

Abstract
Poly(ADP-ribose) polymerase 1 (PARP1, also known as ADPRT1) is a multifunctional human ADP-ribosyltransferase. It plays a role in multiple DNA repair pathways, including the base excision repair (BER), non-homologous end joining (NHEJ), homologous recombination (HR), and Okazaki-fragment processing pathways. In response to DNA strand breaks, PARP1 covalently attaches ADP-ribose moieties to arginine, glutamate, aspartate, cysteine, lysine, and serine acceptor sites on both itself and other proteins. This signal recruits DNA repair proteins to the site of DNA damage. PARP1 binding to these sites enhances ADP-ribosylation via allosteric communication between the distant DNA binding and catalytic domains. In this review, we provide a general overview of PARP1 and emphasize novel potential approaches for pharmacological inhibition. Clinical PARP1 inhibitors bind the catalytic pocket, where they directly interfere with ADP-ribosylation. Some inhibitors may further enhance potency by “trapping” PARP1 on DNA via an allosteric mechanism, though this proposed mode of action remains controversial. PARP1 inhibitors are used clinically to treat some cancers, but resistance is common, so novel pharmacological approaches are urgently needed. One approach may be to design novel small molecules that bind at inter-domain interfaces that are essential for PARP1 allostery. To illustrate these points, this review also includes instructive videos showing PARP1 structures and mechanisms.
Keywords: PARP1, DNA repair, cancer, chemotherapy, drug resistance, review
Graphical Abstract

1. Introduction
Poly(ADP-ribose) polymerases (PARPs)a comprise a family of ADP-ribosyltransferases found in all six major eukaryotic supergroups, as well as several species of prokaryotes and dsDNA virus [1]. PARP proteins transfer ADP-ribose moieties to arginine, glutamate, aspartate, cysteine, lysine, and serine acceptor sites [2–6].
Among the 17 PARP proteins in humans, poly(ADP-ribose) polymerase 1 (PARP1, also known as ADPRT1) was the first identified [7, 8]. PARP1 is a multifunctional protein that initiates DNA repair [9, 10]. Given its biological importance and potential as a cancer drug target, PARP1 has been the subject of intense study (>1,200 publications since 2015 according to PubMed). This review aims to provide a general overview of PARP1, with appropriate references to domain-specific articles for those who wish to learn more. We also include helpful videos [11] illustrating PARP1 structures and mechanisms that others can use in their own presentations and classroom lectures.
We will first provide a broad overview of four DNA repair pathways relevant to PARP1: the base excision repair (BER), non-homologous end joining (NHEJ), homologous recombination (HR), and Okazaki-fragment processing pathways. We will then describe PARP1’s role in recruiting repair pathways to DNA strand breaks, as well as the orthosteric/allosteric mechanisms that ultimately enable DNA repair. Next, we will review the relevant pharmacology, including PARP1’s role in HR-deficient cancer, clinically approved small-molecule PARP1 inhibitors (PARPi), and known PARPi resistance mechanisms. Finally, we will discuss the future of PARP1 inhibition.
2. PARP1 and DNA Damage Repair
At least four DNA repair pathways are influenced, to one degree or another, by PARP1 activity. Collectively, pathways such as these constitute the DNA damage response (DDR) [12–14]. DNA integrity is continuously threatened by environmental agents, reactive oxygen species, UV radiation, and replication errors [14, 15], which introduce base modifications, abasic sites, double strand DNA breaks (DSBs), and single strand DNA breaks (SSBs). During the process of normal DNA replication, unligated Okazaki fragments on the lagging strand also contain transient SSBs [16]. If unrepaired, this damage can result in cancer-causing mutations, cell death, or cellular senescence. Life has evolved multiple strategies for identifying different types of DNA damage, as required to repair or terminate the cell [12, 13]. In humans, prominent DDR mechanisms include the BER, NHEJ, HR, and Okazaki-fragment processing pathways.
2.1. Base Excision Repair (BER)
PARP1 recruits BER repair proteins to DNA strand breaks undergoing processing [17] and so plays a major role in the BER pathway. The BER mechanism repairs many types of DNA lesions, including abasic sites, SSBs, and base alterations caused by oxidation, alkylation, or deamination [17–19]. Unless corrected, these lesions can stall DNA replication and transcription [20], cause genomic instability, progress into DSBs [21], and result in cell death [20]. As its name implies, BER excises small regions of DNA damage, thereby creating an SSB intermediate [19]. Subsequent proteins in the BER pathway protect this intermediate from progressing into a DSB and ensure that the damage is repaired [18, 19].
Several variations of the BER pathway share key proteins in common. All follow five general steps [18]: (1) a damage-specific glycosylase detects and excises the damaged DNA base, producing an apurinic/apyrimidinic (AP), or abasic, site; (2) incise the AP site (creating a SSB) using an AP endonuclease or a glycosylase-associated AP lyase; (3) repair the terminal ends of the sugar backbone using a lyase or a phosphodiesterase; (4) replace the missing region with complementary DNA using a DNA polymerase; and (5) ligate the DNA to remove nicks using a DNA ligase. A more detailed review of the BER pathway can be found in refs. [18] and [22].
2.2. Non-Homologous End Joining (NHEJ)
PARP1 binding and activation also impact NHEJ, which repairs DSBs by direct ligation [23]. Though this repair mechanism preserves DNA integrity generally, NHEJ is error prone because the rejoined ends often share little to no homology. The lack of an effective guide results in lost DNA bases at the joining site [23].
The role of PARP1 in the canonical NHEJ (c-NHEJ) sub-pathway is surprisingly uncertain. c-NHEJ is activated when Ku70 and Ku80 (Ku proteins) bind to sites of DSBs [21]. PARP1 may play a role in recruiting proteins such as these to the locations of DNA strand breaks; PARP1 covalently attaches adenosine diphosphate ribose (ADP-ribose) chains to DNA repair proteins via poly-ADP-ribosylation (PARylation, see below), and Ku proteins and are known to bind these chains [21, 23, 24]. On the other hand, PARP1 and Ku compete with each other for DSB binding, so PARP1 may also inhibit c-NHEJ [25].
The role of PARP1 in the more recently discovered alternative NHEJ (alt-NHEJ) sub-pathway is less ambiguous: its binding and activation stimulates alt-NHEJ [23]. Given that Ku proteins have stronger affinities for DSB sites, PARP1-initiated alt-NHEJ likely serves as a backup to Ku-initiated c-NHEJ [25]. Indeed, this sub-pathway has been called “backup NHEJ,” as well as microhomology mediated end joining (MMEJ) [23]. More detailed reviews of NHEJ are provided in refs. [26] and [27].
2.3. Homologous Recombination (HR)
PARP1 does not directly stimulate the HR pathway, but cells that are HR deficient tend to be hypersensitive to PARP1 inhibition [28–31]. HR uses homologous DNA as a template for DSB repair [21, 26]. Unlike NHEJ, which remains active throughout the cell cycle, HR activity is mostly limited to the S and G2 phases [13, 26, 32]. Additionally, HR repair is less error prone than NHEJ [21, 26] and is therefore critically important for genome stability and repair fidelity, particularly during DNA replication [32].
PARP1 inhibitors have been successfully used to treat patients with HR-deficient cancers [28–31]. Many cancer cells have deficiencies in HR-pathway proteins [13, 32–34], especially the breast-cancer tumor-suppressor proteins BRCA1 and BRCA2 [13, 32]. These proteins are involved in transcription regulation [33], end resection [32, 34], filament promotion [32, 34], DNA-strand protection at stalled forks [32], cell-cycle checkpoints [33], and cellular apoptosis [33]. HR-deficient cells must rely more heavily on other repair pathways such as BER or NHEJ to avoid genomic instability [13, 34, 35]. Therefore, HR-deficient cancers are less able to survive further loss of non-HR repair pathways (e.g., due to PARP1 inhibition) than are HR-proficient cells [13, 34, 35]. More detailed reviews of HR repair and BRCA proteins can be found in refs. [32] and [36].
2.4. Okazaki-Fragment Processing
During DNA replication, the replisome machinery unwinds DNA, separating the two complementary strands at the so-called replication fork [37]. On the leading strand, replication occurs continuously in the 5’ to 3’ direction, towards the progressing fork. But the complementary (lagging) strand runs in the 3’ to 5’ direction, so replication must proceed discontinuously in short segments with RNA primers in the direction opposite the fork. These short segments, called Okazaki fragments [38, 39], must be processed, and the resulting SSBs must then be ligated.
Recent work has found that PARP1 plays a critical role in detecting these SSBs, which occur appropriately during DNA replication rather than as a result of unexpected damage. Indeed, most PARP activity occurs at sites of DNA replication during S phase, where PARP1 likely serves to recruit SSBs repair proteins such as XRCC1 [16].
3. PARP1 Recruits DNA Repair Proteins via ADP-Ribosylation
In this section, we describe how PARP1-mediated ADP-ribosylation recruits DNA repair proteins to strand breaks. We discuss ADP-ribosylation generally, as well as PARP1’s role in ADP-ribose-mediated signaling.
3.1. ADP-Ribosylation, a Form of DNA Repair Signaling
Nicotinamide adenine dinucleotide (NAD+) is an organic small molecule common to all living organisms [2]. It is composed of an adenosine 5’-monophosphate (ADP) and a nicotinamide mononucleotide that are covalently connected by a phosphodiester bond (Figure S1) [2, 40]. Among its many biological roles, NAD+ can be catabolized to ADP-ribose and nicotinamide via cleavage of the N-glycosidic bond (Figure S1) [2].
ADP-ribosyltransferases catalyze this reaction and attach the resulting ADP-ribose molecule(s) to acceptor protein(s) [2]. Both mono-ADP-ribosylation (MARylation) and poly-ADP-ribosylation (PARylation) are possible [2]; the former involves a single ADP-ribose addition, and the latter produces both linear and branching poly(ADP-ribose) (PAR) chains (Figure S1B) [2]. In contrast, proteins such as PAR glycohydrolase and PAR hydrolase [5, 34] rapidly degrade these chains. Given that ADP-ribosylation consumes both ATP and NAD+, a proper balance between PAR-chain synthesis and degradation is critical. Both hyperactivation of PARP1 (leading to increased ADP-ribosylation) and inhibition of PAR glycohydrolase (leading to impaired PAR-chain degradation) can cause ATP and NAD+ depletion, leading to a form of metabolic death known as parthanatos [24, 34, 41, 42].
PAR chains act as scaffolds that recruit DNA repair proteins to the locations of DNA strand breaks [21, 43]. Many DNA repair proteins contain a PAR-binding motif that interacts non-covalently with PAR-chains, including p53, DNA ligase III, XRCC1, DNA-PK(CS), and Ku70 [44]. PAR-binding motifs are typically 20 amino acids long and consist of two conserved regions: a region dense in basic amino acids, and a region consisting of both hydrophobic and basic amino acids [44]. The location of PAR-binding motifs coincides with five different functional domains that are involved in nuclear localization, protein degradation, nuclear export, protein-protein interactions, and DNA binding [44]. Several other structures also facilitate protein-PAR noncovalent interactions, including the PAR-binding zinc fingers found in the NHEJ protein aprataxin and PNK-like factor (APLF) [2, 45], tryptophan and glutamic-acid rich WWE domains found in E3 ligases [2, 46], and macro domains found in PAR glycohydrolase [2, 47, 48].
3.2. PARP1 is Both an ADP-Ribosyltransferase and an ADP-Ribose Acceptor
Multiple members of the PARP family, including PARP1, act both as ADP-ribosyltransferases and PARylation acceptors [2]. Specifically, PARP1 ADP-ribosylates (1) itself as a monomer (in cis modification) [49]; (2) other PARP1 molecules, both as symmetric and asymmetric homodimers (in trans modification) [49]; and (3) other proteins [49] such as XRCC1, XRCC5, XRCC6, DNA topoisomerase 2-alpha (TOP2), replication protein A1 (RPA1), and RPA2 [50].
The primary acceptor of PARP1-mediated PARylation is PARP1 itself [5, 51]. There are at least 102 potential PAR-acceptor sites on PARP1 [3, 52–58] available for autoPARylation (Figure S2) [2, 13, 40, 49]. The PARP1 automodification domain (AD) houses many of the most well-characterized autoPARylation sites [3, 5, 51–58]. This domain includes the BRCA C-terminus (BRCT) subdomain, which interacts with other proteins (e.g., the BER protein XRCC1 [59, 60]) but is not essential for DNA binding or catalytic activity (Figure 1, Video S1) [40, 51]. Aside from serving as a scaffold for recruiting DNA repair proteins to strand breaks [21, 43], negatively charged PAR chains attached to PARP1 may also help PARP1 dissociate from negatively charged DNA [34], allowing recruited DNA repair-proteins to access the damage site [61].
