

Supplemental Digital Content is available in the text.
Keywords: desmosomes, diagnosis, genes, genetic testing, tachycardia
Abstract
Background:
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited disease characterized by ventricular arrhythmias and progressive ventricular dysfunction. Genetic testing is recommended, and a pathogenic variant in an ARVC-associated gene is a major criterion for diagnosis according to the 2010 Task Force Criteria. As incorrect attribution of a gene to ARVC can contribute to misdiagnosis, we assembled an international multidisciplinary ARVC Clinical Genome Resource Gene Curation Expert Panel to reappraise all reported ARVC genes.
Methods:
Following a comprehensive literature search, six 2-member teams conducted blinded independent curation of reported ARVC genes using the semiquantitative Clinical Genome Resource framework.
Results:
Of 26 reported ARVC genes, only 6 (PKP2, DSP, DSG2, DSC2, JUP, and TMEM43) had strong evidence and were classified as definitive for ARVC causation. There was moderate evidence for 2 genes, DES and PLN. The remaining 18 genes had limited or no evidence. RYR2 was refuted as an ARVC gene since clinical data and model systems exhibited a catecholaminergic polymorphic ventricular tachycardia phenotype. In ClinVar, only 5 pathogenic/likely pathogenic variants (1.1%) in limited evidence genes had been reported in ARVC cases in contrast to 450 desmosome gene variants (97.4%).
Conclusions:
Using the Clinical Genome Resource approach to gene-disease curation, only 8 genes (PKP2, DSP, DSG2, DSC2, JUP, TMEM43, PLN, and DES) had definitive or moderate evidence for ARVC, and these genes accounted for nearly all pathogenic/likely pathogenic ARVC variants in ClinVar. Therefore, only pathogenic/likely pathogenic variants in these 8 genes should yield a major criterion for ARVC diagnosis. Pathogenic/likely pathogenic variants identified in other genes in a patient should prompt further phenotyping as variants in many of these genes are associated with other cardiovascular conditions.
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy characterized by fibro-fatty myocardial replacement, frequent ventricular arrhythmias, and slowly progressive ventricular dysfunction. Patients typically present between their second and fifth decades with symptoms associated with ventricular arrhythmias.1 Sudden cardiac death is a common presentation, occurring in up to half of probands.
ARVC clusters in families and its pattern of inheritance is generally autosomal dominant with age-related, reduced penetrance.2 The discovery that Naxos disease, a rare cardiocutaneous autosomal recessive form of ARVC, was caused by pathogenic variants in JUP-encoded plakoglobin3 prompted rapid identification of pathogenic variants in other desmosomal genes (PKP2, DSP, DSG2, and DSC2) in ARVC populations. In contemporary ARVC cohorts meeting 2010 Task Force Criteria (TFC), up to two-thirds of cases have pathogenic/likely pathogenic (P/LP) desmosomal variants.4,5
Exponential growth in sequencing capacity led investigators to sequence ever-growing lists of candidate genes and to undertake exome and genome sequencing in attempts to identify the genetic basis of ARVC in gene-elusive patients. These studies identified variants of interest in numerous genes which were then suggested to be ARVC causative. However, some reports were based on an incomplete understanding of the extent of rare variation in the human genome. Older studies used dated diagnostic criteria for inclusion. Finally, isolated gene-elusive ARVC may be oligogenic,2 calling the assumptions underlying some gene identification studies into question. Despite these limitations, newly reported ARVC genes have been promptly added to diagnostic ARVC sequencing panels which now generally range from 11 to 46 genes. Genetic testing is recommended for patients with ARVC to confirm the diagnosis, inform management, and enable cascade genetic testing.6
The NIH-funded Clinical Genome Resource (ClinGen) created a standardized evidence-based framework to systematically assess gene-disease relationships.7 Recently, evaluation of genes associated with hypertrophic cardiomyopathy,8 long QT syndrome,9 and Brugada syndrome10 called into question causality of many disease genes. Similar weaknesses in conventional understanding of the genetic architecture for ARVC seemed likely. ARVC can be difficult to diagnose, particularly when phenotypic expression is mild or when the patient has biventricular disease raising the possibility that gene:diseases associations may have been erroneously derived from participants with other cardiovascular diseases. The TFC was updated in 2010 and older articles relied on less sensitive and specific 1994 criteria5 making phenotyping in older publications potentially problematic.
Since identification of a P/LP variant constitutes a major criterion in the TFC5 accurate understanding of genetic architecture has direct implications for ARVC diagnosis and management. Incorrect ARVC gene:disease associations may result in (1) a patient’s phenotype incorrectly attributed to a variant leading to potential over-diagnosis and incorrect cascade genetic testing or (2) a variant associated with a different disease incorrectly attributed to ARVC missing the opportunity for correct genotype-specific management of a family. Therefore, an international multidisciplinary ARVC ClinGen Gene Curation Expert Panel (GCEP; Table I in the Data Supplement; https://clinicalgenome.org/affiliation/40003/) with expertise in ARVC research, genetics, and clinical care was assembled to formally reappraise all previously reported ARVC genes using the ClinGen Gene-Disease Clinical Validity Framework. To enhance scientific rigor, we used a dual, blinded, independent curation approach (Figure 1). In this effort, we defined ARVC by fulfillment of the 2010 TFC.5 While arrhythmogenic cardiomyopathy has been suggested as a concept by several groups of authors,6,11,12 at present, there is considerable variability in the breadth of phenotypes covered by the term arrhythmogenic cardiomyopathy and no standard agreed-upon diagnostic criteria that could be applied for gene curation. Here, we report our results.
Figure 1.
