

Abstract
Objective: Due to the tumor immune microenvironment (TIME) complexity and cancer heterogeneity, the clinical outcomes of hepatocellular carcinoma (HCC) are barely elicited from the conventional treatment options, even from the promising anti-cancer immunotherapy. As a suppressive TIME-related marker, the role played by cyclooxygenase-2 (COX-2) in HCC TIME, and its potential effects on anti-cancer T cell immune response remains unknown. In our study, to investigate the COX-2-dependent immune regulation pathway, we verified that the macrophages phenotypes were correlated to COX-2/PGE2 expressions among HCC patients. A multi-cellular co-culture platform containing HCC cells, macrophages, and T cells were established to mimic HCC TIME in vitro and in animal model. M2 macrophage polarization and activated CD8+ T cells exhaustion were observed under high COX-2 levels in HCC cells, with further evaluation using CRISPR/Cas9-based PTGS2 knocking out and COX-2 blockade (celecoxib) treatment controls. PGE2, TGF-β, Granzyme B, and IFN-γ levels were testified by flow cytometry and ELISA to fully understand the mechanism of COX-2 suppressive effects on T cell-based anti-HCC responses. The activation of the TGF-β pathway evaluated by auto-western blot in T cells was confirmed which increased the level of phosphorylated Smad3, phosphorylated Samd2, and FoxP1, leading to T cell de-lymphotoxin. In conclusion, high COX-2-expressing HCC cell lines can induce anti-tumor abilities exhaustion in activated CD8+ T cell through M2 TAMs polarization and TGF beta pathway. COX-2 inhibitors may reduce the inhibitory effect on CD8+ T cells through regulating TAMs in TIME, thus enhance the T cell-based cytotoxicity and improve the prognosis of HCC patients.
Keywords: Hepatocellular carcinoma, adoptive cell therapy, cyclooxygenase-2, M2 macrophage, T lymphocytes
Introduction
With approximately 0.89 million annual deaths, of which hepatocellular carcinoma (HCC) accounts for the majority, liver cancer is the fourth most common cause of cancer-related death globally. According to the projection from World Health Organization, by 2030 there will be 10% more patient deaths worldwide [1]. Despite universal HBV vaccination, increasing cure rates of HCV infection, and numerous advances in early diagnostic techniques, which can hopefully reduce this burden, the conventional treatment options, such as adjuvant therapies after surgical resection or ablation, only achieved limited responses in HCC patients, and barely elicited the clinical outcomes [2]. Compared with the breakthroughs happened among other cancer types, novel personalized anti-cancer strategies to eliminate HCC cells and prevent postoperative recurrence or metastasis need further exploration.
In recent years, numerous strategies for anti-cancer immune system mobilization or adoptive cell immunotherapy have garnered the most attentions in HCC [3,4] and other malignant diseases [5]. Based on new scientific and technological advances in onco-immunology, the ideas to normalizing antitumor immunity, such as immune checkpoint inhibitors (ICI), have achieved huge success in mediating complete, durable antitumor responses in most cancer types [6]. In addition, in spite of hardness finding specific Tumor-associated antigens, many cancer enhancement immunotherapy approaches, especially chimeric antigen receptors (CAR) T cells [7] and DC vaccines [8], are still promising for many treatments of malignances. However, in HCC, ICI5, CAR-T cells [9] and vaccination [10] have similarly failed to elicit clinical responses, and, subsequently, shown no advantage for overall survival. Development of favorable immunotherapeutic approaches for HCC patients is still challenging [11].
To date, it is known that the tumor immune microenvironment (TIME) plays an important role in affecting the anti-cancer response [12]. In contrast to most other malignancies, HCC almost arises in chronic inflammation, while the liver is a central player both in immunomodulation and immunotolerance [13]. The complexity of HCC TIME drives a unique pattern depicted by anti-tumor immune regulators, including tumor-associated macrophages (TAMs) [14], Tregs and myeloid-derived suppressor cells (MSDC) [15], and effectors which mainly represent T cells immunity [16]. As evidenced in many HCC expreimental models, revealing potential novel suppressive TIME-related markers which can impair the T cell cytotoxity and induce exhastion in HCC will create promising targets for improving immunotherapy outcomes [17].
Normally considered as an inflammation related protein, cyclooxygenase-2 (COX-2) overexpression represents poor prognosis in many cancer types and, notably, has been strongly related to TIME formation [18]. In past decades, many studies demonstrated that the elevated COX-2 level can promote HCC tumorigenesis and progression, and subseqently be a mechanism-based therapatic target in severel HCC combined treatment clinial trials [19-21]. However, using mainly mice cells, only one recent study has correlated macrophages with COX-2 in HCC [22]. The relationship between COX-2 and HCC TIME has not been elucidated so far.
In our study, depending on multi-cellular co-culture platform both in vitro and in NOD-Prkdcscid-II2rgnull mice model for TIME mimic, we aimed to investigate that the correlation among the expression of COX-2 in human HCC, activated CD8+ T cell exhaustion and macrophages polarization.
Materials and methods
Study approval
Permission to use the tissues for research purposes was provided by Peking University People’s Hospital Ethics Committee. The ethics committee at the Peking University People’s Hospital (Beijing, China) approved the current study (permission: 2018PHB257). Ethical approval for the use of peripheral blood from healthy donors was obtained from the Peking University People’s Hospital Ethics Committee. All patients signed an informed consent form.
Cell culture and reagents
Human hepatocellular carcinoma cell lines SNU423 and BEL7402, obtained from China Type Culture Collection (Shanghai, China), were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco, 21870076) supplemented with 10% FBS (Gibco, 10270-106) and 100 IU/ml penicillin and streptomycin (Gibco, 1514-122) at 37°C in a 5% CO2 atmosphere. SNU423 cells were transferred to Beijing Sino Biological Co., Ltd. (Beijing, China) for the construction of SNU423COX2-/- cell line. CRISPR/Cas9 was used for PTGS2 knockout. The sgRNA’s sequence (5’-TCAAGACAGATCATAAGCGA-3’) was cloned into pLV vectors. HEK293T cells were co-transfected with pLV vector, pSPAX2, and pMD2. G plasmids, and the lentiviruses were precipitated using PEG8000-NaCl. The HCC cells were transduced with the lentivirus and selected 48 hours later using 3 ug/mL puromycin (MCE, HY-B1743A). The single-cell clone was obtained by repeated pressure culture with puromycin. The PGE2 synthesis inhibitor celecoxib (MCE, HY-14398) was used at a concentration of 25 µM. Additional information is provided in Supplementary Methods.
Peripheral blood mononuclear cell (PBMC) isolation
PBMCs from the healthy donors were isolated by centrifugation over a Ficoll gradient (GE, 17144002), 2000 r, 30 min. CD14+ cells were magnetically labeled with CD14 microbeads and positively selected by stem cell technology (Stem Cell, 17858). CD14+ monocytes were cryopreserved at -80°C with CryoStor CS10 (Stem Cell, 07930) for subsequent coculture.
