

SUMMARY
Abnormal activity of the core cell-cycle machinery is seen in essentially all tumor types and represents a driving force of tumorigenesis. Recent studies revealed that cell-cycle proteins regulate a wide range of cellular functions, in addition to promoting cell division. With the clinical success of CDK4/6 inhibitors, it is becoming increasingly clear that targeting individual cell-cycle components may represent an effective anti-cancer strategy. Here, we discuss the potential of inhibiting different cell-cycle proteins for cancer therapy.
Introduction
The cell-cycle is a highly regulated process enabling cell growth, duplication of genetic material and cell division. Progression from one cell-cycle phase to another is driven by the core cell-cycle machinery operating in cell nucleus. This machinery is composed of proteins called cyclins and their catalytic partners, the cyclin‐dependent kinases (CDKs) (Malumbres and Barbacid, 2009). Different cyclin-CDK complexes become activated in specific phases of the cell-cycle and phosphorylate their target proteins (Box 1, Figure 1).
Box 1. The cell-cycle.
The cell-cycle is composed of four phases: Gap 1 (G1), DNA-synthesis (S), Gap 2 (G2) and mitosis (M) (Figure 1). Mitogenic signals received during G1 phase cause upregulation of D-type cyclins (D1, D2, D3), which bind and activate their catalytic partners, CDK4 or CDK6. The activity of cyclin D-CDK4/6 kinase is negatively regulated by the INK4 family of inhibitors (p16INK4A, p15INKB, p18INK4C, p19INK4D) (Figure 1). The formation of stable cyclin D-CDK4/6 complexes requires KIP/CIP proteins (p21CIP1, p27KIP1, p57KIP2), which serve as ‘assembly factors’ for cyclin D-CDK4/6. In contrast, KIP/CIP proteins are inhibitors of CDK2- and CDK1-containing complexes (Sherr and Roberts, 2004). Cyclin D–CDK4/6 kinases phosphorylate various cellular targets, most importantly, the retinoblastoma protein (RB1), as well as related RBL1 (p107) and RBL2 (p130) (Malumbres and Barbacid, 2001). In its unphosphorylated form, RB1 binds and inactivates or represses E2F transcription factors. Phosphorylation of RB1 by cyclin D-CDK4/6 partially inactivates RB1, leading to release of E2Fs, which then upregulate their transcriptional targets. Among E2F targets are two E-type cyclins (E1 and E2, collectively referred to as ‘cyclin E’), which bind and activate CDK2. Cyclin E-CDK2 is responsible for completing phosphorylation of RB1, thereby unleashing the E2F transcriptional program and resulting in synthesis of proteins required for entry of cells into the S-phase (Hwang and Clurman, 2005). During progression through the G1 phase, several proteins – some of them representing E2F transcriptional targets – assemble on origins of DNA replication and form pre-replication complexes, composed of ORC1–6, CDC6, CDT1 and MCM2–7 DNA helicase. At the onset of the S-phase, DNA replication is triggered by a series of sequential phosphorylations carried out by DBF4-CDC7 and cyclin E-CDK2 (Moiseeva and Bakkenist, 2018) (Figure 1). These phosphorylation events promote binding of CDC45 and GINS to MCM2–7, resulting in formation of the CDC45-MCM2–7-GINS (CMG) complex and activation of DNA helicase (Moiseeva and Bakkenist, 2018). Subsequently, the role of cyclin E-CDK2 in firing of DNA replication origins is taken over by cyclin A-CDK2. Cyclin A can activate CDK1 and CDK2; it plays important roles during S-phase as well as in G2-M. The crucial function of cyclin A during G2/M-transition is to activate cyclin B-CDK1 (Figure 1). Cyclin B accumulates in the nucleus at the onset of mitosis, upon being phosphorylated by PLK1 (Martínez-Alonso and Malumbres, 2020). Cyclin B-CDK1 phosphorylates several proteins, most of them associated with events related to mitotic progression and cell division, such as cytoskeleton reorganization, nuclear envelope breakdown, chromosome condensation, mitotic spindle assembly and function, chromosome segregation, and cytokinesis (Martínez-Alonso and Malumbres, 2020). The CDK-activating kinase (CAK) represents the major driver of cell-cycle progression (Sava et al., 2020). This complex is composed of cyclin H, the catalytic subunit (CDK7) as well as a RING protein called MAT1, which stabilizes the complex. CAK activates cell-cycle CDKs by phosphorylating the critical threonine residue within their T-loops. Moreover, CAK-dependent phosphorylation stabilizes some cyclin-CDK complexes, such as cyclin B-CDK1. In addition to its cell-cycle role, CAK functions as a member of TFIIH, a general transcription factor that acts to recruit RNA polymerase II to gene promoters. Within this complex, CDK7 phosphorylates the C-terminal domain of RNA-polymerase II (Sava et al., 2020).
Figure 1. Overview of the cell-cycle.
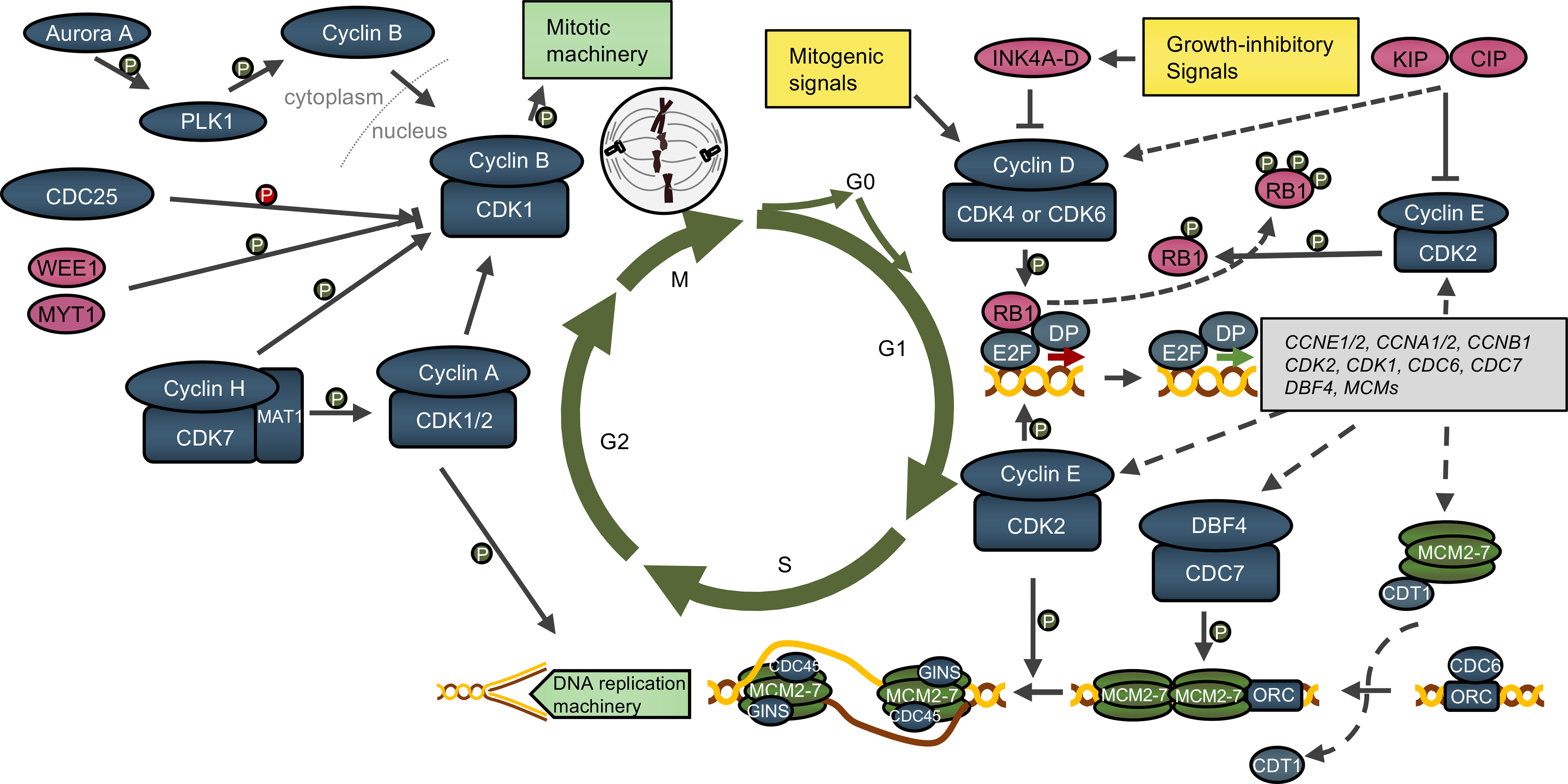
Please see Box 1 for details. Activation of cyclin D-CDK4/6 kinases triggers a cascade of events resulting in activation of E2F-dependent transcriptional program. DP1, the dimerization partner of E2F transcription factors. Examples of E2F targets are provided in a grey rectangle. Multiple proteins (such as ORC, CDC6, MCMs) assemble on DNA replication origins during the G1 phase, resulting in DNA origin licensing. Subsequent phosphorylations (P) by DBF4-CDC7 and cyclin E-CDK2 activate the DNA helicase and trigger origin firing and entry of cells into the S-phase. During S-phase, cyclin A-CDK2 phosphorylates components of the DNA replication machinery. In G2-phase, cyclin A activates cyclin B-CDK1. Cyclin B is localized in the cytoplasm during G2-phase, but it translocates into the nucleus upon being phosphorylated by PLK1. Inhibitory phosphorylations of CDK1 provided by WEE1 and MYT1 kinases are removed by the CDC25 phosphatase family. Activation of cyclin-CDK complexes is also carried out by cyclin H-CDK7-MAT1 (CAK1) complex. Upon activation, cyclin B-CDK1 phosphorylates components of the mitotic machinery. Blue and pink ovals denote, respectively, positive and negative regulators of the cell-cycle.
In normal cells, the activity of cell-cycle proteins is tightly controlled by their cell-cycle-specific transcription, protein degradation (Box 2, Figure 2), as well as by several CDK-inhibitor proteins (Sherr and Roberts, 2004). All these mechanisms are frequently dysregulated in human cancers, resulting in aberrant activation of cell-cycle proteins. Indeed, genetic lesions within the core cell-cycle machinery, resulting in its hyperactivation, play a causative role in development of most tumor types (Malumbres and Barbacid, 2009). For these reasons, targeting cell-cycle proteins seems to represent an effective way of halting tumor growth.
Box 2. Regulation of cell-cycle machinery by proteasomal degradation.
An important aspect of cell-cycle regulation is governed by multi-protein E3 ubiquitin ligase complexes, such as the anaphase-promoting complex or cyclosome (APC/C), Skp-Cullin-F-box containing complex (SCF), and BTB-Cul3-Rbx1 complex (BCR), which sequentially promote degradation of key cell-cycle regulators (Figure 2) (Dang et al., 2020; Rape et al., 2006). Recognition of specific degron motifs by components of E3 ligase complexes, such as CDC20, CDH1, FBXW7, CUL3, Cyclin F, SKP2 and βTrCP, leads to ubiquitination and proteasomal degradation of target proteins. APC/CCDH1 stabilizes G1 phase by maintaining low levels of mitotic and S-phase cyclins, thereby preventing premature initiation of S-phase (Malumbres and Barbacid, 2009). At the end of G1 phase, the APC/CCDH1 becomes inactivated by cyclin E-CDK2; this inactivation is further maintained by early mitotic inhibitor 1 (EMI1). During S phase, cyclin E is degraded via FBXW7-mediated ubiquitination (Otto and Sicinski, 2017), and it can be also degraded by the BCR complex (Singer et al., 1999). SKP2 is thought to act as an oncogene, by contributing to the ubiquitination and degradation of CDK inhibitors from the KIP/CIP family (Dang et al., 2020). βTrCP plays an important role in regulating CDK1 activity. It prevents premature activation of CDK1 by inducing degradation of CDC25A and EMI1, and it can contribute to CDK1 activation by mediating WEE1 degradation in G2 phase. Also during G2 phase, Cyclin F shuts down the activity of E2F, prevents the synthesis of replicative histones, contributes to the regulation of centrosomal duplication and mediates the degradation of CDH1 (Dang et al., 2020). In mitosis, cyclin A is degraded by APC/CCDC20 following nuclear envelope breakdown and before metaphase (Martínez-Alonso and Malumbres, 2020). Subsequently, degradation of cyclin B and securin (responsible for the anaphase onset) is executed by APC/CCDC20 (Martínez-Alonso and Malumbres, 2020). Finally, APC/CCDH1 function starts in anaphase where it modulates the stability of mitotic players such as Aurora A and PLK1; the APC/CCDH1 activity then continues into the next G1-phase (Dang et al., 2020).
Figure 2. Regulation of cell-cycle machinery by proteasomal degradation.
