

Abstract
The SARS‐CoV‐2 pandemic imposed a large burden on health and society. Therapeutics targeting different components and processes of the viral infection replication cycle are being investigated, particularly to repurpose already approved drugs. Spike protein is an important target for both vaccines and therapeutics. Insights into the mechanisms of spike‐ACE2 binding and cell fusion could support the identification of compounds with inhibitory effects. Here, we demonstrate that the integrity of disulfide bonds within the receptor‐binding domain (RBD) plays an important role in the membrane fusion process although their disruption does not prevent binding of spike protein to ACE2. Several reducing agents and thiol‐reactive compounds are able to inhibit viral entry. N‐acetyl cysteine amide, L‐ascorbic acid, JTT‐705, and auranofin prevented syncytia formation, viral entry into cells, and infection in a mouse model, supporting disulfides of the RBD as a therapeutically relevant target.
Keywords: ACE2, disulfides, SARS‐CoV‐2, spike, thiol‐reacting compounds
Abbreviations
- ACE2
angiotensin‐converting enzyme‐2
- BFP
blue fluorescent protein
- DTT
dithiothreitol
- GFP
green fluorescent protein
- NAC
N‐acetylcysteine
- NACA
N‐acetyl cysteine amide
- RBD
receptor‐binding domain
- RBM
receptor‐binding motif
- RFP
red fluorescent protein
- TMPRSS2
transmembrane protease serine 2
1. INTRODUCTION
The COVID‐19 pandemic is one of the most serious medical emergencies since the last century. SARS‐CoV‐2, which was first reported at the end of 2019, has affected the entire world, and there are still very few therapeutic options. One of the fastest ways toward therapy would be the repurposing of already approved drugs and dietary compounds. Several drugs have been shown to inhibit viral replication by targeting either viral components, such as inhibitors of viral RNA polymerases, 1 , 2 or compounds that target the human cell proteins that interact with or process viral components, such as protease or kinase inhibitors [reviewed in 3, 4]. Approved compounds that might interfere with the binding of spike proteins to human angiotensin‐converting enzyme‐2 (ACE2) viral receptors have been proposed. 5 For some compounds, the mechanism of inhibition or molecular target is not clear although the effect on viral replication has been observed. An example is auranofin. 6 Insight into the mechanism of action is crucial, as it can guide the design of more effective inhibitors, combination therapies, or type of therapeutic application.
SARS‐CoV and SARS‐CoV‐2 spike fusion proteins need to interact with the ACE2 receptor to attach to and enter target cells. For some viruses, notably the human immunodeficiency virus (HIV‐1), the redox state of the fusion protein has been shown to be important for fusion. 7 , 8 The RBD of SARS‐CoV comprises two disulfide bridges, but SARS‐CoV‐2 contains four disulfide bridges. Lavillette et al 9 showed the relative redox insensitivity of SARS‐CoV spike protein compared with HIV‐1, as only DTT was able to inhibit viral fusion. However, a recent computational study suggested that the binding affinity of RBD from SARS‐CoV‐2 with ACE2 may become less favorable when all disulfide bridges of RBD are disrupted by a reduction to thiols. 10
There are several known thiol redox compounds that might disrupt the disulfide bonds of the RBD. One compound of this type is the thiol antioxidant N‐acetylcysteine (NAC) and its derivate N‐acetylcysteine amide (NACA), which has better chelating and antioxidant properties than NAC. 11 NAC has already been reported to elicit clinical improvement in COVID‐19 patients. 12 Several publications have also proposed L‐ascorbic acid (vitamin C) as a prophylaxis with other antioxidant compounds for the treatment of COVID‐19. 13 Another interesting thiol‐reactive compound is auranofin, an antirheumatic drug that has been proposed for possible use in the treatment of parasitic, bacterial, and viral infections [reviewed in 14]. Recently, auranofin has been identified as an inhibitor of SARS‐CoV‐2 replication without the identification of its inhibitory mechanism or target. 6 Auranofin targets thiol redox homeostasis and has previously been shown to bind to SH groups via its gold atom. 15 A similar mechanism of action has been shown for JTT‐705, 15 an inhibitor of cholesteryl ester transfer protein (CETP), which entered a phase III clinical trial for hypercholesterolemia but was terminated because of ineffectiveness. 16
Here, we show that several thiol/disulfide‐reactive compounds (NACA, L‐ascorbic acid, JTT‐705, and auranofin) inhibit spike and ACE2 protein‐dependent cell fusion as well as viral entry into cells. Moreover, those compounds also prevented infection in a mouse model. Interestingly, some similar compounds, such as aurothioglucose or NAC, had no effect. To reveal the inhibitory mechanism, a flow cytometry‐based assay was established, which allowed us to differentiate between spike‐ACE2 binding and the membrane fusion process. NACA, L‐ascorbic acid, and JTT‐705 were efficient in preventing cell fusion. The importance of disulfides within the RBD that could be targeted by the compounds was confirmed by mutating selected cysteine residues, which almost completely abrogated the cell fusion process but not spike‐ACE2 binding, most likely by preventing spike conformational changes. We propose that the integrity of disulfides within the RBD of the spike protein is essential for fusion and that their targeting by several approved drugs may represent a therapeutic option to combat the disease.
2. MATERIALS AND METHODS
2.1. Materials
Auranofin and JTT‐705 were purchased from Cayman Chemicals. L‐ascorbic acid, dithiothreitol (DTT), N‐acetylcysteine (NAC), N‐acetylcysteine amide (NACA), aurothiomalate, aurothioglucose, and reduced glutathione were purchased from Sigma.
2.2. Plasmids
Spike expression plasmids for SARS‐CoV‐2 pCG1‐S and human ACE2 expression plasmid pCG1‐hACE2 were a gift from Stefan Pöhlmann (German Primate Center––Germany). 17 Split firefly luciferase expression plasmids were prepared by cloning N7 coiled‐coil‐forming peptide with C‐terminal part of the firefly luciferase (pcDNA3‐cLuc:N7) and N8 coiled‐coil‐forming peptide with N‐terminal part of the firefly luciferase (pcDNA3‐ nLuc:N8). The split GFP reporter was designed by splitting the sequence of sfGFP between the 10th and the 11th β‐strands into two parts: GFP1‐10 and GFP11. 18 Split GFP expression plasmids were prepared by cloning N7 coiled‐coil‐forming peptide to GFP(1‐10) (pcDNA3‐GFP(1‐10):gs:N7) and a chain of N8 coiled‐coil‐forming peptides to GFP(11) (pcDNA3‐3x(N8:gs:GFP11)). 19
The spike mutants Cys336Ser, Cys361Ser, Cys379Ser, Cys391Ser, Cys480Ser, Cys488Ser, and Cys590Ser were prepared from pCG1‐S plasmid using quick change PCR. Mutations were confirmed by sequence analysis.
