

Abstract
Post-translational modifications (PTMs) play roles in both physiological and pathophysiological processes through the regulation of enzyme structure and function. We recently identified a novel PTM, lactoylLys, derived through a non-enzymatic mechanism from the glycolytic by-product, lactoylglutathione. Under physiologic scenarios, glyoxalase 2 prevents the accumulation of lactoylglutathione and thus lactoylLys modifications. What dictates the site-specificity and abundance of lactoylLys PTMs, however, remains unknown. Here, we report sirtuin 2 as a lactoylLys eraser. Using chemical biology and CRISPR-Cas9, we show that SIRT2 controls the abundance of this PTM both globally and on chromatin. These results address a major gap in our understanding of how non-enzymatic PTMs are regulated and controlled.
Keywords: LactoylLys, Sirtuin, Post-Translational Modification, Molecular Modeling, Protein Modification
Graphical Abstract
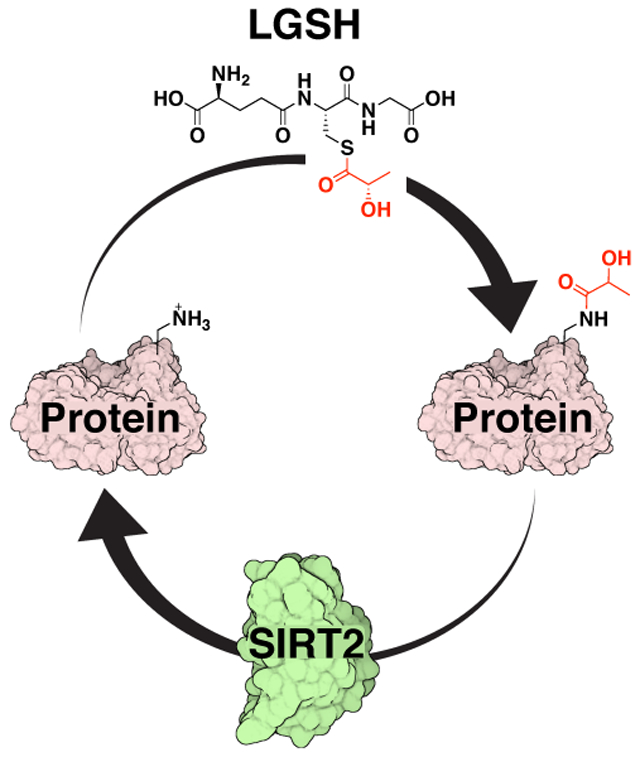
Protein post-translational modifications (PTMs) alter the structure and function of enzymes and are often regulated by enzymatic removal via ‘erasers.’ The non-enzymatically-derived PTM lactoylLys was recently identified in states of elevated glycolytic flux. Herein, we report the deacylase sirtuin 2 as the first known ‘eraser’ for lactoylLys, expanding the knowledge of enzymatic regulation of non-enzymatically-derived protein PTMs.
Post-translational modifications (PTMs) expand the functional proteome by regulating enzyme structure and function.[1] Many PTMs are derived from intermediates produced during cell metabolism and can serve as sensors for metabolic feedback providing proper nutrients to maintain cell growth and homeostasis.[1b, 1c] Due to their critical role in preserving homeostasis, site-specificity and abundance are tightly regulated, often through enzymatic addition (‘writers’) and removal (‘erasers’)[1c]. Mounting evidence, however, points to significant roles for non-enzymatic PTMs in the regulation of transcription, metabolism, and signaling.[2] We recently identified a PTM derived from the glyoxalase cycle intermediate, lactoylglutathione (LGSH) (Figure 1).[2b] When glycolytic flux exceeds the capacity of the glyoxalase cycle, glyoxalase 2 (GLO2), the rate limiting enzyme, becomes overwhelmed resulting in the accumulation of LGSH and subsequent non-enzymatic acylation of protein Lys residues (i.e. lactoylLys) (Figure 1).[2b] This modification is enriched on glycolytic enzymes, serving as a negative feedback mechanism for glycolysis.[2b] Coinciding with our report, Zhang, et al. observed lactoylLys modifications on histones, linking this modification to inflammatory signaling.[3]
Figure 1.