Figure 1. PARP1 domains and subdomains.

The protein schematic (above) was generated with the help of PROSITE [159]. The protein structure (below) shows PARP1 bound to DNA (PDB: 4DQY). The zinc finger 2 (Zn2) and BRCA C-terminus (BRCT) subdomains are not shown, as they are not present in the crystal structure. See Video S1 for an additional review of these PARP1 domains.
4. PARP1 Allosteric and Catalytic Mechanisms
In this section, we will discuss how PARP1 binds to DNA strand breaks, how that binding allosterically activates the catalytic mechanism, and how PARP1 eventually dissociates from the bound DNA. Although we here focus on the PARP1 response to DNA damage, we note that the protein plays other cellular roles as well. Interested readers may wish to consult refs. [62, 63] to learn more about PARP1 and inflammation, as well as refs. [63, 64] to learn more about PARP1 ADP-ribosylation of chromatin.
4.1. Recognizing and Binding to DNA Strand Breaks
The PARP1 N-terminal DNA binding domain (DBD) includes three DNA binding zinc fingers that bind DNA strand breaks: zinc finger 1 (Zn1), zinc finger 2 (Zn2), and zinc finger 3 (Zn3) (Figure 1, Video S1) [51, 64–66]. Though most zinc fingers recognize specific DNA sequences, the PARP1 zinc fingers are unusual in that they recognize DNA structures [5, 67]. Zn1 and Zn2 identify many types of DNA lesions with high affinity and are required for DNA binding [2, 51, 67, 68]. Without Zn1, Zn2, and Zn3, a SSB cannot trigger PARP1 DNA-dependent catalytic activation [68–71]. Similarly, without Zn1 and Zn3, a DSB cannot trigger activation [2, 51, 66–68]. Simultaneous deletion of Zn1 and Zn2 reduces PARP1 binding affinity by over 250-fold, and a fragment containing only Zn1 and Zn2 binds to DNA with nearly the same affinity as wild-type PARP1 [67]. Isolated Zn1 fragments have much weaker binding affinities for DNA than do isolated Zn2 [67]. But Zn2 deletion does not impact the DNA binding affinity of PARP1, and Zn1 deletion results in an approximately three-fold reduction in binding affinity [67]. Despite that fact that Zn2 has a much stronger affinity for DNA, Zn1 combined with the C-terminal region of the DBD can apparently compensate for the loss of Zn2 [67].
Zn3, which has a different structure and function than Zn1 and Zn2 [51, 67, 72], makes crucial inter-domain contacts that are required for PARP1 activation (Figure 1, Video S1) [51, 67]. Similarly, the tryptophan-glycine-arginine-rich (WGR) domain also interacts with DNA and contributes to important inter-domain contacts (Figure 1, Video S1), as described below.
4.2. Allosteric Communication Between the DNA Binding and Catalytic Domains
DNA binding induces cascading conformational changes that ultimately initiate PARP1-mediated ADP-ribosylation [64, 73]. PARP1 catalytic activity is largely DNA dependent. When not bound to DNA, PARP1 has only a low basal level of DNA-independent catalytic activity. In contrast, PARP1 activity is much elevated when it binds DNA [5, 51]. The minimum PARP1 construct with near-wild-type DNA-dependent activity (i.e., activity when bound to DNA) contains the Zn1, Zn3, WGR, and catalytic (CAT) (sub)domains [51].
DNA binding to Zn1, Zn3, and the WGR domain induces conformational changes in the helical domain (HD) and the ADP-ribosyl transferase (ART) subdomain of the CAT domain (Figure 1, Video S1) by way of a network of inter-domain interactions [65, 66, 73]. Several key inter-domain interfaces mediate this allosteric signal [40, 51, 74]: (1) the Zn1-Zn3 interface (Figure 2A and B); (2) the Zn1-WGR- HD interface (Figure 2A and C); (3) the HD-WGR-Zn3 interface (Figure 2A and D); and (4) two HD-ART interfaces (Figure 3A and B). See Videos S2 and S3 for an additional view of these critical interfaces.
Figure 2. Interdomain interfaces.
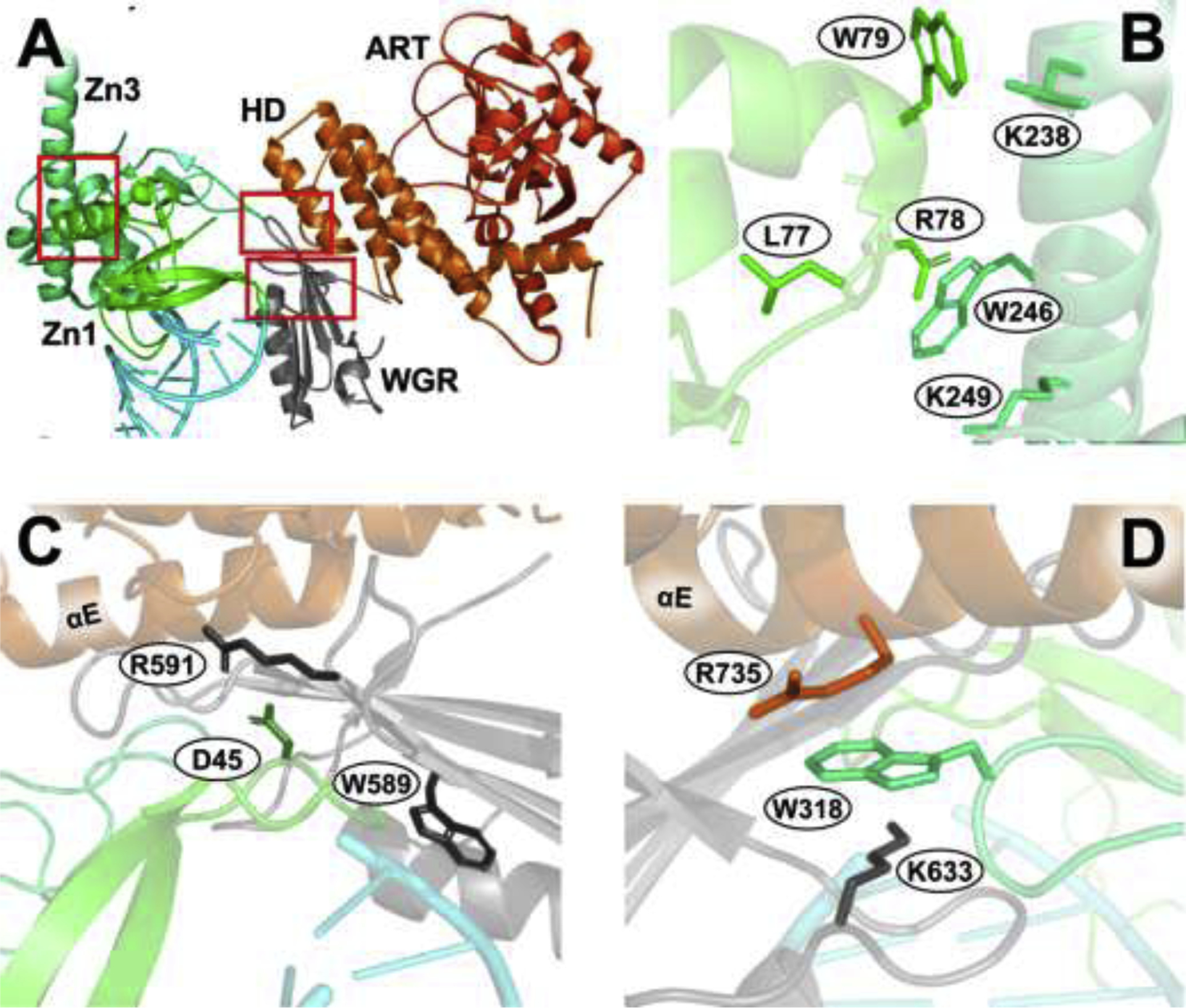
Select residues are shown as sticks. (A) PARP1 (PDB: 4DQY) with red boxes encompassing three critical interdomain interfaces. (B) The Zn1-Zn3 interface. K97 (Zn1) is omitted, as it was not resolved. (C) The Zn1-WGR-HD interface. (D) The HD-WGR-Zn3 interface. See ref. [51] for more details, and Video S2 for an additional review of these PARP1 interfaces.
Figure 3. Critical CAT interfaces (4R6E:A).
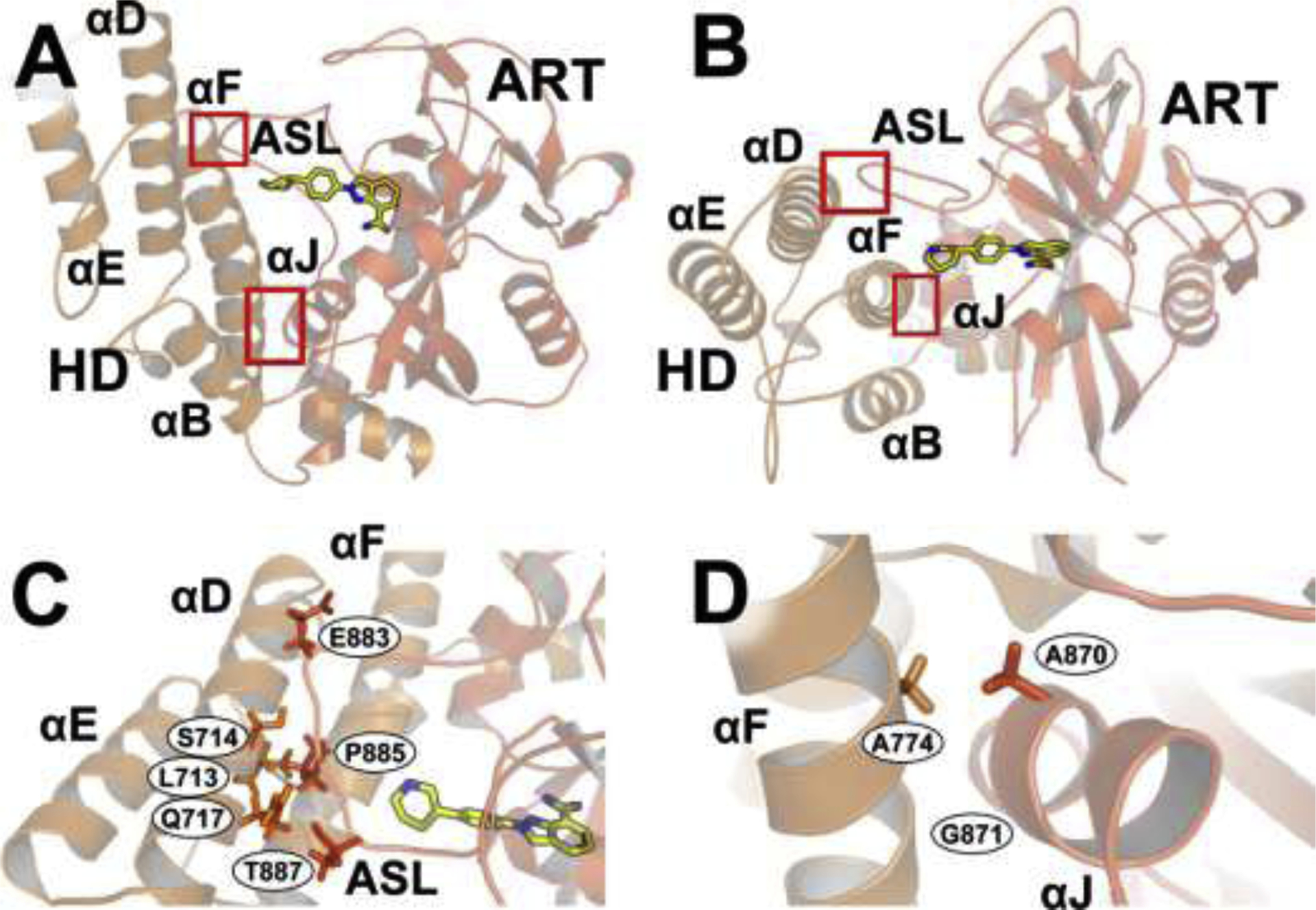
Select residues are shown as sticks. (A) and (B) Two views of the interfaces between the HD (light orange) and ART subdomain (brownish orange), marked with red boxes. The PARPi niraparib (yellow sticks) binds the NI site. (C) The interface between the HD αD helix and the ART ASL. (D) The interface between the HD αF helix and the ART αJ helix. Figure adapted with rights and permissions from ref. [74]. See Video S3 for an additional review of these interfaces.