2.3. Cellular fusion detection assay
HEK293T (seeded at 7.0 × 105/well) cells were transiently transfected in 6‐well plate with different amounts of pCG1‐S wt (from 0.01 to 0.5 µg) and pcDNA3‐cLuc:N7 (1.0 µg) or pCG1‐hACE2 (from 0.01 to 0.5 µg) and pcDNA3‐ nLuc:N8 (1.0 µg) plasmids and pcDNA3 plasmids to reach 2.0 µg of plasmids/transfection using a PEI transfection reagent. After 24 hours, the cells were detached using cold PBS with 2.5 mM EDTA and counted. Ten thousand cells expressing spike protein were mixed with 10 000 cells expressing ACE2 protein and plated to 96‐well plate for 3 hours. Syncytia formation was measured after the addition of 2 mM D‐luciferin (Xenogen) with 0.5 mM ATP on a luminometer Orion (Berthold Technology).
To measure TMPRSS2 contribution to cell fusion, HEK293T cells were transfected with pCG1‐S wt (0.01 µg) and pcDNA3‐cLuc:N7 (1.0 µg) or pCG1‐hACE2 (0.01 µg), pCMV3‐c‐Myc‐TMPRSS2 (0.1 µg) and pcDNA3‐ nLuc:N8 (1.0 µg) and pcDNA3 plasmid to reach 2.0 µg of plasmids/transfection using PEI transfection reagents. Camostat mesylate was added to 10 000 cells expressing ACE protein and mixed with 10 000 cells expressing spike protein and plated to 96‐well plate for 3 hours. Syncytia formation was measured.
To test the efficiency of compounds, HEK293T cells were transiently transfected with either pCG1‐S wt (0.5 µg) and pcDNA3‐cLuc:N7 (1.0 µg) or pCG1‐hACE2 (0.5 µg) and pcDNA3‐ nLuc:N8 (1.0 µg) and pcDNA3 plasmid to reach 2.0 µg of plasmids/transfection using PEI transfection reagents. Ten thousand cells expressing spike protein were preincubated with tested compounds for 30 minutes at 37°C and mixed with 10 000 cells expressing ACE2 protein and plated to 96‐well plate for 3 hours. Syncytia formation was measured.
To measure the fusogenic activity of Cys mutants, HEK293T cells were transiently transfected with either pCG1‐S wt (0.03 µg), pCG1‐S C488S (0.2 µg), pCG1‐S C480S (0.5 µg), or pCG1‐S C379S (1.0 µg) and pcDNA3‐cLuc:N7 (1.0 µg) or pCG1‐hACE2 (0.5 µg) and pcDNA3‐ nLuc:N8 (1.0 µg) and pcDNA3 plasmid to reach 2.0 µg of plasmids/transfection using PEI transfection reagents. Ten thousand cells expressing spike protein were mixed with 10 000 cells expressing ACE2 protein and plated to 96‐well plate for 3 hours. Syncytia formation was measured.
2.4. VSVΔG* pseudoviral assay
A pseudovirus system based on the vesicular stomatitis virus 20 was utilized to follow the effect of the compounds on the entry of the virus. The preparation of a pseudovirus was described previously. 17 , 21 Pseudoviral genome encodes for GFP and firefly luciferase, which are expressed upon infection. For the neutralization assay, HEK293 were seeded (2.5 × 104/well) a day before transfection with pCG1‐hACE2 and phRL‐TK (encoding Renilla luciferase; Invitrogen) plasmids. Compounds were preincubated with spike pseudovirus for 30 minutes before being added to the cells. The next day, the medium was removed and the cells were lysed in Passive lysis buffer (Biotium). Luciferin substrate (Xenogen) was used to detect firefly luciferase activity as a measure of the pseudovirus infection, and coelenterazine H (Xenogen) was used to follow Renilla luciferase activity for the determination of transfection efficiency and normalization on luminometer Orion. For auranofin, GFP fluorescence (excitation 485 nm/emission 530 nm) upon pseudoviral infection was measured on a multiplate reader Synergy Mx (BioTek).
2.5. Western blot
HEK293T cells (seeded at 7.0 × 105/well in a 6‐well plate) were transiently transfected with wt or each Cys mutant plasmid. The next day, cells were lysed and 30 µg of proteins was used for detection. SDS‐PAGE and WB were performed. Spike protein was detected using anti‐S Ab (Abcam; 1:1000) and anti‐rabbit secondary Ab with HRP (1:4000). Bands were visualized using West Pico reagent. Anti‐β‐actin Ab (Cell signaling 1:5000; anti‐mouse sAb, Jackson Immuno Research, 1:3000) was used as a loading control.
2.6. Flow cytometry
For the analysis of surface expression of Cys mutants, HEK293T cells (seeded at 7.0 × 105/well in 6‐well plate) were transiently transfected with wt or each Cys mutant plasmid. Different amounts of Cys mutant and wt spike plasmids were used to reach a similar surface expression. Plasmid pcDNA3 was used to adjust the amount of plasmid DNA. The next day, cells were washed with ice‐cold PBS and incubated for 30 minutes on ice with anti‐Spike Ab (GeneTex; 1:100), washed and further incubated for 30 minutes with anti‐mouse sAb‐AF488 (Invitrogen; 2 µg/mL) on ice. Afterward, cells were washed with PBS and analyzed on a spectral flow cytometer with 405‐nm 100 mW violet, 488‐nm 50 mW blue, and 650‐nm 80 mW red lasers (Aurora, Cytek). Data were analyzed using FlowJo software (Tree Star).
For the analysis of syncytia formation, HEK293T cells (seeded at 7.0 × 105/well in 6‐well plate) were transiently transfected with indicated amounts of wt or each Cys mutant plasmid, pcDNA3‐iRFPNLS (200 ng) and pcDNA3‐GFP(1‐10):gs:N7 (800 ng) or pCG1‐hACE2 (500 ng), pcDNA3‐BFPNLS (200 ng) and pcDNA3‐3x(N8:gs:GFP11) (1300 ng) using a PEI transfection reagent. Plasmid pcDNA3 was used to adjust the amount of plasmid DNA. The next day, the cells were detached using cold PBS with 2.5 mM EDTA, and 1 × 105 of spike‐expressing cells were mixed with 1 × 105 of ACE2‐expressing cells and seeded to 48‐well plate. Syncytia were analyzed 3 hours later after detaching the cells using cold PBS with 2.5 mM EDTA, centrifugation, and resuspension in medium. Cells from three wells were combined and measured on flow cytometer. For testing the inhibitory efficiency of compounds, cells were transfected with either 500 ng of spike plasmid (for NACA and auranofin) or 30 ng for L‐ascorbic acid and JTT‐705. Spike‐expressing cells were incubated for 30 minutes with certain compounds before mixing them with ACE2‐expressing cells.