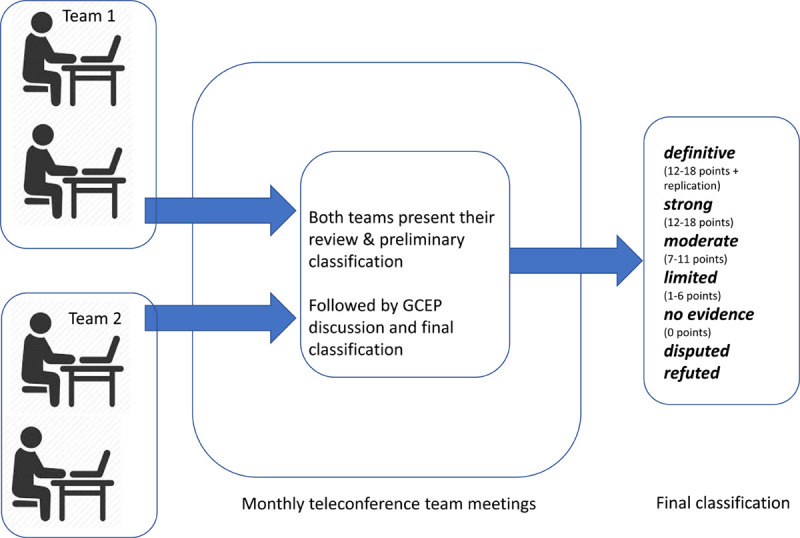
Arrhythmogenic right ventricular cardiomyopathy (ARVC) gene curation approach. Two-member teams conducted blinded independent dual curation using the semiquantitative Clinical Genome Resource (ClinGen) framework with ARVC-specific rules for required minor allele frequency of variants detected in patients and phenotypic evaluation of model systems. Each summarized their analysis in separate presentations for the entire ARVC gene curation expert panel (GCEP) who arrived by consensus at the final gene classification.
Materials and Methods
The data that support the findings of this study are available from the corresponding authors upon reasonable request. The study was approved by the Johns Hopkins Medicine institutional review board (data for variant frequency cutoffs) and subjects gave informed consent. The full methods for this study are available as Data Supplement (Methods in the Data Supplement).
Results
Overview
PubMed/OMIM searches resulted in 26 genes reported to cause human ARVC: ACTC1, CDH2, CTNNA3, LDB3, DES, DSC2, DSG2, DSP, JUP, LMNA, MYBPC3, MYH7, MYL2, MYL3, PKP2, PLN, RYR2, SCN5A, TGFB3, TJP1, TMEM43, TNNI3, TNNC1, TNNT2, TPM1, and TTN (Table 1). As shown in Figure 2, based on initial scoring, 6 genes (PKP2, DSP, DSC2, DSG2, JUP, and TMEM43) had strong evidence (12–18 points) and were judged to be definitive for ARVC causation as each had replication across ARVC cohorts. There was moderate evidence for 2 genes, DES (9.5 points) and PLN (11 points). The remaining genes had only limited or no evidence for ARVC causation (0–6 points). Curation team scores were highly concordant. For every gene both curation teams arrived at the same preliminary classification based on points achieved (Table II in the Data Supplement).
Table 1.
Reported Genes for ARVC
Figure 2.
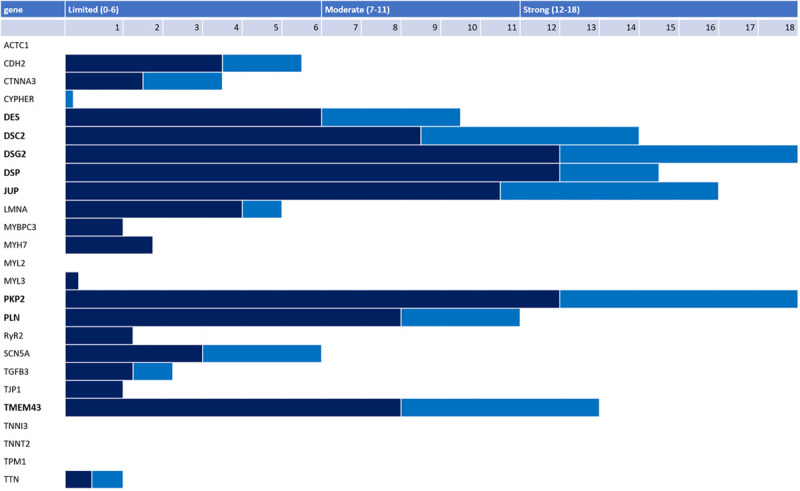
Level of evidence scores for genes reported for arrhythmogenic right ventricular cardiomyopathy (ARVC). Final genetic (dark blue) and experimental (light blue) evidence scores for 26 genes reported in the literature as associated with ARVC. Only 8 genes (bold font) had strong or moderate evidence for ARVC causality. The granular scores for each gene along with a complete list of references used are available in Table IV in the Data Supplement.
Table 2 summarizes the evidence for genes designated to have definitive or moderate evidence for ARVC causation by the GCEP. The evidence used to arrive at these final classifications was predominantly derived from clinical genetic studies. Table III in the Data Supplement shows scoring for genes with limited or no evidence for ARVC. Granular scores for each subcategory of genetic and experimental evidence can be found for each gene in Table IV in the Data Supplement. The most up-to-date curation data for each gene can be accessed at https://clinicalgenome.org/.
Table 2.
Genetic Architecture of ARVC
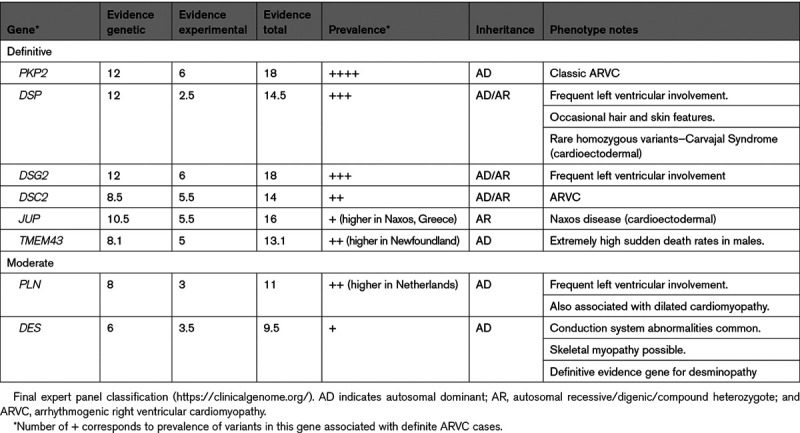
As shown in Table 2, the GCEP classified each gene with a score in the strong range (12–18 points) as having definitive evidence for ARVC. The genes encoding the cardiac desmosome (PKP2, DSP, DSG2, DSC2, and JUP) have been consistently identified across multiple ARVC cohorts. TMEM43 was initially identified due to a founder variant segregating in a well-characterized ARVC population in Newfoundland, and subsequently in the United Kingdom, Denmark, and Germany and recently on a different haplotype in Spain.13–15 The GCEP confirmed the moderate evidence classification of PLN which was likewise initially associated with ARVC due to a segregating founder variant.16 A moderate evidence classification was also confirmed for DES. While not typically considered an ARVC gene, it is frequently associated with a myopathy17 in which arrhythmogenic cardiomyopathy (myofibrillar myopathy-1, desminopathy) is a presenting feature and patients can meet TFC.18
The GCEP concluded the remaining 18 genes—more than two-thirds of the genes analyzed—did not have convincing evidence for ARVC causality. These fell into 2 categories: (1) genes—often newly published—with rare variants detected in small families with a clear ARVC phenotype but for which data was yet limited and (2) genes known to be associated with other cardiomyopathies or arrhythmia syndromes where evidence for ARVC causality was relatively scant. Notably, the panel refuted RYR2 as an ARVC gene, finding P/LP variants in RYR2 typically cause catecholaminergic polymorphic ventricular tachycardia (CPVT) not ARVC. All the literature asserting ARVC causality was judged to be incorrect and based on incomplete phenotyping, wrong interpretation of clinical data (personal communication G. Thiene and C. Basso), use of dated diagnostic criteria, and designation of relatively common RYR2 variants as pathogenic.