Regulation of lactoylLys. The glyoxalase cycle and the non-enzymatic acyl transfer of lactate generating lactoylLys modifications. To date, a dedicated ‘eraser’ has yet to be identified. MGO, methylglyoxal; GSH, glutathione; LGSH, lactoylglutathione; GLO1, glyoxalase 1; GLO2, glyoxalase 2.
Due to the novelty of this PTM, little is known about their regulation. With the non-enzymatic acyl-transfer mechanism defined in our report[2b] lactoylLys modifications would presumably accumulate in the absence of a dedicated ‘eraser’ protein. Thus, we hypothesized that a molecular ‘eraser’ protein(s) must exist for this PTM. LactoylLys modifications contain the necessary acyl-moiety required for the deacylase enzymes, sirtuins.[2b] Sirtuins play a role in both metabolism and inflammation through the regulation of Lys PTMs.[4] The sirtuin family is comprised of seven NAD+-dependent enzymatic deacylases (SIRT1-7), defined broadly by their subcellular localization (Figure 2A).[5] As we previously reported, our proteomic inventory of lactoylLys-modified proteins revealed an enrichment for glycolytic enzymes,[2b] suggesting the cytosolic sirtuins (SIRT1 and SIRT2) as potential candidates for a lactoylLys eraser. SIRT2 recognizes a more diverse group of substrates; furthermore, SIRT2 plays a regulatory role in both glycolysis and inflammation.[4a-c, 5a, 6] While SIRT2 is primarily localized to the cytosol in cells, it can translocate to the nucleus to interact with acylated chromatin.[5, 6b, 7] This substrate promiscuity and subcellular localization make SIRT2 a likely enzymatic deacylase for lactoylLys modifications.
Figure 2.

SIRT2 removes lactoylLys modifications in vitro. (A) Sirtuin localization in cells. (B) Sirtuins 1-7 incubated with lactoylLys modified peptides. N = 3; *p < 0.05; ***p < 0.001, ± S.D. Statistical significance determined via One-Way ANOVA. (C) Kinetic parameters for SIRT2 on acetylated and lactoylated peptides. N = 3; Km ± S.E.; Kcat ± S.E.
To investigate this hypothesis, we screened the activity of recombinant sirtuins (SIRT1-7) against a lactoylated peptide from a known lactoylated protein, pyruvate kinase M2 (PKM2) (Figure 2B).[2b] This Lys residue (EIPAE-K*-VFLAQ) is also reported to be acetylated and regulated by SIRT2.[6a] The acyl transfer from LGSH to Lys residues defined in our previous work occurs at the carbonyl carbon rather than at the chiral carbon; thus, we predict the lactoylLys modification outlined in our report is generated in the D orientation in the same manner as D-lactate formation.[2b] In contrast, the hypothesized mechanism reported by Zhao et al. suggests enzymatic addition through L-lactyl-CoA, thus yielding L-lactoylLys modifications.[3]
As shown in Figure 2B, SIRT1, 2, 3, and 5 removed their canonical acetylated peptide. The L- enantiomer of lactoylLys, however, displayed significantly reduced removal with SIRT1 and 5 while SIRT2 was able to remove the L-enantiomer. In contrast, all SIRTs displayed minimal activity towards the D-enantiomer. To control for non-enzymatic hydrolysis of the acyl moiety, peptides were incubated without recombinant enzyme (CTRL, Figure 2B) or without NAD+ (Figure S1). A kinetic analysis confirms these findings; SIRT2 catalyzes the deacylation of the acetyl peptide at a faster rate than the lactoylated peptides (Figure 2C, S2). In further support of Figure 2B, SIRT2 displayed a greater propensity for the L-enantiomer vs the D-enantiomer, whereby steady-state could not be achieved, resulting in a large Km and standard error (Figure 2C, S2). This data suggests that SIRT2 is capable of removing the L- enantiomer suggesting this to be the predominant form in vivo. Further investigation is needed to determine the enantiomeric species generated from this non-enzymatic acyl transfer from LGSH.
