

Abstract
Copy number variants (CNVs) have been implicated in neuropsychiatric disorders, with rare-inherited and de novo CNVs (dnCNVs) having large effects on disease liability. Recent studies started exploring a class of dnCNVs that occur post-zygotically, and are therefore present in some but not all cells of the body. Analogous to conditional mutations in animal models, the presence of risk mutations in a fraction of cells has the potential to enlighten how damaging mutations affect cell-type/cell-circuit specific pathologies leading to neuropsychiatric manifestations. Although mosaic CNVs appear to contribute to a modest fraction of risk (0.3–0.5%), expanding our insights about them with more sensitive experimental and statistical methods, has the potential to help clarify mechanisms of neuropsychiatric disease.
Keywords: neuropsychiatry, somatic variants, medical genetics, development, genomics
Introduction
The expansion of sequencing technologies and consortia efforts have allowed the characterization of many loci and genes involved with psychiatric disorders. The genetic architecture of these disorders has implicated inherited as well rare de novo variants, where copy number variants (CNVs) tend to have large effect sizes[1–6]. Many of these variants are under evolutionary constraint, such that only viable mutations can be observed when studying germline mutations. De novo variants can also arise post-zygotically, resulting in a mosaic individual with a subset of cells affected by a mutation (Figure 1). These variants are not under the same constraints as germline variants, allowing us to observe mutations that would be embryonically lethal, and yet may illuminate new mechanistic understanding of disease. Somatic variants provide a unique opportunity to study natural experiments akin to conditionally transgenic mouse models, where only a subset of cells is affected. This review focuses on recent discoveries of somatic CNVs (sCNVs), their contribution to neuropsychiatric disorders, and novel insights from somatic CNVs in non-diseased tissue that might illuminate potential mechanisms originating these events and discusses potential future paths to further advance the field.
Figure 1.
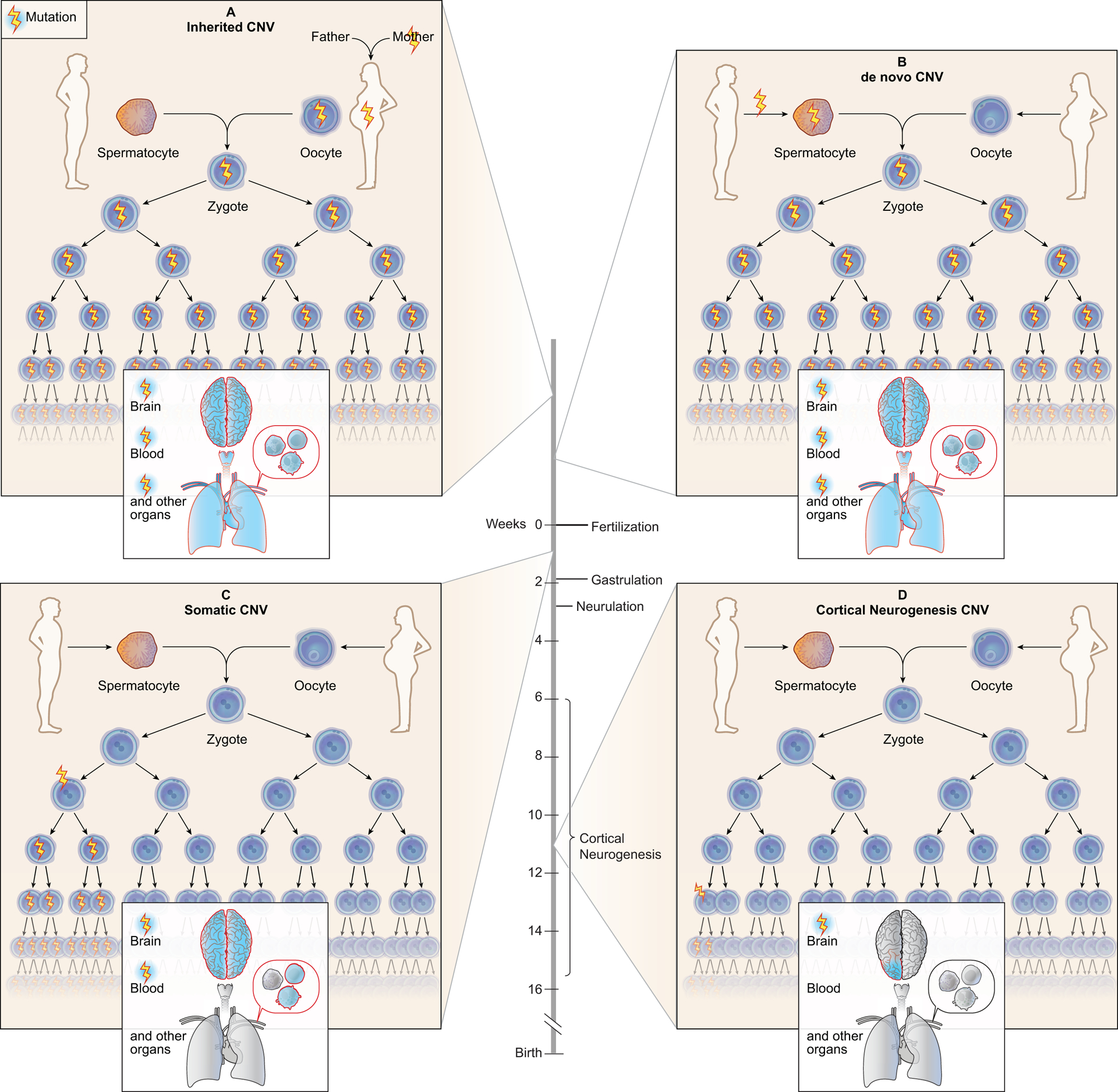
Inherited, de novo, and somatic CNVs affect a different proportion of cells and tissues. (A) Inherited CNVs are present in all cells of the parent and are transmitted to the offspring during fertilization, causing all the child’s cells to carry the variant. (B) De novo CNVs occurring in the parental germ cells can pass the variant to the offspring during fertilization, resulting in all child’s cells carrying the variant. (C) Somatic CNVs can occur before gastrulation and organ cell commitment resulting in these variants to be present in a fraction of cells in several tissues such as brain and blood. (D) If somatic CNVs occur after gastrulation they can be organ specific, with variants arising during cortical neurogenesis present in the brain - later occurring variants can be more focal.