4.2.1. Zn1-Zn3 Interface
The Zn1-Zn3 interface forms when DNA binds Zn1, thereby exposing a Zn1 surface that subsequently binds Zn3 [49, 75]. This interface, which primarily consists of the Zn1 C-terminal helix and the Zn3 N-terminal helix (Figure 2A and B) [51], is close to but not in direct contact with the bound DNA (per the 4DQY crystal structure [51]). Mutagenesis studies suggest that the Zn1-Zn3 interface is critical for the allosteric communication that activates DNA-dependent catalytic activity, but that the interface does not influence DNA binding itself [51, 66, 72, 75]. Mutations that disrupt major contacts between Zn1 and Zn3, such as R78 (Zn1) and W246 (Zn3), W79 (Zn1) and K238 (Zn3), etc. [72], decrease catalytic activity without disrupting DNA binding (Figure 2B) [51, 66, 72, 75]. For example, the conservative mutation K97R (Zn1) and the non-conservative mutations L77P (Zn1) and K249E (Zn3) still bind DNA [76], but with PARP1 catalytic activity <0.5% that of wild-type. Similarly, the R78A (Zn1) and W79A (Zn1) mutations result in markedly decreased catalytic activity, but bind DNA nearly as well as the wild-type [66]. And the W246A (Zn3) mutant has virtually no catalytic activity [51, 75], likely because it cannot form the DNA-dependent Zn1-Zn3 interaction [75].
4.2.2. Zn1-WGR-HD Interface
The Zn1-WGR-HD interface (Figure 2A and C) also regulates DNA-dependent PARP1 catalytic activity [51, 67] through contacts with DNA, Zn1, and the CAT-domain HD αE helix (Figure 2C) [51]. The WGR domain interacts with DNA via the W589 (WGR) residue, which stacks with the ribose sugars of the DNA 5’ strand [51]. Visual inspection suggests that the Zn1 interacts with the WGR domain via an electrostatic interaction between D45 (Zn1) and R591 (WGR). The WGR in turn interacts with the HD αE helix in large part via non-specific hydrophobic interactions involving I562, V560, L641, and Y639 on the WGR, and L739 on the αE. As expected, mutations that disrupt these inter-domain interactions impact catalytic activity. The R591A (WGR) mutation disrupts PARP1 autoPARylation, as do the D45A (Zn1) and Q40A (Zn1) mutations [51, 66, 67].
4.2.3. HD-WGR-Zn3 Interface
The HD-WGR-Zn3 interface also plays an important role in DNA-dependent catalytic activation and inter-domain communication. Aside from its interactions with the WGR domain (mentioned above), the HD αE helix also interacts with the extended loop of the Zn3 zinc-ribbon fold. The W318 (Zn3) residue, located on a loop that interacts with the R735 (HD) and K633 (WGR) residues, is at the center of this interface (Figure 2A and D) [51]. W318R and W318A mutations reduce PARP1 PAR synthesis without impairing localization to DNA strand breaks [51, 66, 74], likely by disrupting inter-domain allosteric communication over the interface [51, 74]. Similarly, the R735A (HD) and K633A (WGR) mutations also reduce PARylation activity [51, 66].
4.2.4. HD-ART Interfaces
Within the CAT domain, two critical interfaces between the HD and the ART subdomain profoundly impact PARP1 catalytic activity [74]: the αD-active site loop (ASL) interface and the αF-αJ interface (Figure 3). In the absence of DNA, the αD-ASL interface anchors the HD and the ART subdomain together [74, 77], and the fully folded HD inhibits catalytic activity by blocking NAD+ access to the catalytic pocket—so much so that PARP1 constructs lacking the HD have DNA-independent activity equal to that of the DNA-bound wild-type protein [74]. When PARP1 binds to a DNA break, the specific assembly of regulatory domains (Zn1, Zn3 and WGR) creates a network of new interactions with the CAT domain that are mediated/propagated through the HD [74]. This allosteric signal causes select HD helical regions (αB, αD, and the C-terminal of αF) to unfold [74], disrupting the otherwise auto-inhibitory influence of the HD on the ART αJ (Figure 3A, B, and D; Video S3) [74]. The disruption opens the catalytic pocket and allows NAD+ to bind [74].
Mutations that disrupt the αF-αJ interface (e.g., A774L and A774S on αF; A870L, A870S, A871L, and A871S on αJ; etc.) partially decouple PARP1 catalytic activity from DNA binding. These mutations result in 3- to 10-fold increases in PARP1 DNA-independent activity (i.e., PARP1 catalytic activity when not bound to DNA), compared to the wild-type [74]. Some mutations that disrupt the αD-ASL interface (e.g., S714A and Q717A on the αD helix; P882G, P885S, P885G, and the quadrupole-substitution mutant E883A/A884S/P885S/V887A on the ASL) reduce DNA-dependent activity, with minimal impact on DNA binding and DNA-independent (basal) activity [74]. Mutations at L713 (L713F/L713A; αD) are notable exceptions that increase DNA-independent activity [51, 74, 78].
4.3. PARP1 Catabolizes NAD+ to Perform ADP-Ribosylation
Having discussed how PARP1 binds DNA and the allosteric communication that links DNA binding to catalysis, we now describe the catalytic mechanism itself. The highly conserved PARP1 CAT domain is located on the C-terminal region of the protein and includes the ART subdomain. This subdomain contains the enzymatic pocket, which binds NAD+ and catalyzes ADP-ribosylation (Figure 1, Video S1) [65, 66]. The catalytic pocket also binds all clinically approved PARP1 inhibitors (Figure 1) [65], and “PARP1 inhibition” is most often measured in terms of PARP1 catalytic activity [65].
4.3.1. PARP1 Catalytic Mechanism
PARP1 ADP-ribosylation begins once NAD+ binds the catalytic pocket (Video S4) [40, 49]. The pocket houses a highly conserved catalytic triad consisting of H862, Y896, and E988 (Figure 4A), which are required for catalytic activity [49]. NAD+ interactions with these residues are well characterized because the CAT domain has been co-crystallized with benzamide adenine dinucleotide (BAD), a non-hydrolyzable NAD+ analogue (PDB: 6BHV; Figure 4A [77]). H862 forms a hydrogen bond with the NAD+ 2’ adenine-ribose hydroxyl group [49, 79–81]; Y896 and Y907 interact with the nicotinamide moiety via π-π stacking and hydrophobic interactions, respectively (Figure 4A) [79]; and E988 forms a hydrogen bond with the 2’-hydroxyl group of the nicotinamide ribose, which orients NAD+ as required for nucleophilic attack (Figure 4A) [79]. This attack both catabolizes NAD+ and covalently attaches the resulting ADP-ribose to the acceptor protein or growing PAR chain [79].
Figure 4. . Catalytic binding site.

(A) BAD (sticks), an NAD+ analog, bound to the catalytic pocket (PDB: 6BHV:A). The catalytic triad (purple) binds to and catabolizes NAD+. Y907 and the catalytic-triad residue Y896 position the benzamide moiety via π-π stacking interactions. NAD+ likely binds the catalytic pocket in a similar orientation. (B) Niraparib (cyan sticks) bound to the catalytic pocket (PDB: 4R6E:A). Niraparib forms contacts with both the HD (light orange) and the ART (reddish orange). Select residues that interact with niraparib are shown as colored sticks.
The literature often divides the catalytic pocket into two subsites (Video S4). The first, called the donor site, includes three subregions that bind to and position the NAD+ “donor” molecule [79]: the nicotinamide-binding (NI), adenine-ribose-binding (ARB), and phosphate-binding (PH) sites. The NI site stabilizes the NAD+ nicotinamide moiety and is defined by the catalytic triad residues Y896 and E988, as well as Y907 [79]. The ARB site stabilizes the NAD+ adenine-ribose moiety and is defined by the catalytic triad residue H862, as well as D770, S864, G876, and R878 [79]. Finally, the PH site, which consists of E763 and D766, stabilizes the NAD+ pyrophosphate group [79]. The majority of all PARP1 inhibitors bind to PARP1’s donor site (e.g., as in Figure 4B) [49, 82].
The second catalytic-pocket subsite, called the acceptor site, binds and positions the pyrophosphate group of the “acceptor” amino acid or PAR chain to be ADP-ribosylated (Video S4) [79]. The acceptor site consists of H826, M890, K903, L985, and Y986 [49, 79] and is thought to influence the branching properties of the PAR chains [49, 82].
4.3.2. Influence of HPF1 on PARP1 Catalysis
Recent work has identified histone PARylation factor 1 (HPF1) as a critical PARP1 binding partner that impacts the PARP1 catalytic mechanism [83]. Histones are among the primary targets for PARylation by PARP1 [83, 84]. The PARP1 C-terminal domains (WGR /CAT) bind to histone H4 in the nucleosome, leading to long-term ADP-ribosylation that “loosens” the chromatin [64]. This loosening improves transcription-machinery access to DNA and enables DNA-damage repair and DNA replication [64].
HPF1 plays an essential role in facilitating PARP1 ADP-ribosylation of histones [83, 84]. Like PARP1, HPF1 is recruited to sites of DNA damage. This recruitment requires the PARP1 protein itself but not PARP1 enzymatic activation, suggesting that HPF1 recruitment is independent of PARylation [85, 86]. When the PARP1 ART subdomain is in an activated state (i.e., the otherwise auto-inhibitory HD subdomain has locally unfolded and NAD+ is bound; see Section 4.2.4), the HPF1 C-terminal region binds to the PARP1 CAT/ART subdomain [84–86]. In this bound state, a portion of HPF1 occupies part of the PARP1 active site, contributing key residues to a new composite binding pocket [84].
The newly formed composite pocket differs from the original PARP1-only site in three fundamental ways. First, though PARP1 normally ADP-ribosylates many amino acids (e.g., aspartate, glutamate), the composite site acts predominantly on serine [83, 85, 87, 88]. This switch in specificity is important because recent evidence suggests PARylation in the context of DNA damage occurs primarily on serine residues [84, 89]. Crystal structures show that HPF1—not PARP1—contributes the active-site catalytic base (HPF1 E284) that deprotonates serine hydroxyl groups [83, 85], thus enabling the subsequent nucleophilic attack required for ADP-ribosylation [84].
Second, the electrostatics of the composite active site may promote histone association [84]. Like histones, the PARP1-only site and surrounding surfaces are positively charged. But the PARP1/HPF1 composite site has an overall negative charge that is arguably more compatible with histone binding [84].
Third, HPF1 binding to PARP1 limits the autoPARylation typical of PARP1 activity [84, 85]. Bound HPF1 occupies roughly the same position relative to PARP1 that would otherwise be occupied by the PARP1 AD domain. The AD domain includes many PARP1 automodification sites, so HPF1 likely limits hyper-automodification by displacing the AD domain from its location near the catalytic site [84, 86]. Reduced autoPARylation may further encourage PARP1 association with DNA and, indirectly, DNA-associated histones, because PAR chains are negatively charged and thus promote PARP1 dissociation from negatively charged DNA [34, 64]. The pharmacological impacts of the HPF1/PARP1 interaction are discussed further in Section 6.2.
4.4. Release from DNA is Regulated by Interdomain Interactions
PARP1 allostery has historically been discussed in terms of the influence that DNA binding has on catalytic activity [29, 51, 65], but recent research has also examined the influence of the PARP1 CAT domain on DNA unbinding [77, 90]. In this section, we discuss PARP1/DNA dissociation and how PARP1 inhibitors might act by interfering with the dissociation mechanism.
Some catalytic-pocket ligands trigger an allosteric mechanism that may immobilize PARP1 on DNA [77, 90], a phenomenon called “trapping” [77, 91–96]. It has been proposed that trapping PARP1 on DNA prevents DNA repair proteins from accessing strand breaks, stalls and/or collapses replication forks, and results in cell-cycle arrest or apoptosis [91]. However, more recent experiments using quantitative live-cell imaging and fluorescence recovery after photo-bleaching suggest that PARP1 molecules in fact undergo rapid exchange at the locations of DNA strand breaks [97], consistent with observations that single PARP1 molecules have increased constrained motion (i.e., DNA sliding) when bound to the inhibitor olaparib [92]. The role that trapping plays in the mechanism of current PARP1 inhibitors thus remains controversial; indeed, among PARP1 inhibitors used clinically, overall toxicity does not correlate clearly with the degree of presumed trapping [98].