Genes With Definitive Evidence for ARVC Causality
Cardiac desmosomes are specialized structures composed of proteins (cadherins, armadillo proteins, and plakins) responsible for cardiomyocyte adhesion. ARVC is classically considered a disease of the desmosome and the desmosomal genes rapidly achieved sufficient evidence for a strong designation with replication across cohorts making the gene:disease relationship definitive. There were several nuances. First, ARVC can be accompanied by skin and hair features. This has long been recognized in families with JUP-associated Naxos disease and DSP-associated Carvajal syndrome, both of which have autosomal recessive inheritance. Several authors have reported skin and hair findings in patients with heterozygous DSP variants which can be subtle.19 Importantly, there is evidence across studies for a relatively high prevalence of patients with multiple desmosomal P/LP variants beyond Naxos disease and Carvajal syndrome. Pedigrees segregating multiple DSC2 and DSG2 variants have been reported and inheritance more consistent with autosomal recessive than autosomal dominant inheritance has been recognized in specific populations.20,21
The TMEM43 (transmembrane protein 43) gene encodes a nuclear membrane protein. One heterozygous pathogenic variant (NM_024334.3(TMEM43):c.1073C>T; p.Ser358Leu) was identified as a founder mutation in a large number of patients and families from Newfoundland, Denmark, and Germany and has also been identified in other populations.15,22 It is associated with a highly penetrant and arrhythmogenic subtype of ARVC in which biventricular involvement can often be appreciated. Evidence of pathogenicity of other TMEM43 variants remains limited.
Moderate Evidence Genes
DES and PLN had moderate evidence for ARVC causality. DES was initially proposed as an ARVC gene based on data from 27 Dutch individuals in 5 families segregating a rare missense variant (NM_001927.4(DES):c.38C>T; p.Ser13Phe).23 Cases had right ventricular involvement consistent with ARVC but also conduction disease which is atypical for ARVC. Additional families carrying DES LP/P variants with a clinical ARVC diagnosis as well as families with left-predominant disease have been described.18,24,25 Experimental evidence including expression systems integrating variants found in these families showed phenotypic alterations consistent with histological examinations of skeletal and cardiac muscle of ARVC cases18 and disruption of cellular adhesion.26 Nonetheless, DES variants associated with ARVC appear to be very rare and have not been observed in some large ARVC cohorts.
The PLN p.Arg14del variant (NM_002667.5(PLN):c.37_39AGA[1]) was first identified in ARVC in a cohort of 12/97 patients fulfilling TFC.27 Histology showed the typical fibro-fatty replacement and interstitial fibrosis yet compared with desmosomal gene-positive patients, PLN p.Arg14del showed significantly more severe fibrotic changes in the left ventricle, underscoring its biventricular character.28
Limited/No Human Evidence Genes
Ten genes had limited evidence for ARVC causality (1–6 points): SCN5A, LMNA, CDH2, CTNNA3, TGFB3, TTN, TJP1, MYH7, MYBPC3, and MYL3. In comparison to the wealth of literature linking desmosomal genes with ARVC, evidence for these gene:disease relationships had been generated by relatively few research groups. For some of these genes, assertions of ARVC causality are relatively new and further data from larger cohorts might lead to an upgraded level of evidence in the future. For others, the assertion of ARVC causality was published some time ago, leading the panel to give additional weight to the failure to confirm the observation across ARVC cohorts. Our review strongly suggested that none of these genes account for a substantial fraction of patients with ARVC.
Genes With Variants Identified in Classic ARVC Families but Yet Limited Evidence
CDH2, CTNNA3, and TJP1 showed evidence for a classic ARVC phenotype and segregation in several relatively small families but with human data as yet limited. CDH2 and CTNNA3 encode the area composita proteins cadherin-2 and α T catenin, respectively. TJP1 encodes a scaffolding protein, tight junction protein 1, which localizes to the intercalated discs in cardiomyocytes.
Evidence for CDH2 included identification of 2 LP rare missense variants in 3 probands meeting TFC.29,30 One variant (NM_001792.4(CDH2): c.1219G>A; p.Asp407Asn) was identified in both a South African and a Norwegian family. Another (NM_001792.4(CDH2): c.686A>C; p.Gln229Pro) segregated among 5 affected family members. A murine knockout model showed disrupted desmosomes, cardiomyopathy, ventricular tachycardia, and sudden death, suggestive of a phenotype compatible with ARVC.
Similarly, for CTNNA3, 2 variants were reported in 2 ARVC probands: one likely de novo missense variant absent from gnomAD (NM_013266.3(CTNNA3): c.281T>A; p.Val94Asp) and one in-frame deletion (NM_013266.3(CTNNA3): c.2296_2298del; p.Leu766del) with limited segregation.31 A germline knockout mouse showed altered PKP2 distribution without affecting other junctional components of the area composita. These mice had progressive dilated cardiomyopathy (DCM), and the GCEP judged the phenotype not completely convincing for ARVC. Furthermore, no CTNNA3 LP/P variants were reported in 2 series of gene-elusive ARVC patients.
Finally, 2 probands with ARVC (as well as 2 probands with DCM) had variants in TJP1.32 Modest segregation data allowed this to be counted as limited human evidence.