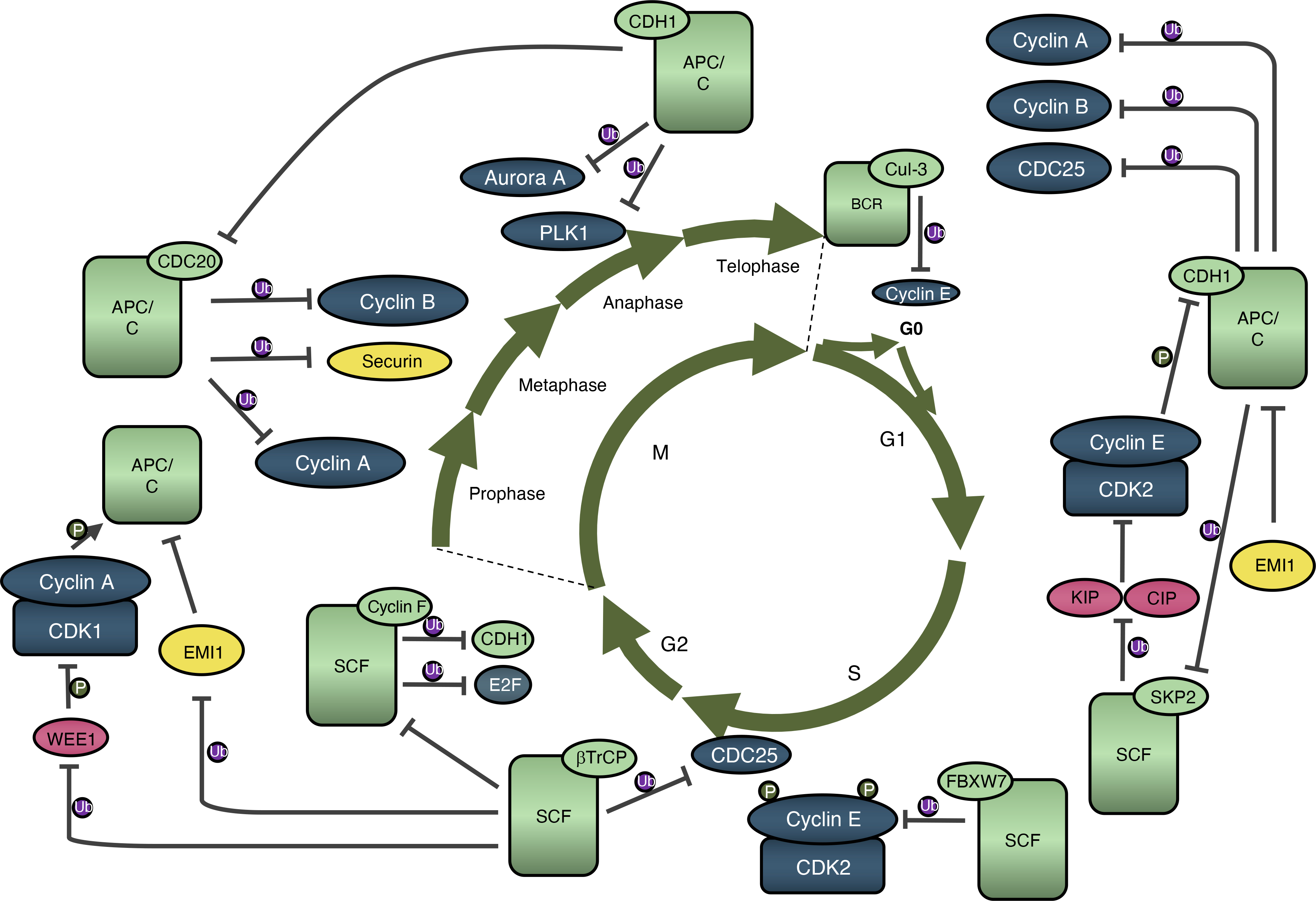
E3 ubiquitin ligases ascertain unperturbed cell cycle progression by governing timely degradation of various cell-cycle proteins. Light green rounded rectangles and ovals represent ubiquitin ligase complexes and their respective activators/f-box proteins. Ub in a purple circle indicates ubiquitination. FBXW7 - F-Box and WD Repeat Domain Containing 7; Cul-3 – Cullin 3; SKP2 – S-Phase Kinase Associated Protein 2; βTrCP - Beta-Transducin Repeat Containing E3 Ubiquitin Protein Ligase.
The enthusiasm for inhibiting cyclin-CDK kinases was initially mitigated by the widely held belief that these proteins are essential for proliferation of normal non-transformed cells. However, genetic experiments revealed that individual cyclins and CDKs are largely dispensable for proliferation of normal tissues (Sherr and Roberts, 2004). In contrast, these proteins are essential for proliferation of specific cancer types, depending on the genetic lesions they carry (Malumbres and Barbacid, 2009). The most clear-cut success of targeting cell-cycle machinery are inhibitors of CDK4 and CDK6. Introduction of these compounds to the clinics represented a breakthrough in the treatment of breast cancers, and will likely have a profound effect on treatment of many other tumor types (Alvarez-Fernandez and Malumbres, 2020). Quite unexpectedly, recent work revealed that cell-cycle proteins play additional roles in tumor development by affecting not only tumor cells, but also its microenvironment, for example by modulating the anti-tumor immune response (Deng et al., 2018; Goel et al., 2017; Zhang et al., 2018). Hence, inhibition of these proteins is likely to affect several aspects of the tumorigenic process.
Despite the current success with CDK4/6 inhibitors (CDK4/6i), targeting cell-cycle proteins for cancer treatment is still at its infancy. In this review, we discuss the current state of affairs and provide our view about the most promising future directions.
D-type Cyclins and CDK4/6 in Cancer
Amplification or rearrangements of genes encoding D-cyclins (CCND1, CCND2, CCND3), CDK4 or CDK6, and overexpression of these proteins was documented in a very large number of tumor types (Cancer Genome Atlas Research, 2013). Also, inactivation of CDK4/6-inhibitor proteins, in particular p16INK4A, represents a frequent feature of many tumor types (Figure 3, (Cancer Genome Atlas Research, 2013)). All these molecular lesions result in hyperactivation of cyclin D-CDK4/6 kinase in tumor cells and abrogation of RB1-mediated suppression of the cell-cycle. This, in turn, drives uncontrolled cell proliferation that becomes uncoupled from the external mitogenic and anti-mitogenic signals (Alvarez-Fernandez and Malumbres, 2020).
Figure 3. The landscape of genetic alterations within the core cell-cycle machinery genes.

Data according to The Cancer Genome Atlas PanCancer (n=10,967 samples). Shown is the frequency of alterations within groups of the indicated cell-cycle genes. Ten most affected cancer types are shown for each group, together with percentages of cases displaying genetic alterations. Tumor type designation: ACC - Adrenocortical Carcinoma; BLCA - Bladder Urothelial Carcinoma; BRCA - Breast Invasive Carcinoma; CESC - Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma; CHOL – Cholangiocarcinoma; COADREAD - Colorectal Adenocarcinoma; DLBC - Diffuse Large B-cell Lymphoma; ESCA - Esophageal Carcinoma, GBM - Glioblastoma Multiforme, HNSC - Head and Neck Squamous Cell Carcinoma, LIHC - Liver Hepatocellular Carcinoma, LUAD – Lung Adenocarcinoma, LUSC- Lung Squamous Cell Carcinoma, MESO – Mesothelioma, OV - Ovarian Serous Cystadenocarcinoma, PAAD - Pancreatic Adenocarcinoma, PRAD - Prostate Adenocarcinoma, SARC – Sarcoma, SKCM - Skin Cutaneous Melanoma, STAD - Stomach Adenocarcinoma, UCEC - Uterine Corpus Endometrial Carcinoma, UCS - Uterine Carcinosarcoma, UVM - Uveal Melanoma.
Targeted overexpression of D-cyclins or CDK4 in transgenic mice resulted in development of tumors, confirming the role of deregulated cyclin D-CDK4/6 kinases in tumorigenesis (Otto and Sicinski, 2017). Conversely, genetic ablation of D-cyclins, CDK4 or CDK6 reduced or prevented formation of tumors. For example, mice lacking cyclin D1, CDK4, or expressing kinase-inactive cyclin D1-CDK4/6 complexes are resistant to development of breast cancers (Alvarez-Fernandez and Malumbres, 2020). Importantly, an acute and global shutdown of cyclin D1 in mice bearing Her2-driven mammary carcinomas halted tumor growth and triggered senescence exclusively in tumor cells, while having no obvious impact on animals’ physiology (Choi et al., 2012). Cell-cycle arrest and senescence of tumor cells was also seen upon ablation of Cdk4 in mice with Kras-driven lung cancers (Puyol et al., 2010). Collectively, these observations documented the requirement for cyclin D-CDK4/6 activity in tumor maintenance. They also revealed a differential reliance of tumor cells - but not of most normal cell types - on individual D-cyclins or CDKs for proliferation.
Intriguingly, analyses of a mouse glioma model revealed that cyclin D1-Cdk4 contributes to progression of glial tumors also by acting in non-tumor cells. Specifically, ablation of cyclin D1 or Cdk4 impeded activation of tumor-associated microglia, thereby inhibiting tumorigenesis (Ciznadija et al., 2011). Others reported increased expression of cyclin D1 in breast cancer-associated stromal cells. Importantly, overexpression of cyclin D1 in stromal fibroblasts promoted breast tumorigenesis, via secretion of pro-inflammatory cytokines and osteopontin (Pestell et al., 2017). These observations, together with a recent demonstration of a role for CDK4/6 in anti-tumor immunity (see below) reveal that cyclin D-CDK4/6 affects tumorigenesis both by tumor-intrinsic mechanisms as well as by affecting tumor microenvironment.
CDK4/6 Inhibitors
The first CDK4/6-specific inhibitor – palbociclib – was synthesized in 2001 and showed efficacy against a wide range of human cancer cell lines and xenografts, including breast cancers (Fry et al., 2004). Finn et al. (2009) demonstrated that breast cancer cell lines representing hormone receptor-positive (HR+), luminal-type mammary carcinomas were most sensitive to CDK4/6 inhibition This evidence, together with mouse genetic experiments described above, led to the use of CDK4/6i (palbociclib, ribociclib and abemaciclib) in clinical trials for breast cancer patients (Table 1, Figure 4).
Table 1.
Inhibitors of cell-cycle proteins in clinical development.
| Drug name (alternative names) | Primary target (IC50) | Off target (IC50) | Clinical trials/clinical utilization | Reference |
|---|---|---|---|---|
| 1st generation (Pan-kinase inhibitors) | ||||
| Flavopiridol (Alvocidib, L86-8275, HMR-1275) | CDK9 (20 nM), CDK1 (30 nM), CDK2 (40 nM), CDK4 (20–40 nM), CDK6 (60 nM) | CDK7 (875 nM), MAPK, PKC, EGFR | 66 phase I or II clinical trials in different malignancies; 5 active trials (MDS/AML); not registered for clinical use in oncology | (Shapiro, 2006) |
| Roscovitine (Seliciclib, CYC202) | CDK5 (0.16 μM), CDK1 (0.65 μM), CDK2 (0.7 μM) | ERK | 3 phase I or II clinical trials in different malignancies; no active trial and no registration for clinical use in oncology | (Ali et al., 2009; Jabbour-Leung et al., 2016) |
| 2nd generation (Multiple-kinase inhibitors) | ||||
| Dinaciclib (SCH727965) | CDK2 (1 nM), CDK5 (1 nM), CDK1 (3 nM), CDK9 (4 nM) | 18 phase I, II and III clinical trials in different malignancies; the only phase 3 trial in CLL (results to be posted); 4 active trials; no registration for clinical use in oncology | (Parry et al., 2010; Ghia et al. 2017) | |
| AT7519 | CDK9 (<10 nM), CDK5 (13 nM), CDK2 (47 nM) | CDK4 (100 nM), CDK6 (170 nM), CDK1 (210 Nm), CDK7 (2400 nM), GSK-3β | 5 phase I or II clinical trials in heamotologic malignancies; 1 active trial; no registration for clinical use in oncology | (Otto and Sicinski, 2017) |
| Milciclib (PHA-848125) | CDK2 (45 nM) | CDK7 (150 nM), CDK4 (160 nM), CDK5 (265 nM), CDK2 (363 nM), CDK1 (398 nM), TrKa | 4 phase I or II clinical trials in thymic and liver carcinomas; 1 active trial; no registration for clinical use in oncology | (Otto and Sicinski, 2017) |
| SB1317 (TG02) | CDK2 (13 nM) | JAK2, FLT3 | 7 phase I or II clinical trials in various solid tumors and hematological malignancies; 3 active trials; no registration for clinical use in oncology | (Otto and Sicinski, 2017) |
| CYC065 | CDK2 (5 nM), CDK9 (26 nM) | 3 phase I active clinical trials in hematological malignancies; no registration for clinical use in oncology | (Otto and Sicinski, 2017) | |
| RGB-286638 | CDK9 (1 nM), CDK1 (2 nM), CDK2 (3 nM), CDK4 (4 nM), CDK3 (5 nM), CDK5 (5 nM) | CDK7 (44 nM), CDK6 (55 nM), GSK-3β, TAK1, JAK2, MEK1 | 1 withdrawn phase I clinical trial | (Otto and Sicinski, 2017) |
| 3rd generation (Specific kinase inhibitors) | ||||
| Palbociclib (PD-0332991, Ibrance) | CDK4 (9–11 nM), CDK6 (15 nM) | registered for clinical use in hormone receptor positive, HeR2-negative advanced breast cancer patients; 215 phase I/II/III/IV clinical trials, including PALOMA series; being tested in various malignancies (173 active) | (Fry et al., 2004) | |
| Ribociclib (LEE011, Kisqali) | CDK4 (10 nM), CDK6 (39 nM) | registered for clinical use in hormone receptor positive, HeR2-negative advanced breast cancer patients; 103 phase I/II/III clinical trials, including MONALEESA series; being tested in various malignancies (72 active) | (Hortobagyi et al., 2016) | |
| Abemaciclib (LY2835219, Verzenio) | CDK4 (2 nM), CDK6 (10 nM) | registered for clinical use in hormone receptor positive, HER2-negative advanced breast cancer patients; 100 phase I/II/III/IV clinical trials, including MONARCH series; being tested in various malignancies (81 active) | (Sledge et al., 2017) | |
| Trilaciclib (G1T28) | CDK4 (1 nM), CDK6 (4 nM) | 4 phase I and II clinical trials in small cell lung cancer and breast cancer; no registration for clinical use in oncology | (Tan et al., 2019) | |
| Lerociclib (G1T38) | CDK4 (1 nM), CDK6 (4 nM) | CDK9 (28 nM), CDK5 (0.8–1.2 μM), CDK2 (1.5–3.6 μM), CDK1 (2.4 μM), CDK7 (2.4 μM) | 3 phase I and II clinical trials in lung and breast cancer; no registration for clinical use in oncology | (Bisi et al., 2017) |
| PHA-767491 (NMS-1116354) | CDC7 (10 nM), CDK9 (34 nM) | CDK2 (240 nM), CDK1 (250 nM), CDK5 (460 nM), GSK-3β, MK2, PLK1, CHK2 | 2 withdrawn phase I clinical trials in advanced solid tumors | (Montagnoli et al., 2008) |
| XL-413 (BMS-863233) | CDC7 (3.4 nM) | PIM1, CK2 | 2 withdrawn phase I and II clinical trials in advanced solid tumors and hematologic malignancies | (Koltun et al., 2012) |
| LY-3143921 | CDC7 (3.3 nM) | 1 active phase I clinical trial in advanced solid cancers | ||
| TAK-931 (Simurosertib) | CDC7 (<0.3 nM) | 3 phase I and II clinical trials in various advanced solid tumors; one on-going | (Iwai et al., 2019) | |
| BS-181 | CDK7 (210 nM) | CDK2 (0.9 μM), CDK5 (3 μM), CDK9 (4.2 μM), CDK1 (8.1 μM), CDK4 (33 μM), CDK6 (47 μM) | not entered clinical studies yet | (Ali et al., 2009) |
| ICEC0942 (CT7001, Samuraciclib) | CDK7 (40 nM) | CDK2 (620 nM), CDK9 (1.2 μM), CDK1 (1.8 μM) | 1 phase I/II on-going clinical trial in advanced solid tumors | (Patel et al., 2018) |
| THZ1 | CDK7 (3.2 nM) | not entered clinical studies yet | (Kwiatkowski et al., 2014) | |
| YKL-5-124 | CDK7 (9.7 nM) | CDK2 (1300 nM) CDK9 (3020 nM) | not entered clinical studies yet | (Olson et al., 2019) |
| SY-5609 | CDK7 (0.6 nM) | CDK2 (2900 nM) CDK9 (970 nM) CDK12 (770 nM), | 1 phase I dose-escalation in select advanced solid tumors | (Sava et al., 2020) |
| SY-1365 | CDK7 (84 nM) | CDK2 (2117 nM) CDK9 (914 nM) CDK12 (204 nM), | 1 phase I dose-escalation/safety advanced solid tumors | (Sava et al., 2020) |
Inhibitors of cell- cycle kinases which entered clinical trials of any phase; MAPK - Mitogen-Activated Protein Kinase; PKC – Protein Kinase C; EGFR - Epidermal Growth Factor Receptor; ERK - Extracellular Signal-Regulated Kinase; JAK2 – Janus Kinase 2; FLT3 - Receptor-type tyrosine-protein kinase FLT3; GSK-3β - Glycogen Synthase Kinase 3 beta; TAK1 - Transforming Growth Factor-β Activated Kinase 1; MEK1 - Dual Specificity Mitogen-Activated Protein Kinase 1; MK2 - MAP Kinase-Activated Protein Kinase 2; PLK1 – Polo-Like Kinase 1; CHK2 - Checkpoint Kinase 2; PIM1 - Proto-oncogene Serine/Threonine-Protein Kinase 1; CK2 – Casein Kinase 2; MDS/AML – Myelodysplastic Syndrome/ Acute Myeloid Leukemia, CLL – Chronic Lymphocytic Leukemia
Figure 4.