To further investigate the interaction of SIRT2 and lactoylLys, we performed molecular docking experiments (Figure 3). This docking provides in silico insight into the enantiomeric preference of SIRT2 for lactoylated substrates. Unsurprisingly, docking of an acetylLys peptide to the catalytic pocket of SIRT2 results in the acetyl group being placed in a catalytically competent conformation. The acetyl oxygen points toward NAD+, able to attack the C1’ of the nicotinamide ribose ring with a distance of 4.3 Å (Figure 3A). Docking of this peptide modified with L-lactoylLys results in the same catalytically favorable conformation for the L-lactoyl group, with the oxygen of the lactoyl carbonyl also oriented toward the nicotinamide ribose ring of NAD+ with a distance of 4.1 Å (Figure 3B). However, molecular docking of the D-lactoylLys peptide results in a different orientation within the catalytic pocket of SIRT2 with the oxygen of the lactoyl carbonyl 6.4 Å from the C1’ of the nicotinamide ribose ring of NAD+. D-lactoylLys is positioned with the lactoyl hydroxyl pointing toward NAD+ and the oxygen of the lactoyl carbonyl pointing away from the nicotinamide ribose ring of NAD+ (Figure 3C), resulting in a conformation that is catalytically incompetent for deacylation. The positioning of the lactoyl group within the catalytic pocket of SIRT2 may explain the enhanced propensity for deacylation of L- over D-lactoylLys by SIRT2.
Figure 3.

Molecular modeling provides insights into enantiomeric specificity of lactoylLys modifications. Carba-NAD+ is shown on the right side of each panel. (A) Molecular modeling of an acetylLys peptide (green) docked to SIRT2 with carba-NAD+ in a catalytically competent conformation. Predicted distance from acyl carbonyl group to C1’ of the nicotinamide ribose ring is 4.3 Å. (B) L-lactoylLys peptide (orange) docked to SIRT2 with carba-NAD+ in a catalytically competent conformation. Predicted distance from acyl carbonyl group to C1’ of the nicotinamide ribose ring is 4.1 Å. (C) D-lactoylLys peptide (cyan) docked to SIRT2 in a catalytically incompetent conformation. Predicted distance from acyl carbonyl group to C1’ of the nicotinamide ribose ring is 6.4 Å. SIRT2 PDB: 5G4C; Peptide sequence: AA-K*-T.
LactoylLys PTMs are dependent on the intracellular concentrations of LGSH.[2b] As GLO2 is the only reported enzyme to hydrolyze LGSH,[8] we generated SIRT2−/− cell lines in wildtype (WT) and GLO2−/− HEK293 cells using CRISPR-Cas9 (Figure 4A).[2b] To evaluate the abundance of lactoylated PTMs in each cohort, we performed reactivity-based protein profiling using an alkyne analog of methylglyoxal (aMGO).[9] Cells were treated with 50 μM aMGO for 6h, which is consistent with our previous report.[2b] An evaluation of whole cell lysates reveals protein labeling in GLO2−/− cells. Cells lacking both GLO2 and SIRT2, however, displayed a marked elevation over GLO2−/− alone (Figure 4B). These effects are more pronounced in chromatin fractions isolated from each cohort, indicative of tighter SIRT2 regulation on chromatin (Figure 4C).
Figure 4.

