambermon E, Wessels MW, Loeys BL, Roos-Hesselink JW, van de Laar IMBH National Working Group on BAV & TAA. Expert consensus recommendations on the cardiogenetic care for patients with thoracic aortic disease and their first-degree relatives. Int J Cardiol. 2018;258:243–248. doi: 10.1016/j.ijcard.2018.01.145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.