4.4.1. Catalytic-Pocket Ligands may Trigger Trapping via Allostery
Some have claimed that different catalytic-pocket ligands impact “trapping” to varying degrees [77, 94, 95]. The degree of chromatin retention appears to be related to each ligand’s ability to (de)stabilize the HD [90]. Those that destabilize the HD (e.g., the NAD+ analog BAD) allosterically strengthen PARP1’s affinity for DNA and consequently have strong trapping capacity [77, 90]. In contrast, those that stabilize the αB and αF helices of the HD weaken DNA binding affinity and release PARP1 from DNA [90].
For example, the catalytic-pocket ligand EB-47 forms contacts with D766 and D770, two residues located on the HD αF helix [90]. These contacts destabilize the helix, possibly leading to PARP1 “trapping” on DNA [90]. But the D766A/D770A double mutant disrupts the contacts, thereby preventing trapping [90]. In contrast, the PARP1 inhibitor veliparib does not form contacts with the HD when bound to the catalytic pocket (Figure 5A) and so is apparently less effective at trapping wild-type PARP1. But UKTT-15, a veliparib-derived inhibitor that does form contacts with the N-terminal portion of the HD αF helix (Figure 5B; Video S3), is more effective at DNA trapping and more toxic to BRCA-deficient cells, even though veliparib and UKTT-15 inhibit PARylation to similar degrees [90].
Figure 5. Differences in veliparib and UKTT-15 binding to the CAT domain.

(A) Veliparib (cyan, 2RD6:A) does not interact with the HD. (B) UKTT-15, a veliparib analog (green, 6VKO:A), displaces the HD αF helix. See Video S3 for further discussion.
Small-molecule (de)stabilization of the HD triggers allosteric inter-domain communication between the CAT domain and the DBD [90], through the HD-WGR-Zn3 and Zn1-WGR-HD interfaces. The W318R (Zn3) and R591C (WGR) mutations, which disrupt the HD-WGR-Zn3 and Zn1-WGR-HD interfaces, respectively, appear to prevent EB-47 trapping activity. Mutations such as these likely have clinical significance. The R591C mutation was recently identified in a patient with ovarian cancer who had developed resistance to the PARP1 inhibitor talazoparib [90, 99].
4.4.2. PARPi Classification System
A recently developed PARPi classification system groups PARP1 inhibitors into three types based on their allosteric effects [90]. This system is based on the hypothesis that some inhibitors can promote PARP1 retention on DNA. Type I inhibitors are those that allegedly promote PARP1 trapping through allosteric regulation, resulting in long DNA-retention times and high toxicity [90]. BAD, UKTT-15, and EB-47 are good examples of this class because they apparently promote PARP1 “trapping” via contacts that disrupt the HD (Figure 5B; Video S3) [90]. Type II inhibitors are neither pro-retention nor pro-release (i.e., relatively allosterically neutral), resulting in moderate DNA-retention times and toxicity [90]. Examples include talazoparib and olaparib, which successfully inhibit PARylation without much impact on DNA trapping [90]. Finally, type III inhibitors promote PARP1 release through allosteric mechanisms, resulting in short DNA-retention times and lower toxicity [90]. Rucaparib, niraparib, and veliparib are good example of type III inhibitors. They stabilize the αB and αF helices of the HD while simultaneously promoting increased αC/αD-helix dynamics [90]. This influence ultimately increases PARP1 dissociation from DNA [90]. Aside from these allosteric considerations, other factors may also contribute to PARP1 trapping in cells [90, 100, 101], including potency of inhibition, PARP-1 affinity, and compound half-life, though there is controversy [96].
5. PARP1 in Cancer and Cancer Treatment
In this section, we will (1) discuss the prevalence and available treatments for patients with HR-deficient cancers; (2) describe the mechanisms that make PARP1 drug inhibition lethal to HR-deficient cancers; (3) overview current clinically approved PARP1 inhibitors; (4) document common PARPi-resistance mechanisms; and (5) describe the shortcomings of current pharmaceutical options as well as strategies for improvement.
5.1. HR-Deficient Cancers: Prevalence and Standard-of-Care Management
Breast, ovarian, prostate, and pancreatic cancers are frequently HR deficient [102–105], often due to mutations in the BRCA1/2 genes [102–104, 106], which encode breast-cancer tumor-suppressor proteins that play critical roles in HR DNA repair. Since the 1970s, surgical cytoreduction together with chemotherapy have been the primary treatments for HR-deficient cancers. But traditional chemotherapies do not leverage the specific biology of HR deficiency to achieve a more targeted approach.
More recent drugs that inhibit PARP1 are effective against many types of cancer [21, 28–31, 42, 93, 107]. These inhibitors tend to be more toxic to HR-deficient cancer cells than to non-cancerous cells and so have the potential for fewer side effects. Several studies have shown that PARP1 inhibitors are effective as monotherapies (i.e., standalone treatments) and co-treatments alongside other chemotherapeutics (i.e., combination therapies) [21, 28–31, 42, 93, 107].
5.2. HR-Deficient Cells and PARPi Sensitivity: Synthetic Lethality
PARP1 inhibition is an excellent strategy for treating multiple cancers, in part because PARP1 plays well-documented roles in multiple DNA repair pathways [28–31]. PARP1 inhibitors are particularly effective against HR-deficient cancer cells because the HR pathway has a synthetically lethal relationship with PARP1 (i.e., a cell can survive a deficiency in HR repair or PARP1 function, but not in both, due to some functional redundancy) [21, 35, 61, 107, 108]. PARP1 inhibition is in fact the first successful example of a synthetic-lethal approach in pharmaceutical treatment [61].
HR-deficient cancer cells lack an important DSB repair pathway, but they remain viable by exploiting the complementary functions of non-HR repair pathways (e.g., the BER and NHEJ pathways) [35]. Consequently, PARP1, which is responsible for recruiting the BER [91] and alt-NHEJ [23] pathways, is often upregulated in HR-deficient cancer cells [34, 109], and HR-deficient cells tend to be hypersensitive to PARP1 inhibition [34, 109]. Impaired HR combined with PARP1-inhibited BER/alt-NHEJ leads to an accumulation of DSB lesions. These lesions are particularly detrimental to actively replicating cells (e.g., tumor cells) because unrepaired breaks often result in daughter cells that lack essential genes. In contrast, wild-type (HR-proficient) cells are insensitive to PARP1 inhibitors because they can still repair DNA damage via high-fidelity HR [21, 35, 61, 107, 110].
5.3. Clinically Approved PARP1 Inhibitors (PARPi)
Currently, there are four FDA-approved PARP1 inhibitors (olaparib, rucaparib, niraparib, and talazoparib) for treating HR-deficient cancers (Figure 6) [21, 28–31, 40, 42, 93, 107], and several more are in varying stages of clinical trials [28–31]. Although some PARP1 inhibitors are used as standalone cancer treatments, they are commonly paired with other treatments such as DNA-damaging chemotherapies or radiation [42, 93, 107]. For example, olaparib, the first FDA-approved PARP1 inhibitor [30], has been used as both a monotherapy and in combination with other therapies [30, 111, 112]. It was first approved in 2014 for treating ovarian cancers with BRCA1/2 mutations [30] but is now also used to treat some HR-defective breast [113], prostate [114], and pancreatic [115] cancers.
Figure 6. The four FDA-approved PARPi.

Chemical structures are shown together with the generic names, companies, and years of FDA approval. Nicotinamide, a NAD+ moiety, is shown to illustrate the chemical similarities between the PARPi and the endogenous ligand.
All current PARP1 inhibitors used in the clinic bind to the PARP1 catalytic pocket and act as competitive inhibitors of NAD+ [90]. They share structural similarities with nicotinamide (Figure 6) [107] and so form similar protein-ligand interactions [65, 74, 116–123]. For instance, many PARP1 inhibitors, including the NAD+ analog BAD and the drug niraparib (Figure 4), form π-π stacking interactions with the PARP1 Y907 residue [65, 74, 117–124].
Aside from impairing PARylation, PARP1 inhibitors may also be lethal to HR-deficient cells because of their varying abilities to “trap” PARP1 on DNA [90, 125, 126], which could potentially cause stalling and/or replication-fork collapse, resulting in cell-cycle arrest or apoptosis [91]. As evidence that competitive inhibition alone cannot explain the toxicity of PARP1 inhibitors, we note that cellular sensitivity to PARPi decreases when PARP1 expression is reduced via siRNA knockdown [96]. If competitive catalytic inhibition were entirely responsible for PARPi toxicity, one would expect that siRNA knockdown/knockout—which similarly reduces or even abolishes that activity—would be similarly toxic. That this is not the case suggests that PARP1 inhibitors confer additional toxicity through secondary mechanisms. DNA trapping is one possible, albeit controversial, explanation.
5.4. PARPi Resistance Mechanisms
Because PARP1 is involved in multiple complex biological pathways, many possible resistance mechanisms enable cancer cells to compensate for PARP1 inhibition. In this discussion we will focus on resistance mechanisms known to affect HR-defective cells: (1) HR reversion/increased HR capacity; (2) altered NHEJ capacity; (3) corrected replication forks; (4) decreased intracellular PARPi concentrations; and (5) modified PARP1 expression, activity, allosteric regulation, PARylation, and/or PARPi binding.
5.4.1. HR Reversion and Increased HR Capacity
Many proposed and clinically observed PARPi resistance mechanisms involve increased HR capacity [35, 127]. HR-deficient cells are sensitive to PARP1 inhibitors precisely because they have an impaired HR DNA repair pathway and so cannot survive the additional inhibition of PARP1-implicated repair pathways [35, 127]. Consequently, partial or complete restoration of HR capacity results in PARPi resistance [35, 127]. This section will discuss three mechanisms that increase HR capacity: (1) restoring HR-proficiency through secondary reversion mutations, (2) increasing HR protein availability, and (3) altering regulators of HR repair.
5.4.1.1. Secondary reversion mutations that restore HR proficiency can cause PARPi resistance [127].
HR-deficiencies are frequently caused by single-nucleotide mutations, short insertions, and frameshift deletions that disrupt the BRCA1/2 genes [102, 103, 127]. Secondary mutations that restore BRCA function can desensitize previously BRCA-deficient cells to PARP1 inhibitors, per multiple clinical observations [35, 127, 128]. The restoration of these genes speaks to the selective pressure that PARP1 inhibitors exert on cancer cells [127]. Unfortunately, reversion mutations in BRCA1/2 are often associated with resistance to platinum-based chemotherapies [129] as well, though treatment with taxanes may still be effective in such circumstances [130].
5.4.1.2. Increased availability of HR proteins can also cause PARPi resistance [35, 131–133].
Several factors influence the expression of these proteins. For instance, PARP1 itself negatively regulates BRCA2 expression by binding to the silencer-binding region of the brca2-promoter [35, 131]. By inactivating and possibly “trapping” PARP1 on DNA, PARP1 inhibitors could thus indirectly increase BRCA2 expression, contributing to PARPi resistance [35, 131]. Other factors also impact BRCA1 expression. For example, reduced levels of the microRNA miR182, which negatively regulates BRCA1 expression, can also increase BRCA1 protein production and so can lead to PARPi resistance [35, 132, 133].
Altered expression of other HR-pathway proteins also contributes to resistance. Overexpression of RAD51, a recombinase and important HR protein, is one example [35, 134, 135]. RAD51 is frequently overexpressed in BRCA1-deficient tumors and is associated with partially restored HR repair [35, 134–137]. RAD51 overexpression has been observed in a rucaparib-resistant high-grade ovarian carcinoma, as well as colon carcinoma cells that were resistant to a PARPi administered with the alkylating chemotherapeutic temozolomide [35, 134, 135].
5.4.1.3. Proteins that regulate HR repair also influence HR capacity.
These proteins include 53BP1 and ataxia telangiectasia mutated (ATM) [35, 127, 136, 138, 139]. 53BP1 is similar to BRCA1 in that it modulates HR and NHEJ repair, but it has the opposite effect on these pathways. Whereas loss of BRCA1 results in decreased HR repair and increased NHEJ repair, loss of 53BP1 results in increased HR repair and decreased NHEJ repair [35, 127, 136, 138]. This increased HR repair can contribute to PARPi resistance in previously HR-deficient cells [35, 136, 138]. 53BP1 acts on the HR-repair pathway via the ATM kinase, which triggers a kinase cascade in response to DNA damage that ultimately promotes HR repair [136, 138]. Increased ATM activity can also confer PARPi resistance [35, 136, 138]. Some have proposed combination therapies that include PARP1 and ATM inhibitors to circumvent these resistance mechanisms [139].