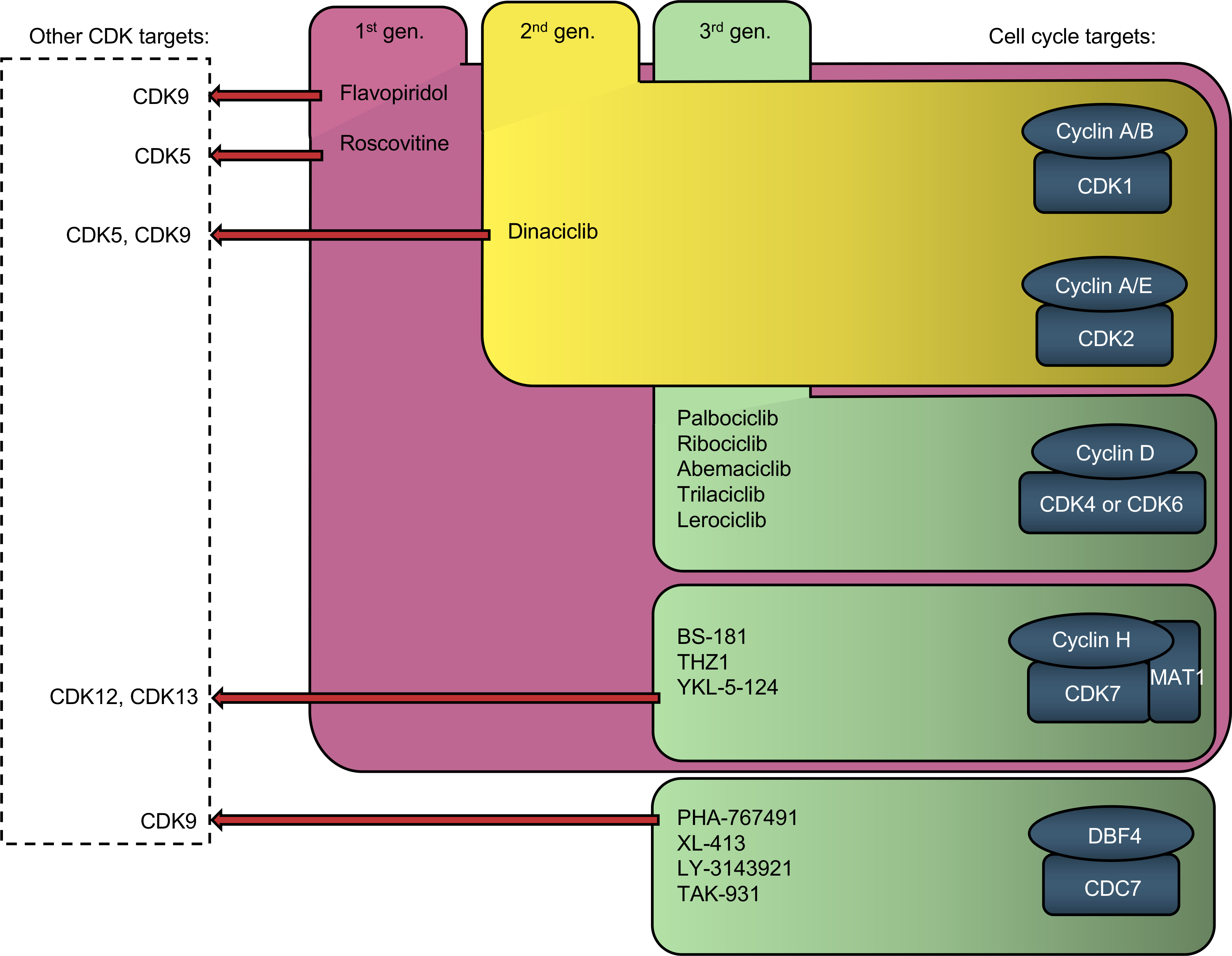
Inhibitors of cell-cycle proteins and their major non-cell-cycle CDK targets.
The very first clinical trial began in 2007 and demonstrated palbociclib’s efficacy against mantle cell lymphoma (Leonard et al., 2012). Starting in 2015, phase II and III clinical trials have been carried out with palbociclib (PALOMA series), ribociclib (MONALEESA) and abemaciclib (MONARCH) (Alvarez-Fernandez and Malumbres, 2020). These trials investigated the utility of combining CDK4/6i with standard endocrine-therapy (an estrogen receptor-antagonist fulvestrant or an aromatase-inhibitor, letrozole) in patients with advanced HR+/HER2− breast cancers. Addition of either of these CDK4/6i dramatically extended progression-free survival and prolonged the overall survival of patients. Moreover, abemaciclib extended progression-free survival when used as monotherapy in women with HR+/HER2− metastatic breast cancer (Finn et al., 2015; Gao et al., 2020). Consequently, all three CDK4/6i were approved by the US Food and Drug Administration (FDA) for treatment of patients with HR+/HER2− metastatic or advanced breast cancer.
Palbociclib and ribociclib represent highly specific inhibitors of CDK4 and CDK6, while abemaciclib also inhibits several other kinases (Klein et al., 2018). For palbociclib and ribociclib, neutropenia, thrombocytopenia and anemia represent the rate-limiting toxicities (Finn et al., 2016; Hortobagyi et al., 2016). This likely represents an on-target effect due to cyclin D3-CDK6 inhibition in hematopoietic cells (Sicinska et al., 2003). In contrast, these side-effects are less pronounced in patients treated with abemaciclib, which may be caused by the higher potency of this compound in inhibiting CDK4 rather than CDK6 (Sledge et al., 2017). For this reason, abemaciclib can be administered continuously, while palbociclib and ribociclib require an intermittent dosing schedule (Klein et al., 2018). The most frequent side-effects in abemaciclib-treated patients are gastrointestinal symptoms (Sledge et al., 2017). While the molecular mechanism is unknown, it was postulated that inhibition of CDK9 by abemaciclib may be responsible for this phenomenon (Chen et al., 2016).
It remains to be seen whether the ability of abemaciclib to inhibit other kinases represents an advantage or disadvantage over the more selective CDK4/6i. A recent PALLAS study revealed that addition of palbociclib to adjuvant endocrine therapy in patients with early-stage HR+/HER2- breast cancer did not improve invasive disease-free survival, compared with adjuvant endocrine therapy alone (Mayer et al., 2021). In contrast, a similar study with abemaciclib reported a significant prolongation of invasive disease-free survival (Johnston et al., 2020). It is possible that the ability of abemaciclib to inhibit other kinases may be responsible for the therapeutic effect. It is also likely that the efficacy of abemaciclib as a single-agent is in part due to its off-target effects.
An important issue for treatment with CDK4/6i it to develop biomarkers of sensitivity and resistance. It has been widely documented that tumor cells which lost RB1 expression do not arrest their proliferation upon CDK4/6 inhibition (O’Leary et al., 2016). Indeed, intact RB1 status represents the best predictive feature for CDK4/6i efficacy. Other predictors are not clear. Cancers with genomic activation of CCND1–3 genes were reported to be particularly susceptible to CDK4/6i (Gong et al., 2017). However, in post hoc analyses from the PALOMA trials, neither CCND1 amplification nor the levels of cyclin D1 mRNA correlated with responsiveness to CDK4/6i (Finn et al., 2020). The predictive value of CDK4 gene amplification and protein overexpression is unclear, with different studies linking these lesions to increased sensitivity or resistance (Alvarez-Fernandez and Malumbres, 2020). Immunohistochemical detection of Thr172-phosphorylated CDK4 (an activating phosphorylation carried out by the CDK-activating kinase, CAK) in breast cancer was shown to predict susceptibility to palbociclib treatment (Raspé et al., 2017). This observation may provide a predictive marker, which can be assessed in tumor samples. On the other hand, high levels of cyclin E1 mRNA in metastatic lesions were shown to correlate with resistance to palbociclib (Turner et al., 2019). Despite the progress, more markers are needed to reliably predict patients’ response to CDK4/6 inhibition.
Consistent with the growth-promoting function of cyclin D-CDK4/6, treatment with CDK4/6i causes cell-cycle arrest of tumor cells. In addition, some tumors respond to CDK4/6 inhibition by undergoing senescence (Alvarez-Fernandez and Malumbres, 2020). Several different mechanisms involving RB1 as well as FOXM1 (which also represents CDK4/6 phosphorylation substrate), were proposed to explain this phenomenon (Alvarez-Fernandez and Malumbres, 2020). Kovatcheva et al., (2017) postulated that expression of chromatin-remodeling enzyme ATRX determines the choice between quiescence and senescence in response to CDK4/6 inhibition.
Inhibition of CDK4/6 was also shown to affect tumor metabolism. Wang et al. (2017) observed that in tumor types expressing high levels of cyclin D3-CDK6 (such as T-ALL), inhibition of CDK4/6 reduces the flow of glucose-derived carbon into the pentose phosphate and serine pathways, thereby depleting antioxidants NADPH and glutathione. This, in turn, increases the levels of reactive oxygen species and causes tumor cell apoptosis. Franco et al. (2016) reported that treatment with CDK4/6i induces tumor cell metabolic reprogramming and increases the rate of oxidative phosphorylation.
Several recent studies revealed that treatment with CDK4/6i modulates the anti-tumor immune response of the host, via multiple mechanisms. First, inhibition of CDK4/6 preferentially impedes proliferation of immuno-inhibitory CD4+FOXP3+ Treg cells, with smaller effect on other T cell subtypes, such as cytotoxic CD8+ T cells (Deng et al., 2018; Goel et al., 2017; Ruscetti et al., 2018; Schaer et al., 2018). This decreases the Treg to CD8+ ratio of intratumoral T cells and facilitates tumor cell killing by the CD8+ cells. Second, inhibition of CDK4/6 awakens expression of endogenous retroviral elements in tumor cells, resulting in increased production of type-III interferons (Goel et al., 2017; Hurvitz et al., 2020; Schaer et al., 2018). This, in turn, increases presentation of tumor antigens. Third, inhibition of CDK4/6 triggers dephosphorylation and nuclear translocation of NFAT transcription factors, resulting in T cell activation, which enhances the anti-tumor immune response (Deng et al., 2018; Schaer et al., 2018). Lastly, inhibition of CDK4/6 causes upregulation of a checkpoint protein PD-L1 in tumor cells (Jerby-Arnon et al., 2018; Jin et al., 2019; Zhang et al., 2018). Importantly, studies using mouse cancer allografts revealed that treatment with CDK4/6i strongly synergizes with anti-PD-1/PD-L1 immune checkpoint blockade (Deng et al., 2018; Goel et al., 2017; Jerby-Arnon et al., 2018; Zhang et al., 2018). Since the action of CDK4/6i on host immune cells does not dependent of tumor’s RB1 status, these findings raise a possibility that CDK4/6i could be used to stimulate host antitumor immunity and to boost the effectiveness of immune checkpoint blockade in case RB1-negative tumors.
Inhibition of CDK4/6 was also shown to impair DNA damage repair through homologous recombination, following cytotoxic chemotherapy. Salvador-Barbero et al. (2020) demonstrated that sequential treatment of pancreatic cancers first with taxanes (or other chemotherapeutic compounds) followed by CDK4/6i strongly potentiates the anti-tumor effect of the former due to repression of DNA repair machinery.
The intrinsic (pre-existing) or acquired resistance to CDK4/6i represents the major obstacle in their clinical effectiveness. Since this issue was the subject of a recent review (Alvarez-Fernandez and Malumbres, 2020), we will only briefly mention the major mechanisms. These include: (i) inactivation of RB1; (ii) overexpression of CDK6 through amplification of the CDK6 gene or via other mechanisms; (iii) activation of cyclin E-CDK2 that by-passes cyclin D-CDK4/6; (iv) activation of FGFR1/2 or EGFR/ERBB2 → RAS-RAF-MEK-ERK or PI3K-AKT signaling (Alvarez-Fernandez and Malumbres, 2020); (v) lysosomal sequestration of CDK4/6i (Fassl et al., 2020; Llanos et al., 2019); (vi) activation of AURKA identified by a recent genome-wide study (Wander et al., 2020).
While CDK4/6i represent a breakthrough in breast cancer treatment, they are now being tested in clinical trials for many different cancer types. These trials are exploring various combination treatments involving CDK4/6i; several additional synergies with CDK4/6 inhibitors have been demonstrated in pre-clinical studies and will likely enter the clinics (Malumbres, 2019). One of the main challenges will be to develop better biomarkers for predicting sensitivity and resistance of tumors to CDK4/6i and the efficacy of combinatorial treatments against tumors carrying defined sets of genomic lesions. Efforts should be made to develop more selective compounds that inhibit only CDK4 or CDK6, as agents that selectively inhibit CDK4 could be potentially used at higher doses without encountering bone marrow toxicity. Another possibility is the use of degrader compounds that would degrade CDK4 and/or CDK6 (Box 3). Such compounds are expected not only to inhibit CDK4/6 kinase, but also to liberate cyclin D-CDK4/6-bound p21CIP1/p27KIP1 inhibitors, which would then inhibit cyclin E-CDK2.
Box 3. Targeted degradation of cell cycle proteins in cancer cells.