2.7. Confocal microscopy
For confocal microscopy, HEK293T acceptor cells were seeded in an 8‐well tissue culture chamber (µ‐Slide 8 well, Ibidi Integrated BioDiagnostics) at 5 × 104 cells/well and transfected with pCG1‐hACE2 (40 ng), pcDNA3‐EGFP (100 ng), and w/o pcDNA3‐BFP:KRf [which anchors plasma membrane, 22] (100 ng) plasmids using a PEI transfection reagent. The HEK293T donor cells were seeded in 24‐well plate at 1 × 105 cells/well. The donor cells were transfected with pCG1‐S wt (25 ng) and pcDNA3‐iRFP (500 ng) plasmid. Plasmid pcDNA3 was used to adjust the amount of plasmid DNA. The next day, the spike‐expressing cells were detached using a cold PBS with 2.5 mM EDTA, and 1 × 105 cells were added to the acceptor cells. Syncytia were analyzed 3 hours later.
Microscopic images were acquired using the Leica TCS SP5 inverted laser‐scanning microscope on a Leica DMI 6000 CS module equipped with an HCX Plane‐Apochromat lambda blue 63× oil‐immersion objectives with NA 1.4 (Leica Microsystems). A 488‐nm laser line of a 100‐mW argon laser with 10% laser power was used for EGFP excitation, and the emitted light was detected between 500 and 540 nm; a 10‐mW 633‐nm HeNe laser was used for iRFP excitation, and emitted light was detected between 580 and 620 nm. A 50‐mW 405‐nm diode laser was used for BFP, and emitted light was detected between 420 and 460 nm. The images were processed with LAS AF software (Leica Microsystems) and ImageJ software (National Institute of Mental Health).
2.8. Inhibition of spike/ACE2 interaction
Recombinant human ACE2‐Fc protein (Genescript) was absorbed to ELISA plates at 2 µg/mL in 50 µL/well of phosphate buffer saline (PBS) at 4°C overnight. The plates were washed five times using PBS + 0.01% Tween and blocked in 0.45 mM BSA in PBS for 1 hour. Recombinant spike‐STREP tag (0.2 µg/mL) was prepared as described in 21.The protein was incubated with the indicated concentrations of compounds: NACA (0.5, 2, 5, and 10 mM), auranofin (0.1, 0.2, 0.5, and 1 mM), L‐ascorbic acid (5, 10, and 20 mM), and JTT‐705 (20, 50, and 200 µM) for 1 hour in PBS at 37°C. Then samples were added to wells and incubated for an additional 2 hours at room temperature. After washing, the plate was incubated with STREP‐Tactin‐HRP (1:10 000) in BSA/PBS for 1 hour at room temperature. After the final wash, TMB substrate was added and the reaction was stopped by the addition of 3 M H3PO4. Absorbance was measured using a Synergy Mx microtiter plate reader (Biotek). DMSO and EtOH backgrounds were added to the auranofin and JTT‐705 results.
2.9. Surrogate assay of the prevention of pseudoviral infections by inhibitors
To test the possible inhibitory effects of various substances, female 8‐ to 10‐week‐old Balb/c OlaHsd mice (Envigo, Italy) were used for experiments. All animal experiments were performed according to the directives of the EU 2010/63 and were approved by the Administration of the Republic of Slovenia for Food Safety, Veterinary and Plant Protection of the Ministry of Agriculture, Forestry and Foods, Republic of Slovenia (Permit Number U34401‐8/2020/9, U34401‐8/2020/15, U34401‐12/2020/6).
Laboratory animals were housed in IVC cages (Techniplast), fed standard chow (Mucedola), and provided with tap water ad libitum. Mice were maintained in a 12‐12 hour dark‐light cycle. All animals used in the study were healthy and accompanied by health certificates from the animal vendor.
Balb/c mice were given inhibitors NACA (3 mg/mouse dissolved in PBS) and auranofin (0.25 mg/mouse dissolved in 2% DMSO, 8.5% EtOH, and 5% PEG‐600) intraperitoneally (200 μL/animal) on days 1 and 2. On day 2, mice were transfected with a 30 μL mixture of in vivo‐jetPEI and plasmid DNA (20 μg pCG1‐hACE2, 1 μg pCMV3‐c‐Myc‐TMPRSS2 per animal) via intranasal administration in inhalation anesthesia. For potential inhibition with L‐ascorbic acid, 3% L‐ascorbic acid suspensions in tap water were given to mice for 6 d. Suspension was changed every 2 days. On day 6 of the treatment, the mice were transfected with hACE2 and TMPRSS2, as described above.
A day after the intranasal transfection, the mice were intranasally infected with 70 μL of VSV‐Spike pseudovirus. Twenty‐four hours later, mice received 150 mg/kg of body weight of D‐luciferin (Xenogen) subcutaneously and were imaged in vivo using IVIS Lumina Series III (Perkin Elmer). Bioluminescence that depicted the state of pseudoviral infection was determined. Values of mice background were subtracted from bioluminescence values. Data were analyzed using Living Image 4.5.2 (Perkin Elmer).
2.10. Viability/metabolic activity XTT assay
Compounds at indicated concentrations were added to HEK293T cells (seeded at 2 × 104/well in 96‐well plate) and incubated for 3 hours. Medium was changed for DMEM + 10% FBS without phenol red (100 µL) and 50 µL of solution of tetrazolium salt (XTT), and phenazine methosulfate (PMS) were added. Upon color development absorbance at 490 nm using a multiplate reader, Synergy Mx (BioTek) was measured.
2.11. Reporter virus strain, stock preparation, plaque assay, and in vitro infection
SARS‐CoV‐2‐GFP (PMID: 32365353) was produced by infecting Vero E6 cells cultured in DMEM medium (10% FCS, 100 µg/mL Streptomycin, and 100 IU/mL Penicillin) for 2 d (MOI 0.01). Viral stock was harvested and spun twice (1000 g/10 min) before storage at −80°C. Titer of viral stock was determined by plaque assay. Confluent monolayers of Vero E6 cells were infected with serial fivefold dilutions of virus supernatants for 1 hour at 37°C. The inoculum was removed and replaced with serum‐free MEM (Gibco, Life Technologies) containing 0.5% carboxymethyl cellulose (Sigma‐Aldrich). Two days post‐infection, cells were fixed for 20 minutes at room temperature with formaldehyde directly added to the medium to a final concentration of 5%. Fixed cells were washed extensively with PBS before staining with H2O containing 1% crystal violet and 10% ethanol for 20 minutes. After rinsing with PBS, the number of plaques was counted and the virus titer was calculated.