5.4.2. Altered NHEJ Capacity
Changes to an HR-defective cell’s NHEJ capacity can also cause resistance to PARP1 inhibitors [21, 26, 35, 127]. Because NHEJ repair is regulated by 53BP1 and BRCA1, alterations to these proteins can impact NHEJ capacity [35, 127, 136, 138]. Factors that alter the expression or function of NHEJ proteins such as DNA-PK and Ku proteins can also impact NHEJ capacity and consequently cellular sensitivity to PARPi [21, 35, 127]. Both the downregulation and upregulation of NHEJ proteins can drive chromosomal instability, resulting in PARPi resistance [26, 35]. The consequences of up/downregulated NHEJ capacity are reviewed in more depth in refs. [26] and [35].
5.4.3. Corrected Replication Forks
Some PARP1 inhibitors cause PARP1 to stall replication forks when bound to DNA [91]. BRCA1/2 proteins are involved in DNA-strand protection at stalled forks [32], so BRCA-deficient cells are sensitive to fork collapse and nascent-strand shortening [127, 140, 141]. Consequently, proteins that stabilize the replication fork can contribute to PARPi resistance [141]. For example, the nucleosome remodeling factor chromodomain-helicase-DNA binding protein 4 (CHD4) is expressed at lower levels in some chemotherapy-resistant BRCA2-mutant cancer cells, even though those cells remain deficient in RAD51-dependent HR repair [141]. BRCA2-deficient cells with reduced CHD4 can better protect nascent replication tracts and are therefore resistant to PARP1 inhibitors [141, 142].
5.4.4. Decreased Intracellular PARPi
Factors that reduce intracellular PARPi concentration can also impact PARPi efficacy. P-glycoprotein, an ATP-binding cassette (ABC) drug efflux transporter, is largely responsible for regulating these concentrations [35, 127, 143–146]. Reduced P-glycoprotein levels tend to increase sensitivity to PARP1 inhibitors in BRCA1-deficient mouse models [143, 144]. Additionally, P-glycoprotein inhibition re-sensitizes some BRCA-deficient, PARPi-resistant cells to PARPi [143, 145]. This resistance mechanism has been studied mainly in animal and cell models, so further research is needed [35, 127, 143–146].
5.4.5. Altered PARP1 Capacity
Factors that impact PARylation and potentially PARP1 “trapping” on DNA can contribute to PARPi cytotoxicity in HR-deficient cells [90, 125, 126]. Alterations to either of these processes, as well as to PARP1 expression levels, can lead to PARPi resistance.
5.4.5.1. Changes to the PARP1 catalytic site (where PARylation takes place) are particularly impactful.
For example, the proto-oncogene receptor tyrosine kinase c-Met, which is commonly overexpressed in many forms of cancer, alters the PARP1 catalytic site by phosphorylating Y907 (Video S4) [147, 148]. Y907 forms π-π stacking interactions with both NAD+, the endogenous substrate, and all clinically approved PARP1 inhibitors [148]. Phosphorylated Y907 (pY907) has increased catalytic activity, but weakened binding affinity for multiple PARP1 inhibitors [147, 148]. c-Met upregulation can therefore confer broad resistance [148]. Indeed, pY907 is known to confer resistance to veliparib, olaparib, and rucaparib [148].
5.4.5.2. Changes to other proteins responsible for regulating PARylation also contribute to PARPi resistance.
PAR glycohydrolase (PARG), a protein that degrades PAR chains, is one such protein [127, 149]. Factors that reduce PARG activity prevent PAR-chain degradation, potentially compensating for the reduced PARylation caused by PARP1 inhibition. This resistance mechanism was first discovered in a screen of BRCA2-deficient mouse cells with partially rescued PARP1-dependent DNA damage singling [127, 149]. Knockdown of PARG by shRNAs in human cancer cells with BRCA1 and BRCA2 deficiencies (SUM149PT and DLD-1, respectively) caused olaparib resistance [149]. Low PARG expression levels have also been measured in a subset of human triple-negative breast tumors and serous ovarian carcinomas [149].
5.4.5.3. Mutations that disrupt the allosteric communication allegedly required for PARP1 “trapping” on DNA may also contribute to PARPi resistance.
At least one mutation—R591C (WGR) at the Zn1-WGR-HD interface—has been identified clinically in a talazoparib-resistant ovarian tumor. This same mutation appears to reduce PARP1 trapping [90, 99]. Many additional lab-generated, interfacial mutations are known to alter PARP1 activity, PARPi inhibition, and/or PARPi trapping (see above), suggesting additional routes to PARPi resistance.
5.4.5.4. PARP1 expression levels and catalytic activity correlate with PARPi sensitivity.
For example, PARPi-resistant colorectal-carcinoma HCT116 cells have markedly low levels of PARP1 expression [35, 134]. Cancer cells with decreased PARylation activity but normal PARP1 expression levels also tend to be more resistant to PARP1 inhibitors [35, 150]. On the other hand, HR-deficient cells with higher levels of PARylation tend to be more sensitive to PARP1 inhibitors [35, 151].
6. Outlook
Because all clinically approved PARP1 inhibitors bind the same catalytic pocket with similar binding modes, they are likely subject to similar resistance mechanisms. Given this general vulnerability, there is a need for next-generation PARP1 inhibitors that can evade common resistance mechanisms. In this section, we will suggest several future pharmaceutical approaches to PARP1 inhibition.
6.1. Targeting Alternative Binding Pockets
Given that many PARPi resistance mechanisms involve changes to the catalytic pocket where PARP1 inhibitors bind, identifying next-generation inhibitors that target other pockets is a promising pharmacological approach. New PARP1 inhibitors that do not rely on the same binding interactions as current inhibitors may overcome resistance mechanisms caused by PARP1 post-translational modifications or mutations.
There is good reason to suppose that allosteric ligands that bind outside the catalytic pocket could be successfully exploited in future drug-discovery campaigns. It is true that all known PARP1 inhibitors act as orthosteric inhibitors of PARylation, but some may also act as allosteric inhibitors that “trap” PARP1 on DNA [94, 96]. The allosteric signal that links the CAT domain and the DBD must traverse several important inter-domain interfaces. Mutagenesis studies show that disrupting these interfaces can decouple catalytic activity from DNA binding (see above) [51, 66, 75]. Small-molecule drugs that similarly disrupt these interfaces by binding to adjacent pockets might also limit catalytic activity while preserving PARP1 trapping on DNA.
To provide further support for this pharmaceutical approach, we applied FTMap [152] to the 4DQY [51] PARP1 structure. FTMap is an algorithm that searches for druggable “hotspots” with favorable ligand-binding properties [152]. It identified five PARP1 hotspots (excluding those positioned on the co-crystallized DNA molecule, Figure 7) [40].
Figure 7. FTMap-detected hot spots (PDB: 4DQY).

(A) Molecular fragments used to identify hot spots are shown as blue spheres. There are two hot spots at the Zn1-Zn3 interface and two in the catalytic pocket. (B) The pocket volumes of the Zn1-Zn3 interface hot spot (top left) and the Zn1-WGR-HD interface hot spot (bottom center) are shown as transparent yellow surfaces, calculated using POVME2 [160]. See Video S4 for further discussion.
6.1.1. Catalytic Pocket.
Two hotspots located in the CAT domain serve as positive controls because both occupy the catalytic binding site, where existing inhibitors are known to bind.
6.1.2. Zn1-WGR-HD Interface.
One hotspot is located near the Zn1-WGR-HD interface (Figure 7A, bottom center; Video S4). PARP1 with mutations at this interface, such as D45A (Zn1) and R591A (WGR), has near-wild-type levels of DNA binding activity despite dramatically reduced catalytic activity [51, 66, 67]. Small molecules that similarly disrupt the interface could in theory have a similar impact.
The apparent volume of the hotspot-associated pocket (Figure 7B, bottom center), as measured by the DoGSiteScorer algorithm [153], is also encouraging. The Zn1-WGR-HD pocket has roughly half the volume of the ligand-bound, 4R6E:A catalytic pocket [65] (595 Å3 vs. 1,262 Å3), but its size is still well within the range of typical ligand-binding pockets (100–1,000 Å3 [154]). This finding is important because larger and deeper pockets are typically considered more druggable [155].
6.1.3. Zn1-Zn3 Interface.
Two hotspots located at the Zn1-Zn3 interface are close to each other and so can be considered a single site (Figure 7A, on the left; Video S4). Disrupting the Zn1-Zn3 interface is also a promising pharmacological strategy, given that PARP1 with the W246A (Zn3) mutation has near-wild-type levels of DNA binding activity but much reduced catalytic activity [51, 66, 75]. Notably, the Zn1-Zn3 pocket (Figure 7B, on the left) is slightly larger than even the Zn1-WGR-HD pocket (634 Å3 vs. 595 Å3), per DoGSiteScorer [153].
6.2. Targeting the PARP1/HPF1 Composite Catalytic Site
The recently identified HPF1 protein (see Section 4.3.2) profoundly impacts the PARP1 catalytic site by contributing key residues that enhance serine ADP-ribosylation activity [83–85, 87, 88]. This new PARP1/HPF1 composite pocket is so radically altered that it is effectively a new pocket. The associated active-site changes present many new opportunities for drug discovery; indeed, recent work has shown that HPF1 has a substantial impact on the binding affinity of some PARP1 inhibitors (e.g., it increases the affinity of olaparib) [83]. On the other hand, it does not appear to affect the affinity of other inhibitors such as veliparib [83]. The role of HPF1 in regulating PARP1 activity has only recently been characterized [86], and the first structures of the PARP1/HPF1 complex were published in 2020 [87, 88]. Consequently, the PARP1/HPF1 interaction has yet to inform the design of any clinically approved PARP1 inhibitor. Designing inhibitors that bind the composite PARP1/HPF1 catalytic site is a promising future direction.
6.3. Combining Treatments to Circumvent PARPi Resistance
Aside from identifying novel PARP1 inhibitors, combining such inhibitors with other treatments will continue to be critical. PARP1 inhibitors are unlikely to overcome some resistance mechanisms, regardless of their specific mode of action (whether orthosteric/competitive or allosteric). As described above, examples of such resistance mechanisms include secondary BRCA1/2 mutations that restore HR function and factors that limit PARP1 expression.
Existing, clinically approved PARP1 inhibitors are often paired with other chemotherapeutics and/or surgical cytoreduction [21, 28–31, 42, 93, 107]. Combination therapies that target proteins/pathways involved in PARPi resistance can in many cases (re)sensitize cells to PARPi. Several studies have examined the synergistic effects of combining PARP1 inhibitors with inhibitors of ataxia-telangiectasia mutated and Rad3-related (ATR) kinase [156], c-Met [148], and histone deacetylase [157]. Interested readers may wish to consult Yi et al. (2019), who recently reviewed several additional PARPi combination therapies [158]. Further research is needed to identify which co-treatment strategies can optimally circumvent the many documented PARPi resistance mechanisms.
7. Conclusions
Though PARP1 has been the subject of intense study, much remains unknown about this complex, multi-domain protein with both orthosteric and allosteric mechanisms. Several clinically approved PARP1 inhibitors target the catalytic pocket, but future small-molecule ligands that act at other sites have the potential to evade known resistance mechanisms. Ligands designed to disrupt the various PARP1 allosteric mechanisms could serve as selective therapeutics for the treatment of PARPi-sensitive cancers.
Supplementary Material
Research highlights.
PARP1 recruits DNA repair proteins to strand breaks undergoing processing.
PARP1 signals repair pathways by attaching ADP-ribose to itself and other proteins.
Both orthosteric and allosteric mechanisms govern this process of ADP-ribosylation.
Clinical PARP1 inhibitors (PARPi) impede ADP-ribosylation and may “trap” PARP1 on DNA, though the latter mechanism is controversial.
PARPi resistance is common, so novel pharmacological approaches are urgently needed.
Acknowledgments
We thank the University of Pittsburgh’s Center for Research Computing (https://crc.pitt.edu/) for providing valuable computer resources. We also thank Pauline Spiegel for help with editing, and Dr. Ke-Yi Lin for helpful feedback on Figure S1.
Funding Sources
BVH is supported by a grant from the National Institute of Environmental Health Sciences, National Institutes of Health, NIH Grant R35ES031638.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors report no declarations of interest.
Common abbreviations: AD: automodification domain. AP: apurinic/apyrimidinic. ART: ADP-ribosyl transferase. ASL: αD-active site loop. BAD: benzamide adenine dinucleotide. BER: base excision repair. BRCT: BRCA C-terminus. CAT: catalytic. DBD: DNA binding domain. DDR: DNA damage response. DSB: double strand DNA break. HD: helical domain. HR: homologous recombination. MARylation: mono-ADP-ribosylation. MMEJ: microhomology mediated end joining. NAD+: nicotinamide adenine dinucleotide. NHEJ: non-homologous end joining. PAR: poly(ADP-ribose). PARP: poly(ADP-ribose) polymerase. PARP1: poly(ADP-ribose) polymerase 1. PARPi: PARP1 inhibitors. PARylation: poly-ADP-ribosylation. SSB: single strand DNA break. WGR: tryptophan-glycine-arginine-rich. Zn1: zinc finger 1. Zn2: zinc finger 2. Zn3: zinc finger 3.