Somatic CNVs in normal brain
The frequency of sCNVs in the normal human brain and pathological conditions remains largely unknown. Studies of sCNVs in the normal human brain have provided relative baseline rate measurements as well as suggested potential mechanisms that might lead to neuropsychiatric disease. One of the first studies using fluorescence in situ hybridization (FISH) for only chromosome 21 in post-mortem brain tissue suggested an aneuploidy rate of ~4% in neuronal as well as non-neuronal cell populations in equal proportions[7]. This initial estimate suggested that somatic aneuploidy might be ubiquitous in the brain, and could potentially serve in the physiological role of generating cellular diversity. However, more recent studies using single cell whole-genome sequencing (scWGS) suggest that the prevalence of chromosomal aneuploidy in the adult human brain is much lower, occurring in perhaps 2–5% of neurons, significantly less than the 20% previously reported using older methods[8–10]. These findings suggest that, as in other tissues, aneuploidy is in general deleterious and rare in the human brain.
While somatic aneuploidy appears to be rare, scWGS studies have revealed that smaller, sub-chromosomal sCNVs in neurons in post-mortem human brain occur in 13–40% of cells[9–11]. The reported events tend to be large (> 1 Mb) - indeed, present scWGS methods have limited ability to accurately detect smaller sCNVs - with deletions being more common than duplications in neurons. In addition, McConnell et al[10] found that these sub-chromosomal sCNVs were not enriched in canonical regions involved in genome stability, nor regions enriched for germline CNVs, which suggests that sCNVs could potentially alter novel regions of the genome whose essential were not previously defined in the germline state. Single neurons were also found to have a higher frequency of complex karyotypes and affect more of the genome compared to non-neuronal cells[11]. Cai et al[9] also found one sCNV in 15q13.2–13.3, a CNV that has been associated with autism and other neuropsychiatric disorders[5,12].
The mechanism by which sCNVs occur in the normal human brain remains largely unknown. A mechanism of post-zygotic CNVs that has gathered much attention in recent years is CNV variants that occur due to chromosomal instability in early embryogenesis, which might result in sCNV shared clonally by many cells[13,14]. The early zygote’s chromosomal instability has been attributed to overexpression of cell-cycle promoting genes, and reduced cell cycle checkpoints, along with relative lack of support from tissue architecture[14,15]. Earlier single-cell genome analyses of blastomeres from human cleavage-stage embryos produced during the process of in vitro fertilization have shown that up to 90% of embryos acquire numerical or structural chromosomal abnormalities, with 39% of them being mosaic[16], while other studies have suggested mosaicism levels as high as 73%[17]. In a recent study Liu et al[18] showed that there are embryos with multiple de novo CNVs that tend to be DNA copy number gains that appear either as tandem duplications or as more complex DNA rearrangements. The breakpoint junctions tend to reveal sequence microhomology, suggesting that these CNVs involve erroneous DNA replicative mechanisms, and are of post-zygotic origin, but surprisingly, tended to be present in all cells of the individuals analyzed rather than exhibiting a mosaic pattern. However, this lack of mosaicism can be attributed to the fact that not all blastomeres of a cleavage-stage embryo necessarily contribute to the fetal cell lineage, as some of them contribute to the trophoblast/placenta, which might have a physiological role at preventing pregnancy loss[19,20]. As further studies characterize the mosaic pattern of these events, questions still remain such as whether there is any genomic context enrichment of these CNVs or whether they tend to affect specific sets of genes that might predispose to disease later in life. The genomic instability of the early zygote could predispose it to obtain somatic structural variants present in a large fraction of cells, which might impact brain function akin to neurodevelopmental disorders, though this remains an area of uncertainty.
While the previous mechanism suggests a way that neural cells could have gained sCNVs through mitotic cell division during early development, a major question is whether non-cycling neurons are subject to ongoing creation of sCNVs, since until recently it has been impossible to study the genome of a single neuron. Studies in mice have suggested that neuronal activity can produce double-stranded breaks in the DNA[21]. These double stranded breaks could potentially result in mosaic CNVs even in the absence of cell division through deletions secondary to non-homologous end joining repair (NHEJ), though this remains unproven. Neuronal sCNVs were shown to occur in locations enriched in neural or synaptic genes using neural progenitor cells[22]. Similar sCNV susceptibility of neuronal genes have been observed in post-mortem human brain using single-cell DNA sequencing[11]. Thus, resolution of these events resulting in sCNVs may negatively affect neuronal function in some cases.