2.12. SARS‐CoV‐2‐GFP reporter virus assay
A549‐ACE2 cells were seeded into 96‐well plate at a density of 7500 cells per well in DMEM medium (10% FCS, 100 µg/mL Streptomycin, and 100 IU/mL Penicillin) 1 day before the assay. Inhibitors or a corresponding amount of vehicle solutions (2.5% PBS for NACA and 0.125% DMSO for auranofin) were added to the cells 3 hours prior to infection. Virus inoculum, corresponding to an MOI 1, was further added to the supernatant of the cells. The cells were incubated at standard culturing conditions before the confluence, and GFP intensity was measured using the IncuCyte S3 Live Cell Analysis System (4x magnification, one image per well, phase and GFP channel). Normalized GFP intensity was computed using IncuCyte S3 Software (Essen Bioscience; version 2019B Rev2) as integrated GFP intensity per well divided by the total area of the cells per well. The GFP intensity of the treated samples was normalized to the matching technical replicate of the vehicle control and multiplied by 100 (%).
2.13. LC‐MS/MS analysis of the disulfides of the spike protein
Purified RBD/spike protein (0.25 µg) was treated with auranofin (20 µM) for 1 hour at 37°C and afterward iodoacetamide (15 µM) treated for 1 hour in dark ST was added in order to alkylate. Later, the samples were digested with trypsin (1 µL of 250 ng/µL) (MS grade, Sigma) overnight and purified over a C18 ZipTip (Merck Millipore). Each eluate was dried and redissolved in 20 µL of 0.1% TFA and 2% acetonitrile. Eight microliters were injected onto nano RP‐HPLC in a line connected to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). Samples were analyzed with a 1 hour LC‐MS/MS gradient in high‐energy collision ion trap (HCD‐IT) mode. Peak list was created with Mascot Distiller (Matrix Science) and searched against the sequences of the S and RBD proteins. By comparing the peptide coverage of each sample bearing the cysteines in mind, we identified cysteines that might have different oxidative states in different samples. The areas of the peptides containing the covered cysteines between samples and the area normalized to non‐cysteine peptides were compared using Skyline software (Sciex). Peptide standards to normalize against are as follows: for RBD (trypsin) RVQPTESIVRF, KSNLKPFERD, and KIADYNYKL; for spike (trypsin) RGVYYPDKV, RVQPTESIVRF, and KVGGNYNYLYRL.
2.14. Statistical analysis
GraphPad Prism 8 was used to prepare the graphs and perform statistical analysis. Bar data from independent experiments are represented as mean ± SEM. Unpaired two‐tailed Student's t test was used for the comparison of the two groups.
3. RESULTS
3.1. A sensitive split luciferase assay enables the measurement of syncytia formation
The interaction of the spike protein of coronaviruses and a human receptor promotes membrane fusion and the formation of multinucleated cells—syncytia. This has been shown for SARS‐CoV as well as for SARS‐CoV‐2. 23 , 24 Previous reports analyzed syncytia formation between two cell types that express either spike protein or ACE‐2 at their membrane using fluorescence labeling. 24 , 25 , 26 In another study Jun/Fos and split reporter system was used, which reconstituted when the syncytium, triggered by the Nipah virus F and G protein that interacted with the Ephrin B2 receptor on other cells, was formed. 27 To increase the sensitivity and streamline the analysis, we developed a split luciferase reporter assay based on our previously designed tightly interacting N7/N8 coiled‐coil peptide pair, 19 where each of the N7/N8 peptide of the pair was attached to one fragment of the split firefly luciferase. Upon syncytium formation, cLuc:N7 and nLuc:N8, which are initially in different cells, are localized to the same compartment and form a coiled‐coil dimer with concomitant luciferase reconstitution. The reporter system was sensitive to the amount of spike and the number of ACE2 proteins expressed on donor and acceptor cells, respectively (Figure 1A). Although spike protein and ACE2 are sufficient for syncytia formation (Figure 1B), transmembrane protease serine 2 (TMPRSS2) co‐expression on ACE2 cells further promoted syncytia formation (Figure 1C), thus increasing the sensitivity of the system, which was validated by inhibition with the camostat mesylate, a TMPRSS2 inhibitor.
FIGURE 1.
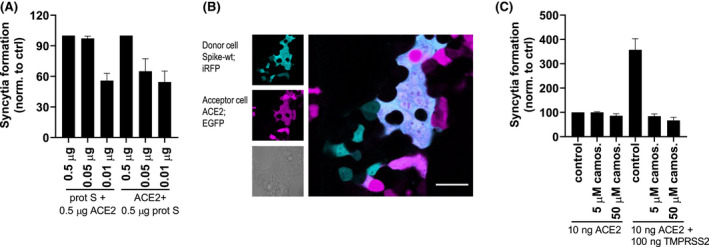
A sensitive split luciferase assay enables the measurement of cell fusion. A, HEK239T were transfected with different amounts of spike and cLuc:N7 or ACE2 and nLuc:N8 plasmids. After 24 hours, the cells were mixed and seeded. After 3 hours, cell fusion was detected by measuring luciferase activity. B, Instead of split luciferase, EGFP and iRFP were transfected along with spike and ACE2 plasmids. Formation of syncytia was observed. C, HEK239T were transfected with spike and cLuc:N7 or ACE2 (and/or TMPRSS2) and nLuc:N8 plasmids. Camostat mesylate was added to ACE2‐expressing cells prior to mixing with spike‐expressing cells. After 3 hours, cell fusion was detected by measuring luciferase activity. Combined means from three (A and C) independent experiments are shown as mean ± SEM. Data from (B) are a representative of three independent experiments
3.2. Thiol/disulfide‐reactive compounds inhibit syncytia formation
Since the RBD of spike contains several disulfide bridges or cysteine residues in the proximity that could form disulfides that have in some viruses been shown to be sensitive to the reduction (Fenouillet et al, 2007), we wanted to investigate whether cysteine residues could represent a target for the inhibition of viral cell entry. Several thiol‐reactive compounds among the registered drugs were tested for their effects on cell fusion (Figure 2). NAC did not inhibit cell fusion; however, its derivate NACA, with more potent antioxidant properties, strongly inhibited the formation of syncytia (Figure 3A). L‐ascorbic acid also efficiently inhibited cell fusion (Figure 3B), but no inhibition was observed with reduced glutathione (Figure 3C). We showed before that auranofin and JTT‐705 can interact with the SH group and inhibit the functioning of MD‐215; therefore, we reasoned that those compounds might interact with the cysteine residues of RBD. Preincubation of spike protein‐expressing cells with either of those compounds efficiently prevented syncytia formation (Figure 3D,E) without affecting cell metabolic activity at concentrations used (Figure S1). Interestingly, two other auranofin‐related gold‐containing compounds, aurothiomalate and aurothioglucose, that also have antirheumatic activity, were tested, but none of them exhibited inhibitory activity (Figure 3F).