References
- [1].Perina D, Mikoč A, Ahel J, Ćetković H, Žaja R, Ahel I, Distribution of protein poly(ADP-ribosyl)ation systems across all domains of life, DNA Repair (Amst), 23 (2014) 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lin K-Y, Molecular Mechanism of Poly(ADP-ribosyl)ation Catalyzed by Human Poly(ADP-ribose) Polymerase-1.
- [3].Daniels CM, Ong SE, Leung AKL, The Promise of Proteomics for the Study of ADP-Ribosylation, Molecular Cell, 58 (2015) 911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wei H, Yu X, Functions of PARylation in DNA Damage Repair Pathways, Genomics, Proteomics and Bioinformatics, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].D’Amours D, Desnoyers S, D’Silva I, Poirier GG, Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions, Biochemical Journal, 342 (1999) 249–268. [PMC free article] [PubMed] [Google Scholar]
- [6].Palazzo L, Leidecker O, Prokhorova E, Dauben H, Matic I, Ahel I, Serine is the major residue for ADP-ribosylation upon DNA damage, eLife, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Amé JC, Spenlehauer C, De Murcia G, The PARP superfamily, BioEssays, (2004). [DOI] [PubMed] [Google Scholar]
- [8].Héberlé E, Amé J-C, Illuzzi G, Dantzer F, Schreiber V, Discovery of the PARP Superfamily and Focus on the Lesser Exhibited But Not Lesser Talented Members, in.
- [9].Luo X, Kraus WL, On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1, Genes & Development, 26 (2012) 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kamaletdinova T, Fanaei-Kahrani Z, Wang Z-Q, The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers, Cells, 8 (2019) 1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Durrant JD, BlendMol: Advanced Macromolecular Visualization in Blender, Bioinformatics, 35 (2018) 2323–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Van Houten B, Santa-Gonzalez GA, Camargo M, DNA repair after oxidative stress: Current challenges, Current Opinion in Toxicology, 7 (2018) 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bitler BG, Watson ZL, Wheeler LJ, Behbakht K, PARP inhibitors: Clinical utility and possibilities of overcoming resistance, Gynecologic Oncology, 147 (2017) 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Eisemann T, Pascal JM, Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity, Cellular and Molecular Life Sciences, 77 (2020) 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stern HR, Sefcikova J, Chaparro VE, Beuning PJ, Mammalian DNA Polymerase Kappa Activity and Specificity, Molecules, 24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, Caldecott KW, The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication, Mol Cell, 71 (2018) 319–331 e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hegde ML, Hazra TK, Mitra S, Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells, Cell Research, 18 (2008) 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim Y-J, Wilson III DM, Overview of Base Excision Repair Biochemistry, Current Molecular Pharmacology, 5 (2011) 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Caldecott KW, Single-strand break repair and genetic disease, Nature Reviews Genetics, 9 (2008) 619–631. [DOI] [PubMed] [Google Scholar]
- [20].Chatterjee N, Walker GC, Mechanisms of DNA damage, repair, and mutagenesis, Environmental and Molecular Mutagenesis, 58 (2017) 235–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Patel AG, Sarkaria JN, Kaufmann SH, Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells, Proceedings of the National Academy of Sciences of the United States of America, 108 (2011) 3406–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Beard WA, Horton JK, Prasad R, Wilson SH, Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism, Annu Rev Biochem, 88 (2019) 137–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chiruvella KK, Liang Z, Wilson TE, Repair of double-strand breaks by end joining, Cold Spring Harbor Perspectives in Biology, 5 (2013) 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gassman NR, Stefanick DF, Kedar PS, Horton JK, Wilson SH, Hyperactivation of PARP Triggers Nonhomologous End-Joining in Repair-Deficient Mouse Fibroblasts, PLoS ONE, 7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G, PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways, Nucleic Acids Research, 34 (2006) 6170–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sishc BJ, Davis AJ, The role of the core non-homologous end joining factors in carcinogenesis and cancer, Cancers, 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chang HHY, Pannunzio NR, Adachi N, Lieber MR, Non-homologous DNA end joining and alternative pathways to double-strand break repair, Nat Rev Mol Cell Biol, 18 (2017) 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Balasubramaniam S, Beaver JA, Horton S, Fernandes LL, Tang S, Horne HN, Liu J, Liu C, Schrieber SJ, Yu J, Song P, Pierce W, Robertson KJ, Palmby TR, Chiu HJ, Lee EY, Philip R, Schuck R, Charlab R, Banerjee A, Chen XH, Wang X, Goldberg KB, Sridhara R, Kim G, Pazdur R, FDA approval summary: Rucaparib for the treatment of patients with deleterious BRCA mutation–associated advanced ovarian cancer, Clinical Cancer Research, 23 (2017) 7165–7170. [DOI] [PubMed] [Google Scholar]
- [29].Ison G, Howie LJ, Amiri-Kordestani L, Zhang L, Tang S, Sridhara R, Pierre V, Charlab R, Ramamoorthy A, Song P, Li F, Yu J, Manheng W, Palmby TR, Ghosh S, Horne HN, Lee EY, Philip R, Dave K, Chen XH, Kelly SL, Janoria KG, Banerjee A, Eradiri O, Dinin J, Goldberg KB, Pierce WF, Ibrahim A, Kluetz PG, Blumenthal GM, Beaver JA, Pazdur R, FDA approval summary: Niraparib for the maintenance treatment of patients with recurrent ovarian cancer in response to platinum-based chemotherapy, Clinical Cancer Research, 24 (2018) 4066–4071. [DOI] [PubMed] [Google Scholar]
- [30].Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, Chiu HJ, Ricks TK, Palmby T, Russell AM, Ladouceur G, Pfuma E, Li H, Zhao L, Liu Q, Venugopal R, Ibrahim A, Pazdur R, FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy, Clinical Cancer Research, 21 (2015) 4257–4261. [DOI] [PubMed] [Google Scholar]
- [31].Hoy SM, Talazoparib: First Global Approval, Drugs, 78 (2018) 1939–1946. [DOI] [PubMed] [Google Scholar]
- [32].Chen C-C, Feng W, Lim PX, Kass EM, Jasin M, Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer, Annual Review of Cancer Biology, 2 (2018) 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Deng CX, BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution, Nucleic Acids Research, 34 (2006) 1416–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ko HL, Ren EC, Functional Aspects of PARP1 in DNA Repair and Transcription, Biomolecules, 2 (2012) 524–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Montoni A, Robu M, Pouliot É, Shah GM, Resistance to PARP-Inhibitors in Cancer Therapy, Frontiers in Pharmacology, 4 (2013) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wright WD, Shah SS, Heyer W-D, Homologous recombination and the repair of DNA double-strand breaks, Journal of Biological Chemistry, 293 (2018) 10524–10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Balakrishnan L, Bambara RA, Okazaki fragment metabolism, Cold Spring Harb Perspect Biol, 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Okazaki R, Okazaki T, Sakabe K, Sugimoto K, Sugino A, Mechanism of DNA chain growth. I. Possible discontinuity and unusual secondary structure of newly synthesized chains, Proc Natl Acad Sci U S A, 59 (1968) 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sakabe K, Okazaki R, A unique property of the replicating region of chromosomal DNA, Biochim Biophys Acta, 129 (1966) 651–654. [DOI] [PubMed] [Google Scholar]
- [40].Spiegel JO, Targeting the Poly (ADP-Ribose) Polymerase-1 Catalytic Pocket Using AutoGrow4, a Genetic Algorithm for De Novo Design, University of Pittsburgh, ProQuest Dissertations Publishing, 2020. [Google Scholar]
- [41].David KK, Andrabi SA, Dawson TM, Dawson VL, Parthanatos A messenger of death, Frontiers in Bioscience, 14 (2009) 1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG, PARP inhibition: PARP1 and beyond, Nature Reviews Cancer, 10 (2010) 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Javle M, Curtin NJ, The role of PARP in DNA repair and its therapeutic exploitation, British Journal of Cancer, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR, Poly(ADP-ribose) Binds to Specific Domains in DNA Damage Checkpoint Proteins, Journal of Biological Chemistry, 275 (2000) 40974–40980. [DOI] [PubMed] [Google Scholar]
- [45].Ahel I, Ahel D, Matsusaka T, Clark AJ, Pines J, Boulton SJ, West SC, Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins, Nature, 451 (2008) 81–85. [DOI] [PubMed] [Google Scholar]
- [46].Wang Z, Michaud GA, Cheng Z, Zhang Y, Hinds TR, Fan E, Cong F, Xu W, Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly (ADP-ribosyl)ation-dependent ubiquitination, Genes and Development, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kim I-K, Kiefer JR, Ho CMW, Stegeman RA, Classen S, Tainer JA, Ellenberger T, Structure of mammalian poly(ADP-ribose) glycohydrolase reveals a flexible tyrosine clasp as a substrate-binding element, Nature Structural & Molecular Biology, 19 (2012) 653–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, Ahel I, The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase, Nature, 477 (2011) 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Alemasova EE, Lavrik OI, Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins, Nucleic Acids Research, 47 (2019) 3811–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML, Proteome-wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses, Molecular Cell, 52 (2013) 272–285. [DOI] [PubMed] [Google Scholar]
- [51].Langelier M-F, Planck JL, Roy S, Pascal JM, Structural Basis for DNA Damage-Dependent Poly(ADP-ribosyl)ation by Human PARP-1, Science, 336 (2012) 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO, Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites, Nucleic Acids Research, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tao Z, Gao P, Liu HW, Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: Analysis and implications, Journal of the American Chemical Society, (2009). [DOI] [PubMed] [Google Scholar]
- [54].Sharifi R, Morra R, Denise Appel C, Tallis M, Chioza B, Jankevicius G, Simpson MA, Matic I, Ozkan E, Golia B, Schellenberg MJ, Weston R, Williams JG, Rossi MN, Galehdari H, Krahn J, Wan A, Trembath RC, Crosby AH, Ahel D, Hay R, Ladurner AG, Timinszky G, Scott Williams R, Ahel I, Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease, EMBO Journal, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chapman JD, Gagné JP, Poirier GG, Goodlett DR, Mapping PARP-1 auto-ADP-ribosylation sites by liquid chromatography-tandem mass spectrometry, Journal of Proteome Research, (2013). [DOI] [PubMed] [Google Scholar]
- [56].Zhang Y, Wang J, Ding M, Yu Y, Site-specific characterization of the Asp-and Glu-ADP-ribosylated proteome, Nature Methods, (2013). [DOI] [PubMed] [Google Scholar]
- [57].Daniels CM, Ong SE, Leung AKL, Phosphoproteomic approach to characterize protein mono- and poly(ADP-ribosyl)ation sites from cells, Journal of Proteome Research, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gagné JP, Ethier C, Defoy D, Bourassa S, Langelier M-F, Riccio AA, Pascal JM, Moon KM, Foster LJ, Ning Z, Figeys D, Droit A, Poirier GG, Quantitative site-specific ADP-ribosylation profiling of DNA-dependent PARPs, DNA Repair, (2015). [DOI] [PubMed] [Google Scholar]
- [59].Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G, XRCC1 Is Specifically Associated with Poly(ADP-Ribose) Polymerase and Negatively Regulates Its Activity following DNA Damage, Molecular and Cellular Biology, (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Beernink PT, Hwang M, Ramirez M, Murphy MB, Doyle SA, Thelen MP, Specificity of protein interactions mediated by BRCT domains of the XRCC1 DNA repair protein, Journal of Biological Chemistry, (2005). [DOI] [PubMed] [Google Scholar]