The dynamics of sCNVs through aging remain mostly unknown. While neuropsychiatric disorders have relatively early disease onset, which might suggest a genetic mechanism that occurs in early development, somatic mutations could also be acquired with aging after development. This differential rate of somatic mutation acquisition is an attractive hypothesis for the heterogeneity of disease onset of neuropsychiatric disorders. Somatic single nucleotide variants (sSNVs) have been shown to accumulate with aging in non-diseased neurons[23]. In contrast, in a recent study using scWGS of 474 neuronal nuclei from 5 non-diseased individuals along with previous published data of 458 neurons from 11 additional individuals showed a surprising inverse correlation between the fraction of cells with CNVs and age, ranging from ~37% in individuals younger than 25 years old to ~5% for individuals older than 75 years old[11]. In this study the number of sCNVs per neuron had a general trend towards more CNVs per neuron in older individuals, however there was large within individual variability of the sCNV burden per neuron, suggesting this trend should be interpreted with caution. In addition, sCNV calling from scWGS tends to be biased towards larger events, implying that these trends if true might be relevant to larger events that have a greater chance to be deleterious. Understanding the dynamics of sCNVs accumulation on neural cells will likely require exploring a wide range of neuronal types and conditions. Nevertheless, these data might suggest a deleterious role of sCNVs on neuronal survival since older individuals who are considered healthy tend to have a lesser fraction of neurons with sCNVs.
Exploring the idea that age-related sCNV might contribute to disease, Lee et al[24] performed a series of RT-PCR and DNA in situ hybridization (DISH) experiments on normal and Alzheimer’s disease (AD) brain. Their data suggested that a variety of structural rearrangements at the APP gene involved in AD were enriched in the brains of individuals with sporadic AD and increased with aging, suggesting that sporadic recombination events leading to sCNVs in this locus could contribute to disease risk. However, initial attempts to replicate these findings found that key experiments in Lee et al showed artifactual vector contamination, making it uncertain whether any sort of amplification or recombination at the APP locus indeed occurs with age in vivo[25]. Consequently, even though deleterious effect of sCNVs are an interesting avenue to explain later onset neuropsychiatric phenotypes, the field would benefit from further characterization sCNVs of the aging dynamics of sCNVs to fully assess their impact.
Somatic CNVs in neurodevelopmental disorders
Somatic mutations including sCNVs have been shown to play an important pathogenic role in several neurodevelopmental disorders such as focal cortical disorders known as focal cortical dysplasia, hemimegaloencephaly (HMG), and polymicrogyria[9,26–28]. These CNVs can result in lesions observable with clinical imaging such as MRI as in the case of HMG, which is characterized as a brain overgrowth syndrome. Mosaic 1q gains has been shown to play a causative role in this disorder using SNP arrays[26] and more recently scWGS[9], which is consistent with results implicating gain of function mutations in the AKT3 gene which lies within the 1q region that is involved[26,27,29,30]. In the scWGS study it was shown that the chr1 gain was actually tetrasomy, not trisomy, which could reflect a mosaic isodicentric 1q-1q event, as previously described in pediatric nasopharyngeal cancer[31]. The gain of 1q was present in neuronal and non-neuronal cells, suggesting that it arose early in development, yet these sCNVs are generally limited to brain and produced a brain specific phenotype.
Large-scale studies using peripheral tissue SNP arrays have also been carried out to assess the burden of sCNVs in developmental disorders (DD). A study of sCNVs usin ~7,000 SNP arrays in an ascertained cohort of subjects with developmental disorders estimated an event frequency of ~0.9% of cases tested, which represented a 40-fold enrichment in cases compared to control[32]. In this study they focused on events that were present in at least 10% of cells, and larger than 2Mb. They found sCNVs that were consistent with the phenotypes of each child, using the assumption that they expected the phenotype of the events to match that of the germline state albeit less extensive. These results suggest the potential of early developmental sCNVs to alter brain function and behavioral phenotypes.
Somatic CNVs in Neuropsychiatric Disorders
Some neuropsychiatric disorders such as autism spectrum disorder (ASD) tend to have an early onset of disease, suggesting that somatic mutations acquired during development might contribute to disease liability. Studies of sSNVs have implicated somatic variation in the genomic landscape of ASD[33–38]. A recent study of somatic mosaicism in ASD used a novel sCNV calling algorithm that leverages haplotype-phasing information from SNP array data to detect mosaic events present in as low as 1% of cells[33,39]. This algorithm was applied to the blood derived SNP array data from the Simon Simplex Cohort and the SPARK datasets, totaling 12,077 probands and 5,500 siblings. In this study, the percent of probands carrying a sCNV event was 0.4% compared to the 0.2% of siblings. A statistically significant enrichment was of observed in events >4Mb in ASD. Larger sCNVs were positively correlated with ASD clinical severity as measured by SCQ summary score. Surprisingly, previously implicated CNVs loci such as 16p11.2 had no detectable phenotypic effects in the mosaic state, and were if anything more common in normal individuals than ASD probands. This result led the authors to conclude that mosaic and germline CNVs could result in autism through different mechanisms, requiring some events to be present in all cells to create a phenotype. In the mosaic state, the events might be embedded in larger CNVs that could be more toxic, thus restricted to a fraction of cells. In WGS of 59 post-mortem brain samples from ASD, one case showed 10.3 Mb complex CNV gain of 2pcen-2q11.2 present in 26% of cells, considered likely to be contributory, since it involved a region where germline CNV gain is associated with severe developmental disability, and since the CNV included two genes associated with ASD (MAL and NPAS2)[33]. This case was used to show that the sCNV was present in both neuronal and non-neuronal cells in brain, suggesting that better methods might allow deeper analysis of the distribution of somatic events in postmortem brain. Together these data suggest a modest but consistent contribution of sCNVs to ASD and neurodevelopmental disorders. The likely contribution of sCNV to ASD (0.4%) appears to somewhat smaller than the contribution to more severe DDs (0.9%), though this difference might still represent technical or ascertainment differences.