Genes Associated With Other Cardiomyopathies/Arrhythmia Syndromes
The remaining genes curated were each strongly associated with other cardiomyopathies or arrhythmia syndromes. The GCEP concluded each had limited or no evidence for ARVC causality. The gene with the most evidence (6 points) was SCN5A which encodes the Nav 1.5 sodium channel and previously curated as definitive for both Brugada syndrome and long QT syndrome.9,10 The most robust evidence for SCN5A as an ARVC gene comes from identification of a variant (NM_198056.2(SCN5A): c.5693G>A; p.Arg1898His) in a gene-elusive ARVC patient via exome sequencing followed by derivation of an induced pluripotent stem cell-derived cardiomyocyte model.33 This model showed a one-third reduction in peak sodium current and reduced abundance of both SCN5A and CDH2 clusters at the intercalated disk which normalized in a CRISPR/Cas9 corrected line. The authors subsequently identified 5 SCN5A variants among 281 ARVC probands. One variant was excluded for being too common and 2 were found in probands who also had pathogenic desmosomal variants. One proband had an in-frame deletion (NM_198056.2(SCN5A): c.2184-2186del; p.Leu729del) which segregated with the phenotype, but most family members did not fulfill definite TFC.
Potentially pathogenic variants in LMNA have been published in several ARVC cohorts.34,35 While evidence was sufficient to merit a limited evidence classification, the GCEP noted that while most probands did meet TFC, many of their affected family members did not. The phenotypes observed overlapped with DCM and were characterized by prominent conduction system abnormalities and atrial arrhythmias.
Eleven articles were reviewed to assess the relationship of TGFB3 with ARVC, the majority from the same research group. TGFB3 emerged as a candidate gene based on linkage to 14q23-q24 in several Italian families. Sequencing found variants in the 3′ untranslated region in one family and a second regulatory noncoding variant in an unrelated family.36 These variants were both associated with increased activity compared with wild type in an expression assay. However, 2 of the initial families with significant linkage to the candidate region had no P/LP variants in the TGFB3 coding sequences, UTRs, and promoter regions. No definitively pathogenic variants have been subsequently reported in ARVC probands. Nowadays, TGFB3 is believed to underlie Loeys-Dietz syndrome type 5, a connective tissue disease phenotype with features of Marfan syndrome, including aortic abnormalities.37 The GCEP concluded that while TGFB3 merited a limited evidence classification for ARVC, a variant detected in TGFB3 should be treated with great caution and is unlikely the cause of a patient’s ARVC.
TTN, encoding titin, is a frequent cause of familial DCM. Nine papers were evaluated for the role of TTN variants in ARVC causation. Many reported missense variants which were relatively common in gnomAD. Furthermore, in study reporting 11/35 ARVC probands with TTN missense variants, relatives carrying the variant had no evidence of disease leading them to conclude these TTN variants had very low penetrance or negligible pathogenicity.38 One article did describe a rare missense variant NM_133378.4 (TTN): c.8678C>T; p.Thr2896Ile that segregated among 9 family members—6 of whom met TFC.39 An in vitro functional assay by 2 independent groups found that the variant introduced aberrant function. Further evidence has shown that TTN-associated DCM is not particularly associated with an arrhythmogenic phenotype.40 The GCEP thus concluded there was very limited evidence for TTN as ARVC causative.
The sarcomere genes have been considered by several research groups as a potential cause of ARVC.41 MYH7, MYBPC3, and MYL3 were scored as having very limited evidence. The other sarcomere genes had no evidence. These genes are well-established as causative for hypertrophic cardiomyopathy.8
Refuted and Disputed Genes—RYR2 Is Not an ARVC Gene
The GCEP refuted the association of RYR2 with ARVC. A thorough review, including 57 articles, showed that the assertion of ARVC causality was initially derived from 3 publications from the same research group who first established linkage to chromosome 1q42-43, and subsequently to RYR2 in families with a phenotype called arrhythmogenic right ventricular dysplasia 2 described as CPVT with fibro-fatty replacement of the right ventricle.42 The clinical features described in these manuscripts reflect CPVT rather than ARVC with cases not meeting TFC. This was confirmed by collaborators from the original research group (C. Basso and G. Thiene personal communication). A mouse model with one of the variants also showed a CPVT-like phenotype with no evidence of fibro-fatty infiltration or structural alterations characteristic of ARVC.43 In articles reporting RYR2 missense variants in possible ARVC probands the minor allele frequency was often too high, cases did not have a clear ARVC diagnosis, segregation information was often not informative, and in several cases, CPVT was said to also be present in the family. Rare cases of RYR2 deletions associated with DCM with CPVT-like arrhythmias have been described,44 but none associated with ARVC. Taken together, the evidence is convincing that pathogenic variants in RYR2 do not cause ARVC, rather they cause CPVT.
LDB3 was disputed as a cause for ARVC. The only variant reported in an ARVC family had a minor allele frequency higher than the cutoff established, particularly in the relevant ethnic population (Europeans).
Prevalence of Variants Reported in ClinVar for Each Gene
Figure 3 shows the distribution of variants reported to ClinVar associated with ARVC (Figure 3A) and the proportion of P/LP variants found in the genes with definitive or moderate evidence for ARVC in comparison to the other genes (Figure 3B). As can be appreciated, ARVC-associated P/LP variants were nearly exclusively reported in the desmosomal genes (450/462, 97.4%) with the established founder variants in PLN and TMEM43 also reported. Notably, only 5 P/LP variants (1.1%) were reported in genes with limited evidence, including one variant in CTNNA3, 3 in LMNA, and one in TGFB3.
Figure 3.