Amplification and activation of CD8+ T cells
The CD3/CD28 monoclonal antibody (Invitrogen, 16-0037-85/16-0289-35) was used to coat the bottom of T25 flasks at 4°C overnight (5 ml PBS+25 ul CD3+10 ul CD28). After isolating CD14+ monocytes from PBMCs, the remaining cells were plated in T25 flasks coated with the CD3/CD28 mAb, and IL-2 (Peprotech, 200-02, 10 U/ml) was added to the medium. After 7 days, CD8+ T cells were magnetically labeled with CD8 microbeads and positively selected by stem cell technology (Stem Cell, 17853).
Establishment of the co-culture model
CD14+ monocytes were sorted out from PBMC and cryopreserved at -80°C, and the remaining cells were placed in a T25 culture flask coated with the CD3/CD28 monoclonal antibody. On day 5, cryopreserved CD14+ monocytes were recovered, and the co-expression of CD163/CD206 in CD14+ monocytes was detected by flow cytometry. CD14+ monocytes (0.1 million) were cocultured with hepatocellular carcinoma cell lines (0.5 million) at a ratio of 1:5 in 12-well plate. Hepatocellular carcinoma cell lines were placed between two layers of rat tail collagen by a sandwich three-dimensional culture method, and monocytes were placed in the upper layer of a transwell co-culture chamber (Corning, 3460), and cocultured with hepatocellular carcinoma cell lines for 2 days. On day 7, CD8+ T cells were sorted out from cells in the T25 culture flask. The expression of CD28, CD25, CD107A in CD8+ T cells was detected by flow cytometry. At this time, CD8+ T cells were added into the co-culture system for 24 hours. After 24 hours, the expression of Granzyme B and IFN-γ in CD8+ T cells and the medium were detected. During the whole co-culture process, the co-culture system contained 0.5 million hepatocellular carcinoma cell lines, 0.1 million primary CD14+ monocytes, and 0.1 million primary CD8+ T cells. Additional information is provided in Supplementary Methods and Figures.
Flow cytometry
Ex vivo CD14+ monocytes and CD8+ T cells cocultured with hepatocellular carcinoma cell lines were analyzed by validated flow cytometry methods, with a Beckman flow cytometer (Beckman). Surface marker expression was analyzed with flow cytometry using the following fluorochrome-labeled monoclonal antibodies: anti-CD14-APC (BioLegend, 325607), anti-CD163-PE-Cy7 (BioLegend, 326514), anti-CD206-PE (BioLegend, 321105), anti-CD8-APC-CY7 (BioLegend, 344714), anti-CD28-Percp-CY5.5 (BioLegend, 302921), anti-CD25-PE (BioLegend, 302605), anti-CD107A-APC (BioLegend, 328619), anti-Granzyme B-APC (Biolegend, 396407) and anti-IFN-γ-PE (BioLegend, 506506). Equivalent amounts of isotype-matched control antibodies and unstained cells were included in all experiments as negative and autofluorescence controls. Data were analyzed with CytExpert software (Beckman), after gating on the lymphoid and myeloid populations in the FSC/SSC plot. Values were expressed as the percent ratio of the marker of interest over that in the unstained cells. IFN-γ and Granzyme B in the medium were detected by the flow cytometry kit (BioLegend), and all operation steps were performed according to the product instructions. LEGENDplex v8.0 software (BioLegend) was used to analyze the results of the flow cytometry assay.
ELISA
A PGE2 ELISA assay kit (R&D system, KGE004B) was used to detect the level of PGE2 in hepatocellular carcinoma tissue lysate and culture medium, TGF-β ELISA assay kit (Thermo, BMS249-4) was used to detect the content of TGF-β in culture medium, and IFN-γ ELISA kit (Thermo, BMS228) and Granzyme B ELISA assay kit (Thermo, BMS2027) were used to detect the levels of IFN-γ and Granzyme B in CD8+ T cells lysates. All procedures followed the instructions of the kit.
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNA of normal liver tissue, BEL7402 cells, SNU423 cells and SNU423COX2-/- cells were extracted by the Trizol method, and the high quality of RNA was verified by UV analysis. RNA (1 µg) was reversed transcribed with Invitrogen SuperScript III Reverse Transcriptase to obtain cDNA. PCR primers were designed and synthetized by Shanghai Invitrogen Biotechnology Co., Ltd. (Shanghai, China). GAPDH was used as the internal reference. The following primers were used for PCR analyses: human PTGS2 forward, 5’-CTGGCGCTCAGCCATACAG-3’; human PTGS2 reverse, 5’-CACCTCGGTTTTGACATGGGT-3’; human GAPDH forward, 5’-CCATGGGTGGAATCATATTGGA-3’; human GAPDH reverse, 5’-TCAACGGATTTGGTCGTATTGG-3’. PCR amplification conditions were as follows: pre-denaturation at 95°C for 3 min, denaturation at 95°C for 15 s, annealing at 60°C for 1 minute, and extension at 72°C for 1 min for a total of 40 cycles, and finally extension at 72°C for 10 min. The PCR results were analyzed by iQ5 Real-Time PCR Systems (Bio-Rad). The threshold was manually selected at the lowest point of the logarithmic amplification curve to obtain the Ct value of all reaction tubes. The data were analyzed by the 2^ΔΔCt method, represented as the ratio of the target gene expression between the experimental group and the control group and calculated by the formula ΔΔCt = [Ct (target gene) - Ct (reference gene)] experimental group - [Ct (target gene) - Ct (reference gene)] control group. Real-time PCR was set up with the SYBR Green PCR Master Mix (Promega Corporation). The experiment was conducted three times and the mean value was calculated.
Immunofluorescence analysis
Sections (4 µm) and cells were stained using standard immunofluorescence techniques for the expression of COX-2 (Abcam, ab15191), CD163 (Abcam, ab156769) and CD206 (Abcam, ab64693). All the stained slides were scanned using a Leica confocal microscope. The size limit for particle analysis was carefully chosen to include only macrophages. Damaged samples were excluded from the analysis. The data were analyzed in a double-blinded fashion. Additional information is provided in Supplementary Methods.
Immunohistochemical analysis
The tissue samples of hepatocellular carcinoma patients were collected and made into a 4 um tissue chip. Tissue chip were stained using standard immunohistochemical techniques for the expression of COX-2 (DAB, Abcam, ab64238), CD163 (DAB, Abcam, ab183159) and CD206 (AP-red, Abcam, ab183159). All the stained slides were scanned using automatic digital slice scanning system (KFBIO, KF-PRO-120). The size limit for particle analysis was carefully chosen to include only macrophages. Damaged samples were excluded from the analysis. The data were analyzed in a double-blinded fashion. Additional information is provided in Supplementary Methods.