The great overlap in the genetic architecture of neurodevelopmental disorders such as ASD and other neuropsychiatric disorders suggests that sCNVs could similarly contribute to their genomic landscape[40–42]. Whereas the familiar role of de novo germline CNV in SCZ (see other articles in this issue) has been best demonstrated by large-scale studies, similarly large studies on sCNV have not yet been carried out. One of the first reports attempting to implicate sCNVs in a neuropsychiatric disorder used multicolor FISH on post-mortem brain samples of six subjects with SCZ. They observed aneuploidy in up to 0.5% to 4% of neurons involving chromosomes X and 18 in two subjects with SCZ[43]. More recent studies combining multicolor FISH and quantitative FISH estimated an aneuploidy rate of 0.31–0.46% in controls and 1.09–2.73% in SCZ in autosomes 1,9,15,16, and 18[44–46]. These aneuploidy estimates obtained through FISH need further replication due to the small sample sizes analyzed. Methods that incorporate next generation sequencing data at the single cell level to detect aneuploidies could potentially provide more accurate estimates due to the analysis of larger sample sizes, and genome-wide measurements.
Smaller intra-chromosomal sCNVs have been observed using deep WGS data on SCZ post-mortem brain samples[47]. In one study, Kim et al used an integrated somatic deletion calling pipeline to identify brain specific sCNVs in three SCZ cases. They reported deletions in the genes CBX3, PRKRA, MRPL4, SUCLG2 and TDG in SCZ, ranging from 466bp to 5.6Kb[47]. Deletions of PRKRA and MIR548N were observed in two independent cases, which they interpreted as evidence that these genes might be in region susceptible to structural variation. It will be interesting to see the potential relevance of these intriguing results to disease biology as larger sample sizes are analyzed.
Twin studies have also been used to study sCNVs in schizophrenia[48,49]. In a recent study by Castellani et al[49], they used next-generation sequencing from blood and cheek swab samples of two pairs of monozygotic twins discordant for schizophrenia (MZD) and their families to characterize post-zygotic somatic variation. They found multiple sequence differences between the twins from SNVs to CNVs and structural variants. Events involving glutamate receptor signaling and dopamine feedback in cAMP signaling pathways were enriched in the affected twin in both families using an ingenuity pathway analysis, which aligns with previously studies suggesting the importance of synaptic machinery and the glutamate pathway in this disease[1,2,50]. The authors suggest that these post-zygotic differences in MZD twins might increase the liability to SCZ for a given genomic background, which could account for MZ twin discordance for these disorders[49]. However, some studies have not been able to replicate such discordance in schizophrenia[51,52], but did find discordance of somatic mutations between MZD for gender dysphoria[51]. These discrepancies might be due to limitations in validating low-frequency allele mutations with Sanger sequencing[51], but of greater concern is the small sample sizes of these studies (2 pairs of monozygotic twins), which preclude strong conclusions about the role of sCNVs in disease liability. Finally, it is worth mentioning that care must be taken when performing gene set enrichment analyses in CNVs, especially in neurological and neuropsychiatric studies, since spurious enrichments of neuronal related genes might be found based on the background distribution of these genes across the genome[53]. Thus, standard enrichment methods might provide false positive associations if covariates such as event length, number of genes overlapped, and length of the genes are not accounted for.
While evidence of a role of sCNVs in the genetic architecture of neuropsychiatric disorders also comes from large scale blood derived studies, these studies must be performed cautiously due to the recently recognized tendency for blood cells themselves to undergo unequal clonal expansion with age. Causative somatic mutations have been identified in peripheral tissue samples from patients with brain malformations or Rett syndrome at allele fractions of 1 to 43%, which translates to presence in 2–86% of cells[30,54]. These allele fraction suggest that these mutations occurred early in development. Thus, analysis of peripheral tissues can provide a unique opportunity to analyze large sample sizes when the relevant brain tissue is not available and can provide insight into those mutations that occurred in the early stages of development. However, care must be taken to exclude sCNVs in peripheral blood cells that arose secondary to clonal hematopoiesis (CHIP), which tend to be observed in individuals older than 40 years-old[39,55], and are uncommon in younger individuals[32,33]. In a study aiming to characterize the burden of sSNVs in individuals with SCZ and controls from blood derived WES, the investigators were not able to find any enrichments in cases vs controls due to the large effect of clonal sSNV accumulation with age as a result of CHIP events[55]. This study provides a cautionary tale on the limitations of estimating somatic mutational burden in neuropsychiatric disorders from blood samples. As CHIP derived sCNVs are characterized further[39,56,57], it may be possible to develop filtering criteria that produce mosaicism distributions that are statistically different from those expected from a CHIP event. A study from the International Schizophrenia Consortium used blood derived SNP-array data from 3,518 SCZ cases and 4,238 controls. Ruderfer et al[58] estimated the rate of sCNVs to be nominally increased in cases (0.42% vs 0.26% in controls), though the sample size prevented this from reaching statistical significance. Their CNV events ranged from 10Mb to complete chromosomal copy number changes, they thus concluded that these events were somatic since such large alterations are unlikely to be viable with life. However, upon quantitative PCR and nanostring validation of these events, they found that they were present in all the lymphocytes, which might indicate that their method was not able to detect lower mosaic variants, and that these events might have resulted from clonal hematopoietic expansion. Further studies using more sensitive sCNV detection pipelines, larger sample sizes, and more stringent filtering criteria to dissect early developmental sCNVs from CHIP events should eventually allow more accurate estimates of the role of sCNVs in SCZ.