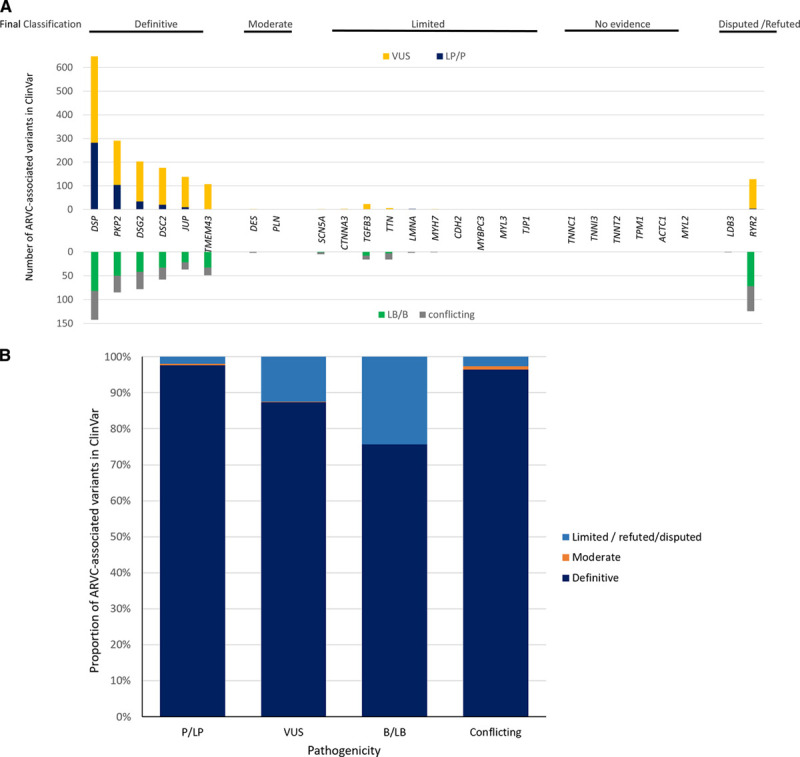
Variants in ClinVar in arrhythmogenic right ventricular cardiomyopathy (ARVC) gene curation expert panel (GCEP) curated genes. A, Distribution of variants in each gene curated—pathogenic and likely pathogenic (P/LP) variants (blue) were reported primarily in genes encoding the cardiac desmosome. B, Nearly all P/LP variants reported in ClinVar for ARVC are in the genes categorized as definitive or moderate evidence ARVC genes while refuted/disputed/limited evidence genes account for a higher proportion of variant of uncertain significance (VUS) and benign/likely benign (B/LB) variants.
Discussion
The data presented here, derived from a rigorous, international evaluation of 26 genes published as ARVC causing using the ClinGen framework, confirm that ARVC is primarily a disease of the cardiac desmosome, with PKP2, DSP, DSC2, DSG2, and JUP, definitively associated with ARVC and these genes accounting for nearly all reported ARVC-associated P/LP variants. PLN and TMEM43 contribute to disease pathogenesis, particularly in geographic regions with well-characterized founder variants. This study also demonstrates that the majority of published ARVC genes had only limited (N=10) or no (N=8) evidence and contribute little to the classic ARVC phenotype. While there has been extensive discussion of the genetic heterogeneity of ARVC and overlap syndromes, P/LP variants in genes with strong/definitive evidence for another cardiovascular disease do not substantially contribute to ARVC causation. In particular, this analysis disqualified RYR2 as an ARVC gene, finding cases and model systems instead had CPVT. Taken together, these findings call into question the extent of genetic heterogeneity truly contributing to classic ARVC as defined by the TFC.
This reappraisal of ARVC genes is strengthened by our methodological approach that included an extensive literature review, dual, blinded, independent curation, and final adjudication of evidence by an international multidisciplinary panel with substantial experience with ARVC. The semiquantitative ClinGen framework for evidence classification was effective. The independent curation teams had a high degree of uniformity in applying the framework with 100% concordance of preliminary classification of the level of evidence and no disagreement between the curation teams’ conclusions and the final opinion of the other GCEP members in arriving at the final level of evidence.
Eighteen of the 26 published ARVC genes had either limited or no evidence for ARVC causation. Excluded genes fell into 2 categories: (1) recently published genes with rare variants detected in small families with clear ARVC but for which data was limited and (2) genes known to be associated with other cardiomyopathies or arrhythmia syndromes where a thorough literature review showed limited or no evidence for ARVC causality. For the genes in the first category (CDH2, CTNNA3, and TJP1) multicenter studies, some currently underway, will address whether the identification of segregating P/LP variants in ARVC families will be replicated and if so in what proportion of gene-elusive ARVC cases. These data could lead to the level of evidence for these genes being upgraded in the future.
This misattribution of the latter group of genes to ARVC illustrates the well-known challenges of ascertaining, and then attributing, clinical data to a specific cardiac disease. This challenge, particularly before 2010 when the ARVC diagnostic criteria were refined, was the primary reason for the erroneous assertion that RYR2 caused ARVC, with our review revealing affected individuals in published pedigrees segregating RYR2 variants had clinical characteristics consistent with CPVT rather than ARVC. Diagnostic challenges also emerged in papers that described associations of LMNA, TTN, SCN5A, and even the moderate evidence gene DES with ARVC. In most, while several cases met 2010 TFC, others did not and the pedigrees often included clinical features not typically seen in ARVC.
Older genetic/genomic methodologies also contributed to incorrect assertions of ARVC causality. Some articles identified variants of interest as LP based on small control cohorts. Reassessment revealed the minor allele frequency of quite a few variants was too high given current understanding of the frequency of rare variants in the general population.
Clinical Implications
Genetic testing is recommended for patients with ARVC and genetic test results are part of the 2010 TFC.5,6 Optimal genetic testing requires both wise genetic test selection and accurate interpretation of results. By defining the genetic architecture of ARVC, this study informs both.
Interpretation and Utilization of Genetic Test Results for Diagnosis and Cascade Testing
A pathogenic variant categorized as associated or probably associated with ARVC constitutes a major ARVC diagnostic criterion.5 Based on our results, we recommend that only P/LP variants in genes with definitive and moderate evidence for ARVC causation (PKP2, DSP, DSC2, DSG2, JUP, TMEM43, PLN, and DES) should yield a major criterion for ARVC diagnosis.
We found that genes with strong or definitive evidence for other cardiovascular diseases had at-most moderate (DES) and usually limited or no evidence for ARVC causation. This suggests skepticism is warranted when a P/LP variant in one of these genes is identified in a putative ARVC patient. The American College of Medical Genetics and Genomics explicitly warns against relying on their guidelines for interpretation of pathogenicity in genes of unknown significance.45 Reevaluation of the patient and family for features suggesting an alternate clinical diagnosis may be useful and the full associated clinical spectrum (eg, desminopathy, laminopathy) should inform medical care and familial cascade screening. In the absence of evidence suggesting an alternate diagnosis, reevaluating the pathogenicity of the variant may be warranted.
A few percent of patients with ARVC have multiple P/LP variants in strong/moderate evidence ARVC genes, leading to earlier and more severe ARVC manifestations. The rare patients with definite ARVC and also definitively pathogenic variants in limited evidence genes typically associated with other cardiovascular diseases are particularly likely to harbor additional genetic variants. Real harm can be done by cascade testing of a variant which does not (fully) explain the disease in a family. These second variants in other cardiomyopathy or arrhythmia related genes may also drive the phenotype towards an atypical yet severe, manifestation of ARVC. This underscores that while ARVC is not a condition with substantial genetic heterogeneity, the paradigm of one gene-one disease is challenged.46 Evidence suggests oligogenic inheritance with multiple additional variants that may or may not reach the LP/P status (or in genes that are not definitive for ARVC) could also contribute to disease expression.