Automated western blot analysis
CD8+ T cells were lysed with protein lysis buffer (CST) and stored at -80°C. The Pierce BCA Protein Assay Kit was used to detect protein concentration. Then, a multifunction automatic western blot quantitative analysis system Jess (proteinsimple) was used to detect the expression of target proteins in CD8+ T cells. Antibodies against the following target proteins were used: Smad2 (Abcam, ab40855), Smad3 (Abcam, ab40854), pSmad2 (Abcam, ab188334), pSmad3 (Abcam, ab52903), and FoxP1 (Abcam, ab134055). β-Actin (CST, 4970T) was used as the internal reference. The entire experiment was performed three times. Compass for Simple Western software (proteinsimple) was used to analyze the results of the automated Western blotting.
Establishment of the in vivo model
5 million SNU423 hepatocellular carcinoma cell lines were implanted under the skin of NPG mice (NOD, Cg-Prkdcscid Il2rgtmlVst/Vst, one of a series of high-immunodeficiency mouse models independently developed by Beijing Weitong Biotechnology Co., LTD., and the obtained Il2rg gene knockout mice were returned to the high-immunodeficiency model established under the background of NOD-SCID) at age of 7 weeks old. During this period, PBMCs from healthy donors were collected, and CD14+ monocytes were sorted out and stored at -80°C. After 2 weeks, the activated and amplified CD8+ T cells were sorted out and mixed with the recoverd CD14+ monocytes at a ratio of 1:1 (the total number of cells was 5 million), and then the mixture was transferred back into the tumor-forming NPG mice through the tail vein. During this period, celecoxib was injected into mice by intraperitoneal injection. Celecoxib was dissolved in DMSO at 5 mg/ml, further diluted 10 times in 0.5% methylcellulose with 0.025% Tween 80 and injected (IP) daily at a dose of 5 mg/kg body weight for 2 weeks. After 2 weeks, some mice subcutaneous tumors were digested into single-cell suspension, and the ratio of CD163/CD206 double-positive cells in the tissues was analyzed by flow cytometry. The human CD8+ T cells in the single cell suspension were sorted out, and the content of IFN-γ and Granzyme B in the cells was analyzed by ELISA technology, and the expression of TGF-β related signal pathways was analyzed by automated western blot technology. ELISA technology was used to analyze the content of PGE2 and TGF-β in the remaining tumor tissues.
Statistical analysis
All data were evaluated using Prism software version 8 (GraphPad). Measurement data were presented as the mean ± standard error of mean. A linear correlation between 2 continuous variables was tested with the R2 coefficient of determination. Data consistent with normal distribution were analyzed using a t-test, and comparisons among multiple groups were analyzed using one-way ANOVA. P<0.05 was considered statistically significant. The data from representative experiments were obtained from triplicate cultures with cells from individual donors. Each independent experiment was replicated at least 3 times.
Results
In tissue chips and paraffin sections of HCC patients, the expression of COX-2 was correlated with M2 macrophage markers-CD163/CD206, and identification of three HCC cell lines with different expression of COX-2
To investigate the expression of COX-2 in HCC patients and its correlation with the number of M2 macrophages, we stained tissue chips of 30 HCC patients by immunohistochemical staining (Figure S3). Meanwhile we stained paraffin sections of one normal liver and 5 HCC patient livers by immunofluorescence staining. Patients with high COX-2 expression were found to have significantly higher numbers of M2 macrophages (cells with co-expression of CD163 and CD206) than patients with low COX-2 expression (Figure 1A, 1B, 1D-F). Moreover, we used ELISA to detect the levels of PGE2 in the tissues of the 35 HCC patients, and the results showed that the levels of PGE2 in HCC patients with high COX-2 expression were significantly higher than those of patients with low COX-2 expression (Figure 1C and 1G). Finally, immunofluorescence staining and RT-PCR were used to identify two HCC cell lines and one PTGS2 knockout cell line with different COX-2 expression, namely, BEL7402 with low COX-2 expression, SNU423 with high COX-2 expression, and SNU423COX2-/- with low COX-2 expression (Figure 1H-J). The ELISA results of the culture medium of the three HCC cell lines showed that the level of PGE2 in the supernatant of SNU423 cells was significantly higher than that of BEL7402 cells and SNU423COX2-/- cells (Figure 1K).
Figure 1.

Analysis of clinical samples of HCC patients and HCC cell lines. A. Immunohistochemical staining of COX-2 and M2 macrophages (CD163/CD206) on tissue array of 30 patients with hepatocellular carcinoma. Scale bars = 200 µm. B. Linear analysis of immunohistochemical staining of COX-2 and M2 macrophages (CD163/CD206) in tissue chips of 30 patients with hepatocellular carcinoma. C. Linear analysis of COX-2 immunohistochemical staining on tissue array of 30 hepatocellular carcinoma patients and PGE2 ELISA results in the corresponding tissue. D. Immunofluorescence staining of COX-2 and M2 macrophages (CD163/CD206) in paraffin sections of a normal liver and five patients’ tissue with hepatocellular carcinoma. Scale bars = 100 µm. E-G. Histogram and statistical analysis of COX-2 and M2 macrophages (CD163/CD206) in paraffin sections of 5 hepatocellular carcinoma patients, and PGE2 ELISA results in the corresponding tissue. H and I. Immunofluorescence staining and histogram of COX-2 in three hepatocellular carcinoma cell lines (BEL7402, SNU423, SNU423COX2-/-). Scale bars = 100 µm. J. COX-2 gene expression in two hepatocellular carcinoma cell lines (BEL7402, SNU423, SNU423COX2-/-) relative to that in normal liver and the corresponding statistical analysis. K. Histogram and statistical analysis of ELISA detection of PGE2 secreted by three liver cancer cell lines (BEL7402, SNU423, SNU423COX2-/-). Values are the mean ± SEM of a minimum of 3 independent experiments. *P<0.05; **P<0.005; ***P<0.0005; ****P<0.0001.
HCC cell lines with high COX-2 expression could induce more human primary CD14+ monocytes to polarize to M2 macrophages
Using flow cytometry, it can be clearly seen that after co-culture of human primary CD14+ monocytes with BEL7402, SNU423 cells, or SNU423COX2-/- cells for 48 h, the number of M2 macrophages (polarized from CD14+ monocytes) induced by SNU423 cells was higher (Figure 2A and 2B). The ELISA results of the co-culture media also showed that the level of PGE2 was higher in the medium of SNU423+M0 group (Figure 2C), and the induced M2 macrophages secreted more TGF-β (Figure 2D). Then, in order to further demonstrate that COX-2 and downstream PGE2 were responsible for the polarization of monocytes into M2 macrophages, we added the COX-2 selective inhibitor celecoxib to the co-culture system of SNU423 cells. The results of flow cytometry showed that after the addition of celecoxib (25 µM), the proportion of monocytes polarized into M2 macrophages was significantly reduced (Figure 2E and 2F), and the levels of PGE2 in the co-culture system and TGF-β secreted by M2 macrophages were significantly reduced (Figure 2G and 2H).