Conclusions and future directions
While initial estimates of the burden of sCNVs in neuropsychiatric disorders are starting to emerge, there remains many unanswered questions about the role of these mutations in disease liability (Figure 2). Most published studies to-date have focused on sCNVs in ASD and Schizophrenia, while other disorders such as Bipolar, Attention Deficit Hyperactivity Disorders (ADHD), and Tourette’s syndrome remain largely unexplored. The largest studies estimating the burden of sCNVs in neuropsychiatric disorders come from SNP array data, and they suggest that these events are likely to contribute to a consistent, albeit modest, proportion of cases < 0.5%[33,58]. However, these are likely to be underestimates since they are based on blood samples, restricting their sensitivity to very early developmental disorders, as these studies require filtering of low mosaic fraction events that might be blood specific. To reduce CHIP contamination these analyses have also filtered out regions of the genome that are involved in immune function such as the MHC complex since those events are difficult to differentiate from clonal hematopoietic events. However, these loci might contain important regions involved in neuropsychiatric disorders such as the C4 locus which has been strongly implicated in SCZ[59,60]. Sequencing studies of brain tissue might provide further resolution of the effects of sCNVs later in development in neuropsychiatric disorders.
Figure 2.
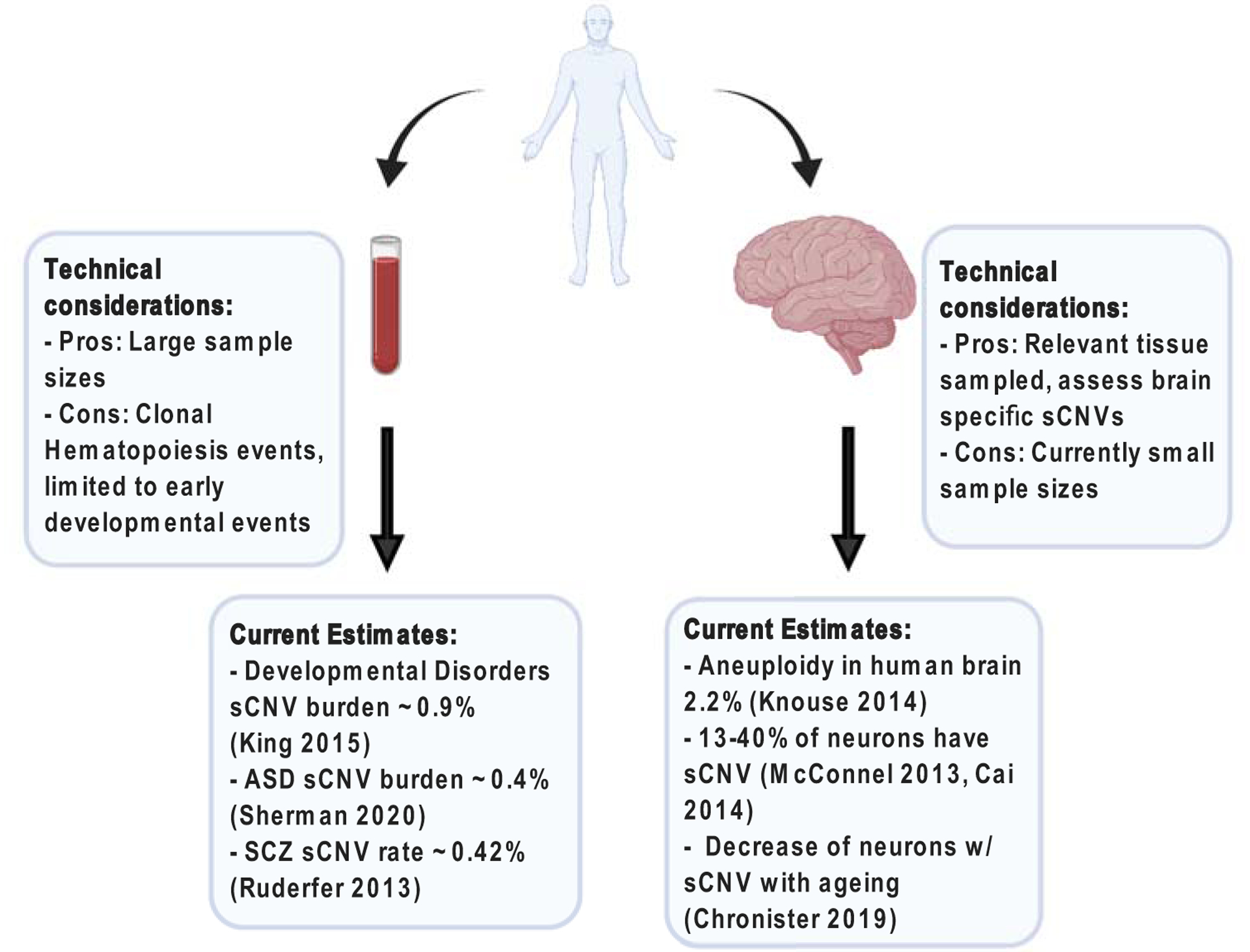
Approaches to study somatic CNVs in neuropsychiatric disorders. The most used tissues for the study of somatic CNVs are blood and post-mortem brain tissue. These approaches have technical considerations to have favored/limited their use to answer specific question. Most of the current somatic CNV burden estimates for neuropsychiatric disorders come from blood studies, however these are likely to be underestimates as there are limited to early events. While the study of post-mortem brain tissue has provided insights into somatic CNV rates in the non-diseased brain, the small sample sizes of neuropsychiatric studies limits the generalizability of their findings in a neuropsychiatric disease context.