FIGURE 2.
Thiol‐reducing antioxidant compounds. Several compounds with thiol‐reducing and antioxidant properties NAC, NACA, L‐ascorbic acid, reduced glutathione, auranofin, JTT‐705, aurothiomalate, and aurothioglucose were selected to inhibit spike and ACE2 interaction and fusion
FIGURE 3.
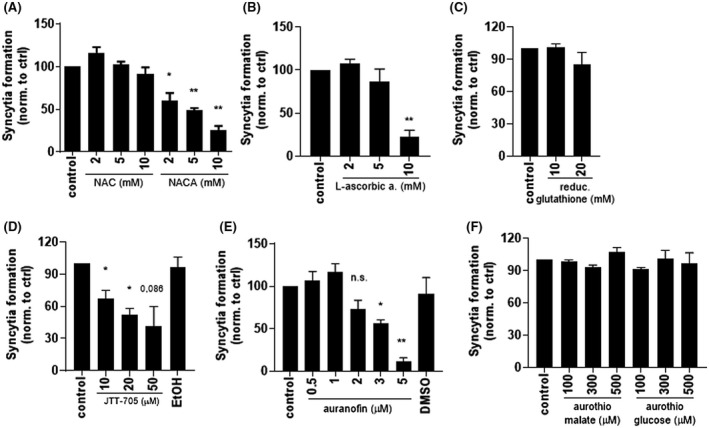
Thiol/disulfide‐reacting compounds inhibit syncytia formation. A‐F, HEK293T spike‐expressing cells were preincubated for 30 minutes with the indicated concentrations of compounds. ACE2‐expressing cells were added, mixed, and seeded. After 3 hours, cell fusion was detected by measuring luciferase activity. Combined means from two (F) or three (A‐E) independent experiments are shown as mean ± SEM P values of <.05 (*), <.01 (**) are indicated; n.s.‐not significant. See also Figure S1
3.3. Mutations of cysteine residues of RBD diminish the fusogenicity of the spike protein
A previous computational study suggested that the binding affinity of SARS‐CoV‐2 spike RBD to ACE2 becomes less favorable if the disulfides of RBD are reduced. 10 The impact of the reduction of disulfides on spike‐ACE2 interaction has been reported as relatively weak for SARS‐CoV, 10 as only millimolar concentrations of DTT prevented fusion. 9 To test the effect of reducing agents on SARS‐CoV‐2 binding, we incubated cells expressing spike protein with various concentrations of DTT (0.2, 0.5, and 1 mM) for 30 minutes before mixing them with ACE2‐expressing cells. DTT efficiently inhibited syncytia formation at 0.5 mM (Figure 4A), implying that the reduction of disulfides of spike protein might be effective in inhibiting the interaction. To investigate the effect of disulfide formation, several cysteine residues in cysteine pairs 336‐361, 391‐525 and 379‐432, which help to stabilize the β sheet structure of the RBD, and 480‐488, which connects the loop in the receptor‐binding motif (RBM) 28 (Figure 4B), were mutated. Although all six mutants were detected in the cell lysate at similar amounts (Figure S2A), some mutants were presented in lower amounts at the cell membrane compared with the wt protein. As cysteine pairs 379‐432 and 480‐488 lie at the border or within RBM (residues 438‐506), 28 we reasoned that those residues might affect the binding of spike to ACE2 and were further evaluated. To compare their ability to interact with ACE2 and promote cell fusion, the amounts of transfection plasmids were increased to achieve comparable presentation of Cys379Ser, Cys480Ser, and Cys488Ser mutants at the cell membrane as the wt protein. At comparable membrane amounts of wt and mutated spike protein (Figure S2B), formation of syncytia by Cys488Ser was inhibited by ~70% compared with wt protein, while Cys480Ser and especially Cys379Ser lost almost all ability to support syncytia formation (Figure 4C), demonstrating the importance of disulfides within RBD.
FIGURE 4.
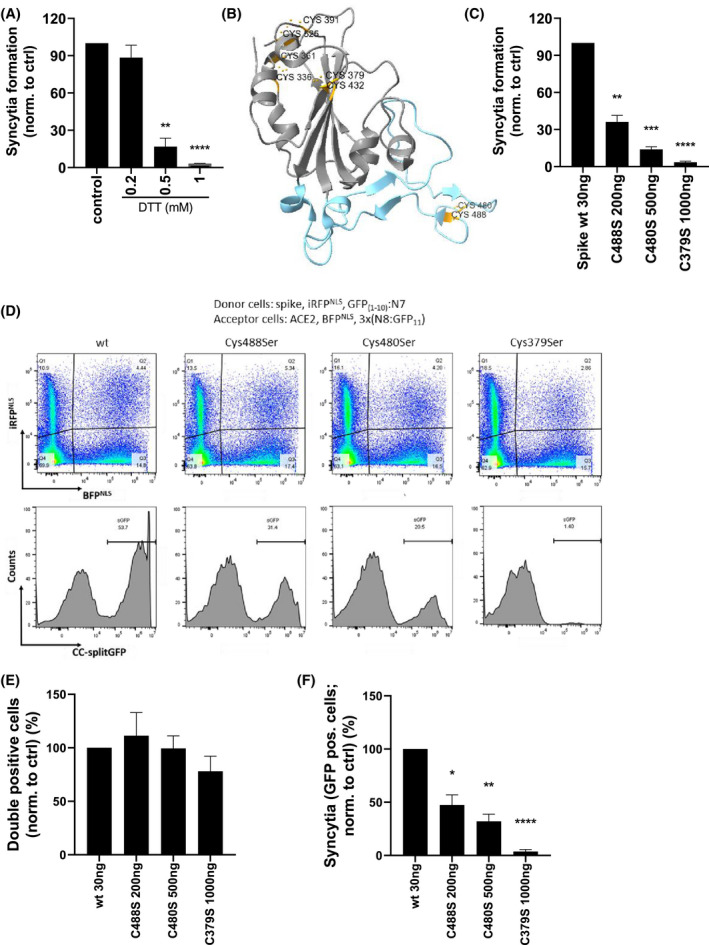
Mutation of cysteine residues in RBM decreases the fusogenic ability of spike protein. A, HEK293T spike‐expressing cells were preincubated for 30 minutes with the indicated concentrations of DTT. ACE2‐expressing cells were added, mixed, and seeded. After 3 hours, cell fusion was detected by measuring luciferase activity. B, Structure of SARS‐CoV‐2 spike RBD with indicated cysteine pairs (orange) and RBM (blue). C, HEK239T were transfected with the indicated amounts of spike wt or cysteine mutants and cLuc:N7 or ACE2 and nLuc:N8 plasmids. After 24 hours, the cells were mixed and seeded. After 3 hours, cell fusion was detected by measuring luciferase activity. D‐F, HEK239T were transfected with the indicated amounts of spike wt or cysteine mutants, iRFPNLS and split GFP(1‐10):N7 or ACE2, BFPNLS, and split 3x(N8:GFP11) plasmids. The fusogenic ability of the spike was determined by syncytia formation after mixing with ACE2‐expressing cells and measuring on a flow cytometer after 3 hours. The percentage of double‐positive cells (E) and syncytia (F) normalized to spike wt‐expressing cells is shown. Combined means from three (A, C) or pooled data from four (E, F) independent experiments are shown as mean ± SEM P values of <.05 (*), <.01 (**), <.001 (***), or .0001 (****) are indicated; (D) is representative of four independent experiments. See also Figures S2 and S3