Figure 2.

Co-culture of hepatocellular carcinoma cell lines with human primary monocytes. A and B. Flow cytometric detection results and statistical analysis of human primary CD14+ monocytes after co-culture with three hepatocellular carcinoma cell lines for 2 days. C and D. ELISA detection of PGE2 and TGF-β in culture medium after co-culture of three hepatocellular carcinoma cell lines with human primary CD14+ monocytes for 2 days. E and F. Flow cytometric results and statistical analysis of CD14+ monocytes after adding celecoxib to the co-culture system of SNU423 cells and primary human CD14+ monocytes. G and H. ELISA detection of PGE2 and TGF-β in culture medium after adding celecoxib to the co-culture system of SNU423 cells and primary human CD14+ monocytes. Values are the mean ± SEM of a minimum of 3 independent experiments.
M2 macrophages induced by HCC cell lines can inhibit the production of IFN-γ and Granzyme B by activated CD8+ T cells
The CD3/CD28 monoclonal antibody was applied to activate and amplify human primary CD8+ T cells for one week. The flow cytometry results showed that the expression levels of CD25, CD28 and CD107A were significantly increased, indicating that CD8+ T cells were in an activated state (Figure 3A and 3B). After the activated CD8+ T cells were cocultured in the co-culture system for 24 h, the expression levels of IFN-γ and Granzyme B in the Tu+T+M0 and Tu+T groups were determined by flow cytometry. The flow cytometry results showed that BEL7402, SNU423 and SNU423COX2-/- cells could inhibit the production of Granzyme B and IFN-γ in activated CD8+ T cells through the induced M2 macrophages, but the decreasing trend was more obvious in the SNU423 group than the other groups (Figure 3C and 3D). At the same time, flow cytometry was used to compare the expression of IFN-γ and Granzyme B in the BEL7402-T+M0, SNU423-T+M0 and SNU423COX2-/--T+M0 groups. SNU423-induced M2 macrophages had a stronger inhibitory effect on the production of Granzyme B and IFN-γ by activated CD8+ T cells (Figure 3C and 3D). Granzyme B and IFN-γ levels in CD8+ T cells of each group were detected by ELISA, and the results showed the same trend as the results of flow cytometry (Figure 3H and 3I). We also used ELISA to detect TGF-β in the co-culture medium of each group. By comparing the Tu+T+M0 and Tu+T groups, it was found that the levels of TGF-β in the co-culture medium of the Tu+T+M0 group was higher (Figure 3E). By comparing the BEL7402-T+M0, SNU423-T+M0 and SNU423COX2-/--T+M0 groups, it was found that M2 macrophages induced by SNU423 secreted more TGF-β (Figure 3E). Finally, Granzyme B and IFN-γ in the co-culture medium of each group were detected by flow microarray technology. The results showed the same trend as the results of flow cytometry and ELISA (Figure 3F and 3G).
Figure 3.

Activation and amplification of CD8+ T cells and analysis of co-culture system in vitro. A and B. Flow cytometry and statistical analysis of human primary CD8+ T cells before and after activation and amplification. C and D. Flow cytometric detection and statistical analysis of IFN-γ and Granzyme B in CD8+ T cells 24 hours after adding CD8+ T cells to the co-culture system. E. ELISA detection and statistical analysis of TGF-β in the culture medium after adding CD8+ T cells to the co-culture system for 24 hours. F and G. Flow cytometric detection and statistical analysis of IFN-γ and Granzyme B in the culture medium after adding CD8+ T cells to the co-culture system for 24 hours. H and I. ELISA detection and statistical analysis of IFN-γ and Granzyme B in CD8+ T cells after adding CD8+ T cells to the co-culture system for 24 hours. Values are the mean ± SEM of a minimum of 3 independent experiments. *P<0.05; **P<0.005; ***P<0.0005; ****P<0.0001.
M2 macrophages induced by HCC cell lines inhibit the production of IFN-γ and Granzyme B by activated CD8+ T cells through the TGF-β pathway
Since there were too few CD8+ T cells in each co-culture system for conventional western detection, we used the automatic western technology of Raybiotech Company to analyze proteins associated with the TGF-β pathway in each group. The results of the comparison among the BEL7402-T+M0, SNU423-T+M0 and SNU423COX2-/--T+M0 groups showed no significant difference in the amount of total Smad2 and Smad3 protein, but the amounts of pSmad2 and pSmad3 in the SNU423-T+M0 group were significantly higher than that of BEL7402-T+M0 and SNU423COX2-/--T+M0 group, and the related protein-FoxP1 was also significantly higher than the other two groups (Figure 4A-F). Related signal pathway illustration is shown in Figure S4.
Figure 4.
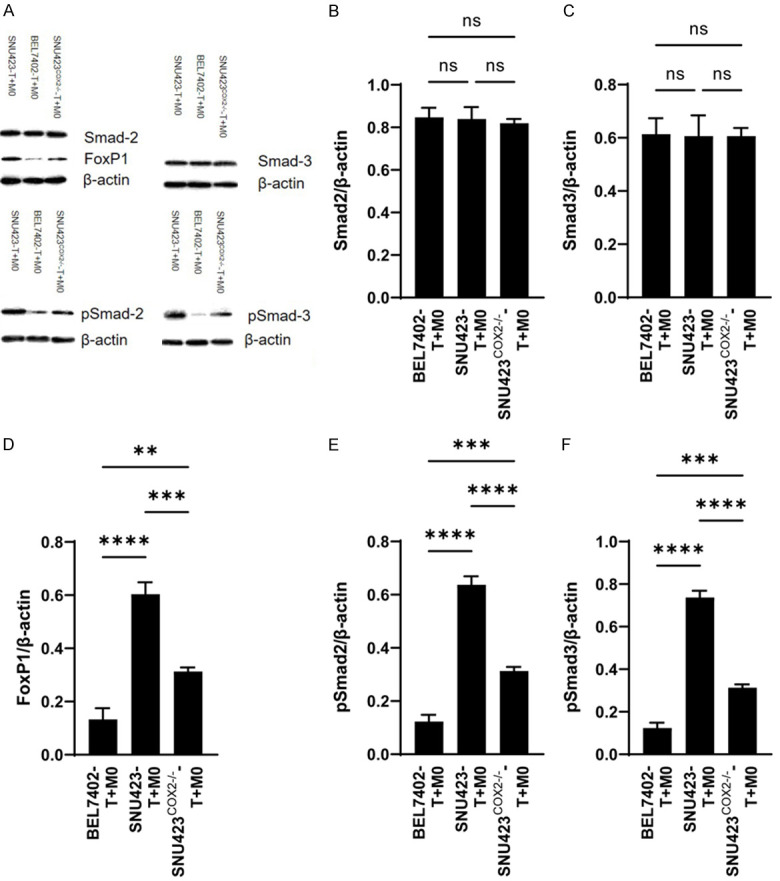
Validation of the TGF-β pathway. A. Western results for members of the TGF-β pathway. B-F. Statistical analysis of TGF-β pathway data. Values are the mean ± SEM of a minimum of 3 independent experiments. *P<0.05; **P<0.005; ***P<0.0005; ****P<0.0001.