Traditionally, targeting the cell cycle machinery involves inhibition of the catalytic activity of the core enzymes. In recent years, an array of approaches involving targeted protein degradation (TPD) has been developed (reviewed in (Wu et al., 2020)). In principle, TPD can be achieved by endowing the target protein with a degradation domain or by adapting the existing chemical inhibitors. The most popular approach is called PROteolysis TArgeting Chimeras (PROTAC). This approach involves bi-functional molecules, or “molecular glues”, in which one domain binds the target protein and the other interacts with a component of an E3 ubiquitin ligase, such as CRBN, pVHL or β-TCRP. This recruits the E3 ligase to the target protein, thereby triggering its degradation by the ubiquitin-proteasome system. TPD has proven to represent a powerful tool in basic research and it holds promise in targeting of currently “undruggable” proteins. Several TPD approaches are being developed and applied to target members of the cell cycle machinery in the context of cancer therapy. Selective CDK4/6 inhibitors have been used to synthesize PROTACs that target these kinases (Brand et al., 2019; Jiang et al., 2019; Rana et al., 2019). For example, Brand et al. (2019) developed a compound in which palbociclib has been conjugated to pomalidomide, a phthalimide-based moiety that is expected to recruit CRL4CRBN E3 ligase complex. Surprisingly, this compound selectively depleted CDK6, but not CDK4 in acute myeloid leukemia cells, thereby allowing interrogation of CDK6-dependent pathways and functions. Jiang et al. (2019) described a panel of imide-based degrader molecules capable of targeting both CDK4 and CDK6, or selectively degrading either of these kinases. These molecules were further adapted to additionally degrade lymphoid transcription factors Ikaros (IKZF1) and Aiolos (IKZF3), known targets of imides and of some imide-based degraders. The authors reported that in mantle cell lymphoma cell lines, degradation of IKZF1/3 coupled with dual CDK4/6 degradation had a stronger anti-proliferative effect than degradation of CDK4/6 or IKZF1/3 alone, or a selective CDK4/6i. De Dominici et al. (2020) identified a CDK6-selective PROTAC that suppressed tumor burden of Philadelphia chromosome-positive acute lymphoblastic leukemia in vivo more effectively than CDK4/6i. These findings are consistent with the observations that CDK6 plays kinase-independent, transcriptional functions (Kollmann et al., 2013). Degradation of CDK6 is expected to remove both catalytic and transcriptional CDK6 functions, while CDK6 inhibitors block only its enzymatic activity. Hence, in addition to reduced toxicity stemming from their excellent sensitivity, degraders can help to exploit cancer cell dependencies on non-catalytic functions of target proteins. Teng et al. (2020) reported the development of a CDK2 and CDK5 dual degrader, which displayed a considerable selectivity over CDK1, CDK4/6 as well as transcriptional CDKs (CDK7 and CDK9). The authors demonstrated that this compound, through its ability to degrade CDK2, inhibited proliferation of cyclin E1-overexpressing human ovarian cancer cells. These observations point to CDK2-degradation as a potentially valuable therapeutic strategy in ovarian and other cancers that overexpress cyclin E1.
Cyclin E
Overexpression of E-cyclins and hyperactivation of cyclin E-CDK2 kinase have been documented in many human tumors (Figure 3). Upregulation of cyclin E can be driven by CCNE1 or CCNE2 gene amplification, increased activity of c-MYC or E2F transcription factors (which transcriptionally induce cyclin E), or inactivation of the F-box protein, FBXW7 (CDC4), which mediates cyclin E degradation (Hwang and Clurman, 2005). Moreover, several tumor types express proteolytically cleaved, ‘low-molecular weight’ (LMW) cyclin E (Caruso et al., 2018). LMW-cyclin E binds CDK2 with an increased affinity and forms hyperactive cyclin E-CDK2 complexes that are resistant to inhibition by p21CIP1 and p27KIP1 (Porter et al., 2001; Wingate et al., 2005).
In most cases studied, overexpression of cyclin E conferred poor clinical outcome (Hwang and Clurman, 2005) and was linked to resistance to several therapeutic compounds. For instance, elevated cyclin E levels represent a mechanism of resistance against trastuzumab in HER2+ breast cancers (Scaltriti et al., 2011). In HR+ breast cancers, the presence of cytoplasmic cyclin E correlates with resistance to aromatase inhibitors as well as to treatment with anti-estrogens plus CDK4/6i (Alvarez-Fernandez and Malumbres, 2020). Amplification of CCNE1 confers resistance to platinum-based chemotherapy in high-grade serous ovarian cancer (Etemadmoghadam et al., 2009). Also, overexpression of cyclin E2 was implicated as a mechanism of resistance to the endocrine therapy (Caldon et al., 2012).
Analyses of mouse cancer models confirmed the causative role of cyclin E-overexpression in tumorigenesis. Transgenic mice overexpressing cyclin E1 in T cells develop clonal lymphomas upon treatment with N-methylnitrosourea, or when crossed with p27Kip1-null mice (Geisen et al., 2003; Karsunky et al., 1999). Mice engineered to overexpress cyclin E1 in mammary glands develop mammary gland hyperplasias that progress at a low rate to mammary carcinomas (Bortner and Rosenberg, 1997). Mammary-specific expression of LMW-cyclin E1 triggers breast cancer development in a much higher fraction of mice than that of full-length cyclin E1. Importantly, the tumorigenic function of LMW cyclin E critically depends on Cdk2, as genetic ablation of Cdk2 completely blocked breast cancer occurrence in LMW-cyclin E transgenic mice (Akli et al., 2011)
While overexpression of cyclin E causes tumorigenesis, genetic ablation of E-cyclins was shown to impede tumor formation. Constitutive knockout of cyclin E prevented development of hepatocellular carcinomas, while an acute shutdown of E-cyclins in tumor-bearing mice blocked liver cancer progression (Geng et al., 2018; Sonntag et al., 2018). Moreover, mice with reduced levels of E-cyclins (cyclin E1+/−E2−/− and E1−/−E2+/−) show decreased susceptibility to Myc-driven breast cancers (Zhou et al., 2020).
It is not entirely clear how overexpressed cyclin E contributes to tumorigenesis. Given its well-established role in cell-cycle progression, a likely scenario is that overexpressed cyclin E drives uncontrolled cell proliferation. However, depletion of cyclin E in cyclin E1-amplified ovarian cancer cell lines triggered apoptosis, suggesting that cyclin E may play a pro-survival role in these cells (Etemadmoghadam et al., 2010). High levels of cyclin E in tumor cells were also shown to lead to genomic and chromosomal instability (Teixeira and Reed, 2017). Overexpressed cyclin E causes replication stress in S phase and chromosome segregation errors in M phase, resulting in genomic instability. Human mammary epithelial cells overexpressing cyclin E were shown to enter mitosis with short unreplicated genomic segments at a small number of specific loci, leading to anaphase anomalies and ultimately deletions. Interestingly, incompletely replicated regions were preferentially located at late-replicating domains and regions characterized by a paucity of replication origins (Teixeira and Reed, 2017). Other studies implicated overexpressed cyclin E in inhibiting APCCDH1 activity, resulting in the abnormal accumulation of APCCDH1 substrates as cells enter mitosis. This, in turn, impairs progression through mitosis, thereby contributing to polyploidy (Caldon et al., 2013). Overexpression of cyclin E was also implicated in erroneous MCM-loading, interference with nucleotide biosynthesis, induction of DNA-damage in stalled forks, uncontrolled DNA-replication firing and replication-transcription collisions, and all these mechanisms may contribute to genome instability (Mazumder et al., 2007; Teixeira and Reed, 2017). Another possible role of cyclin E in promoting tumorigenesis was provided by the observations that cyclin E-CDK2 phosphorylates c-MYC, and that this event prevents oncogene-induced senescence (Campaner et al., 2010; Hydbring et al., 2010).
All these functions of cyclin E are carried out in the cell nucleus, consistent with nuclear localization of full-length cyclin E. Intriguingly, proteolytically cleaved LMW-cyclin E is localized in the cytoplasm (Delk et al., 2009), suggesting that cyclin E may promote tumorigenesis by acting in this compartment. Lucenay et al. (2016) reported that cytoplasmic cyclin E physically interacts with ATP citrate lyase, an enzyme in de novo lipogenesis pathway, and enhances its enzymatic activity.
It is currently assumed that cyclin E exerts its oncogenic functions by activating its catalytic partner, CDK2. However, some groups proposed that cyclin E might play a kinase-independent role in driving proliferation of tumor cells. Sweeney et al. (1998) reported that in breast cancer cells, there is no evident relationship between CDK2 activity and cyclin E expression, with most of cyclin E residing in inactive complexes containing CDK-inhibitors p21CIP1 and p27KIP1. In another study, cyclin E mutants which were unable to activate CDK2, transformed rat embryonic fibroblasts in collaboration with H-Ras (Geisen et al., 2003). Cyclin E was also shown to localize to centrosomes and to promote S phase entry in a CDK2-independent fashion (Matsumoto and Maller, 2004). Ectopic expression of cyclin E mutant deficient in activating CDK2 but retaining the centrosomal localization sequence accelerated S phase entry and promoted polyploidy (Matsumoto and Maller, 2004). Recent studies of mouse liver cancer models revealed that while cyclin E is essential for tumor progression, Cdk2 is not (Geng et al., 2018; Sonntag et al., 2018). Consistent with these findings, in vitro cultured human liver cancer cell lines require cyclin E protein, but not cyclin E-associated kinase, for tumor cell proliferation (Geng et al., 2018). These observations raise a possibility that cyclin E might play a kinase-independent role in tumorigenesis. However, the molecular basis of such possible function remains unknown.
Targeting cyclin E may represent an attractive therapeutic strategy. Indeed, depletion of cyclin E arrested proliferation of cancer cell lines representing many tumor types (Hwang and Clurman, 2005). Importantly, mouse genetic experiments revealed that E-type cyclins are dispensable for proliferation of normal, non-transformed tissues of postnatal mice (Geng et al., 2007). Collectively, these observations indicate that targeting of cyclin E may selectively inhibit the proliferation of tumor cells, while sparing normal tissues. One possible approach to target cyclin E could employ ‘degrader’ compounds (Lai and Crews, 2017) (Box 3), which would selectively deplete E-cyclins. Such approach would extinguish all cyclin E functions, thereby assuring the therapeutic effect.
CDK2
While the role of cyclin E in tumorigenesis is well established, the requirement for CDK2 in tumor growth is still the subject of debate. Mouse knockout experiments revealed that CDK2 is not required for normal development and physiologic cell proliferation, except for meiotic divisions in male and female germ cells (Berthet et al., 2003; Ortega et al., 2003).
Analyses of several human cancer cell lines revealed that CDK2 is dispensable for their proliferation (Tetsu and McCormick, 2003). In contrast, CDK2 is required for proliferation of melanoma cells, where its expression is regulated by the melanocyte lineage transcription factor MITF (Desai et al., 2013). CDK2 also plays an essential function in ovarian cancer cells with CCNE1 amplification. Depletion of CDK2 or inhibition of CDK2 kinase in these cells arrested tumor cell proliferation and in several instances triggered tumor cell apoptosis (Au-Yeung et al., 2017; Etemadmoghadam et al., 2013; Yang et al., 2015). Inhibition of CDK2 also impeded ovarian cancer tumorigenesis in vivo (Au-Yeung et al., 2017), which was attributed to suppression of metastatic colonization (Yang et al., 2015). In diffuse large B-cell lymphomas, CDK2 was shown to promote tumor cell survival by maintaining the expression of the anti-apoptotic factor MCL1. Depletion of CDK2 resulted in down-regulation of MCL1 and triggered apoptosis of tumor cells (Faber and Chiles, 2007). Silencing of CDK2 also caused p53-dependent apoptosis of neuroblastoma cells with MYCN-amplification and overexpression (Molenaar et al., 2009). High levels of CDK2 were observed in glioblastoma multiforme, where CDK2 was shown to be essential for cell proliferation and confer resistance to radiation, as inhibition of CDK2 increased tumor cell radiosensitivity (Wang et al., 2016).
Hyperactivation of CDK2 – through different molecular mechanisms – was shown to represent one of the mechanisms of breast cancer resistance to CDK4/6i (Alvarez-Fernandez and Malumbres, 2020). Hence, inhibition of CDK2 may represent a therapeutic strategy to block proliferation of CDK4/6i-resistant tumors. Indeed, depletion or inhibition of CDK2 restored the sensitivity of resistant breast cancer cells to CDK4/6i (Fassl et al., 2020; Herrera-Abreu et al., 2016).
Several studies suggested the therapeutic value of combining CDK2 inhibition with other treatments. For example, in RB1-negative cyclin E-overexpressing triple-negative breast cancers (TNBC), inhibition of CDK2 was shown to act synergistically with chemotherapy (Rao et al., 2017). In medulloblastoma, CDK2 inhibition combined with targeting of MYC using BET bromodomain inhibitors synergistically suppressed tumor progression by causing cell-cycle arrest and apoptosis (Bolin et al., 2018). Combined inhibition of phosphatidylinositol 3-kinase (PI3K) and CDK2 induced cell death and enhanced antitumor activity in colorectal cancer xenografts (Beale et al., 2016). Treatment with CDK2 inhibitors led to a dramatic decrease in proliferation and enhanced apoptosis of cyclin E-overexpressing trastuzumab-resistant breast cancer cell lines and significantly reduced tumor growth of trastuzumab-resistant xenografts (Scaltriti et al., 2011). Depletion of CDK2 blocked cell-cycle progression, increased cell death and reduced colony formation in tamoxifen-resistant breast cancer cell lines (Johnson et al., 2010). Azimi et al. (2018) reported that upregulation of CDK2 confers resistance to BRAF and Hsp90 inhibitors in melanoma cells, and that CDK2 depletion sensitizes cells to these inhibitors. Since no CDK2-specific inhibitors are available, several of the studies utilized non-specific compounds that target other kinases. Other studies depleted CDK2 (using siRNAs or sRNAs), which may result different outcomes than kinase inhibition (Horiuchi et al., 2012a).
In addition to driving cell-cycle progression, CDK2 was shown to play other functions, which may be relevant in the context of therapeutic CDK2 inhibition. A recent study demonstrated that CDK2 phosphorylates proteins that regulate histone modifications, chromatin, transcription, and RNA/DNA metabolism (Chi et al., 2020). It remains to be seen how inhibition of CDK2 would affect these processes in cancer cells. Moreover, CDK2 was shown to phosphorylate and negatively regulate the stability of FOXP3, the master regulator of Tregs (Morawski et al., 2013). Hence, inhibition of CDK2 may increase the number of immunosuppressive Treg cells, which would impede the anti-tumor immune response. Despite this concern, the demonstrated essential role of CDK2 in certain tumor types justifies the efforts to develop specific CDK2 inhibitors. Such inhibitors would need to selectively target CDK2, but not a related kinase CDK1, given the essential function of the latter in cell proliferation (see below).