To elucidate whether spike‐ACE2 binding or the cell fusion process was impaired, a flow cytometric analysis that is able to distinguish between those two processes was established. Spike wt‐ or mutant‐expressing cells cotransfected with iRFPNLS and split GFP(1‐10):N7, ACE2‐expressing cells cotransfected with BFPNLS, and split 3x(N8:GFP11) were mixed and incubated for 3 hours. The formation of either double‐positive (iRFP and BFP) cells, mainly representing cells where spike and ACE2 interact, or GFP‐positive cells, representing syncytia as a subset within the double‐positive population, was detected (Figure S3A,B and Figure 4D). Cys379Ser, Cys480Ser, and Cys488Ser interacted with ACE2 to a similar extent as wt spike (Figure 4E), implying that a single cysteine residue mutation does not affect spike‐ACE2 interaction. Moreover, the formation of syncytia was severely impaired for all three mutants (Figure 4F) in agreement with split luciferase assay results, suggesting that mutated cysteine residues destabilize spike protein and prevent cell fusion. In the Cys379Ser mutant, no syncytia were observed by confocal microscopy as well (Figure S3C). Prevention of cell fusion by a single cysteine mutant in RBD, while maintaining binding to ACE2, implies that transition to a post‐fusion conformation may require an RBD with intact integrity, while a lack of a single disulfide decreases its stability.
3.4. Thiol/disulfide‐reactive compounds either inhibit spike‐ACE2 interaction or prevent cell fusion
The selected compounds were tested for inhibition of binding of spike to ACE2. NACA, L‐ascorbic acid, and JTT‐705 efficiently inhibited this interaction in spike‐ACE2 binding assay (Figure 5A), implying this group of compounds can affect direct interaction between the spike and ACE2. Auranofin, moreover, did not prevent the binding of spike to ACE2 in this assay; therefore, we further checked by mass spectrometric analysis whether auranofin can bind or modify cysteine residues within RBD. We could not detect covalent binding of auranofin to any of the cysteine residues; however, the redox state of specific cysteines was altered, especially Cys379 and Cys525 (Figure S4), which was, however, not sufficient to prevent spike and ACE2 interaction.
FIGURE 5.
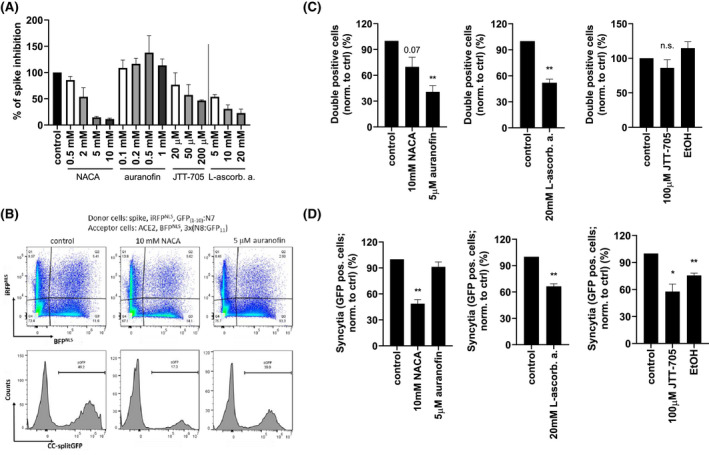
Compounds inhibit spike and ACE2 interaction as well as cell fusion. A, Spike protein was incubated with the indicated concentrations of compounds or buffer for 1 hour and added to human ACE2‐coated plates for an additional 2 hours. Bound spike protein was detected with streptactin‐HRP by measuring absorbance. B‐D, HEK239T were transfected with spike, iRFPNLS and split GFP(1‐10):N7 or ACE2, BFPNLS and split 3x(N8:GFP11) plasmids. Spike‐expressing cells were incubated for 30 minutes with the indicated concentrations of compounds. ACE2‐expressing cells were added, mixed, and seeded. After 3 hours, cell fusion was detected using flow cytometer. Percentage of double‐positive cells (C) and syncytia (D) normalized to untreated cells (control) is shown. Combined means from two (A) and pooled data from four (C, D) independent experiments are shown as mean ± SEM P values of <.05 (*), <.01 (**) are indicated; n.s.‐not significant. (B) is representative of four independent experiments. See also Figure S4
The same flow cytometry experiment as for disulfide mutants was also performed for the compounds. The compounds were preincubated with spike‐expressing cells for 30 minutes, followed by incubation with ACE2‐expressing cells (Figure 5B). All compounds reduced the number of double‐positive cells (Figure 5C). Additionally, NACA, L‐ascorbic acid, and JTT‐705, but not auranofin, inhibited the cell fusion process (Figure 5D), suggesting that reduction of disulfides prevented binding as well as fusion, while auranofin is likely targeting a different process.