M2 macrophages induced by HCC cell lines inhibit the production of IFN-γ and Granzyme B by activated CD8+ T cells in vivo
NPG mice were conducted as animal models for in vivo studies. As can be seen from the results of the study, the tumor growth volume varies with different treatment regimens: the tumor volume of the PBS treatment group was the largest, and that of the CD8+ T+M0+celecoxib group was the smallest, and significantly smaller than that of the CD8+ T+M0 and celecoxib groups (Figure 5A and 5B). Flow cytometry of single cell suspension derived from tumor tissues revealed that celecoxib can inhibit the polarization of CD14+ monocytes into M2 macrophages in vivo (Figure 5C and 5D). ELISA results of tumor tissues showed that the content of PGE2 and TGF-β were significantly higher in the CD8+ T+M0 group than CD8+ T+M0+celecoxib group (Figure 5E and 5F). Meanwhile, ELISA results of CD8+ T cells sorted from tumor tissues showed that CD8+ T cells in the CD8+ T+M0+celecoxib group contained more IFN-γ and Granzyme B than that of the CD8+ T+M0 group (Figure 5G and 5H). The Automated Western blot results of the CD8+ T+M0 and CD8+ T+M0+celecoxib groups showed no significant difference in the amount of total Smad2 and Smad3 protein, but the amounts of pSmad2, pSmad3 and FoxP1 in the CD8+ T+M0 group were significantly higher than that of CD8+ T+M0+celecoxib group (Figure 5I-N).
Figure 5.

Verification and analysis of in vivo models. A and B. Results and statistical analysis of different treatment regimes in NPG mouse after tumor formation. C and D. Flow cytometry results and statistical analysis of M2 macrophages (CD163/CD206) derived from human CD14+ monocytes in tumor tissues given different treatment regimes. E. ELISA detection and statistical analysis of PGE2 in in tumor tissues given different treatment methods. F. ELISA detection and statistical analysis of TGF-β in in tumor tissues given different treatment methods. G and H. ELISA detection and statistical analysis of IFN-γ and Granzyme B in human CD8+ T cells after sorting from tumor tissues. I. Western results for members of the TGF-β pathway. J-N. Statistical analysis of TGF-β pathway data. Values are the mean ± SEM of a minimum of 3 independent experiments. *P<0.05; **P<0.005; ***P<0.0005; ****P<0.0001.
Discussion
Promising advances have been achieved in the field of cancer immunotherapy as a result of great research work on the mechanisms that regulate antitumor T cell responses, including eventual translation of these concepts to the clinic. This has attracted more attentions to effective cytotoxicity and the different tumor-cell-intrinsic and -extrinsic factors that would result in resistance to T cell-based immunotherapy. Here, depending on multicellular co-culture models, we successfully demonstrate the exhaustion of anti-tumor abilities in activated CD8+ T cell caused by high COX-2-expressing HCC cell lines through M2 TAMs polarization and TGF beta pathway.
According to previous studies, COX-2, which is frequently expressed in many types of cancers and associated with poor prognosis, is a stimulator of cancer stem cell-like activity, apoptotic resistance, proliferation, angiogenesis, inflammation, invasion, and metastasis [18], thus, leading to the clinical application of COX-2 blockade as an effective enhancement of cancer chemoprevention [23]. A previous study demonstrated the important role of COX2 overexpression in promoting HCC carcinogenesis [24]. Furthermore, a recent report suggested that high COX2-expressing HCC patients can have worse prognosis and COX2 blockade (celecoxib) can suppress HCC cell growth and invasion [25]. To date, one of the most common effects from COX-2 during carcinogenesis and tumor prognosis is mediated through overproduction of PGs, especially PGE2, which have diverse functions in favor of tumor promotion18. Interestingly, one previous report showed that PGE2 can induce M2 polarization of macrophages through regulation of the CREB-KLF4 pathway [26]. To further investigate the role of COX-2 in HCC TIME, using immunofluorescence staining, we firstly confirmed that the number of M2-polarized TAMs (co-expressing CD163 and CD206) increased correspondingly in high COX2-expressing HCC patients. As an important TIME component, M2-polarized TAMs can affect T cell cytotoxicity, thus promote cellular exhaustion both in adaptive and innate anti-tumor responses [27]. Noteworthly, most facets of this are still incompletely understood in HCC TIME, and further experimental data are needed to elucidate its potential role played in HCC immunotherapy.
In our study, a multilayer co-culture system which integrated HCC cells with CD14+ monocytes and activated CD8+ T cells both in vitro and in NOD-Prkdcscid-II2rgnull mice model was established. To avoid any nonspecific immune responses caused by MHC mismatching while using T cells from different donors, HCC cells were separated from the immune cells via a 3D collagen sandwich configuration, according to our previous HCC tumoroid formation protocol with some modifications [28]. We found that the high COX-2 expression HCC cell line SNU423 could induce more M2 macrophages from CD14+ monocytes than the negative control HCC cell lines-BEL7402 and SNU423COX2-/-, with M2 polarization ratio greater than 80%, most likely as a result of a significant PGE2 overproduction detected in the medium from SNU423 cells. As expected, after treatment with a selective COX-2 blockade (celecoxib), our data further verified that COX2/PGE2-dependent M2 TAMs polarization occurred in the HCC TIME.
Our multi-cellular TIME co-culture and animal platform also provided the first evidence that the high COX2-expressing HCC cell lines showed a greater ability to exhaust CD8+ T cells mainly mediated by M2 TAMs polarization. Following 2 days of co-culture, the production of tumor cells lysing proteins in T cells cultured with SNU423 (high COX-2 expression) and M0 cells revealed the greatest declining patterns in comparison with BEL7402, SNU423COX2-/- and other control groups. In addition, despite of not being correlated with COX-2 expression, our study also found the suppression of T cells cytotoxicity in Tumor-T cell co-culture groups, and demonstrated that the function of T cells can be impaired not only by M2 TAMs but also possibly by other unknown mechanism from HCC cells themselves which needed further investigation.
The results of our in vivo experiments are basically consistent with those of in vitro experiments. In hepatocellular carcinoma tumor tissues with high COX-2 expression (SNU423), celecoxib inhibits the polarization of CD14+ monocytes into M2 macrophages, thereby reducing its depletion effect on CD8+ T cells. In addition, according to relevant studies, celecoxib can inhibit the growth and metastasis of colon cancer cells by inhibiting the synthesis of PGE2 [29]. It can also be found from our study that the tumor growth rate of the group treated with celecoxib alone was lower than that of the group treated with PBS, but the tumor growth rate of the group treated with CD8+ T+M0+celecoxib was lowest, and significantly lower than that of the CD8+ T+M0 and celecoxib groups. According to our in vivo experiment results, celecoxib may affect the direct effect of tumor cells on T cells through other unknown mechanisms besides affecting the polarization of M2 macrophages, which need further verification.