Cyclin A
Two A-type cyclins operate in mammalian cells, the ubiquitously expressed cyclin A2 and testes-specific cyclin A1 (Martínez-Alonso and Malumbres, 2020). Cyclin A2 expression is upregulated in many tumor types (Figure 3). In contrast, high levels of cyclin A1 were documented in a much narrower set of tumors, mainly in myeloid leukemias and in testicular and endometrial cancers (Horie et al., 2019; Yasmeen et al., 2003). Increased levels of cyclin A2 were shown to correlate with early relapse and poor overall survival in hepatocellular carcinoma and endometrioid adenocarcinoma (Santala et al., 2014; Yam et al., 2002). In contrast, other authors reported that cancers with low levels of cyclin A2 were more aggressive than those with high cyclin A2 expression (Aaltomaa et al., 1999; Bendris et al., 2014; Mashal et al., 1996). Arsic et al. (2012) observed that depletion of cyclin A2 enhanced the invasiveness of transformed cells via down-regulation of RhoA activity. Consistent with these observations, colorectal cancer metastases were found to express decreased levels of cyclin A2, as compared to primary tumors.
Bayard et al. (2018) reported the presence of a distinct subgroup of hepatocellular carcinomas containing genomic activation of the CCNA2 gene driven by diverse mechanisms. These include structural rearrangements resulting in CCNA2 fusion with several different genes, as well as viral (HBV or AAV2) insertions within the CCNA2 gene. Interestingly, viral insertions induce the expression of abnormal CCNA2 transcripts, predicted to generate a truncated cyclin A2 protein. Hepatocellular carcinomas displaying CCNA2 activation share the molecular features with liver cancers overexpressing cyclin E1, and together they define a distinct subgroup of tumors characterized by a unique gene expression profile indicative of E2F and ATR pathway activation. Cyclin A2/E1-overexpressing tumors display a specific structural rearrangement signature. These rearrangements are enriched at active chromatin regions and were shown to activate TERT promoter, which likely contributes to tumorigenesis (Bayard et al., 2018). A recent analysis of gastric cancer cell lines revealed that cyclin A2 expression is elevated in KRAS-mutant gastric tumors, causing an increased sensitivity to PLK1-inhibitors due to mitotic catastrophe and apoptosis upon PLK1-inhibition. These results suggest that cyclin A2 levels may serve as biomarker of PLK1-inhibitor sensitivity (Lee et al., 2020).
Analyses of most mouse cancer models support a pro-tumorigenic role of overexpressed cyclin A2. Transgenic mice overexpressing cyclin A2 in mammary glands exhibited hyperplasia and nuclear abnormalities suggestive of pre-neoplastic alterations (Yam et al., 2002). Conversely, deletion of cyclin A2 in oncogene-transformed mouse embryonic fibroblasts suppressed their tumorigenic potential (Gopinathan et al., 2014). Moreover, hepatocyte-specific ablation of cyclin A2 significantly delayed incidence of liver cancers triggered by expression of oncogenic Ras plus p53-silencing (Gopinathan et al., 2014). Transgenic mice overexpressing cyclin A1 in the myeloid lineage displayed abnormal myelopoiesis; however cyclin A1 overexpression alone was not sufficient to induce myeloid leukemia (Yam et al., 2002). In contrast to these findings, mice with hypomorphic CCNA2 alleles were more susceptible to spontaneous and induced tumor formation, particularly in the lung. Splenocytes and fibroblasts derived from these animals displayed increased chromosomal instability and chromosome segregation defects (Kanakkanthara et al., 2016). These results indicate that cyclin A2-insufficiency may promote neoplastic transformation.
The full range of molecular functions of cyclin A in tumorigenesis remains unclear. Elevated cyclin A levels may accelerate cell-cycle progression. Moreover, cyclin A2-CDK2 directly phosphorylates AKT, promoting its oncogenic function (Liu et al., 2014). Cyclin A2 and its associated CDK activity are required for the induction of progesterone receptor target genes in breast cancer cells (Moore et al., 2014). A tumor-suppressive role for cyclin A was suggested by the observations that cyclin A2-CDK2 phosphorylates p53 protein and stimulates its sequence-specific DNA-binding (Wang and Prives, 1995). Cyclin A2-CDK1 and A2-CDK2 can also phosphorylate MDM2, which weakens its inhibitory interaction with p53 (Zhang and Prives, 2001). These two mechanisms are expected to increase p53 activity in cyclin A2-overexpressing cells.
It is not clear whether targeting cyclin A2 would represent a therapeutic option, given the essential requirement for cyclin A in adult organisms. Conditional ablation of cyclin A in mouse hematopoietic lineage resulted in a rapid lethality, indicating that a global inhibition of cyclin A would likely have life-threatening consequences (Kalaszczynska et al., 2009). Given that cyclin A can activate both CDK2 and CDK1, it is possible that a specific inhibition of cyclin A2-CDK2 (or A2-CDK1) may have a more selective effect on cancer cells.
DBF4–CDC7
Overexpression of CDC7 was documented in a wide range of human malignancies (Figure 3). In most of the tumor types studied, CDC7 overexpression correlates with poor clinical outcome and aggressive tumor phenotype (Montagnoli et al., 2010). Intriguingly, overexpression of CDC7 was found predominantly in tumors or tumor cell lines with mutant p53 (Bonte et al., 2008). Importantly, human cancer cells with mutated p53 were reported to be particularly sensitive to CDC7-inhibition. Depletion/inhibition of CDC7 using si/shRNAs or chemical CDC7i triggered apoptosis of p53-mutant tumor cells. In contrast, cells expressing wild-type p53 responded to CDC7 depletion/inhibition by undergoing a transient cell-cycle arrest (Montagnoli et al., 2004; Rodriguez-Acebes et al., 2010). It has been argued that inhibition of CDC7 in p53 wild-type cells triggers p53-dependent cell-cycle arrest. In contrast, p53-mutant cancer cells attempt to continue DNA synthesis upon CDC7-inhibition, which results DNA-damage and ultimately cell death (Sawa and Masai, 2009). However, several other investigators concluded that inhibition of CDC7 is equally effective against tumors regardless of p53 status, with some studies reporting cell-cycle arrest, and other apoptosis as the main outcome (Iwai et al., 2019; Wang et al., 2019).
Wang et al., (2019) demonstrated that treatment of p53-mutant liver cancer cells with a CDC7i, XL-413, induces tumor cell senescence. Furthermore, the senescent cells can be killed by inhibition of the mTOR pathway. Others reported that simultaneous treatment with another CDC7i, PHA-767491 and 5-fluorouracil (5-FU) elicited a synergistic antitumor effect in hepatocellular carcinoma (Li et al., 2015). However, PHA-767491 is not CDC7-specific, as it also has a considerable activity against CDK9 (Natoni et al., 2011)(Figure 4) and other CDKs. Indeed, it was shown that PHA-767491 enhances the efficacy of 5-FU by inhibiting CHK1 phosphorylation (through CDC7-inhibition) (Yang et al., 2019) and by down-regulating MCL1 (a target of CDK9) (Cidado et al., 2020). In another study, administration of PHA-767491 synergized with multiple EGFR tyrosine kinase-inhibitors to overcome the resistance to EGFR-targeted therapy in various TNBC cell lines. Again, the synergistic effect was in large part dependent on CDK9 inhibition (McLaughlin et al., 2019). PHA-767491/NMS-1116354 and XL-413/BMS-863233 have been used in phase I clinical trials (Table 1). These compounds did not move into phase II, suggesting that they elicited significant toxicity. Phase I clinical trials involving two other compounds, LY-3143921 and TAK-931 are either in progress or have been completed but results have not been disclosed.
In summary, the utility of targeting CDC7 in cancer treatment remains inconclusive. It is likely that the toxic effects of anti-CDC7 compounds observed in the clinics represented an off-target effect of inhibiting other kinases, such as CDK9. It remains to be demonstrated whether CDC7 inhibition would selectively kill p53-mutant tumor cells in an in vivo setting, while having a transient and reversible effect on normal tissues.
Cyclin B and CDK1
High levels of cyclins B1 and B2 were observed in a wide spectrum of tumors (Figure 3) and linked to tumor aggressiveness and poor prognosis (Martínez-Alonso and Malumbres, 2020). Suzuki et al. (2007) reported that in breast cancers, the presence of nuclear cyclin B1 is associated poor clinical prognosis, whereas cytoplasmic cyclin B1 has no predictive value. Hassan et al. (2002) demonstrated that overexpression of cyclin B1 in head and neck squamous cell carcinomas renders tumors resistant to radiation therapy, as evidenced by significantly increased recurrence of cyclin B1-overexpressing tumors following radiotherapy. It was proposed that overexpressed cyclin B1 expedites G2/M transition, a stage during which cancer cells are most sensitive to this treatment.
Cyclin B1 has been proposed to represent a tumor antigen. Yu et al. (2002) observed that in contrast to cell-cycle-regulated expression of cyclin B1 in normal cells, in tumor cells cyclin B1 is constitutively expressed at high levels in the cytoplasm. This leads to recognition of cyclin B by the host immune system (Vella et al., 2009; Yu et al., 2002). Indeed, it was reported that during lung tumorigenesis, aberrant expression of cyclin B1 in premalignant lesions correlates with T cell-dependent antibody responses against cyclin B1 (Egloff et al., 2006). Anti-cyclin B1 antibody responses were also reported in patients with prostate, breast, colorectal and hepatocellular cancers (Egloff et al., 2006). Intriguingly, anti-cyclin B1 antibodies and anti-cyclin B1 memory T-cells were also found in healthy individuals with no cancer history, suggesting that events other than cancer may lead to aberrant cyclin B1 expression and anti-cyclin B1 immunity. Importantly, vaccine-elicited anti-cyclin B1 immunity was shown to protect mice against cyclin B1-overexpressing tumors (Vella et al., 2009).
Studies on colorectal cancers and pituitary adenomas indicated that cyclin B may have tumor-suppressive properties. The authors postulated that low levels of cyclin B1 are associated with poor survival of patients, and that overexpression of this protein correlates with decreased invasion and metastasis of cancer cells. This phenomenon was attributed to ability of cyclin B1 to regulate EMT markers, such as E-cadherin (Fang et al., 2015; Li et al., 2019).
Transgenic mice engineered to overexpress cyclins B1 and B2 are prone to formation of tumors (e.g. colon, lung cancer), which has been attributed to defects in chromosome segregation (Nam and van Deursen, 2014). While both cyclins B1 and B2 can induce aneuploidy when overexpressed, they do it through distinct mechanisms. In vitro analysis of MEFs from cyclin B-overexpressing mice revealed that cyclin B1 inhibits separase activation, leading to anaphase bridges, whereas cyclin B2 triggers Aurora-A-mediated Plk1 hyperactivation, which results in accelerated centrosome separation and lagging chromosomes (Nam and van Deursen, 2014). Furthermore, cyclin B2–Cdk1 acts antagonistically to p53 to control Aurora-A-mediated centrosome disjunction, with p53 keeping Aurora A protein levels under control through transcriptional and post-translational inhibitory mechanisms, and cyclin B2–Cdk1 activating the residual Aurora-A pool (Nam and van Deursen, 2014).
Deregulated CDK1 activity in tumorigenesis is typically associated with aberrant expression of B-type cyclins (Nam and van Deursen, 2014) rather than CDK1-overexpression. Liver-specific ablation of Cdk1 was shown to confer resistance against tumorigenesis induced by activated Ras and silencing of p53 (Diril et al., 2012). However, analyses of knockout mice indicated that Cdk1 is essential for proliferation of normal cells (Santamaría et al., 2007). Hence, inhibition of CDK1 is expected to block proliferation of non-transformed tissues. Intriguingly, it was reported that cells transformed with c-MYC oncogene as well as c-MYC-overexpressing TNBC cells undergo apoptosis when treated with CDK1-inhibitors (Goga et al., 2007; Kang et al., 2014). A similar effect was observed using mouse models of MYC-driven lymphomas or liver tumors, as well as TNBC xenografts (Goga et al., 2007; Horiuchi et al., 2012b; Kang et al., 2014). This pro-survival function of CDK1 was attributed to the ability of CDK1 to phosphorylate BIRC5 (survivin), an inhibitor of apoptosis. Indeed, inhibition of CDK1 downregulated survivin expression (Goga et al., 2007). Another proposed mechanism was p53-independent upregulation of pro-apoptotic BIM protein upon CDK1-inhibition (Horiuchi et al., 2012b; Kang et al., 2014). These observations raise a possibility that a transient inhibition of CDK1 may kill MYC-overexpressing tumor cells, while causing only reversible cell-cycle arrest in normal tissues. However, inhibition of Cdk1 was also shown to cause apoptosis of embryonic stem cells (Huskey et al., 2015). Hence, the impact of CDK1-inhibition on survival of stem cell compartments (such as hematopoietic stem cells) within the adult organism needs to be evaluated.
Cyclin H–CDK7
Cyclin H-CDK7 kinase was shown to play two distinct roles, by serving as the CDK-activating kinase (CAK), as well as a component of a general transcription factor TFIIH (Box 1).
Amplification of the CDK7 gene was detected in several cancer types and shown to correlate with aggressive pathological features and poor prognosis (Figure 3) (Sava et al., 2020). The CDK7 protein is overexpressed in oral squamous cell carcinomas and in TNBC, and correlates with reduced overall and disease-free survival (Sava et al., 2020). On the other hand, increased CDK7 levels observed in estrogen receptor-positive breast cancers inversely correlate with tumor grade and size, and were hence described as a favorable prognostic marker (Sava et al., 2020).
The first ‘CDK7-specific’ inhibitor, BS-181 had a significantly higher potency towards CDK7 than to other CDKs; however, it demonstrated some off-target activity (Table 1). BS-181 was shown to arrest the growth of multiple breast and gastric cancer cell lines through cell-cycle inhibition and induction of apoptosis (Sava et al., 2020). In xenograft models, BS-181 significantly reduced the growth of MCF-7 breast and BGC823 gastric tumors, with no detected overall toxicity. Importantly, treatment with BS-181 led to a reduction of RNA-polymerase II CTD phosphorylation, suggestive of stalling of mRNA transcription (Sava et al., 2020). While BS-181 demonstrated low bioavailability, administration of its improved analog, ICEC0942 (CT7001), resulted in a significant decrease in tumor growth concomitant with the reduction in RNA-polymerase II CTD phosphorylation in vivo (Patel et al., 2018). However, it was not until the development of the first covalent CDK7-inhibitor, THZ1 (Kwiatkowski et al., 2014), that the transcriptional function of CDK7 in cancer became intensely studied. While THZ1 showed a similar antiproliferative potency to that of BS-181, cancer cell line profiling indicated that some cell lines (e.g. T-ALL) were particularly sensitive to THZ1-treatment (Kwiatkowski et al., 2014). A genome-wide analysis in T-ALL cells demonstrated that treatment with THZ1 disproportionally affects transcription of the RUNX1 gene. Importantly, RUNX1 - whose high expression in T-ALL cells is driven by a large super-enhancer domain - plays a key role in the core transcriptional regulatory circuitry and is required for growth and survival of leukemic cells (Kwiatkowski et al., 2014).