3.5. Thiol/disulfide‐reactive compounds inhibit viral entry
NACA, L‐ascorbic acid, JTT‐705, and auranofin were tested for the inhibition of spike protein‐pseudotyped virus infection of ACE2‐expressing HEK293 cells. NACA, L‐ascorbic acid and, to a lower extent, JTT‐705 inhibited pseudoviral entry (Figure 6A‐C). The assay uses Renilla luciferase expression in acceptor cells for normalization. As auranofin affected Renilla measurements, this assay could not be used for this compound. Nevertheless, inhibition by auranofin was detectable through the expression of GFP in acceptor cells, as GFP is also encoded by the pseudovirus (Figure 6D), further indicating that NACA and L‐ascorbic acid inhibit viral entry at millimolar concentrations, while auranofin and JTT‐705 possess antiviral activity in the micromolar range. Inhibitory activity of the selected compounds was further tested using SARS‐CoV‐2 GFP reporter virus by infecting lung‐derived A549 cells stably expressing ACE2 at an MOI of 1, which were pre‐treated for 3 hours by indicated concentrations of the compounds. 29 We measured GFP reporter fluorescence and cell confluence post‐infection as a measure of reporter virus infection and cytotoxicity of the compounds, respectively, using a live cell fluorescent microscope. Efficient inhibition of viral entry after 24 hours of exposure without cytotoxicity was observed for NACA and auranofin (Figure 6E,F).
FIGURE 6.
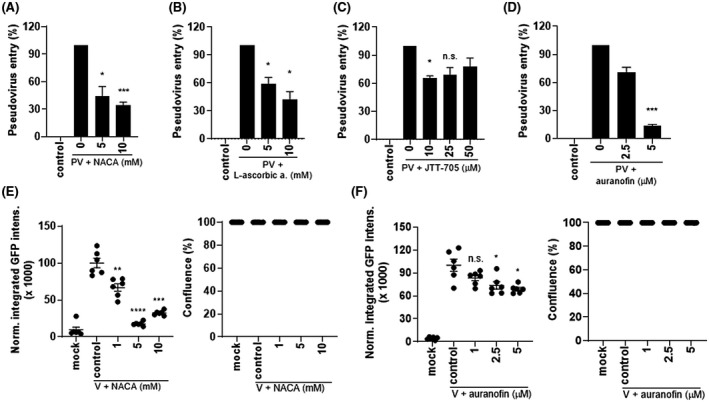
Compounds inhibited pseudoviral and viral entry into cells. A‐D, Pseudoviruses encoding firefly luciferase and GFP were incubated with the indicated concentrations of compounds for 30 minutes and added to ACE2‐expressing HEK293 cells. After 24 hours, cells were lysed and a dual luciferase test was performed (A‐C) or GFP was measured (D). E, F, SARS‐CoV‐2‐GFP reporter virus signal upon 24 hours infection of A549‐ACE2 cells, pre‐treated for 3 hours with the indicated concentrations of (E) NACA and (F) auranofin. An experiment of two independent experiments is shown as mean ± SEM. Combined means from two (C), three (B, D), and four (A) independent experiments are shown as mean ± SEM P values of <.05 (*), <.01 (**), <.001 (***), or .0001 (****) are indicated; n.s.‐not significant
Finally, inhibition was also tested in an animal system, where mice were intranasally transfected with plasmids coding for the human ACE2 receptor and TMPRSS2 to make mouse cells in the respiratory tract susceptible to viral infection. Pseudoviral infection was detected after intranasal application of spike protein‐pseudotyped viruses coding for firefly luciferase. Mice were either injected two times i.p. with 3.0 mg/mouse of NACA, 0.25 mg/mouse of auranofin in a buffered solution or they were offered to drink 3% L‐ascorbic acid in tap water for 6 days. On day 2 or 6, mice were transfected, and after 24 hours, pseudoviruses were delivered intranasally. After 24 hours, luciferase expression was detected in the mice after iv injection of D‐luciferin. L‐ascorbic acid and auranofin significantly and NACA moderately inhibited pseudoviral infection (Figure 7 and Figure S5), supporting the in vivo relevance and potential therapeutic value of the compounds.
FIGURE 7.
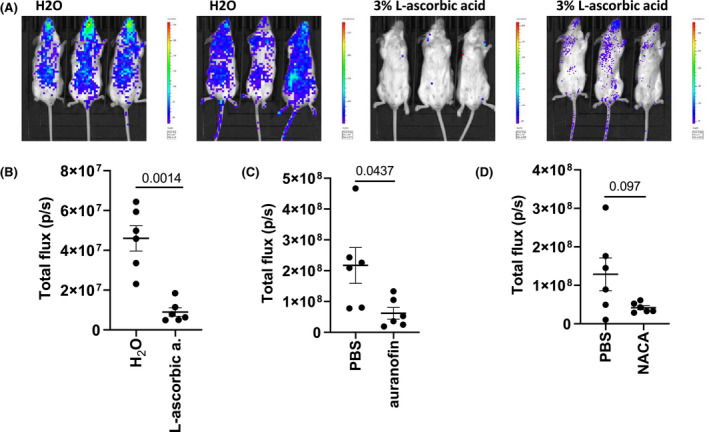
Inhibition of pseudoviral contagion in mice by thiol‐reducing compounds. A, B, Mice were drinking 3% L‐ascorbic acid in tap water or tap water for six consecutive days. (C, D) NACA in PBS (3.0 mg/mouse) and auranofin in buffer (0.25 mg/mouse) or buffer were injected i.p. for two consecutive days. On day 2 (NACA and auranofin) or 6 (L‐ascorbic acid) mice were transfected with human ACE2 and TMPRSS2, and after 24 hours, pseudovirus‐carrying firefly luciferase were intranasally applied. After 24 hours, luciferase activity was measured. Data from one (A, B) or two (C, D) independent experiments are shown. See also Figure S5
4. DISCUSSION
Spike proteins represent the key to viral recognition and entry. Virtually all vaccines and antibodies target this process, while most chemical inhibitors have focused on viral enzymes. The tertiary structure of the spike protein and its dynamics need to be conserved as the complex transition of the spike protein leads to the conversion into the post‐fusion conformation and membrane fusion. In the influenza virus, these structural changes depend on acidic pH; moreover, in HIV‐1 or murine hepatitis virus (MHV), the disulfides play an important role [reviewed in 30, 31] as virus‐receptor interactions and viral entry were inhibited by thiol‐reducing agents. 7 , 8 , 32 In the SARS‐CoV spike protein, disulfides in S2 subunit are maintained during the transition from pre‐ to the post‐fusion conformation. 33 Also, for RBD binding to ACE2, low sensitivity to thiol reduction has been proposed, 9 although the importance of some cysteine residues for ACE2 binding has been determined. 34 Our results suggest that the integrity of disulfides is not essential for binding to ACE2. However, they are required for the conformational transition to the post‐fusion conformation, most likely because this transition imposes a high degree of structural stress on protein domains. The importance of disulfide integrity in the RBD of the SARS‐CoV‐2 spike has been proposed in the receptor binding and fusion process in computational studies. 10 , 35 Meirson et al showed by structural analysis based on X‐ray crystallography and cryo‐EM structures that the binding of RBD to ACE2 facilitates conversion to the active form of the spike. These changes culminate at the hinge of RBD and are driven by an allosteric switching mechanism between cysteine pairs 336‐361 and 391‐525, 35 thus influencing the pre‐ to post‐fusion transition. Additionally, reduction of disulfides in RBD of SARS‐CoV‐2 markedly reduced binding in a molecular simulation study. 10 We show that all disulfides within the RBD domain need to be conserved, as mutations of cysteines within this domain limited the surface expression of spike protein. There are two disulfides at the border or within the RBM (residues 438‐506), namely 480‐488 and 379‐432. Using a flow cytometric assay that distinguishes between two coupled processes, a spike‐ACE2 interaction and a cell fusion process, we showed that mutations of cysteine residues in both pairs reduced cell fusion, although cell interactions still formed. The results imply that some disulfides indeed play a role in the conformational transition of the spike, thus contributing to cell fusion. Interestingly, although Cys480 and Cys488 form a disulfide pair, the Cys480 mutation impaired fusion more effectively than Cys488. Molecular dynamics simulations of the RBD up position observed three salt bridges and two hydrogen bonds (among them N370‐C480) that probably transiently stabilize this position, 36 which might explain the more prominent effect of Cys480 for fusion than Cys488.