- [61].Helleday T, The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings, Molecular Oncology, 5 (2011) 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Pazzaglia S, Pioli C, Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases, Cells, 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Mangerich A, Burkle A, Pleiotropic cellular functions of PARP1 in longevity and aging: genome maintenance meets inflammation, Oxid Med Cell Longev, 2012 (2012) 321653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Thomas C, Ji Y, Wu C, Datz H, Boyle C, MacLeod B, Patel S, Ampofo M, Currie M, Harbin J, Pechenkina K, Lodhi N, Johnson SJ, Tulin AV, Hit and run versus long-term activation of PARP-1 by its different domains fine-tunes nuclear processes, Proc Natl Acad Sci U S A, 116 (2019) 9941–9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Thorsell A-G, Ekblad T, Karlberg T, Löw M, Pinto AF, Trésaugues L, Moche M, Cohen MS, Schüler H, Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors, J Med Chem, 60 (2017) 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Steffen JD, Tholey RM, Langelier M-F, Planck JL, Schiewer MJ, Lal S, Bildzukewicz NA, Yeo CJ, Knudsen KE, Brody JR, Pascal JM, Targeting PARP-1 allosteric regulation offers therapeutic potential against cancer, Cancer Research, 74 (2014) 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Langelier M-F, Planck JL, Roy S, Pascal JM, Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: Structural and functional insights into DNA-dependent PARP-1 activity, Journal of Biological Chemistry, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ikejima M, Noguchi S, Yamashita R, Ogura T, Sugimura T, Gill DM, Miwa M, The zinc fingers of human poly(ADP-ribose) polymerase are differentially required for the recognition of DNA breaks and nicks and the consequent enzyme activation. Other structures recognize intact DNA, The Journal of biological chemistry, 265 (1990) 21907–21913. [PubMed] [Google Scholar]
- [69].Gradwohl G, Menissier de Murcia JM, Molinete M, Simonin F, Koken M, Hoeijmakers JHJ, De Murcia G, The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA, Proceedings of the National Academy of Sciences of the United States of America, 87 (1990) 2990–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Langelier M-F, Servent KM, Rogers EE, Pascal JM, A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation, Journal of Biological Chemistry, (2008). [DOI] [PubMed] [Google Scholar]
- [71].Langelier M-F, Ruhl DD, Planck JL, Kraus WL, Pascal JM, The Zn3 domain of human poly(ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly(ADP-ribose) synthesis activity and chromatin compaction, Journal of Biological Chemistry, 285 (2010) 18877–18887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Langelier M-F, Pascal JM, PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis, Current Opinion in Structural Biology, 23 (2013) 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Mansoorabadi SO, Wu M, Tao Z, Gao P, Pingali SV, Guo L, Liu H.-w., Conformational activation of poly(ADP-ribose) polymerase-1 upon DNA binding revealed by small-angle X-ray scattering, Biochemistry, 53 (2014) 1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dawicki-McKenna JM, Langelier M-F, DeNizio JE, Riccio AA, Cao CD, Karch KR, McCauley M, Steffen JD, Black BE, Pascal JM, PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain, Mol Cell, 60 (2015) 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Eustermann S, Wu W-F, Langelier M-F, Yang J-C, Easton LE, Riccio AA, Pascal JM, Neuhaus D, Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1, Molecular Cell, 60 (2015) 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Trucco C, Flatter E, Fribourg S, de Murcia G, Ménissier-de Murcia J, Mutations in the amino-terminal domain of the human poly(ADP-ribose) polymerase that affect its catalytic activity but not its DNA binding capacity, FEBS Letters, 399 (1996) 313–316. [DOI] [PubMed] [Google Scholar]
- [77].Langelier M-F, Zandarashvili L, Aguiar PM, Black BE, Pascal JM, NAD+ analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains, Nature Communications, 9 (2018) 844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Miranda EA, Dantzer F, O’Farrell M, de Demurcia G, De murcia JM, Characterization of a gain-of-function mutant of poly(ADP-Ribose) polymerase, Biochemical and Biophysical Research Communications, (1995). [DOI] [PubMed] [Google Scholar]
- [79].Steffen JD, Brody JR, Armen RS, Pascal JM, Structural implications for selective targeting of PARPs, Frontiers in Oncology, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Barkauskaite E, Jankevicius G, Ahel I, Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation, Molecular Cell, (2015). [DOI] [PubMed] [Google Scholar]
- [81].Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, Ahel I, Chang P, Family-wide analysis of poly(ADP-ribose) polymerase activity, Nature Communications, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ruf A, Rolli V, De Murcia G, Schulz GE, The mechanism of the elongation and branching reaction of Poly(ADP-ribose) polymerase as derived from crystal structures and mutagenesis, Journal of Molecular Biology, (1998). [DOI] [PubMed] [Google Scholar]
- [83].Rudolph J, Roberts G, Luger K, Histone Parylation factor 1 contributes to the inhibition of PARP1 by cancer drugs, Nat Commun, 12 (2021) 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sun FH, Zhao P, Zhang N, Kong LL, Wong CCL, Yun CH, HPF1 remodels the active site of PARP1 to enable the serine ADP-ribosylation of histones, Nat Commun, 12 (2021) 1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Alemasova EE, Lavrik OI, Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins, Nucleic Acids Res, 47 (2019) 3811–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Gibbs-Seymour I, Fontana P, Rack JGM, Ahel I, HPF1/C4orf27 Is a PARP-1-Interacting Protein that Regulates PARP-1 ADP-Ribosylation Activity, Mol Cell, 62 (2016) 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Suskiewicz MJ, Zobel F, Ogden TEH, Fontana P, Ariza A, Yang JC, Zhu K, Bracken L, Hawthorne WJ, Ahel D, Neuhaus D, Ahel I, HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation, Nature, 579 (2020) 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Bilokapic S, Suskiewicz MJ, Ahel I, Halic M, Bridging of DNA breaks activates PARP2-HPF1 to modify chromatin, Nature, 585 (2020) 609–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Bonfiglio JJ, Leidecker O, Dauben H, Longarini EJ, Colby T, San Segundo-Acosta P, Perez KA, Matic I, An HPF1/PARP1-Based Chemical Biology Strategy for Exploring ADP-Ribosylation, Cell, 183 (2020) 1086–1102 e1023. [DOI] [PubMed] [Google Scholar]
- [90].Zandarashvili L, Langelier M-F, Velagapudi UK, Hancock MA, Steffen JD, Billur R, Hannan ZM, Wicks AJ, Krastev DB, Pettitt SJ, Lord CJ, Talele TT, Pascal JM, Black BE, Structural basis for allosteric PARP-1 retention on DNA breaks, Science, 368 (2020) eaax6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Horton JK, Stefanick DF, Prasad R, Gassman NR, Kedar PS, Wilson SH, Base excision repair defects invoke hypersensitivity to PARP inhibition, Molecular Cancer Research, 12 (2014) 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Liu L, Kong M, Gassman NR, Freudenthal BD, Prasad R, Zhen S, Watkins SC, Wilson SH, Van Houten B, PARP1 changes from three-dimensional DNA damage searching to one-dimensional diffusion after auto-PARylation or in the presence of APE1, Nucleic Acids Research, 45 (2017) 12834–12847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Turk AA, Wisinski KB, PARP Inhibition in BRCA-Mutant Breast Cancer, Cancer, 124 (2018) 2498–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, Di Giammarino EL, Panchal SC, Wilsbacher JL, Gao W, Olson AM, Stolarik DF, Osterling DJ, Johnson EF, Maag D, Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors, Molecular Cancer Research, 13 (2015) 1465–1477. [DOI] [PubMed] [Google Scholar]
- [95].Hopkins TA, Ainsworth WB, Ellis PA, Donawho CK, DiGiammarino EL, Panchal SC, Abraham VC, Algire MA, Shi Y, Olson AM, Johnson EF, Wilsbacher JL, Maag D, PARP1 trapping by PARP inhibitors drives cytotoxicity in both cancer cells and healthy bone marrow, Molecular Cancer Research, (2019). [DOI] [PubMed] [Google Scholar]
- [96].Murai J, Huang S.-y.N., Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y, Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors, Cancer Research, 72 (2012) 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Shao Z, Lee BJ, Rouleau-Turcotte E, Langelier MF, Lin X, Estes VM, Pascal JM, Zha S, Clinical PARP inhibitors do not abrogate PARP1 exchange at DNA damage sites in vivo, Nucleic Acids Res, 48 (2020) 9694–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E, PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance, Front Cell Dev Biol, 8 (2020) 564601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, Frankum J, Ranes M, Pemberton HN, Rafiq R, Fenwick K, Swain A, Guettler S, Lee J-M, Swisher EM, Stoynov S, Yusa K, Ashworth A, Lord CJ, Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance, Nature Communications, 9 (2018) 1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, DiGiammarino EL, Panchal SC, Wilsbacher JL, Gao W, Olson AM, Stolarik DF, Osterling DJ, Johnson EF, Maag D, Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors, Mol Cancer Res, 13 (2015) 1465–1477. [DOI] [PubMed] [Google Scholar]
- [101].Owonikoko TK, Zhang G, Deng X, Rossi MR, Switchenko JM, Doho GH, Chen Z, Kim S, Strychor S, Christner SM, Beumer J, Li C, Yue P, Chen A, Sica GL, Ramalingam SS, Kowalski J, Khuri FR, Sun SY, Poly (ADP) ribose polymerase enzyme inhibitor, veliparib, potentiates chemotherapy and radiation in vitro and in vivo in small cell lung cancer, Cancer Med, 3 (2014) 1579–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Nicolas E, Bertucci F, Sabatier R, Gonçalves A, Targeting BRCA deficiency in breast cancer: What are the clinical evidences and the next perspectives?, Cancers, 10 (2018) 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD, Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer, Cancer Discovery, 5 (2015) 1137–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Sztupinszki Z, Diossy M, Krzystanek M, Borcsok J, Pomerantz MM, Tisza V, Spisak S, Rusz O, Csabai I, Freedman ML, Szallasi Z, Detection of Molecular Signatures of Homologous Recombination Deficiency in Prostate Cancer with or without BRCA1/2 Mutations, Clinical Cancer Research, (2020) 2673–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Zhu H, Wei M, Xu J, Hua J, Liang C, Meng Q, Zhang Y, Liu J, Zhang B, Yu X, Shi S, PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications, Molecular Cancer, 19 (2020) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK, Cancers associated with BRCA 1 and BRCA 2 mutations other than breast and ovarian, Cancer, 121 (2015) 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ferraris VD, Evolution of Poly(ADP-ribose) Polymerase-1 (PARP-1) Inhibitors. From Concept to Clinic, Journal of Medicinal Chemistry, 53 (2010) 4561–4584. [DOI] [PubMed] [Google Scholar]
- [108].Nijman SMB, Synthetic lethality: General principles, utility and detection using genetic screens in human cells, FEBS Letters, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM, Upregulation of poly (ADP-Ribose) polymerase-1 (PARP1) in triple-negative breast cancer and other primary human tumor types, Genes and Cancer, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A, Resistance to therapy caused by intragenic deletion in BRCA2, Nature, (2008). [DOI] [PubMed] [Google Scholar]
- [111].Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RHJ, Sonke GS, Colombo N, Špaček J, Vuylsteke P, Hirte H, Mahner S, Plante M, Schmalfeldt B, Mackay H, Rowbottom J, Lowe ES, Dougherty B, Barrett JC, Friedlander M, Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial, The Lancet Oncology, (2015). [DOI] [PubMed] [Google Scholar]
- [112].Veneris JT, Matulonis UA, Liu JF, Konstantinopoulos PA, Choosing wisely: Selecting PARP inhibitor combinations to promote anti-tumor immune responses beyond BRCA mutations, Gynecologic Oncology, (2020). [DOI] [PubMed] [Google Scholar]
- [113].FDA approves olaparib for germline BRCA-mutated metastatic breast cancer, in, FDA, 2018. [Google Scholar]
- [114].FDA approves olaparib for HRR gene-mutated metastatic castration-resistant prostate cancer, in, 2020.
- [115].FDA approves olaparib for gBRCAm metastatic pancreatic adenocarcinoma, in, 2019.