LactoylLys abundance increases with the loss of SIRT2. (A) Generation of GLO2−/−, SIRT2−/−, and SIRT2−/−/GLO2−/− HEK293 cells with CRISPR/Cas9. (B) LactoylLys modifications are observed in GLO2−/− whole cell lysates and are further elevated in SIRT2−/−/GLO2−/− whole cell lysates when treated with 50 μM aMGO and subjected to click chemistry. (C) LactoylLys modifications are observed in SIRT2−/−/GLO2−/− chromatin when treated with 50 μM aMGO and subjected to click chemistry. (D) Overview of SILAC QuARKMod performed with GLO2−/− and SIRT2−/−/GLO2−/− HEK293 cell lines. (E) Light to heavy ratio of lactoylLys and MGO modifications in SIRT2−/−/GLO2−/− (light) and GLO2−/− (heavy) lysates. Statistical significance was determined via t-test, N = 4, ***p < 0.001, ± S.D. CEL, carboxyethyllysine; MG-H, methylglyoxal-hydroimidazalone; CEA, carboxyethylarginine.
Lastly, to quantify elevated abundance of lactoylLys in the SIRT2−/−/GLO2−/− cell line we used stable isotope labeling of amino acids in cell culture (SILAC) and QuARKMod (a method for the quantitative analysis of arginine (R) and lysine (K) modifications)[2b, 10] in cells treated with high glucose (25 mM, Figure 4D). GLO2−/− cells were cultured in ‘heavy’ media containing 13C615N2 Lys and 13C615N4 Arg and SIRT2−/−/GLO2−/− cells were cultured in ‘light’ media containing natural abundance isotope amino acids. Both cell lines were treated with high glucose to evaluate the impact of lactoylLys PTMs in the context of increased glycolytic flux. As shown in Figure 4E, SIRT2−/−/GLO2−/− cells have a 5.4-fold elevation in lactoylLys modifications and no significant changes in the MGO derived modifications CEL, MG-H, and CEA compared to GLO2−/− cells indicating SIRT2’s role as an eraser of lactoylLys PTMs.
Here, we present the first report of a lactoylLys eraser. LactoylLys modifications play regulatory roles in situations of elevated glycolytic flux, which are found in both hyperglycemic and inflammatory states.[2b, 3] SIRT2 regulates a diverse array of physiological and pathophysiological processes, including inflammation, response to infections, cell metabolism, ageing, cell structure, oxidative stress, and neurological disorders.[4, 11] Consistent with a lack of lactoylLys modifications on histones in our published proteomic inventory,[2b] our results show only SIRT2−/−/GLO2−/− chromatin have dramatically elevated levels of labeled proteins, indicating tighter regulation by SIRT2 on chromatin. In addition to acetylation, SIRT2 regulates Lys benzoylation and 4-oxo-2-nonenal modifications on histones, further indicating a role for SIRT2 PTM regulation at the chromatin level.[12] Collectively, these data provide evidence for the enzymatic regulation of non-enzymatically-derived protein PTMs. Future work will be dedicated to identifying the site-specificity of this regulation in vitro and in vivo.
Supplementary Material
Acknowledgements
aMGO was a generous gift from Dr. Mogens Johannsen, Aarhus University, Aarhus Denmark. Financial support was provided by National Institutes of Health Grants (T32 ES007091 for E.Q.J.; 2T32GM06754 3-17 for J.D.R.; T32 GM008804 for C.J.Z.; NIEHS R01ES031463 for E.C.; R35 GM137910 for J.J.G.) and the ALSAM Skaggs Scholars Program (J.J.G.).