According to Li et al. results that TGF-β mainly derived from macrophages will damage the function of T cells, and study, demonstrated by Stephen et al., that TGF-β inhibits the function of antitumor T cells by regulating the expression of FoxP1, to validate the mechanism of T cell exhaustion in our study, A HCC cell pre-treated TAMs and T cells co-culture (BEL7402- T+M0, SNU423-T+M0 and SNU423COX2-/--T+M0) model was conducted which can also avoid the interference caused by HCC cells [30,31]. The expression of TGF-β pathway related proteins and FoxP1 in CD8+ T cells detected using automated western technology showed phosphorylated Smad2, phosphorylated Smad3 and FoxP1 was significantly elevated in high COX-2 HCC cell line treated group, indicating that COX-2 induced M2 TAMs polarization increased the expression of FoxP1 through the TGF-β pathway, thus inducing activated CD8+ T cells exhaustion.
In this study, hepatocellular carcinoma cell lines were mainly used for relevant verification and analysis, so the results of this study could not fully reflect the actual clinical situation. In order to make the results of research closer to the clinic, in the further process of research, we can consider the application of primary hepatocellular carcinoma cells for verification and analysis, so as to better apply the results of research to the clinic.
In conclusion, our research date provides the rational that, for HCC patients with high COX-2 expression, COX-2 inhibitors may reduce the inhibitory effect on CD8+ T cells through regulating TAMs in TIME, thus enhance the efficacy of T cell-based immunotherapy and improve the prognosis of patients with hepatocellular carcinoma.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81570590 and 81872508), UMHS-PUHSC Joint Institute for Translational and Clinical Research (JZ, TW) (BMU2017JI006) and Peking University People’s Hospital Research and Development Funds (RDY2018-03). We thank Dr. Dingbao Chen for help with patient sample collection and pathology analysis.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Global Burden of Disease Cancer Collaboration. Fitzmaurice C, Abate D, Abbasi N, Abbastabar H, Abd-Allah F, Abdel-Rahman O, Abdelalim A, Abdoli A, Abdollahpour I, Abdulle ASM, Abebe ND, Abraha HN, Abu-Raddad LJ, Abualhasan A, Adedeji IA, Advani SM, Afarideh M, Afshari M, Aghaali M, Agius D, Agrawal S, Ahmadi A, Ahmadian E, Ahmadpour E, Ahmed MB, Akbari ME, Akinyemiju T, Al-Aly Z, AlAbdulKader AM, Alahdab F, Alam T, Alamene GM, Alemnew BTT, Alene KA, Alinia C, Alipour V, Aljunid SM, Bakeshei FA, Almadi MAH, Almasi-Hashiani A, Alsharif U, Alsowaidi S, Alvis-Guzman N, Amini E, Amini S, Amoako YA, Anbari Z, Anber NH, Andrei CL, Anjomshoa M, Ansari F, Ansariadi A, Appiah SCY, Arab-Zozani M, Arabloo J, Arefi Z, Aremu O, Areri HA, Artaman A, Asayesh H, Asfaw ET, Ashagre AF, Assadi R, Ataeinia B, Atalay HT, Ataro Z, Atique S, Ausloos M, Avila-Burgos L, Avokpaho EFGA, Awasthi A, Awoke N, Ayala Quintanilla BP, Ayanore MA, Ayele HT, Babaee E, Bacha U, Badawi A, Bagherzadeh M, Bagli E, Balakrishnan S, Balouchi A, Bärnighausen TW, Battista RJ, Behzadifar M, Behzadifar M, Bekele BB, Belay YB, Belayneh YM, Berfield KKS, Berhane A, Bernabe E, Beuran M, Bhakta N, Bhattacharyya K, Biadgo B, Bijani A, Bin Sayeed MS, Birungi C, Bisignano C, Bitew H, Bjørge T, Bleyer A, Bogale KA, Bojia HA, Borzì AM, Bosetti C, Bou-Orm IR, Brenner H, Brewer JD, Briko AN, Briko NI, Bustamante-Teixeira MT, Butt ZA, Carreras G, Carrero JJ, Carvalho F, Castro C, Castro F, Catalá-López F, Cerin E, Chaiah Y, Chanie WF, Chattu VK, Chaturvedi P, Chauhan NS, Chehrazi M, Chiang PP, Chichiabellu TY, Chido-Amajuoyi OG, Chimed-Ochir O, Choi JJ, Christopher DJ, Chu DT, Constantin MM, Costa VM, Crocetti E, Crowe CS, Curado MP, Dahlawi SMA, Damiani G, Darwish AH, Daryani A, das Neves J, Demeke FM, Demis AB, Demissie BW, Demoz GT, Denova-Gutiérrez E, Derakhshani A, Deribe KS, Desai R, Desalegn BB, Desta M, Dey S, Dharmaratne SD, Dhimal M, Diaz D, Dinberu MTT, Djalalinia S, Doku DT, Drake TM, Dubey M, Dubljanin E, Duken EE, Ebrahimi H, Effiong A, Eftekhari A, El Sayed I, Zaki MES, El-Jaafary SI, El-Khatib Z, Elemineh DA, Elkout H, Ellenbogen RG, Elsharkawy A, Emamian MH, Endalew DA, Endries AY, Eshrati B, Fadhil I, Fallah Omrani V, Faramarzi M, Farhangi MA, Farioli A, Farzadfar F, Fentahun N, Fernandes E, Feyissa GT, Filip I, Fischer F, Fisher JL, Force LM, Foroutan M, Freitas M, Fukumoto T, Futran ND, Gallus S, Gankpe FG, Gayesa RT, Gebrehiwot TT, Gebremeskel GG, Gedefaw GA, Gelaw BK, Geta B, Getachew S, Gezae KE, Ghafourifard M, Ghajar A, Ghashghaee A, Gholamian A, Gill PS, Ginindza TTG, Girmay A, Gizaw M, Gomez RS, Gopalani SV, Gorini G, Goulart BNG, Grada A, Ribeiro Guerra M, Guimaraes ALS, Gupta PC, Gupta R, Hadkhale K, Haj-Mirzaian A, Haj-Mirzaian A, Hamadeh RR, Hamidi S, Hanfore LK, Haro JM, Hasankhani M, Hasanzadeh A, Hassen HY, Hay RJ, Hay SI, Henok A, Henry NJ, Herteliu C, Hidru HD, Hoang CL, Hole MK, Hoogar P, Horita N, Hosgood HD, Hosseini M, Hosseinzadeh M, Hostiuc M, Hostiuc S, Househ M, Hussen MM, Ileanu B, Ilic MD, Innos K, Irvani SSN, Iseh KR, Islam SMS, Islami F, Jafari Balalami N, Jafarinia M, Jahangiry L, Jahani MA, Jahanmehr N, Jakovljevic M, James SL, Javanbakht M, Jayaraman S, Jee SH, Jenabi E, Jha RP, Jonas