- [116].Ye N, Chen CH, Chen T, Song Z, He JX, Huan XJ, Song SS, Liu Q, Chen Y, Ding J, Xu Y, Miao ZH, Zhang A, Design, synthesis, and biological evaluation of a series of benzo[de][1,7]naphthyridin-7(8 H)-ones bearing a functionalized longer chain appendage as novel PARP1 inhibitors, Journal of Medicinal Chemistry, 56 (2013) 2885–2903. [DOI] [PubMed] [Google Scholar]
- [117].Patel MR, Bhatt A, Steffen JD, Chergui A, Murai J, Pommier Y, Pascal JM, Trombetta LD, Fronczek FR, Talele TT, Discovery and structure-activity relationship of novel 2,3-dihydrobenzofuran-7-carboxamide and 2,3-dihydrobenzofuran-3(2 h)-one-7-carboxamide derivatives as poly(ADP-ribose)polymerase-1 Inhibitors, Journal of Medicinal Chemistry, 57 (2014) 5579–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Aoyagi-Scharber M, Gardberg AS, Yip BK, Wang B, Shen Y, Fitzpatrick PA, Structural basis for the inhibition of poly(ADP-ribose) polymerases 1 and 2 by BMN 673, a potent inhibitor derived from dihydropyridophthalazinone, Acta Crystallographica Section:F Structural Biology Communications, 70 (2014) 1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Papeo G, Posteri H, Borghi D, Busel AA, Caprera F, Casale E, Ciomei M, Cirla A, Corti E, D’Anello M, Fasolini M, Forte B, Galvani A, Isacchi A, Khvat A, Krasavin MY, Lupi R, Orsini P, Perego R, Pesenti E, Pezzetta D, Rainoldi S, Riccardi-Sirtori F, Scolaro A, Sola F, Zuccotto F, Felder ER, Donati D, Montagnoli A, Discovery of 2-[1-(4,4-Difluorocyclohexyl)piperidin-4-yl]-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole-4-carboxamide (NMS-P118): A Potent, Orally Available, and Highly Selective PARP-1 Inhibitor for Cancer Therapy, Journal of Medicinal Chemistry, 58 (2015) 6875–6898. [DOI] [PubMed] [Google Scholar]
- [120].Velagapudi UK, Langelier M-F, Delgado-Martin C, Diolaiti ME, Bakker S, Ashworth A, Patel BA, Shao X, Pascal JM, Talele TT, Design and Synthesis of Poly(ADP-ribose) Polymerase Inhibitors: Impact of Adenosine Pocket-Binding Motif Appendage to the 3-Oxo-2,3-dihydrobenzofuran-7-carboxamide on Potency and Selectivity, Journal of Medicinal Chemistry, 62 (2019) 5330–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Chen X, Huan X, Liu Q, Wang Y, He Q, Tan C, Chen Y, Ding J, Xu Y, Miao Z, Yang C, Design and synthesis of 2-(4,5,6,7-tetrahydrothienopyridin-2-yl)-benzoimidazole carboxamides as novel orally efficacious Poly(ADP-ribose)polymerase (PARP) inhibitors, European Journal of Medicinal Chemistry, 145 (2018) 389–403. [DOI] [PubMed] [Google Scholar]
- [122].Kinoshita T, Nakanishi I, Warizaya M, Iwashita A, Kido Y, Hattori K, Fujii T, Inhibitor-induced structural change of the active site of human poly(ADP-ribose) polymerase, FEBS Letters, 556 (2004) 43–46. [DOI] [PubMed] [Google Scholar]
- [123].Hattori K, Kido Y, Yamamoto H, Ishida J, Kamijo K, Murano K, Ohkubo M, Kinoshita T, Iwashita A, Mihara K, Yamazaki S, Matsuoka N, Teramura Y, Miyake H, Rational approaches to discovery of orally active and brain-penetrable quinazolinone inhibitors of poly(ADP-ribose)polymerase, Journal of Medicinal Chemistry, 47 (2004) 4151–4154. [DOI] [PubMed] [Google Scholar]
- [124].Spiegel JO, Durrant JD, AutoGrow4: an open-source genetic algorithm for de novo drug design and lead optimization, Journal of Cheminformatics, 12 (2020) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Murai J, Huang SYN, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, Pommier Y, Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib, Molecular Cancer Therapeutics, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Shen Y, Aoyagi-Scharber M, Wang B, Trapping poly(ADP-Ribose) polymerase, Journal of Pharmacology and Experimental Therapeutics, (2015). [DOI] [PubMed] [Google Scholar]
- [127].Noordermeer SM, van Attikum H, PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells, Trends in Cell Biology, 29 (2019) 820–834. [DOI] [PubMed] [Google Scholar]
- [128].Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, Perez-Lopez R, Dolling D, Robinson DR, Sandhu S, Fowler G, Ebbs B, Flohr P, Seed G, Rodrigues DN, Boysen G, Bertan C, Atkin M, Clarke M, Crespo M, Figueiredo I, Riisnaes R, Sumanasuriya S, Rescigno P, Zafeiriou Z, Sharp A, Tunariu N, Bianchini D, Gillman A, Lord CJ, Hall E, Chinnaiyan AM, Carreira S, De Bono JS, Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition, Cancer Discovery, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM, Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas, Journal of Clinical Oncology, 29 (2011) 3008–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Akashi-Tanaka S, Watanabe C, Takamaru T, Kuwayama T, Ikeda M, Ohyama H, Mori M, Yoshida R, Hashimoto R, Terumasa S, Enokido K, Hirota Y, Okuyama H, Nakamura S, BRCAness predicts resistance to taxane-containing regimens in triple negative breast cancer during neoadjuvant chemotherapy, Clinical Breast Cancer, (2015). [DOI] [PubMed] [Google Scholar]
- [131].Wang J, Bian C, Li J, Couch FJ, Wu K, Zhao RC, Poly(ADP-ribose) polymerase-1 down-regulates BRCA2 expression through the BRCA2 promoter, Journal of Biological Chemistry, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K, Weinstock DM, Gorospe M, Harris AL, Helleday T, Chowdhury D, MiR-182-Mediated Downregulation of BRCA1 Impacts DNA Repair and Sensitivity to PARP Inhibitors, Molecular Cell, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].De Summa S, Pinto R, Sambiasi D, Petriella D, Paradiso V, Paradiso A, Tommasi S, BRCAness: A deeper insight into basal-like breast tumors, Annals of Oncology, 24 (2013) viii13–viii21. [DOI] [PubMed] [Google Scholar]
- [134].Liu X, Han EK, Anderson M, Shi Y, Semizarov D, Wang G, McGonigal T, Roberts L, Lasko L, Palma J, Zhu GD, Penning T, Rosenberg S, Giranda VL, Luo Y, Leverson J, Johnson EF, Shoemaker AR, Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways, Molecular Cancer Research, (2009). [DOI] [PubMed] [Google Scholar]
- [135].Kondrashova O, Nguyen M, Shield-Artin K, Tinker VA, Teng NNH, Harrell MI, Kuiper MJ, Ho GY, Barker H, Jasin M, Prakash R, Kass EM, Sullivan MR, Brunette GJ, Bernstein KA, Coleman RL, Floquet A, Friedlander M, Kichenadasse G, O’Malley DM, Oza A, Sun J, Robillard L, Maloney L, Bowtell D, Giordano H, Wakefield MJ, Kaufmann SH, Simmons AD, Harding TC, Raponi M, McNeish IA, Swisher EM, Lin KK, Scott CL, Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma, Cancer Discovery, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Bunting SF, Callén E, Wong N, Chen H-T, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, Xu X, Deng C-X, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A, 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks, Cell, 141 (2010) 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Martin RW, Orelli BJ, Yamazoe M, Minn AJ, Takeda S, Bishop DK, RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors, Cancer Research, (2007). [DOI] [PubMed] [Google Scholar]
- [138].Aly A, Ganesan S, BRCA1, PARP, and 53BP1: conditional synthetic lethality and synthetic viability, Journal of Molecular Cell Biology, 3 (2011) 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Yazinski SA, Comaills V, Buisson R, Genois M-M, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, Ramaswamy S, Benes CH, Haber DA, Maheswaran S, Birrer MJ, Zou L, ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells, Genes & Development, 31 (2017) 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Schlacher K, Wu H, Jasin M, A Distinct Replication Fork Protection Pathway Connects Fanconi Anemia Tumor Suppressors to RAD51-BRCA1/2, Cancer Cell, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, John S, Day A, Crespo AV, Shen B, Starnes LM, De Ruiter JR, Daniel JA, Konstantinopoulos PA, Cortez D, Cantor SB, Fernandez-Capetillo O, Ge K, Jonkers J, Rottenberg S, Sharan SK, Nussenzweig A, Replication fork stability confers chemoresistance in BRCA-deficient cells, Nature, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Cong K, Kousholt AN, Peng M, Panzarino NJ, Lee WTC, Nayak S, Krais J, Calvo J, Bere M, Rothenberg E, Johnson N, Jonkers J, Cantor SB, PARPi synthetic lethality derives from replication-associated single-stranded DNA gaps, bioRxiv, (2019). [Google Scholar]
- [143].Mweempwa A, Wilson MK, Mechanisms of resistance to PARP inhibitors - an evolving challenge in oncology, Cancer Drug Resistance, (2019) 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Jaspers JE, Kersbergen A, Boon U, Sol W, Van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, Doroshow JH, Cranston A, Martin NMB, Lau A, O’Connor MJ, Ganesan S, Borst P, Jonkers J, Rottenberg S, Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors, Cancer Discovery, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Rottenberg S, Jaspers JE, Kersbergen A, Van Der Burg E, Nygren AOH, Zander SAL, Derksen PWB, De Bruin M, Zevenhoven J, Lau A, Boulter R, Cranston A, O’Connor MJ, Martin NMB, Borst P, Jonkers J, High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs, Proceedings of the National Academy of Sciences of the United States of America, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Durmus S, Sparidans RW, Van Esch A, Wagenaar E, Beijnen JH, Schinkel AH, Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699), Pharmaceutical Research, 32 (2015) 37–46. [DOI] [PubMed] [Google Scholar]
- [147].Boccaccio C, Comoglio PM, Invasive growth: A MET-driven genetic programme for cancer and stem cells, Nature Reviews Cancer, (2006). [DOI] [PubMed] [Google Scholar]
- [148].Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang H-LL, Hsu Y-HH, Lin W-CC, Yu W-HH, Leonard PG, Lee GR, Chen M-KK, Nakai K, Hsu M-CC, Chen TC-HHC-T, Sun Y, Wu Y, Chang W-CC, Huang W-CC, Liu C-LL, Chang Y-CC, Park M, Jones P, Hortobagyi GN, Hung M-CC, Blocking c-Met–mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors, Nature Medicine, 22 (2016) 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, James DI, Guerrero Llobet S, Vis DJ, Annunziato S, van den Broek B, Barazas M, Kersbergen A, van de Ven M, Tarsounas M, Ogilvie DJ, van Vugt M, Wessels LFA, Bartkova J, Gromova I, Andújar-Sánchez M, Bartek J, Lopes M, van Attikum H, Borst P, Jonkers J, Rottenberg S, Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality, Cancer Cell, 33 (2018) 1078–1093.e1012. [DOI] [PubMed] [Google Scholar]
- [150].Oplustilova L, Wolanin K, Mistrik M, Korinkova G, Simkova D, Bouchal J, Lenobel R, Bartkova J, Lau A, O’Connor MJ, Lukas J, Bartek J, Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment, Cell Cycle, 11 (2012) 3837–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, Issaeva N, Sleeth K, Sharma RA, Helleday T, Poly(ADP-Ribose) Polymerase Is Hyperactivated in Homologous Recombination–Defective Cells, Cancer Research, 70 (2010) 5389–5398. [DOI] [PubMed] [Google Scholar]
- [152].Ngan CH, Bohnuud T, Mottarella SE, Beglov D, Villar EA, Hall DR, Kozakov D, Vajda S, FTMAP: Extended protein mapping with user-selected probe molecules, Nucleic Acids Research, 40 (2012) 271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Volkamer A, Kuhn D, Rippmann F, Rarey M, DoGSiteScorer: a web server for automatic binding site prediction, analysis and druggability assessment, Bioinformatics, 28 (2012) 2074–2075. [DOI] [PubMed] [Google Scholar]
- [154].Liang J, Edelsbrunner H, Woodward C, Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design, Protein Sci, 7 (1998) 1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Loving KA, Lin A, Cheng AC, Structure-Based Druggability Assessment of the Mammalian Structural Proteome with Inclusion of Light Protein Flexibility, PLoS Computational Biology, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Lloyd RL, Wijnhoven PWG, Ramos-Montoya A, Wilson Z, Illuzzi G, Falenta K, Jones GN, James N, Chabbert CD, Stott J, Dean E, Lau A, Young LA, Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells, Oncogene, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Marijon H, Lee DH, Ding LW, Sun H, Gery S, de Gramont A, Koeffler HP, Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells, Biomedicine and Pharmacotherapy, (2018). [DOI] [PubMed] [Google Scholar]
- [158].Yi M, Dong B, Qin S, Chu Q, Wu K, Luo S, Advances and perspectives of PARP inhibitors, Experimental Hematology & Oncology, 8 (2019) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Sigrist CJA, De Castro E, Langendijk-Genevaux PS, Le Saux V, Bairoch A, Hulo N, ProRule: a new database containing functional and structural information on PROSITE profiles, Bioinformatics, 21 (2005) 4060–4066. [DOI] [PubMed] [Google Scholar]
- [160].Durrant JD, Votapka L, Sorensen J, Amaro RE, POVME 2.0: An Enhanced Tool for Determining Pocket Shape and Volume Characteristics, Journal of Chemical Theory and Computation, 10 (2014) 5047–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.