References
- [1].a) Karve TM, Cheema AK, J Amino Acids 2011, 2011, 207691; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ducker GS, Rabinowitz JD, Cell Metab 2017, 25, 27–42; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li X, Egervari G, Wang Y, Berger SL, Lu Z, Nat Rev Mol Cell Biol 2018, 19, 563–578; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Spoel SH, J Exp Bot 2018, 69, 4499–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) James AM, Hoogewijs K, Logan A, Hall AR, Ding S, Fearnley IM, Murphy MP, Cell Rep 2017, 18, 2105–2112; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gaffney DO, Jennings EG, Anderson CC, Marentette JO, Shi T, Schou Oxvig AM, Streeter MD, Johannsen M, Spiegel DA, Chapman E, Roede JR, Galligan JJ, Cell Chem Biol 2020, 27, 206–213 e206; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Moellering RE, Cravatt BF, Science 2013, 341, 549–553; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zheng G, Prescott NA, Maksimovic I, David Y, Chem Res Toxicol 2019, 32, 796–807; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wagner GR, Payne RM, J Biol Chem 2013, 288, 29036–29045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, Ding J, Czyz D, Hu R, Ye Z, He M, Zheng YG, Shuman HA, Dai L, Ren B, Roeder RG, Becker L, Zhao Y, Nature 2019, 574, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Liu T, Zhang L, Joo D, Sun SC, Signal Transduct Target Ther 2017, 2; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gomes P, Fleming Outeiro T, Cavadas C, Trends Pharmacol Sci 2015, 36, 756–768; [DOI] [PubMed] [Google Scholar]; c) Rothgiesser KM, Erener S, Waibel S, Luscher B, Hottiger MO, J Cell Sci 2010, 123, 4251–4258; [DOI] [PubMed] [Google Scholar]; d) Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE, J Biol Chem 2011, 286, 9856–9864; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mendes KL, Lelis DF, Santos SHS, Cytokine Growth Factor Rev 2017, 38, 98–105; [DOI] [PubMed] [Google Scholar]; f) Vachharajani VT, Liu T, Wang X, Hoth JJ, Yoza BK, McCall CE, J Immunol Res 2016, 2016, 8167273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Houtkooper RH, Pirinen E, Auwerx J, Nat Rev Mol Cell Biol 2012, 13, 225–238; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) North BJ, Verdin E, Genome Biol 2004, 5, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Park SH, Ozden O, Liu G, Song HY, Zhu Y, Yan Y, Zou X, Kang HJ, Jiang H, Principe DR, Cha YI, Roh M, Vassilopoulos A, Gius D, Cancer Res 2016, 76, 3802–3812; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li T, Garcia-Gomez A, Morante-Palacios O, Ciudad L, Ozkaramehmet S, Van Dijck E, Rodriguez-Ubreva J, Vaquero A, Ballestar E, Nucleic Acids Res 2020, 48, 665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D, Genes Dev 2006, 20, 1256–1261; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tanabe K, Liu J, Kato D, Kurumizaka H, Yamatsugu K, Kanai M, Kawashima SA, Sci Rep 2018, 8, 2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rabbani N, Xue M, Thornalley PJ, Biochem Soc Trans 2014, 42, 419–424. [DOI] [PubMed] [Google Scholar]
- [9].a) Sibbersen C, Schou Oxvig AM, Bisgaard Olesen S, Nielsen CB, Galligan JJ, Jorgensen KA, Palmfeldt J, Johannsen M, ACS Chem Biol 2018, 13, 3294–3305; [DOI] [PubMed] [Google Scholar]; b) Kold-Christensen R, Jensen KK, Smedegard-Holmquist E, Sorensen LK, Hansen J, Jorgensen KA, Kristensen P, Johannsen M, Redox Biol 2019, 26, 101252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Galligan JJ, Wepy JA, Streeter MD, Kingsley PJ, Mitchener MM, Wauchope OR, Beavers WN, Rose KL, Wang T, Spiegel DA, Marnett LJ, Proc Natl Acad Sci U S A 2018, 115, 9228–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang Y, Yang J, Hong T, Chen X, Cui L, Ageing Res Rev 2019, 55, 100961. [DOI] [PubMed] [Google Scholar]
- [12].a) Huang H, Zhang D, Wang Y, Perez-Neut M, Han Z, Zheng YG Hao G, Zhao Y, Nat Commun 2018, 9, 3374; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jin J, He B, Zhang X, Lin H, Wang Y, J Am Chem Soc 2016, 138, 12304–12307; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Galligan JJ, Rose KL, Beavers WN, Hill S, Tallman KA, Tansey WP, Marnett LJ, J Am Chem Soc 2014, 136, 11864–11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.