JB, Jonnagaddala J, Joo T, Jungari SB, Jürisson M, Kabir A, Kamangar F, Karch A, Karimi N, Karimian A, Kasaeian A, Kasahun GG, Kassa B, Kassa TD, Kassaw MW, Kaul A, Keiyoro PN, Kelbore AG, Kerbo AA, Khader YS, Khalilarjmandi M, Khan EA, Khan G, Khang YH, Khatab K, Khater A, Khayamzadeh M, Khazaee-Pool M, Khazaei S, Khoja AT, Khosravi MH, Khubchandani J, Kianipour N, Kim D, Kim YJ, Kisa A, Kisa S, Kissimova-Skarbek K, Komaki H, Koyanagi A, Krohn KJ, Bicer BK, Kugbey N, Kumar V, Kuupiel D, La Vecchia C, Lad DP, Lake EA, Lakew AM, Lal DK, Lami FH, Lan Q, Lasrado S, Lauriola P, Lazarus JV, Leigh J, Leshargie CT, Liao Y, Limenih MA, Listl S, Lopez AD, Lopukhov PD, Lunevicius R, Madadin M, Magdeldin S, El Razek HMA, Majeed A, Maleki A, Malekzadeh R, Manafi A, Manafi N, Manamo WA, Mansourian M, Mansournia MA, Mantovani LG, Maroufizadeh S, Martini SMS, Mashamba-Thompson TP, Massenburg BB, Maswabi MT, Mathur MR, McAlinden C, McKee M, Meheretu HAA, Mehrotra R, Mehta V, Meier T, Melaku YA, Meles GG, Meles HG, Melese A, Melku M, Memiah PTN, Mendoza W, Menezes RG, Merat S, Meretoja TJ, Mestrovic T, Miazgowski B, Miazgowski T, Mihretie KMM, Miller TR, Mills EJ, Mir SM, Mirzaei H, Mirzaei HR, Mishra R, Moazen B, Mohammad DK, Mohammad KA, Mohammad Y, Darwesh AM, Mohammadbeigi A, Mohammadi H, Mohammadi M, Mohammadian M, Mohammadian-Hafshejani A, Mohammadoo-Khorasani M, Mohammadpourhodki R, Mohammed AS, Mohammed JA, Mohammed S, Mohebi F, Mokdad AH, Monasta L, Moodley Y, Moosazadeh M, Moossavi M, Moradi G, Moradi-Joo M, Moradi-Lakeh M, Moradpour F, Morawska L, Morgado-da-Costa J, Morisaki N, Morrison SD, Mosapour A, Mousavi SM, Muche AA, Muhammed OSS, Musa J, Nabhan AF, Naderi M, Nagarajan AJ, Nagel G, Nahvijou A, Naik G, Najafi F, Naldi L, Nam HS, Nasiri N, Nazari J, Negoi I, Neupane S, Newcomb PA, Nggada HA, Ngunjiri JW, Nguyen CT, Nikniaz L, Ningrum DNA, Nirayo YL, Nixon MR, Nnaji CA, Nojomi M, Nosratnejad S, Shiadeh MN, Obsa MS, Ofori-Asenso R, Ogbo FA, Oh IH, Olagunju AT, Olagunju TO, Oluwasanu MM, Omonisi AE, Onwujekwe OE, Oommen AM, Oren E, Ortega-Altamirano DDV, Ota E, Otstavnov SS, Owolabi MO, P A M, Padubidri JR, Pakhale S, Pakpour AH, Pana A, Park EK, Parsian H, Pashaei T, Patel S, Patil ST, Pennini A, Pereira DM, Piccinelli C, Pillay JD, Pirestani M, Pishgar F, Postma MJ, Pourjafar H, Pourmalek F, Pourshams A, Prakash S, Prasad N, Qorbani M, Rabiee M, Rabiee N, Radfar A, Rafiei A, Rahim F, Rahimi M, Rahman MA, Rajati F, Rana SM, Raoofi S, Rath GK, Rawaf DL, Rawaf S, Reiner RC, Renzaho AMN, Rezaei N, Rezapour A, Ribeiro AI, Ribeiro D, Ronfani L, Roro EM, Roshandel G, Rostami A, Saad RS, Sabbagh P, Sabour S, Saddik B, Safiri S, Sahebkar A, Salahshoor MR, Salehi F, Salem H, Salem MR, Salimzadeh H, Salomon JA, Samy AM, Sanabria J, Santric Milicevic MM, Sartorius B, Sarveazad A, Sathian B, Satpathy M, Savic M, Sawhney M, Sayyah M, Schneider IJC, Schöttker B, Sekerija M, Sepanlou SG, Sepehrimanesh M, Seyedmousavi S, Shaahmadi F, Shabaninejad H, Shahbaz M, Shaikh MA, Shamshirian A, Shamsizadeh M, Sharafi H, Sharafi Z, Sharif M, Sharifi A, Sharifi H, Sharma R, Sheikh A, Shirkoohi R, Shukla SR, Si S, Siabani S, Silva DAS, Silveira DGA, Singh A, Singh JA, Sisay S, Sitas F, Sobngwi E, Soofi M, Soriano JB, Stathopoulou V, Sufiyan MB, Tabarés-Seisdedos R, Tabuchi T, Takahashi K, Tamtaji OR, Tarawneh MR, Tassew SG, Taymoori P, Tehrani-Banihashemi A, Temsah MH, Temsah O, Tesfay BE, Tesfay FH, Teshale MY, Tessema GA, Thapa S, Tlaye KG, Topor-Madry R, Tovani-Palone MR, Traini E, Tran BX, Tran KB, Tsadik AG, Ullah I, Uthman OA, Vacante M, Vaezi M, Varona Pérez P, Veisani Y, Vidale S, Violante FS, Vlassov V, Vollset SE, Vos T, Vosoughi K, Vu GT, Vujcic IS, Wabinga H, Wachamo TM, Wagnew FS, Waheed Y, Weldegebreal F, Weldesamuel GT, Wijeratne T, Wondafrash DZ, Wonde TE, Wondmieneh AB, Workie HM, Yadav R, Yadegar A, Yadollahpour A, Yaseri M, Yazdi-Feyzabadi V, Yeshaneh A, Yimam MA, Yimer EM, Yisma E, Yonemoto N, Younis MZ, Yousefi B, Yousefifard M, Yu C, Zabeh E, Zadnik V, Moghadam TZ, Zaidi Z, Zamani M, Zandian H, Zangeneh A, Zaki L, Zendehdel K, Zenebe ZM, Zewale TA, Ziapour A, Zodpey S, Murray CJL. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: a systematic analysis for the global burden of disease study. JAMA Oncol. 2019;5:1749–1768. doi: 10.1001/jamaoncol.2019.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–1462. doi: 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 3.Buonaguro L, Petrizzo A, Tagliamonte M, Tornesello ML, Buonaguro FM. Challenges in cancer vaccine development for hepatocellular carcinoma. J Hepatol. 2013;59:897–903. doi: 10.1016/j.jhep.2013.05.031. [DOI] [PubMed] [Google Scholar]
- 4.Mizukoshi E, Kaneko S. Immune cell therapy for hepatocellular carcinoma. J Hematol Oncol. 2019;12:52. doi: 10.1186/s13045-019-0742-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling TH Rd, Meyer T, Kang YK, Yeo W, Chopra A, Anderson J, Dela Cruz C, Lang L, Neely J, Tang H, Dastani HB, Melero I. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175:313–26. doi: 10.1016/j.cell.2018.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez CR, De Palma M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun. 2019;10:5408. doi: 10.1038/s41467-019-13368-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spear TT, Callender GG, Roszkowski JJ, Moxley KM, Simms PE, Foley KC, Murray DC, Scurti GM, Li M, Thomas JT, Langerman A, Garrett-Mayer E, Zhang Y, Nishimura MI. TCR gene-modified T cells can efficiently treat established hepatitis C-associated hepatocellular carcinoma tumors. Cancer Immunol Immunother. 2016;65:293–304. doi: 10.1007/s00262-016-1800-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Z, Zhu Y, Xia J, Sawakami T, Kokudo N, Zhang N. Status of and prospects for cancer vaccines against hepatocellular carcinoma in clinical trials. Biosci Trends. 2016;10:85–91. doi: 10.5582/bst.2015.01128. [DOI] [PubMed] [Google Scholar]