One of the unique aspects of studying mosaic mutations is that they provide natural experiments equivalent to conditional knockout models that we see in the animal model literature. In the case of somatic mutations in HMG and FCDs we see some evidence of specificity to excitatory neuronal cells, resulting in an epileptic phenotype and localizable radiological findings[29,61–63] at mosaic fractions as small as 1%. Performing analyses of large cohorts of postmortem brain samples would in principle allow the dissection of cell types that might be differentially affected by sCNVs, and measurement of the mosaic fraction that would produce a neuropsychiatric phenotype. Single-cell WGS of healthy human neurons estimate that sub-chromosomal sCNVs are present in 13–40% of neurons, which tend to decrease with age[10,11], suggesting a deleterious effect on neuronal survival of these events. However, it is still unknown what these rates would be for non-neuronal brain cell populations such as glial and microglial cells. Expanding the study of sCNV from scWGS to other brain cell types offers the opportunity to further understand how early developmental sCNVs might impact normal lineage development, which might have a role in neural circuit architecture. Current efforts by consortia such as the Brain Somatic Mosaicism Network (BSMN)[64] aim to generate next-generation sequencing data of various neuropsychiatric disorders to explore some of these questions.
Acknowledgements
E.A.M. is supported by the Harvard/MIT MD-PhD program (T32GM007753), and the Biomedical Informatics and Data Science Training Program (T15LM007098). C.A.W is supported by the NIMH (grant U01MH106883) through the Brain Somatic Mosaicism Network (BSMN), and the Allen Frontiers Program through the Allen Discovery Center for Human Brain Evolution. C.A.W is an Investigator of the Howard Hughes Medical Institute.
Funding
Funding was received for this work.
All of the sources of funding for the work described in this publication are acknowledged below:
E.A.M. is supported by the Harvard/MIT MD-PhD program (T32GM007753), and the Biomedical Informatics and Data Science Training Program (T15LM007098). C.A.W is supported by the NIMH grant (U01MH106883) through the Brain Somatic Mosaicism Network (BSMN), and the Allen Frontiers Program through the Allen Discovery Center for Human Brain Evolution. C.A.W is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None
Intellectual Property
We confirm that we have given due consideration to the protection of intellectual property associated with this work and that there are no impediments to publication, including the timing of publication, with respect to intellectual property. In so doing we confirm that we have followed the regulations of our institutions concerning intellectual property.
References
- 1.Consortium SWG of the PG, Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans PA, Lee P, Bulik-Sullivan B, Collier DA, et al. : Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, et al. : Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 2017, 49:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGrath LM, Yu D, Marshall C, Davis LK, Thiruvahindrapuram B, Li B, Cappi C, Gerber G, Wolf A, Schroeder FA, et al. : Copy number variation in obsessive-compulsive disorder and tourette syndrome: a cross-disorder study. J Am Acad Child Adolesc Psychiatry 2014, 53:910–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. : De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. : Strong association of de novo copy number mutations with autism. Science (80-) 2007, 316:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kataoka M, Matoba N, Sawada T, Kazuno AA, Ishiwata M, Fujii K, Matsuo K, Takata A, Kato T: Exome sequencing for bipolar disorder points to roles of de novo loss-of-function and protein-altering mutations. Mol Psychiatry 2016, 21:885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehen SK, Yung YC, McCreight MP, Kaushal D, Yang AH, Almeida BSV, Kingsbury MA, Cabral KMS, McConnell MJ, Anliker B, et al. : Constitutional aneuploidy in the normal human brain. J Neurosci 2005, 25:2176–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knouse KA, Wu J, Whittaker CA, Amon A: Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci U S A 2014, 111:13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai X, Evrony GDD, Lehmann HSS, Elhosary PCC, Mehta BKK, Poduri A, Walsh CAA: Single-Cell, Genome-wide Sequencing Identifies Clonal Somatic Copy-Number Variation in the Human Brain. Cell Rep 2014, 8:1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, et al. : Mosaic copy number variation in human neurons. Science (80-) 2013, 342:632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chronister WD, Burbulis IE, Wierman MB, Weinberger DR, Bekiranov S, Mcconnell Correspondence MJ: Neurons with Complex Karyotypes Are Rare in Aged Human Neocortex. CellReports 2019, 26:825–835.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study suggests that the number with neurons with a sCNV event decreases with age.
- 12.Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. : Multiple Recurrent De Novo CNVs, Including Duplications of the 7q11.23 Williams Syndrome Region, Are Strongly Associated with Autism. Neuron 2011, 70:863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison RH, Kuo HC, Scriven PN, Handyside AH, Mackie Ogilvie C: Lack of cell cycle checkpoints in human cleavage stage embryos revealed by a clonal pattern of chromosomal mosaicism analysed by sequential multicolour FISH. Zygote 2000, 8:217–224. [DOI] [PubMed] [Google Scholar]
- 14.Vlismas A, Bletsa R, Mavrogianni D, Mamali G, Pergamali M, Dinopoulou V, Partsinevelos G, Drakakis P, Loutradis D, Kiessling AA: Microarray Analyses Reveal Marked Differences in Growth Factor and Receptor Expression between 8-Cell Human Embryos and Pluripotent Stem Cells. Stem Cells Dev 2016, 25:160–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knouse KA, Lopez KE, Bachofner M, Amon A: Chromosome Segregation Fidelity in Epithelia Requires Tissue Architecture. Cell 2018, 175:200–211.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study is one of the first ones to use scWGS to characterize aneuplody in normal neurons, suggesting that these events might be deleterious.
- 16.Vanneste E, Voet T, Le Caignec C, Ampe M, Konings P, Melotte C, Debrock S, Amyere M, Vikkula M, Schuit F, et al. : Chromosome instability is common in human cleavage-stage embryos. Nat Med 2009, 15:577–583. [DOI] [PubMed] [Google Scholar]
- 17.van Echten-Arends J, Mastenbroek S, Sikkema-Raddatz B, Korevaar JC, Jan Heineman M, van der Veen F, Repping S: Chromosomal mosaicism in human preimplantation embryos: a systematic review. Hum Reprod Update 2011, 17:620–627. [DOI] [PubMed] [Google Scholar]
- 18.Liu P, Yuan B, Carvalho CMB, Patel A, Hurles ME, Lupski JR: An Organismal CNV Mutator Phenotype Restricted to Early Human Development. Cell 2017, 168:830–842.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study explores how early developmental genomic instability might lead to sCNVs post-zygotically.