Several other studies reported high THZ1 efficacy in cancers addicted to expression of other super-enhancer-driven genes. Chipumuro et al. (2014) reported that THZ1 disrupted the transcription of amplified MYCN gene in neuroblastoma cells, which led to significant tumor regression in an in vivo model. Such effect was highly selective for MYCN-overexpressing cells and correlated with preferential downregulation of super-enhancer-associated genes. Christensen et al. (2014) found that in small-cell lung cancer, expression of super-enhancer-associated transcription factor genes, including MYC, is highly vulnerable to THZ1-treatment. Finally, Wang et al. (2015) identified an “Achilles cluster” of TNBC-specific genes that are especially sensitive to CDK7-inhibition and are frequently associated with super-enhancers. The authors proposed that CDK7 mediates the transcriptional addiction of TNBC cells to these genes, and hence its inhibition triggers apoptotic cell death of tumor cells.
Collectively, these observations indicated that inhibition of CDK7 specifically affects expression of super-enhancer-driven genes. However, the emergence of an even more selective CDK7-inhibitor, YKL-5–124, has challenged this model. Treatment of cancer cell lines with YKL-5–124 prevented CDK1 and CDK2 T-loop phosphorylation by CAK, and led to cell-cycle arrest at the G1/S boundary and inhibition of E2F-driven gene expression. Strikingly, YKL-5–124-treatment had no effect on RNA-polymerase II CTD phosphorylation (Olson et al., 2019). At this time, it became evident that the effects of the THZ1 compound were primarily due to its off-target inhibition of other transcriptional CDKs, namely CDK12 and CDK13, which contributed to the dramatic inhibition of super-enhancer-associated genes (Olson et al., 2019). In the revised, current view CDK7-inhibition is thought to mainly affect cell-cycle progression. Unexpectedly, a recent observation showed that YKL-5–124-treatment activates immune-response signaling in small-cell lung cancer cells, leading to secretion of essential pro-inflammatory cytokines/chemokines, which in turn can activate CD8+ T-cells (Zhang et al., 2020). It remains to be seen whether this phenomenon truly represents an on-target effect of CDK7-inhibition, or it is again due to inhibition of another kinase. Nevertheless, treatment with YKL-5–124 was shown to inhibit small-cell lung cancer growth in vivo and to enhance tumor response to anti-PD-1 immunotherapy (Zhang et al., 2020). These results indicate that more studies are needed to decipher the full range of normal, physiological functions of CDK7.
Currently, there are three active phase I clinical trials involving CDK7-inhibitors SY-5609, SY-1365 and CT7001 in patients with breast, ovarian and advanced solid malignancies (Table 1). The newest generation of most specific CDK7-inhibitors has not yet been included in clinical trials. It remains to be seen whether these selective CDK7-inhibitors perform better than the compounds which also inhibit CDK12 and CDK13. The clinical efficacy of CDK12/13-inhibitors (which do not target CDK7) also need to be explored.
Non-specific pan-CDK inhibitors
A large number of clinical trials conducted to date utilized non-specific pan-CDKi which target several CDKs (Table 1). The first-generation pan-CDKi included flavopiridol/alvocidib and R-roscovitine/seliciclib (Table 1, Figure 4). Of these inhibitors, flavopiridol has been most extensively studied in clinics (Otto and Sicinski, 2017). Flavopiridol, which inhibits CDK1, CDK2, CDK4, CDK6, CDK7, and CDK9, was shown to cause cell-cycle arrest of tumor cells in G1 and G2 phases (Asghar et al., 2015).Administration of flavopiridol induced apoptosis in several mouse tissues, leading to organ atrophy, an effect attributed to inhibition of CDK9 (Otto and Sicinski, 2017). Despite these limitations, flavopiridol was tested in phase I clinical trials. While in these trials flavopiridol was active as a single agent against some solid malignancies, clinical phase II studies reported insufficient efficacy. However, some evidence of clinical activity was observed in hematological malignancies (Otto and Sicinski, 2017), and flavopiridol received orphan drug designation from the FDA. On the other hand, R-roscovitine (inhibitory activity against CDK1, CDK2, CDK5, and CDK7) failed to show antitumor activities in preclinical and clinical studies as a monotherapy (Asghar et al., 2015). However, it sensitized various cancers to DNA-damaging agents and radiation, which is consistent with its ability to block DNA double stranded break end-resection and to impair DNA repair via homologous recombination (Lin et al., 2018). Moreover, sequential treatment with R-roscovitine followed by doxorubicin was shown to kill TNBC cells. This effect was attributed to the ability of R-roscovitine to arrest tumor cells in the G2/M phase, thereby priming them for DNA-damage. Inactivation of the p53 pathway seems to be required for such combination-induced cytotoxicity, as this treatment was shown to specifically affect p53-mutant TNBC (Jabbour-Leung et al., 2016).
The second-generation of pan-CDKi with increased potency, such as dinaciclib, AT7519, milciclib, TG02, CYC065 and RGB-286638, has been developed (Otto and Sicinski, 2017). Dinaciclib (inhibitory activity against CDK1, CDK2, CDK5, and CDK9) (Figure 4) demonstrated superior (over 100-fold higher) potency in inhibiting RB1 phosphorylation and a more than 10-fold higher therapeutic index than flavopiridol (Otto and Sicinski, 2017). Additionally, dinaciclib inhibited cell-cycle progression in over 100 tumor cell lines of various tumor types and induced the regression of established solid tumors in a range of mouse xenograft models (Asghar et al., 2015). Phase I clinical trials reported partial responses and tolerable toxicity (Mitri et al., 2015; Nemunaitis et al., 2013). However, results from randomized phase II trials of dinaciclib in solid tumors have been mostly disappointing (Mita et al., 2014; Stephenson et al., 2014). On the other hand, promising clinical data obtained in patients with chronic lymphocytic leukemia (CLL) led to a randomized phase III study of dinaciclib in refractory CLL (Ghia et al., 2017).
Dinaciclib-treatment may provide an effective strategy in MYC-overexpressing TNBC and MYC-driven B cell lymphomas. Administration of dinaciclib caused tumor regression in mouse models of these tumor types (Otto and Sicinski, 2017). This effect may be due to the synthetic lethal interaction between CDK1-inhibition and MYC-overexpression (see above). Currently, a phase I study is investigating the use of dinaciclib in patients with MYC-overexpressing breast cancers. Dinaciclib also demonstrated synergistic activity when combined with the AKT-inhibitor MK-2206, and caused strong tumor growth inhibition in pancreatic cancer xenografts (Otto and Sicinski, 2017).
A recent pre-clinical study revealed that dinaciclib treatment induces type I interferon gene signature within the tumor and elicits immunogenic cell death. Moreover, administration of dinaciclib strongly augmented the efficacy of anti-PD-1 immune checkpoint blockade in syngeneic mouse tumor models. Animals treated with anti-PD1 antibody and dinaciclib showed increased T-cell infiltration and dendritic cell activation within the tumor, indicating that this combination improves the anti-tumor immune response (Hossain et al., 2018).
Since these pan-CDKi compounds inhibit multiple CDKs, they cause significant toxicities in normal tissues, thereby severely narrowing their therapeutic window. These shortcomings further highlight the urgent need to develop highly selective CDK-inhibitors, or inhibitors that target defined CDK subsets.
Future Outlook
The success of CDK4/6-inhibitors in the clinics represents only the beginning of targeting cell-cycle proteins in cancer treatment. In the next years, compounds which inhibit other cell-cycle proteins will be tested in pre-clinical studies and ultimately in cancer patients. One of the main challenges will be to identify tumor subsets that are dependent on particular cell-cycle proteins.
Another important challenge will be to delineate the full range of functions of cell-cycle proteins, which may be relevant in the context of cancer therapy. The role of cell-cycle proteins in anti-tumor immune response, acting both within tumor cells as well in the immune system is only beginning to be appreciated, and will be the subject of intense studies. Several other properties of tumor cells as well as of the surrounding stroma were postulated to be regulated by cell-cycle proteins, and further studies are needed to support these findings in vivo in the context of human cancers.
An open question is the advantage of highly specific inhibitors, versus compounds that inhibit several kinases. As we discussed, selective CDK4i may show less on-target toxicity than currently used CDK4/6i. However, since upregulation of CDK6 (and CDK2) represents mechanisms of CDK4/6i resistance (Alvarez-Fernandez and Malumbres, 2020), such narrowly acting compounds may enable acquisition of resistance. It is therefore plausible that less specific compounds, targeting for instance CDK4/6 and CDK2 may result in better clinical outcomes. The same principle applies to ‘CDK7-specific’ inhibitors, where at least some of the therapeutic effects were mediated by inhibition of CDK12 and CDK13 (Olson et al., 2019). When CDK2i become available, it will be of interest to compare compounds that inhibit only cyclin E-CDK2, versus ones that target all CDK2-containing complexes.
It will also be important to design inhibitors with altered chemical properties. It was recently shown that all currently approved CDK4/6i become ‘trapped’ in the lysosomes of some TNBC due to their weakly basic structure (Fassl et al., 2020; Llanos et al., 2019), preventing them from reaching their targets in the nucleus. Importantly, less basic derivatives of a CDK4/6i ribociclib were shown to escape the lysosomal sequestration and to arrest proliferation of ribociclib-resistant TNBC (Fassl et al., 2020). Hence, a new generation of CDK4/6i may extend the use of these compounds to tumor types that are currently resistant to CDK4/6i therapy.
The emerging technologies will also allow to convert the existing inhibitors into degrader compounds (Box 3). Degradation of the kinase may result in a more sustained effect than its inhibition. On the other hand, removal of the kinase subunit, while leaving the cyclin protein intact, may trigger compensatory mechanisms, such as binding of a related CDK to the intact cyclin. Such an outcome may mitigate the effects of CDK degradation. For instance, it was shown that small-molecule inhibition of CDK2 kinase has more profound effects on cell proliferation than CDK2 depletion (Horiuchi et al., 2012a). These different scenarios need to be tested experimentally in vivo using mouse cancer models.
In summary, this exciting field will remain an area of intense investigation for several years to come. It will undoubtedly yield many new therapeutic modalities which will have a major impact on the treatment of cancer patients.
ACKNOWLEDGEMENTS
Supported by grants R01CA202634, R01CA236226, P50CA168504 and R01CA247375 from NIH (to PS). MB was supported by the Polish National Agency for Academic Exchange (PPN/WAL/2019/1/00023) and by the Foundation for Polish Science (START Programme).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aaltomaa S, Lipponen P, Ala-Opas M, Eskelinen M, Syrjanen K, and Kosma VM (1999). Expression of cyclins A and D and p21(waf1/cip1) proteins in renal cell cancer and their relation to clinicopathological variables and patient survival. Br J Cancer 80, 2001–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akli S, Van Pelt CS, Bui T, Meijer L, and Keyomarsi K (2011). Cdk2 is required for breast cancer mediated by the low-molecular-weight isoform of cyclin E. Cancer Res 71, 3377–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S, Heathcote DA, Kroll SH, Jogalekar AS, Scheiper B, Patel H, Brackow J, Siwicka A, Fuchter MJ, Periyasamy M, et al. (2009). The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res 69, 6208–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Fernandez M, and Malumbres M (2020). Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell 37, 514–529. [DOI] [PubMed] [Google Scholar]
- Arsic N, Bendris N, Peter M, Begon-Pescia C, Rebouissou C, Gadea G, Bouquier N, Bibeau F, Lemmers B, and Blanchard JM (2012). A novel function for Cyclin A2: control of cell invasion via RhoA signaling. J Cell Biol 196, 147–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghar U, Witkiewicz AK, Turner NC, and Knudsen ES (2015). The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 14, 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au-Yeung G, Lang F, Azar WJ, Mitchell C, Jarman KE, Lackovic K, Aziz D, Cullinane C, Pearson RB, Mileshkin L, et al. (2017). Selective Targeting of Cyclin E1-Amplified High-Grade Serous Ovarian Cancer by Cyclin-Dependent Kinase 2 and AKT Inhibition. Clin Cancer Res 23, 1862–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimi A, Caramuta S, Seashore-Ludlow B, Boström J, Robinson JL, Edfors F, Tuominen R, Kemper K, Krijgsman O, Peeper DS, et al. (2018). Targeting CDK2 overcomes melanoma resistance against BRAF and Hsp90 inhibitors. Mol Syst Biol 14, e7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayard Q, Meunier L, Peneau C, Renault V, Shinde J, Nault JC, Mami I, Couchy G, Amaddeo G, Tubacher E, et al. (2018). Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat Commun 9, 5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beale G, Haagensen EJ, Thomas HD, Wang LZ, Revill CH, Payne SL, Golding BT, Hardcastle IR, Newell DR, Griffin RJ, and Cano C (2016). Combined PI3K and CDK2 inhibition induces cell death and enhances in vivo antitumour activity in colorectal cancer. Br J Cancer 115, 682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendris N, Cheung CT, Leong HS, Lewis JD, Chambers AF, Blanchard JM, and Lemmers B (2014). Cyclin A2, a novel regulator of EMT. Cell Mol Life Sci 71, 4881–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet C, Aleem E, Coppola V, Tessarollo L, and Kaldis P (2003). Cdk2 knockout mice are viable. Curr Biol 13, 1775–1785. [DOI] [PubMed] [Google Scholar]
- Bisi JE, Sorrentino JA, Jordan JL, Darr DD, Roberts PJ, Tavares FX, and Strum JC (2017). Preclinical development of G1T38: A novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with CDK4/6 sensitive tumors. Oncotarget 8, 42343–42358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolin S, Borgenvik A, Persson CU, Sundström A, Qi J, Bradner JE, Weiss WA, Cho YJ, Weishaupt H, and Swartling FJ (2018). Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 37, 2850–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonte D, Lindvall C, Liu H, Dykema K, Furge K, and Weinreich M (2008). Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia 10, 920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortner DM, and Rosenberg MP (1997). Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol Cell Biol 17, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M, Jiang B, Bauer S, Donovan KA, Liang Y, Wang ES, Nowak RP, Yuan JC, Zhang T, Kwiatkowski N, et al. (2019). Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem Biol 26, 300–306.e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldon CE, Sergio CM, Burgess A, Deans AJ, Sutherland RL, and Musgrove EA (2013). Cyclin E2 induces genomic instability by mechanisms distinct from cyclin E1. Cell Cycle 12, 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldon CE, Sergio CM, Kang J, Muthukaruppan A, Boersma MN, Stone A, Barraclough J, Lee CS, Black MA, Miller LD, et al. (2012). Cyclin E2 overexpression is associated with endocrine resistance but not insensitivity to CDK2 inhibition in human breast cancer cells. Mol Cancer Ther 11, 1488–1499. [DOI] [PubMed] [Google Scholar]