- 11.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, Vonderheide RH, Pittet MJ, Jain RK, Zou W, Howcroft TK, Woodhouse EC, Weinberg RA, Krummel MF. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19:222–232. doi: 10.1038/s41590-018-0044-z. [DOI] [PubMed] [Google Scholar]
- 14.Tian Z, Hou X, Liu W, Han Z, Wei L. Macrophages and hepatocellular carcinoma. Cell Biosci. 2019;9:79. doi: 10.1186/s13578-019-0342-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lacotte S, Slits F, Orci LA, Meyer J, Oldani G, Delaune V, Gonelle-Gispert C, Morel P, Toso C. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunolog. 2016;5:e1234565. doi: 10.1080/2162402X.2016.1234565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zamora AE, Crawford JC, Thomas PG. Hitting the target: how T cells detect and eliminate tumors. J Immunol. 2018;200:392–399. doi: 10.4049/jimmunol.1701413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, Liu J, Freeman GJ, Brown MA, Wucherpfennig KW, Liu XS. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashemi Goradel N, Najafi M, Salehi E, Farhood B, Mortezaee K. Cyclooxygenase-2 in cancer: a review. J Cell Physiol. 2019;234:5683–5699. doi: 10.1002/jcp.27411. [DOI] [PubMed] [Google Scholar]
- 19.Lv X, Chen Z, Li S, Xie H. Knockdown of cyclooxygenase-2 leads to growth inhibition and cell cycle arrest in hepatocellular carcinoma cells. Onco Targets Ther. 2019;12:4341–4349. doi: 10.2147/OTT.S196822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tong H, Wei B, Chen S, Xie YM, Zhang MG, Zhang LH, Huang ZY, Tang CW. Adjuvant celecoxib and lanreotide following transarterial chemoembolisation for unresectable hepatocellular carcinoma: a randomized pilot study. Oncotarget. 2017;8:48303–48312. doi: 10.18632/oncotarget.15684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiu Z, Zhang C, Zhou J, Hu J, Sheng L, Li X, Chen L, Li X, Deng X, Zheng G. Celecoxib alleviates AKT/c-Met-triggered rapid hepatocarcinogenesis by suppressing a novel COX-2/AKT/FASN cascade. Mol Carcinog. 2019;58:31–41. doi: 10.1002/mc.22904. [DOI] [PubMed] [Google Scholar]
- 22.Ye Y, Xu Y, Lai Y, He W, Li Y, Wang R, Luo X, Chen R, Chen T. Long non-coding RNA cox-2 prevents immune evasion and metastasis of hepatocellular carcinoma by altering M1/M2 macrophage polarization. J Cell Biochem. 2018;119:2951–2963. doi: 10.1002/jcb.26509. [DOI] [PubMed] [Google Scholar]
- 23.Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, DuBois RN. Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. J. Clin. Oncol. 2005;23:254–266. doi: 10.1200/JCO.2005.09.112. [DOI] [PubMed] [Google Scholar]
- 24.Chen H, Cai W, Chu ESH, Tang J, Wong CC, Wong SH, Sun W, Liang Q, Fang J, Sun Z, Yu J. Hepatic cyclooxygenase-2 overexpression induced spontaneous hepatocellular carcinoma formation in mice. Oncogene. 2017;36:4415–4426. doi: 10.1038/onc.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tai Y, Zhang LH, Gao JH, Zhao C, Tong H, Ye C, Huang ZY, Liu R, Tang CW. Suppressing growth and invasion of human hepatocellular carcinoma cells by celecoxib through inhibition of cyclooxygenase-2. Cancer Manag Res. 2019;11:2831–2848. doi: 10.2147/CMAR.S183376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A. 2015;112:15642–15647. doi: 10.1073/pnas.1519644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miao J, Lu X, Hu Y, Piao C, Wu X, Liu X, Huang C, Wang Y, Li D, Liu J. Prostaglandin E2 and PD-1 mediated inhibition of antitumor CTL responses in the human tumor microenvironment. Oncotarget. 2017;8:89802–89810. doi: 10.18632/oncotarget.21155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Takeishi K, Li Z, Cervantes-Alvarez E, Collin de l’Hortet A, Guzman-Lepe J, Cui X, Zhu J. Microenvironment of a tumor-organoid system enhances hepatocellular carcinoma malignancy-related hallmarks. Organogenesis. 2017;13:83–94. doi: 10.1080/15476278.2017.1322243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology. 2015;149:1884–1895. e4. doi: 10.1053/j.gastro.2015.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Yang L, Wang L, Wang F, Zhang Z, Li J, Yue D, Chen X, Ping Y, Huang L, Zhang B, Zhang Y. Impaired T cell function in malignant pleural effusion is caused by TGF-β derived predominantly from macrophages. Int J Cancer. 2016;139:2261–2269. doi: 10.1002/ijc.30289. [DOI] [PubMed] [Google Scholar]
- 31.Stephen TL, Rutkowski MR, Allegrezza MJ, Perales-Puchalt A, Tesone AJ, Svoronos N, Nguyen JM, Sarmin F, Borowsky ME, Tchou J, Conejo-Garcia JR. Transforming growth factor β-mediated suppression of antitumor T cells requires FoxP1 transcription factor expression. Immunity. 2014;41:427–439. doi: 10.1016/j.immuni.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.