- 19.Kasak L, Rull K, Sõber S, Laan M: Copy number variation profile in the placental and parental genomes of recurrent pregnancy loss families. Sci Rep 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasak L, Rull K, Vaas P, Teesalu P, Laan M: Extensive load of somatic CNVs in the human placenta. Sci Rep 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suberbielle E, Sanchez PE, Kravitz AV., Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, Mucke L: Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat Neurosci 2013, 16:613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei PC, Chang AN, Kao J, Du Z, Meyers RM, Alt FW, Schwer B: Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 2016, 164:644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava CN, et al. : Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 359:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee M-H, Siddoway B, Kaeser GE, Segota I, Rivera R, Romanow WJ, Liu CS, Park C, Kennedy G, Long T, et al. : Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, doi: 10.1038/s41586-018-0718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, Zhao B, Huang AY, Miller MB, Lodato MA, Walsh CA, Lee EA: APPgene copy number changes reflect exogenous contamination. Nature 2020, 584:E20–E28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poduri A, Evrony GD, Cai X, Christina Elhosary P, Lehtinen MK, Benjamin Hills L, Heinzen EL, Hill A, Sean Hill R, Barry BJ, et al. : Somatic Activation of AKT3 Causes Hemispheric Developmental Brain Malformations. Neuron 2012, 74:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conti V, Pantaleo M, Barba C, Baroni G, Mei D, Buccoliero AM, Giglio S, Giordano F, Baek ST, Gleeson JG, et al. : Focal dysplasia of the cerebral cortex and infantile spasms associated with somatic 1q21.1-q44 duplication including the AKT3 gene. Clin Genet 2015, 88:241–247. [DOI] [PubMed] [Google Scholar]
- 28.Kobow K, Jabari S, Pieper T, Kudernatsch M, Polster T, Woermann FG, Kalbhenn T, Hamer H, Rössler K, Mühlebner A, et al. : Mosaic trisomy of chromosome 1q in human brain tissue associates with unilateral polymicrogyria, very early-onset focal epilepsy, and severe developmental delay. Acta Neuropathol 2020, 1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Gama AM, Woodworth MB, Hossain AA, Bizzotto S, Hatem NE, LaCoursiere CM, Najm I, Ying Z, Yang E, Barkovich AJ, et al. : Somatic Mutations Activating the mTOR Pathway in Dorsal Telencephalic Progenitors Cause a Continuum of Cortical Dysplasias. Cell Rep 2017, 21:3754–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jamuar SS, Lam ATN, Kircher M, D’Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A, et al. : Somatic mutations in cerebral cortical malformations. N Engl J Med 2014, 371:733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beverstock GC, Mollevanger P, Baaij M, Lind J, Van Ieperen L, Bartelings MM, Teunissen K, Brandenburg H, Van Opstal D, Los F: Nasopharyngeal teratoma and mosaic tetrasomy 1q detected at amniocentesis: A case report and review of the literature. Cancer Genet Cytogenet 1999, 115:11–18. [DOI] [PubMed] [Google Scholar]
- 32.King DA, Jones WD, Crow YJ, Dominiczak AF, Foster NA, Gaunt TR, Harris J, Hellens SW, Homfray T, Innes J, et al. : Mosaic structural variation in children with developmental disorders. Hum Mol Genet 2015, 24:2733–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Largest study to-date of sCNVs in children with developmental disorders from blood derived samples. Estimate burden of ~0.9%.
- 33.Sherman MA, Rodin RE, Genovese G, Dias C, Barton AR, Mukamel RE, Berger B, Park PJ, Walsh CA, Loh P-R: Large mosaic copy number variations confer autism risk. [date unknown], doi: 10.1101/2020.01.22.20017624. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Large study of blood derived samples of individuals with ASD. Estimated sCNV burden 0.4%.
- 34.Rodin RE, Dou Y, Kwon M, Sherman MA, D’Gama AM, Doan RN, Rento LM, Girskis KM, Bohrson CL, Kim SN, et al. : The Landscape of Mutational Mosaicism in Autistic and Normal Human Cerebral Cortex. bioRxiv 2020, doi: 10.1101/2020.02.11.944413. [DOI] [Google Scholar]
- 35.Lim ET, Uddin M, De Rubeis S, Chan Y, Kamumbu AS, Zhang X, D’Gama AM, Kim SN, Hill RS, Goldberg AP, et al. : Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci 2017, 20:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krupp DR, Barnard RA, Duffourd Y, Evans SA, Mulqueen RM, Bernier R, Rivière JB, Fombonne E, O’Roak BJ: Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am J Hum Genet 2017, 101:369–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dou Y, Yang X, Li Z, Wang S, Zhang Z, Ye AY, Yan L, Yang C, Wu Q, Li J, et al. : Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum Mutat 2017, 38:1002–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freed D, Pevsner J: The Contribution of Mosaic Variants to Autism Spectrum Disorder. PLOS Genet 2016, 12:e1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loh P-R, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, Birmann BM, Talkowski ME, Bakhoum SF, McCarroll SA, et al. : Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 2018, 559:350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study characterises patterns age related clonal hematopoisis sCNVs. Use of these patterns can be taken into account when studying early developmental sCNVs from blood derived samples.