- Campaner S, Doni M, Hydbring P, Verrecchia A, Bianchi L, Sardella D, Schleker T, Perna D, Tronnersjö S, Murga M, et al. (2010). Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat Cell Biol 12, 54–59; sup pp 51–14. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, and Stuart JM (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso JA, Duong MT, Carey JPW, Hunt KK, and Keyomarsi K (2018). Low-Molecular-Weight Cyclin E in Human Cancer: Cellular Consequences and Opportunities for Targeted Therapies. Cancer Res 78, 5481–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Lee NV, Hu W, Xu M, Ferre RA, Lam H, Bergqvist S, Solowiej J, Diehl W, He YA, et al. (2016). Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol Cancer Ther 15, 2273–2281. [DOI] [PubMed] [Google Scholar]
- Chi Y, Carter JH, Swanger J, Mazin AV, Moritz RL, and Clurman BE (2020). A novel landscape of nuclear human CDK2 substrates revealed by in situ phosphorylation. Sci Adv 6, eaaz9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al. (2014). CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159, 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer H, and Sicinski P (2012). The requirement for cyclin D function in tumor maintenance. Cancer Cell 22, 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, et al. (2014). Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 26, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, Pop-Damkov P, Su N, Roamio Franklin VN, Sekhar Reddy Chilamakuri C, et al. (2020). AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin Cancer Res 26, 922–934. [DOI] [PubMed] [Google Scholar]
- Ciznadija D, Liu Y, Pyonteck SM, Holland EC, and Koff A (2011). Cyclin D1 and cdk4 mediate development of neurologically destructive oligodendroglioma. Cancer Res 71, 6174–6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang F, Nie L, and Wei W (2020). Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Dominici M, Porazzi P, Xiao Y, Chao A, Tang HY, Kumar G, Fortina P, Spinelli O, Rambaldi A, Peterson LF, et al. (2020). Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 135, 1560–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delk NA, Hunt KK, and Keyomarsi K (2009). Altered subcellular localization of tumor-specific cyclin E isoforms affects cyclin-dependent kinase 2 complex formation and proteasomal regulation. Cancer Res 69, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, Chhabra S, Huang W, Liu H, Aref AR, et al. (2018). CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov 8, 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai BM, Villanueva J, Nguyen TT, Lioni M, Xiao M, Kong J, Krepler C, Vultur A, Flaherty KT, Nathanson KL, et al. (2013). The anti-melanoma activity of dinaciclib, a cyclin-dependent kinase inhibitor, is dependent on p53 signaling. PLoS One 8, e59588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, Tessarollo L, and Kaldis P (2012). Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci U S A 109, 3826–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff AM, Vella LA, and Finn OJ (2006). Cyclin B1 and other cyclins as tumor antigens in immunosurveillance and immunotherapy of cancer. Cancer Res 66, 6–9. [DOI] [PubMed] [Google Scholar]
- Etemadmoghadam D, Au-Yeung G, Wall M, Mitchell C, Kansara M, Loehrer E, Batzios C, George J, Ftouni S, Weir BA, et al. (2013). Resistance to CDK2 inhibitors is associated with selection of polyploid cells in CCNE1-amplified ovarian cancer. Clin Cancer Res 19, 5960–5971. [DOI] [PubMed] [Google Scholar]
- Etemadmoghadam D, deFazio A, Beroukhim R, Mermel C, George J, Getz G, Tothill R, Okamoto A, Raeder MB, Harnett P, et al. (2009). Integrated genome-wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clin Cancer Res 15, 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemadmoghadam D, George J, Cowin PA, Cullinane C, Kansara M, Australian Ovarian Cancer Study, G., Gorringe KL, Smyth GK, and Bowtell DD (2010). Amplicon-dependent CCNE1 expression is critical for clonogenic survival after cisplatin treatment and is correlated with 20q11 gain in ovarian cancer. PLoS One 5, e15498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber AC, and Chiles TC (2007). Inhibition of cyclin-dependent kinase-2 induces apoptosis in human diffuse large B-cell lymphomas. Cell Cycle 6, 2982–2989. [DOI] [PubMed] [Google Scholar]
- Fang Y, Liang X, Jiang W, Li J, Xu J, and Cai X (2015). Cyclin b1 suppresses colorectal cancer invasion and metastasis by regulating e-cadherin. PLoS One 10, e0126875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassl A, Brain C, Abu-Remaileh M, Stukan I, Butter D, Stepien P, Feit AS, Bergholz J, Michowski W, Otto T, et al. (2020). Increased lysosomal biomass is responsible for the resistance of triple-negative breast cancers to CDK4/6 inhibition. Sci Adv 6, eabb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et al. (2015). The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 16, 25–35. [DOI] [PubMed] [Google Scholar]
- Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al. (2009). PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 11, R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Liu Y, Zhu Z, Martin M, Rugo HS, Diéras V, Im SA, Gelmon KA, Harbeck N, Lu DR, et al. (2020). Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naïve Metastatic Breast Cancer. Clin Cancer Res 26, 110–121. [DOI] [PubMed] [Google Scholar]
- Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, Harbeck N, Lipatov ON, Walshe JM, Moulder S, et al. (2016). Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 375, 1925–1936. [DOI] [PubMed] [Google Scholar]
- Franco J, Balaji U, Freinkman E, Witkiewicz AK, and Knudsen ES (2016). Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep 14, 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, and Toogood PL (2004). Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 3, 1427–1438. [PubMed] [Google Scholar]
- Gao JJ, Cheng J, Bloomquist E, Sanchez J, Wedam SB, Singh H, Amiri-Kordestani L, Ibrahim A, Sridhara R, Goldberg KB, et al. (2020). CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: a US Food and Drug Administration pooled analysis. Lancet Oncol 21, 250–260. [DOI] [PubMed] [Google Scholar]
- Geisen C, Karsunky H, Yucel R, and Moroy T (2003). Loss of p27(Kip1) cooperates with cyclin E in T-cell lymphomagenesis. Oncogene 22, 1724–1729. [DOI] [PubMed] [Google Scholar]
- Geng Y, Lee YM, Welcker M, Swanger J, Zagozdzon A, Winer JD, Roberts JM, Kaldis P, Clurman BE, and Sicinski P (2007). Kinase-independent function of cyclin E. Mol Cell 25, 127–139. [DOI] [PubMed] [Google Scholar]
- Geng Y, Michowski W, Chick JM, Wang YE, Jecrois ME, Sweeney KE, Liu L, Han RC, Ke N, Zagozdzon A, et al. (2018). Kinase-independent function of E-type cyclins in liver cancer. Proc Natl Acad Sci U S A 115, 1015–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghia P, Scarfò L, Perez S, Pathiraja K, Derosier M, Small K, McCrary Sisk C, and Patton N (2017). Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 129, 1876–1878. [DOI] [PubMed] [Google Scholar]
- Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O, et al. (2017). CDK4/6 inhibition triggers anti-tumour immunity. Nature 548, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goga A, Yang D, Tward AD, Morgan DO, and Bishop JM (2007). Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med 13, 820–827. [DOI] [PubMed] [Google Scholar]
- Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, Dowless M, Dempsey J, Zeng Y, Torres R, et al. (2017). Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 32, 761–776.e766. [DOI] [PubMed] [Google Scholar]
- Gopinathan L, Tan SL, Padmakumar VC, Coppola V, Tessarollo L, and Kaldis P (2014). Loss of Cdk2 and cyclin A2 impairs cell proliferation and tumorigenesis. Cancer Res 74, 3870–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan KA, Ang KK, El-Naggar AK, Story MD, Lee JI, Liu D, Hong WK, and Mao L (2002). Cyclin B1 overexpression and resistance to radiotherapy in head and neck squamous cell carcinoma. Cancer Res 62, 6414–6417. [PubMed] [Google Scholar]
- Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, et al. (2016). Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res 76, 2301–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie K, Yamamoto H, Karube K, Takebayashi K, Yoshino H, Yoshioka H, and Watanabe J (2019). Cyclin A is a reliable proliferation marker in endometrial cancer cell lines. Oncol Lett 17, 4455–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Huskey NE, Kusdra L, Wohlbold L, Merrick KA, Zhang C, Creasman KJ, Shokat KM, Fisher RP, and Goga A (2012a). Chemical-genetic analysis of cyclin dependent kinase 2 function reveals an important role in cellular transformation by multiple oncogenic pathways. Proc Natl Acad Sci U S A 109, E1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, Creasman KJ, Bazarov AV, Smyth JW, Davis SE, et al. (2012b). MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med 209, 679–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Blackwell KL, André F, Winer EP, et al. (2016). Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med 375, 1738–1748. [DOI] [PubMed] [Google Scholar]
- Hossain DMS, Javaid S, Cai M, Zhang C, Sawant A, Hinton M, Sathe M, Grein J, Blumenschein W, Pinheiro EM, and Chackerian A (2018). Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J Clin Invest 128, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurvitz SA, Martin M, Press MF, Chan D, Fernandez-Abad M, Petru E, Rostorfer R, Guarneri V, Huang CS, Barriga S, et al. (2020). Potent Cell-Cycle Inhibition and Upregulation of Immune Response with Abemaciclib and Anastrozole in neoMONARCH, Phase II Neoadjuvant Study in HR. Clin Cancer Res 26, 566–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huskey NE, Guo T, Evason KJ, Momcilovic O, Pardo D, Creasman KJ, Judson RL, Blelloch R, Oakes SA, Hebrok M, and Goga A (2015). CDK1 inhibition targets the p53-NOXA-MCL1 axis, selectively kills embryonic stem cells, and prevents teratoma formation. Stem Cell Reports 4, 374–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HC, and Clurman BE (2005). Cyclin E in normal and neoplastic cell cycles. Oncogene 24, 2776–2786. [DOI] [PubMed] [Google Scholar]
- Hydbring P, Bahram F, Su Y, Tronnersjö S, Högstrand K, von der Lehr N, Sharifi HR, Lilischkis R, Hein N, Wu S, et al. (2010). Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci U S A 107, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K, Nambu T, Dairiki R, Ohori M, Yu J, Burke K, Gotou M, Yamamoto Y, Ebara S, Shibata S, et al. (2019). Molecular mechanism and potential target indication of TAK-931, a novel CDC7-selective inhibitor. Sci Adv 5, eaav3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbour-Leung NA, Chen X, Bui T, Jiang Y, Yang D, Vijayaraghavan S, McArthur MJ, Hunt KK, and Keyomarsi K (2016). Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol Cancer Ther 15, 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997.e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, and Gray NS (2019). Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew Chem Int Ed Engl 58, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Ding D, Yan Y, Li H, Wang B, Ma L, Ye Z, Ma T, Wu Q, Rodrigues DN, et al. (2019). Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-κB Activation and PD-L1 Expression. Mol Cell 73, 22–35.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N, Bentley J, Wang LZ, Newell DR, Robson CN, Shapiro GI, and Curtin NJ (2010). Pre-clinical evaluation of cyclin-dependent kinase 2 and 1 inhibition in anti-estrogen-sensitive and resistant breast cancer cells. Br J Cancer 102, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SRD, Harbeck N, Hegg R, Toi M, Martin M, Shao ZM, Zhang QY, Martinez Rodriguez JL, Campone M, Hamilton E, et al. (2020). Abemaciclib Combined With Endocrine Therapy for the Adjuvant Treatment of HR+, HER2−, Node-Positive, High-Risk, Early Breast Cancer (monarchE). J Clin Oncol 38, 3987–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaszczynska I, Geng Y, Iino T, Mizuno S, Choi Y, Kondratiuk I, Silver DP, Wolgemuth DJ, Akashi K, and Sicinski P (2009). Cyclin A is redundant in fibroblasts but essential in hematopoietic and embryonic stem cells. Cell 138, 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanakkanthara A, Jeganathan KB, Limzerwala JF, Baker DJ, Hamada M, Nam HJ, van Deursen WH, Hamada N, Naylor RM, Becker NA, et al. (2016). Cyclin A2 is an RNA binding protein that controls Mre11 mRNA translation. Science 353, 1549–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Sergio CM, Sutherland RL, and Musgrove EA (2014). Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer 14, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsunky H, Geisen C, Schmidt T, Haas K, Zevnik B, Gau E, and Moroy T (1999). Oncogenic potential of cyclin E in T-cell lymphomagenesis in transgenic mice: evidence for cooperation between cyclin E and Ras but not Myc. Oncogene 18, 7816–7824. [DOI] [PubMed] [Google Scholar]
- Klein ME, Kovatcheva M, Davis LE, Tap WD, and Koff A (2018). CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 34, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmann K, Heller G, Schneckenleithner C, Warsch W, Scheicher R, Ott RG, Schäfer M, Fajmann S, Schlederer M, Schiefer AI, et al. (2013). A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 24, 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltun ES, Tsuhako AL, Brown DS, Aay N, Arcalas A, Chan V, Du H, Engst S, Ferguson K, Franzini M, et al. (2012). Discovery of XL413, a potent and selective CDC7 inhibitor. Bioorg Med Chem Lett 22, 3727–3731. [DOI] [PubMed] [Google Scholar]
- Kovatcheva M, Liao W, Klein ME, Robine N, Geiger H, Crago AM, Dickson MA, Tap WD, Singer S, and Koff A (2017). ATRX is a regulator of therapy induced senescence in human cells. Nat Commun 8, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, and Crews CM (2017). Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Lee CE, Oh S, Kim H, Lee J, Kim SB, and Kim HS (2020). Pharmacogenomic Analysis Reveals CCNA2 as a Predictive Biomarker of Sensitivity to Polo-Like Kinase I Inhibitor in Gastric Cancer. Cancers (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JP, LaCasce AS, Smith MR, Noy A, Chirieac LR, Rodig SJ, Yu JQ, Vallabhajosula S, Schoder H, English P, et al. (2012). Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 119, 4597–4607. [DOI] [PubMed] [Google Scholar]
- Li B, Zhu HB, Song GD, Cheng JH, Li CZ, Zhang YZ, and Zhao P (2019). Regulating the CCNB1 gene can affect cell proliferation and apoptosis in pituitary adenomas and activate epithelial-to-mesenchymal transition. Oncol Lett 18, 4651–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhao XL, Shang SQ, Shen HQ, and Chen X (2015). Dual Inhibition of Cdc7 and Cdk9 by PHA-767491 Suppresses Hepatocarcinoma Synergistically with 5-Fluorouracil. Curr Cancer Drug Targets 15, 196–204. [DOI] [PubMed] [Google Scholar]
- Lin ZP, Zhu YL, and Ratner ES (2018). Targeting Cyclin-Dependent Kinases for Treatment of Gynecologic Cancers. Front Oncol 8, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al. (2014). Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llanos S, Megias D, Blanco-Aparicio C, Hernández-Encinas E, Rovira M, Pietrocola F, and Serrano M (2019). Lysosomal trapping of palbociclib and its functional implications. Oncogene 38, 3886–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucenay KS, Doostan I, Karakas C, Bui T, Ding Z, Mills GB, Hunt KK, and Keyomarsi K (2016). Cyclin E Associates with the Lipogenic Enzyme ATP-Citrate Lyase to Enable Malignant Growth of Breast Cancer Cells. Cancer Res 76, 2406–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M (2019). CDK4/6 Inhibitors: What Is the Best Cocktail? Clin Cancer Res 25, 6–8. [DOI] [PubMed] [Google Scholar]