Furthermore, recent publications show at least one-third of ARVC cases are gene elusive,12 and these patients are disproportionately high-level athletes with no family history of ARVC, pointing to exercise as contributing to cause.47 Among relatives of patients with ARVC with P/LP desmosomal variants, exercise increases penetrance and risk of incident arrhythmias, but not all athletic relatives develop ARVC.48. Additionally, a recent study49 showed 0.23% of a general clinical population harbored a loss of function desmosomal variant. These patients had extremely low ARVC penetrance (estimated at 6%) and were no more likely than controls to have ECG or echocardiography findings that met TFC. Taken together this evidence strongly suggests a threshold model of ARVC pathogenesis in which multiple hits, both environmental and genetic, are required for disease expression.2 Thus, while detection of a P/LP variant in a definitive or moderate evidence gene in a patient with ARVC features is highly indicative of disease, it does not fully predict the clinical features or course of individual patients. The fact that these different aspects influence disease expression, and are not accounted for in the ClinGen framework which is built around true penetrant Mendelian disease, can be considered a limitation and are important to keep in mind when using the results of this analysis to interpret genetic tests.
In summary, substantial caution is required for the interpretation of variants in limited evidence genes which are unlikely to be the sole cause of disease in a patient with ARVC. Our results suggest such variants should not be used to assign the patient a major TFC criterion.
Panel Selection for Genetic Testing
This study also challenges the inclusion of several genes frequently present in ARVC panels—foremost among them RYR2. Our results identify genes with definitive and moderate evidence for ARVC and show that most P/LP variants in patients with ARVC occur in these genes. However, a clinical ARVC diagnosis can be challenging, particularly in early stages of disease where structural abnormalities can be subtle but arrhythmic risk is nonetheless significant.1 Careful use of a larger panel can, therefore, facilitate correct genetic diagnosis of a family. Using a large panel responsibly in this context requires multidisciplinary expertise.6
Limitations
Genes were curated for ARVC per 2010 TFC. Updated diagnostic criteria are being considered, particularly for the left-dominant form of ARVC,50 thus a need to update ARVC curation is anticipated.
Although genes accounting for most familial ARVC have been identified, expanded sequencing efforts and new analytic approaches will identify rare or family specific variants in novel putative ARVC genes that will require adjudication (foremost among these FLNC). Recuration of limited and moderate evidence genes will be done per ClinGen procedures as new data emerges (https://clinicalgenome.org/site/assets/files/2164/clingen_standard_gene-disease_validity_recuration_procedures_v1.pdf).
Conclusions
This evidence-based reevaluation of published ARVC genes by experts in the field shows that only a small number of genes (PKP2, DSP, DSG2, DSC2, JUP, TMEM43, PLN, and DES) are definitively or moderately associated with ARVC and these genes account for the overwhelming majority of P/LP variants in patients with ARVC. We recommend only P/LP variants in these 8 genes should yield a major criterion for ARVC diagnosis by TFC. This analysis is expected to further refine the utility of genetic data in caring for families with ARVC by assisting the clinician in determining what test to order and also by quantifying the strength of evidence underlying the gene:disease relationship relevant to a genetic result.
Sources of Funding
This work was financially supported by grants from the National Institutes of Health (NIH; U41HG009650) and by the Netherlands Cardiovascular Research Initiative (Dr van Tintelen) an initiative supported by the Dutch Heart Foundation (CVON2018-30 PREDICT2 and CVON 2015-12 eDETECT). The Johns Hopkins ARVD/C Program (Dr James and B. Murray) is supported by the Leonie-Wild Foundation, the Leyla Erkan Family Fund for ARVD Research, the Dr Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins, the Bogle Foundation, the Healing Hearts Foundation, the Campanella family, the Patrick J. Harrison Family, the Peter French Memorial Foundation, and the Wilmerding Endowments. Dr Syrris was supported by Fondation Leducq Transatlantic Networks of Excellence Program grant no 14CVD03 and the National Institute for Health Research University College London Hospitals Biomedical Research Centre (United Kingdom). Dr Protonotarios is supported by a British Heart Foundation clinical research training fellowship grant (FS/18/82/34024).
Disclosures
B. Murray and E. Brown are consultants for MyGeneCounsel. The other authors report no conflicts.
Supplemental Materials
Supplemental Methods
Supplemental Tables I–IV
References 51,52
Supplementary Material
Footnotes
Nonstandard Abbreviations and Acronyms
- ARVC
- arrhythmogenic right ventricular cardiomyopathy
- ClinGen
- Clinical Genome Resource
- CPVT
- catecholaminergic polymorphic ventricular tachycardia
- DCM
- dilated cardiomyopathy
- GCEP
- gene curation expert panel
- P/LP
- pathogenic or likely pathogenic
- TFC
- 2010 Task Force Criteria
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.120.003273.
For Sources of Funding and Disclosures, see page 282.