- 40.Sebat J, Levy DL, McCarthy SE: Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet 2009, 25:528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kushima I, Aleksic B, Nakatochi M, Mori D, Iwata N, Ozaki N: Comparative Analyses of Copy-Number Variation in Autism Spectrum Disorder and Schizophrenia Reveal Etiological Overlap and Biological Insights Etiological overlap Orange: Intellectual disability + Patients without pathogenic CNVs. 2018, doi: 10.1016/j.celrep.2018.08.022. [DOI] [PubMed] [Google Scholar]
- 42.McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, Mistry M, Pavlidis P, Solomon R, Ghiban E, et al. : De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry 2014, 19:652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yurov YB, Vostrikov VM, Vorsanova SG, Monakhov VV, Iourov IY: Multicolor fluorescent in situ hybridization on post-mortem brain in schizophrenia as an approach for identification of low-level chromosomal aneuploidy in neuropsychiatric diseases. Brain Dev 2001, 23 Suppl 1:S186–90. [DOI] [PubMed] [Google Scholar]
- 44.Yurov YB, Vorsanova SG, Demidova IA, Kolotii AD, Soloviev IV., Iourov IY: Mosaic Brain Aneuploidy in Mental Illnesses: An Association of Low-level post-zygotic Aneuploidy with Schizophrenia and Comorbid Psychiatric Disorders. Curr Genomics 2017, 19:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yurov YB, Iourov IY, Vorsanova SG, Demidova IA, Kravetz VS, Beresheva AK, Kolotii AD, Monakchov VV., Uranova NA, Vostrikov VM, et al. : The schizophrenia brain exhibits low-level aneuploidy involving chromosome 1. Schizophr Res 2008, 98:139–147. [DOI] [PubMed] [Google Scholar]
- 46.Sakai M, Watanabe Y, Someya T, Araki K, Shibuya M, Niizato K, Oshima K, Kunii Y, Yabe H, Matsumoto J, et al. : Assessment of copy number variations in the brain genome of schizophrenia patients. Mol Cytogenet 2015, 8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J, Shin JY, Kim J Il, Seo JS, Webster MJ, Lee D, Kim S: Somatic deletions implicated in functional diversity of brain cells of individuals with schizophrenia and unaffected controls. Sci Rep 2014, 4:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maiti S, Kumar KHBG, Castellani CA, O’Reilly R, Singh SM: Ontogenetic De Novo Copy Number Variations (CNVs) as a Source of Genetic Individuality: Studies on Two Families with MZD Twins for Schizophrenia. PLoS One 2011, 6:e17125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castellani CA, Melka MG, Gui JL, Gallo AJ, O’Reilly RL, Singh SM: Post-zygotic genomic changes in glutamate and dopamine pathway genes may explain discordance of monozygotic twins for schizophrenia. Clin Transl Med 2017, 6:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, et al. : De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 2012, 17:142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morimoto Y, Ono S, Imamura A, Okazaki Y, Kinoshita A, Mishima H, Nakane H, Ozawa H, Yoshiura KI, Kurotaki N: Deep sequencing reveals variations in somatic cell mosaic mutations between monozygotic twins with discordant psychiatric disease. Hum Genome Var 2017, 4:17032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lyu N, Guan LL, Ma H, Wang XJ, Wu BM, Shang FH, Wang D, Wen H, Yu X: Failure to identify somatic mutations in monozygotic twins discordant for schizophrenia by whole exome sequencing. Chin Med J (Engl) 2016, 129:690–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raychaudhuri S, Korn JM, McCarroll SA, Altshuler D, Sklar P, Purcell S, Daly MJ, Daly MJ: Accurately Assessing the Risk of Schizophrenia Conferred by Rare Copy-Number Variation Affecting Genes with Brain Function. PLoS Genet 2010, 6:e1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clayton-Smith J, Watson P, Ramsden S, Black GCM: Somatic mutation in MECP2 as a non-fatal neurodevelopmental disorder in males. Lancet 2000, 356:830–832. [DOI] [PubMed] [Google Scholar]
- 55.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Purcell SM, et al. : Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. 2014, doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loh PR, Genovese G, McCarroll SA: Monogenic and polygenic inheritance become instruments for clonal selection. Nature 2020, doi: 10.1038/s41586-020-2430-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Terao C, Suzuki A, Momozawa Y, Akiyama M, Ishigaki K, Yamamoto K, Matsuda K, Murakami Y, McCarroll SA, Kubo M, et al. : Chromosomal alterations among age-related haematopoietic clones in Japan. Nature 2020, 584:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruderfer DM, Chambert K, Moran J, Talkowski M, Chen ES, Gigek C, Gusella JF, Blackwood DH, Corvin A, Gurling HM, et al. : Mosaic copy number variation in schizophrenia. Eur J Hum Genet 2013, 21:1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sekar A, Bialas AR, De Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, et al. : Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, Taylor KE, Whelan CW, Tombleson P, Loohuis LMO, et al. : Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 2020, 582:577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lim JS, Kim W Il, Kang HC, Kim SH, Park AH, Park EK, Cho YW, Kim S, Kim HM, Kim JA, et al. : Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015, 21:395–400. [DOI] [PubMed] [Google Scholar]
- 62.Mirzaa GM, Campbell CD, Solovieff N, Goold CP, Jansen LA, Menon S, Timms AE, Conti V, Biag JD, Olds C, et al. : Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol 2016, 73:836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, et al. : De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 2012, 44:941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McConnell MJ, Moran JV., Abyzov A, Akbarian S, Bae T, Cortes-Ciriano I, Erwin JA, Fasching L, Flasch DA, Freed D, et al. : Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science (80-) 2017, 356:eaal1641. [DOI] [PMC free article] [PubMed] [Google Scholar]