- Malumbres M, and Barbacid M (2001). To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1, 222–231. [DOI] [PubMed] [Google Scholar]
- Malumbres M, and Barbacid M (2009). Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- Martínez-Alonso D, and Malumbres M (2020). Mammalian cell cycle cyclins. Semin Cell Dev Biol. [DOI] [PubMed] [Google Scholar]
- Mashal RD, Lester S, Corless C, Richie JP, Chandra R, Propert KJ, and Dutta A (1996). Expression of cell cycle-regulated proteins in prostate cancer. Cancer Res 56, 4159–4163. [PubMed] [Google Scholar]
- Matsumoto Y, and Maller JL (2004). A centrosomal localization signal in cyclin E required for Cdk2-independent S phase entry. Science 306, 885–888. [DOI] [PubMed] [Google Scholar]
- Mayer EL, Dueck AC, Martin M, Rubovszky G, Burstein HJ, Bellet-Ezquerra M, Miller KD, Zdenkowski N, Winer EP, Pfeiler G, et al. (2021). Palbociclib with adjuvant endocrine therapy in early breast cancer (PALLAS): interim analysis of a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. [DOI] [PubMed] [Google Scholar]
- Mazumder S, Plesca D, and Almasan A (2007). A jekyll and hyde role of cyclin E in the genotoxic stress response: switching from cell cycle control to apoptosis regulation. Cell Cycle 6, 1437–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin RP, He J, van der Noord VE, Redel J, Foekens JA, Martens JWM, Smid M, Zhang Y, and van de Water B (2019). A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res 21, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita MM, Joy AA, Mita A, Sankhala K, Jou YM, Zhang D, Statkevich P, Zhu Y, Yao SL, Small K, et al. (2014). Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer 14, 169–176. [DOI] [PubMed] [Google Scholar]
- Mitri Z, Karakas C, Wei C, Briones B, Simmons H, Ibrahim N, Alvarez R, Murray JL, Keyomarsi K, and Moulder S (2015). A phase 1 study with dose expansion of the CDK inhibitor dinaciclib (SCH 727965) in combination with epirubicin in patients with metastatic triple negative breast cancer. Invest New Drugs 33, 890–894. [DOI] [PubMed] [Google Scholar]
- Moiseeva TN, and Bakkenist CJ (2018). Regulation of the initiation of DNA replication in human cells. DNA Repair (Amst) 72, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar JJ, Ebus ME, Geerts D, Koster J, Lamers F, Valentijn LJ, Westerhout EM, Versteeg R, and Caron HN (2009). Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proc Natl Acad Sci U S A 106, 12968–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A, Moll J, and Colotta F (2010). Targeting cell division cycle 7 kinase: a new approach for cancer therapy. Clin Cancer Res 16, 4503–4508. [DOI] [PubMed] [Google Scholar]
- Montagnoli A, Tenca P, Sola F, Carpani D, Brotherton D, Albanese C, and Santocanale C (2004). Cdc7 inhibition reveals a p53-dependent replication checkpoint that is defective in cancer cells. Cancer Res 64, 7110–7116. [DOI] [PubMed] [Google Scholar]
- Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C, et al. (2008). A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat Chem Biol 4, 357–365. [DOI] [PubMed] [Google Scholar]
- Moore NL, Edwards DP, and Weigel NL (2014). Cyclin A2 and its associated kinase activity are required for optimal induction of progesterone receptor target genes in breast cancer cells. J Steroid Biochem Mol Biol 144 Pt B, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawski PA, Mehra P, Chen C, Bhatti T, and Wells AD (2013). Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J Biol Chem 288, 24494–24502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HJ, and van Deursen JM (2014). Cyclin B2 and p53 control proper timing of centrosome separation. Nat Cell Biol 16, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoni A, Murillo LS, Kliszczak AE, Catherwood MA, Montagnoli A, Samali A, O’Dwyer M, and Santocanale C (2011). Mechanisms of action of a dual Cdc7/Cdk9 kinase inhibitor against quiescent and proliferating CLL cells. Mol Cancer Ther 10, 1624–1634. [DOI] [PubMed] [Google Scholar]
- Nemunaitis JJ, Small KA, Kirschmeier P, Zhang D, Zhu Y, Jou YM, Statkevich P, Yao SL, and Bannerji R (2013). A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J Transl Med 11, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary B, Finn RS, and Turner NC (2016). Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13, 417–430. [DOI] [PubMed] [Google Scholar]
- Olson CM, Liang Y, Leggett A, Park WD, Li L, Mills CE, Elsarrag SZ, Ficarro SB, Zhang T, Düster R, et al. (2019). Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem Biol 26, 792–803.e710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martín A, Dubus P, Sotillo R, Barbero JL, Malumbres M, and Barbacid M (2003). Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 35, 25–31. [DOI] [PubMed] [Google Scholar]
- Otto T, and Sicinski P (2017). Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, et al. (2010). Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther 9, 2344–2353. [DOI] [PubMed] [Google Scholar]
- Patel H, Periyasamy M, Sava GP, Bondke A, Slafer BW, Kroll SHB, Barbazanges M, Starkey R, Ottaviani S, Harrod A, et al. (2018). ICEC0942, an Orally Bioavailable Selective Inhibitor of CDK7 for Cancer Treatment. Mol Cancer Ther 17, 1156–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestell TG, Jiao X, Kumar M, Peck AR, Prisco M, Deng S, Li Z, Ertel A, Casimiro MC, Ju X, et al. (2017). Stromal cyclin D1 promotes heterotypic immune signaling and breast cancer growth. Oncotarget 8, 81754–81775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DC, Zhang N, Danes C, McGahren MJ, Harwell RM, Faruki S, and Keyomarsi K (2001). Tumor-specific proteolytic processing of cyclin E generates hyperactive lower-molecular-weight forms. Mol Cell Biol 21, 6254–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, Guerra C, Santamaria D, and Barbacid M (2010). A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 18, 63–73. [DOI] [PubMed] [Google Scholar]
- Rana S, Bendjennat M, Kour S, King HM, Kizhake S, Zahid M, and Natarajan A (2019). Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg Med Chem Lett 29, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Stoehr J, Dokic D, Wan L, Decker JT, Konopka K, Thomas AL, Wu J, Kaklamani VG, Shea LD, and Jeruss JS (2017). Synergistic effect of eribulin and CDK inhibition for the treatment of triple negative breast cancer. Oncotarget 8, 83925–83939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rape M, Reddy SK, and Kirschner MW (2006). The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell 124, 89–103. [DOI] [PubMed] [Google Scholar]
- Raspé E, Coulonval K, Pita JM, Paternot S, Rothé F, Twyffels L, Brohée S, Craciun L, Larsimont D, Kruys V, et al. (2017). CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol Med 9, 1052–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Acebes S, Proctor I, Loddo M, Wollenschlaeger A, Rashid M, Falzon M, Prevost AT, Sainsbury R, Stoeber K, and Williams GH (2010). Targeting DNA replication before it starts: Cdc7 as a therapeutic target in p53-mutant breast cancers. Am J Pathol 177, 2034–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, Chen CC, Ho YJ, Sanchez-Rivera FJ, Feucht J, et al. (2018). NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362, 1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador-Barbero B, Álvarez-Fernández M, Zapatero-Solana E, El Bakkali A, Menéndez MDC, López-Casas PP, Di Domenico T, Xie T, VanArsdale T, Shields DJ, et al. (2020). CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 37, 340–353.e346. [DOI] [PubMed] [Google Scholar]
- Santala S, Talvensaari-Mattila A, Soini Y, Honkavuori-Toivola M, and Santala M (2014). High expression of cyclin A is associated with poor prognosis in endometrial endometrioid adenocarcinoma. Tumour Biol 35, 5395–5399. [DOI] [PubMed] [Google Scholar]
- Santamaría D, Barrière C, Cerqueira A, Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M, and Barbacid M (2007). Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815. [DOI] [PubMed] [Google Scholar]
- Sava GP, Fan H, Coombes RC, Buluwela L, and Ali S (2020). CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev 39, 805–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa M, and Masai H (2009). Drug design with Cdc7 kinase: a potential novel cancer therapy target. Drug Des Devel Ther 2, 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti M, Eichhorn PJ, Cortés J, Prudkin L, Aura C, Jiménez J, Chandarlapaty S, Serra V, Prat A, Ibrahim YH, et al. (2011). Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci U S A 108, 3761–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer DA, Beckmann RP, Dempsey JA, Huber L, Forest A, Amaladas N, Li Y, Wang YC, Rasmussen ER, Chin D, et al. (2018). The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep 22, 2978–2994. [DOI] [PubMed] [Google Scholar]
- Shapiro GI (2006). Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 24, 1770–1783. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, and Roberts JM (2004). Living with or without cyclins and cyclin-dependent kinases. Genes Dev 18, 2699–2711. [DOI] [PubMed] [Google Scholar]
- Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H, and Sicinski P (2003). Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 4, 451–461. [DOI] [PubMed] [Google Scholar]
- Singer JD, Gurian-West M, Clurman B, and Roberts JM (1999). Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev 13, 2375–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledge GW, Toi M, Neven P, Sohn J, Inoue K, Pivot X, Burdaeva O, Okera M, Masuda N, Kaufman PA, et al. (2017). MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J Clin Oncol 35, 2875–2884. [DOI] [PubMed] [Google Scholar]
- Sonntag R, Giebeler N, Nevzorova YA, Bangen JM, Fahrenkamp D, Lambertz D, Haas U, Hu W, Gassler N, Cubero FJ, et al. (2018). Cyclin E1 and cyclin-dependent kinase 2 are critical for initiation, but not for progression of hepatocellular carcinoma. Proc Natl Acad Sci U S A 115, 9282–9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson JJ, Nemunaitis J, Joy AA, Martin JC, Jou YM, Zhang D, Statkevich P, Yao SL, Zhu Y, Zhou H, et al. (2014). Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer 83, 219–223. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Urano T, Miki Y, Moriya T, Akahira J, Ishida T, Horie K, Inoue S, and Sasano H (2007). Nuclear cyclin B1 in human breast carcinoma as a potent prognostic factor. Cancer Sci 98, 644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney KJ, Swarbrick A, Sutherland RL, and Musgrove EA (1998). Lack of relationship between CDK activity and G1 cyclin expression in breast cancer cells. Oncogene 16, 2865–2878. [DOI] [PubMed] [Google Scholar]
- Tan AR, Wright GS, Thummala AR, Danso MA, Popovic L, Pluard TJ, Han HS, Vojnovic Z, Vasev N, Ma L, et al. (2019). Trilaciclib plus chemotherapy versus chemotherapy alone in patients with metastatic triple-negative breast cancer: a multicentre, randomised, open-label, phase 2 trial. Lancet Oncol 20, 1587–1601. [DOI] [PubMed] [Google Scholar]
- Teixeira LK, and Reed SI (2017). Cyclin E Deregulation and Genomic Instability. Adv Exp Med Biol 1042, 527–547. [DOI] [PubMed] [Google Scholar]
- Teng M, Jiang J, He Z, Kwiatkowski NP, Donovan KA, Mills CE, Victor C, Hatcher JM, Fischer ES, Sorger PK, et al. (2020). Development of CDK2 and CDK5 Dual Degrader TMX-2172. Angew Chem Int Ed Engl 59, 13865–13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O, and McCormick F (2003). Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3, 233–245. [DOI] [PubMed] [Google Scholar]
- Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, DeMichele A, Harbeck N, André F, Bayar MA, et al. (2019). Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J Clin Oncol 37, 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella LA, Yu M, Fuhrmann SR, El-Amine M, Epperson DE, and Finn OJ (2009). Healthy individuals have T-cell and antibody responses to the tumor antigen cyclin B1 that when elicited in mice protect from cancer. Proc Natl Acad Sci U S A 106, 14010–14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, Kim D, Luo F, Mao P, Helvie K, et al. (2020). The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor positive metastatic breast cancer. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Vegna S, Jin H, Benedict B, Lieftink C, Ramirez C, de Oliveira RL, Morris B, Gadiot J, Wang W, et al. (2019). Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 574, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, Gao H, Yang G, Williams JA, Suski JM, et al. (2017). The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 546, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yang T, Xu G, Liu H, Ren C, Xie W, and Wang M (2016). Cyclin-Dependent Kinase 2 Promotes Tumor Proliferation and Induces Radio Resistance in Glioblastoma. Transl Oncol 9, 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Prives C (1995). Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin-dependent kinases. Nature 376, 88–91. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. (2015). CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingate H, Zhang N, McGarhen MJ, Bedrosian I, Harper JW, and Keyomarsi K (2005). The tumor-specific hyperactive forms of cyclin E are resistant to inhibition by p21 and p27. J Biol Chem 280, 15148–15157. [DOI] [PubMed] [Google Scholar]
- Wu T, Yoon H, Xiong Y, Dixon-Clarke SE, Nowak RP, and Fischer ES (2020). Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nat Struct Mol Biol 27, 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam CH, Fung TK, and Poon RY (2002). Cyclin A in cell cycle control and cancer. Cell Mol Life Sci 59, 1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CC, Kato H, Shindo M, and Masai H (2019). Cdc7 activates replication checkpoint by phosphorylating the Chk1-binding domain of Claspin in human cells. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Fang D, Chen H, Lu Y, Dong Z, Ding HF, Jing Q, Su SB, and Huang S (2015). Cyclin-dependent kinase 2 is an ideal target for ovary tumors with elevated cyclin E1 expression. Oncotarget 6, 20801–20812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasmeen A, Berdel WE, Serve H, and Muller-Tidow C (2003). E- and A-type cyclins as markers for cancer diagnosis and prognosis. Expert Rev Mol Diagn 3, 617–633. [DOI] [PubMed] [Google Scholar]
- Yu M, Zhan Q, and Finn OJ (2002). Immune recognition of cyclin B1 as a tumor antigen is a result of its overexpression in human tumors that is caused by non-functional p53. Mol Immunol 38, 981–987. [DOI] [PubMed] [Google Scholar]
- Zhang H, Christensen CL, Dries R, Oser MG, Deng J, Diskin B, Li F, Pan Y, Zhang X, Yin Y, et al. (2020). CDK7 Inhibition Potentiates Genome Instability Triggering Anti-tumor Immunity in Small Cell Lung Cancer. Cancer Cell 37, 37–54.e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, et al. (2018). Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, and Prives C (2001). Cyclin a-CDK phosphorylation regulates MDM2 protein interactions. J Biol Chem 276, 29702–29710. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Geng Y, Zhang Y, Chu C, Sharma S, Fassl A, Butter D, and Sicinski P (2020). The requirement for cyclin E in c-Myc overexpressing breast cancers. Cell Cycle, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]