References
- 1.Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376:1489–1490. doi: 10.1056/NEJMc1701400 [DOI] [PubMed] [Google Scholar]
- 2.James CA, Syrris P, van Tintelen JP, Calkins H. The role of genetics in cardiovascular disease: arrhythmogenic cardiomyopathy. Eur Heart J. 2020;41:1393–1400. doi: 10.1093/eurheartj/ehaa141 [DOI] [PubMed] [Google Scholar]
- 3.McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355:2119–2124. doi: 10.1016/S0140-6736(00)02379-5 [DOI] [PubMed] [Google Scholar]
- 4.Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld AC, Sawant AC, Kassamali B, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. 2015;8:437–446. doi: 10.1161/CIRCGENETICS.114.001003 [DOI] [PubMed] [Google Scholar]
- 5.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16:e301–e372. doi: 10.1016/j.hrthm.2019.05.007 [DOI] [PubMed] [Google Scholar]
- 7.Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, Goldstein J, Ghosh R, Seifert BA, Sneddon TP, et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am J Hum Genet. 2017;100:895–906. doi: 10.1016/j.ajhg.2017.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, Dougherty K, Harrison SM, McGlaughon J, Milko LV, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med. 2019;12:e002460. doi: 10.1161/CIRCGEN.119.002460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adler A, Novelli V, Amin AS, Abiusi E, Care M, Nannenberg EA, Feilotter H, Amenta S, Mazza D, Bikker H, et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation. 2020;141:418–428. doi: 10.1161/CIRCULATIONAHA.119.043132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, Jamal SM, Szybowska M, Morel CF, Bowdin S, et al. ; National Institutes of Health Clinical Genome Resource Consortium. Reappraisal of reported genes for sudden arrhythmic death: evidence-based evaluation of gene validity for Brugada syndrome. Circulation. 2018;138:1195–1205. doi: 10.1161/CIRCULATIONAHA.118.035070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elliott PM, Anastasakis A, Asimaki A, Basso C, Bauce B, Brooke MA, Calkins H, Corrado D, Duru F, Green KJ, et al. Definition and treatment of arrhythmogenic cardiomyopathy: an updated expert panel report. Eur J Heart Fail. 2019;21:955–964. doi: 10.1002/ejhf.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corrado D, Perazzolo Marra M, Zorzi A, Beffagna G, Cipriani A, Lazzari M, Migliore F, Pilichou K, Rampazzo A, Rigato I, et al. Diagnosis of arrhythmogenic cardiomyopathy: the Padua criteria. Int J Cardiol. 2020;319:106–114. doi: 10.1016/j.ijcard.2020.06.005 [DOI] [PubMed] [Google Scholar]
- 13.Milting H, Klauke B, Christensen AH, Müsebeck J, Walhorn V, Grannemann S, Münnich T, Šarić T, Rasmussen TB, Jensen HK, et al. The TMEM43 Newfoundland mutation p.S358L causing ARVC-5 was imported from Europe and increases the stiffness of the cell nucleus. Eur Heart J. 2015;36:872–881. doi: 10.1093/eurheartj/ehu077 [DOI] [PubMed] [Google Scholar]
- 14.Dominguez F, Zorio E, Jimenez-Jaimez J, Salguero-Bodes R, Zwart R, Gonzalez-Lopez E, Molina P, Bermúdez-Jiménez F, Delgado JF, Braza-Boïls A, et al. Clinical characteristics and determinants of the phenotype in TMEM43 arrhythmogenic right ventricular cardiomyopathy type 5. Heart Rhythm. 2020;17:945–954. doi: 10.1016/j.hrthm.2020.01.035 [DOI] [PubMed] [Google Scholar]
- 15.Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–821. doi: 10.1016/j.ajhg.2008.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Zwaag PA, van Rijsingen IA, de Ruiter R, Nannenberg EA, Groeneweg JA, Post JG, Hauer RN, van Gelder IC, van den Berg MP, van der Harst P, et al. Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth Heart J. 2013;21:286–293. doi: 10.1007/s12471-013-0401-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, van Langen IM, van den Berg MP, van Tintelen JP. Desmin-related myopathy. Clin Genet. 2011;80:354–366. doi: 10.1111/j.1399-0004.2010.01512.x [DOI] [PubMed] [Google Scholar]
- 18.Klauke B, Kossmann S, Gaertner A, Brand K, Stork I, Brodehl A, Dieding M, Walhorn V, Anselmetti D, Gerdes D, et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet. 2010;19:4595–4607. doi: 10.1093/hmg/ddq387 [DOI] [PubMed] [Google Scholar]
- 19.Maruthappu T, Posafalvi A, Castelletti S, Delaney PJ, Syrris P, O’Toole EA, Green KJ, Elliott PM, Lambiase PD, Tinker A, et al. Loss-of-function desmoplakin I and II mutations underlie dominant arrhythmogenic cardiomyopathy with a hair and skin phenotype. Br J Dermatol. 2019;180:1114–1122. doi: 10.1111/bjd.17388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerull B, Kirchner F, Chong JX, Tagoe J, Chandrasekharan K, Strohm O, Waggoner D, Ober C, Duff HJ. Homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ Cardiovasc Genet. 2013;6:327–336. doi: 10.1161/CIRCGENETICS.113.000097 [DOI] [PubMed] [Google Scholar]
- 21.Chen L, Rao M, Chen X, Chen K, Ren J, Zhang N, Zhao Q, Yu W, Yuan B, Song J. A founder homozygous DSG2 variant in East Asia results in ARVC with full penetrance and heart failure phenotype. Int J Cardiol. 2019;274:263–270. doi: 10.1016/j.ijcard.2018.06.105 [DOI] [PubMed] [Google Scholar]
- 22.Baskin B, Skinner JR, Sanatani S, Terespolsky D, Krahn AD, Ray PN, Scherer SW, Hamilton RM. TMEM43 mutations associated with arrhythmogenic right ventricular cardiomyopathy in non-Newfoundland populations. Hum Genet. 2013;132:1245–1252. doi: 10.1007/s00439-013-1323-2 [DOI] [PubMed] [Google Scholar]
- 23.van Tintelen JP, Van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, van den Wijngaard A, Kuks JB, van Spaendonck-Zwarts KY, Notermans N, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009;6:1574–1583. doi: 10.1016/j.hrthm.2009.07.041 [DOI] [PubMed] [Google Scholar]
- 24.Lorenzon A, Beffagna G, Bauce B, De Bortoli M, Li Mura IE, Calore M, Dazzo E, Basso C, Nava A, Thiene G, et al. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111:400–405. doi: 10.1016/j.amjcard.2012.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hedberg C, Melberg A, Kuhl A, Jenne D, Oldfors A. Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur J Hum Genet. 2012;20:984–985. doi: 10.1038/ejhg.2012.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bermúdez-Jiménez FJ, Carriel V, Brodehl A, Alaminos M, Campos A, Schirmer I, Milting H, Abril BÁ, Álvarez M, López-Fernández S, et al. Novel desmin mutation p.Glu401Asp impairs filament formation, disrupts cell membrane integrity, and causes severe arrhythmogenic left ventricular cardiomyopathy/dysplasia. Circulation. 2018;137:1595–1610. doi: 10.1161/CIRCULATIONAHA.117.028719 [DOI] [PubMed] [Google Scholar]
- 27.van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, Cox MG, van Lochem LT, de Boer RA, Hofstra RM, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14:1199–1207. doi: 10.1093/eurjhf/hfs119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sepehrkhouy S, Gho JMIH, van Es R, Harakalova M, de Jonge N, Dooijes D, van der Smagt JJ, Buijsrogge MP, Hauer RNW, Goldschmeding R, et al. Distinct fibrosis pattern in desmosomal and phospholamban mutation carriers in hereditary cardiomyopathies. Heart Rhythm. 2017;14:1024–1032. doi: 10.1016/j.hrthm.2017.03.034 [DOI] [PubMed] [Google Scholar]
- 29.Mayosi BM, Fish M, Shaboodien G, Mastantuono E, Kraus S, Wieland T, Kotta M, Chin A, Laing N, Ntusi NBA, et al. Identification of Cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2017;10:e001605. [DOI] [PubMed] [Google Scholar]
- 30.Turkowski KL, Tester DJ, Bos JM, Haugaa KH, Ackerman MJ. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit Heart Dis. 2017;12:226–235. doi: 10.1111/chd.12462 [DOI] [PubMed] [Google Scholar]
- 31.van Hengel J, Calore M, Bauce B, Dazzo E, Mazzotti E, De Bortoli M, Lorenzon A, Li Mura IE, Beffagna G, Rigato I, et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:201–210. doi: 10.1093/eurheartj/ehs373 [DOI] [PubMed] [Google Scholar]
- 32.De Bortoli M, Postma AV, Poloni G, Calore M, Minervini G, Mazzotti E, Rigato I, Ebert M, Lorenzon A, Vazza G, et al. Whole-exome sequencing identifies pathogenic variants in TJP1 gene associated with arrhythmogenic cardiomyopathy. Circ Genom Precis Med. 2018;11:e002123. doi: 10.1161/CIRCGEN.118.002123 [DOI] [PubMed] [Google Scholar]
- 33.Te Riele AS, Agullo-Pascual E, James CA, Leo-Macias A, Cerrone M, Zhang M, Lin X, Lin B, Sobreira NL, Amat-Alarcon N, et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc Res. 2017;113:102–111. doi: 10.1093/cvr/cvw234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, Muir A, Pantazis A, McKenna WJ, Elliott PM. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;33:1128–1136. doi: 10.1093/eurheartj/ehr451 [DOI] [PubMed] [Google Scholar]
- 35.Forleo C, Carmosino M, Resta N, Rampazzo A, Valecce R, Sorrentino S, Iacoviello M, Pisani F, Procino G, Gerbino A, et al. Clinical and functional characterization of a novel mutation in lamin a/c gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS One. 2015;10:e0121723. doi: 10.1371/journal.pone.0121723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005 [DOI] [PubMed] [Google Scholar]
- 37.Marsili L, Overwater E, Hanna N, Baujat G, Baars MJH, Boileau C, Bonneau D, Brehin AC, Capri Y, Cheung HY, et al. Phenotypic spectrum of TGFB3 disease-causing variants in a Dutch-French cohort and first report of a homozygous patient. Clin Genet. 2020;97:723–730. doi: 10.1111/cge.13700 [DOI] [PubMed] [Google Scholar]
- 38.Chen K, Song J, Wang Z, Rao M, Chen L, Hu S. Absence of a primary role for TTN missense variants in arrhythmogenic cardiomyopathy: from a clinical and pathological perspective. Clin Cardiol. 2018;41:615–622. doi: 10.1002/clc.22906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 2011;124:876–885. doi: 10.1161/CIRCULATIONAHA.110.005405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, Stolfo D, Haywood ME, Dal Ferro M, Altinier A, et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2019;74:1480–1490. doi: 10.1016/j.jacc.2019.06.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murray B, Hoorntje ET, Te Riele ASJM, Tichnell C, van der Heijden JF, Tandri H, van den Berg MP, Jongbloed JDH, Wilde AAM, Hauer RNW, et al. Identification of sarcomeric variants in probands with a clinical diagnosis of arrhythmogenic right ventricular cardiomyopathy (ARVC). J Cardiovasc Electrophysiol. 2018;29:1004–1009. doi: 10.1111/jce.13621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 2001;10:189–194. doi: 10.1093/hmg/10.3.189 [DOI] [PubMed] [Google Scholar]
- 43.Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, De Biasi M, Wehrens XH, et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci USA. 2006;103:12179–12184. doi: 10.1073/pnas.0600268103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhuiyan ZA, van den Berg MP, van Tintelen JP, Bink-Boelkens MT, Wiesfeld AC, Alders M, Postma AV, van Langen I, Mannens MM, Wilde AA. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation. 2007;116:1569–1576. doi: 10.1161/CIRCULATIONAHA.107.711606 [DOI] [PubMed] [Google Scholar]
- 45.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cerrone M, Remme CA, Tadros R, Bezzina CR, Delmar M. Beyond the one gene-one disease paradigm: complex genetics and pleiotropy in inheritable cardiac disorders. Circulation. 2019;140:595–610. doi: 10.1161/CIRCULATIONAHA.118.035954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sawant AC, Bhonsale A, te Riele AS, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H, et al. Exercise has a disproportionate role in the pathogenesis of arrhythmogenic right ventricular dysplasia/cardiomyopathy in patients without desmosomal mutations. J Am Heart Assoc. 2014;3:e001471. doi: 10.1161/JAHA.114.001471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang W, Tichnell C, Murray BA, Agafonova J, Cadrin-Tourigny J, Chelko S, Tandri H, Calkins H, James CA. Exercise restriction is protective for genotype-positive family members of arrhythmogenic right ventricular cardiomyopathy patients. Europace. 2020;22:1270–1278. doi: 10.1093/europace/euaa105 [DOI] [PubMed] [Google Scholar]
- 49.Carruth ED, Young W, Beer D, James CA, Calkins H, Jing L, Raghunath S, Hartzel DN, Leader JB, Kirchner HL, et al. Prevalence and electronic health record-based phenotype of loss-of-function genetic variants in arrhythmogenic right ventricular cardiomyopathy-associated genes. Circ Genom Precis Med. 2019;12:e002579. doi: 10.1161/CIRCGEN.119.002579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corrado D, van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, Basso C, Bauce B, Brunckhorst C, Bucciarelli-Ducci C, et al. ; International Experts. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41:1414–1429. doi: 10.1093/eurheartj/ehz669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Lint FHM, Murray B, Tichnell C, Zwart R, Amat N, Lekanne Deprez RH, Dittmann S, Stallmeyer B, Calkins H, van der Smagt JJ, et al. Arrhythmogenic right ventricular cardiomyopathy-associated desmosomal variants are rarely de novo. Circ Genom Precis Med. 2019;12:e002467. doi: 10.1161/CIRCGEN.119.002467 [DOI] [PubMed] [Google Scholar]
- 52.Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, Barton PJR, Funke B, Cook SA, MacArthur D, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19:1151–1158. doi: 10.1038/gim.2017.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.