By establishing a sensitive split luciferase assay with a low detection threshold of syncytia formation, we demonstrated that several but not all thiol‐reactive compounds are able to inhibit the formation of syncytia mediated by SARS‐CoV‐2 spike and ACE2. NACA and L‐ascorbic acid inhibited syncytia formation at millimolar concentrations; however, these are compounds with an established safety track record at comparable concentrations and available as food supplements. Moreover, JTT‐705 inhibited syncytia formation at micromolar concentrations. Auranofin was more potent than other Au‐containing compounds and JTT‐705, but it seems that although it can affect cysteine residues, other targets are likely responsible for the observed inhibition. Results of cell fusion assays were also recapitulated in viral entry assays. Importantly, we showed that these compounds can inhibit the infection of mice‐expressing human ACE2 receptors, either when compounds were added to drinking water or intraperitoneally. Noticeably, the effects were observed already after 2 d.
All fusion inhibitory compounds, except auranofin, inhibited the binding of the spike protein to ACE2, as shown by the spike/ACE2 binding assay. The compounds decreased the percentage of double‐positive cells, but NACA, L‐ascorbic acid, and JTT‐705 also prevented membrane fusion, likely by preventing the rearrangement of the spike protein to post‐fusion conformation. Viral binding and entry are not the only processes that could be disrupted by the reduction of disulfides. SARS‐CoV‐2 infection pathogenesis is related to oxidative stress [reviewed in 31, 37]. Oxidative stress relates to several steps such as viral interaction with ACE2 and viral infection as a result of disulfide‐thiol balance. It also disturbs intracellular disulfide‐thiol equilibrium, which affects cytokine and antioxidant response. Importantly, the ACE2 has a role in lowering oxidative stress. 31 Herein, we were only able to detect the influence of the tested compounds on spike‐ACE2 interaction and fusion, which however does not exclude other effects of tested compounds in ameliorating oxidative stress. It has been proposed that the mechanism of action of auranofin may be through the inhibition of reduction/oxidation (redox) enzymes 6 that are essential for maintaining intracellular levels of reactive oxygen species, 14 which could additionally ameliorate the course of the disease. L‐ascorbic acid has pleiotropic effects and a safety record [reviewed in 13]. While most studies have proposed other mechanisms, here we suggest that it might contribute through the inhibition of a viral entry. This is in agreement with studies in which it was used as an efficient prophylaxis 38 but was not efficient for COVID‐19 treatment. 39 NAC has also been proposed for the treatment of COVID‐19. 40 While NAC did not inhibit cell fusion in our assays, the efficiency of NAC in COVID‐19 treatment has been shown, 12 implying that antioxidant and anti‐inflammatory properties, which reduce oxidative stress at the cellular level, and not only thiol‐reducing efficiency, contribute to mitigate the disease. Our results on NAC derivative NACA suggest that it might be replaced by NACA for further trials, as it might exhibit better efficiency in targeting viral infection.
CONFLICT OF INTEREST
The authors declare no competing financial interest.
AUTHOR CONTRIBUTIONS
M. Manček‐Keber, together with R. Jerala, designed the study, and M. Manček‐Keber performed/contributed to most of the experiments and analyzed the data. I. Hafner‐Bratkovič performed pseudoviral assay. D. Lainšček performed mouse experiments. M. Benčina performed confocal experiments and designed the flow cytometry assay. T. Govednik performed ACE‐spike binding assay. S. Orehek prepared pseudoviruses. V. Jazbec prepared samples for MS. T. Plaper prepared split luciferase and GFP constructs. V. Bergant, V. Grass, and A. Pichlmair prepared SARS‐CoV‐2 viruses and performed virus assay. M. Manček‐Keber and R. Jerala wrote the manuscript. R. Jerala supervised the study. All authors discussed the results and commented on the manuscript.
Supporting information
Fig S1‐S5
ACKNOWLEDGMENTS
This study was financially supported by the Slovenian Research Agency (project no. V4‐2038, research core no. P4‐0176 to RJ and project no. J3‐9257 to MMK). The authors thank Van Thai Ha and Peter Pečan for the technical support, Francis Impens from VIB core facility for performing MS analysis, and Bruno Correia from EPFL for recombinant spike protein.
Manček‐Keber M, Hafner‐Bratkovič I, Lainšček D, et al. Disruption of disulfides within RBD of SARS‐CoV‐2 spike protein prevents fusion and represents a target for viral entry inhibition by registered drugs. The FASEB Journal. 2021;35:e21651. 10.1096/fj.202100560R
REFERENCES
- 1. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid‐19—final report. N Engl J Med. 2020;383:1813‐1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Joshi S, Parkar J, Ansari A, et al. Role of favipiravir in the treatment of COVID‐19. Int J Infect Dis. 2020;102:501‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaur U, Chakrabarti SS, Ojha B, et al. Targeting host cell proteases to prevent SARS‐CoV‐2 invasion. Curr Drug Targets. 2020;22:192‐201. [DOI] [PubMed] [Google Scholar]
- 4. Seif F, Aazami H, Khoshmirsafa M, et al. JAK inhibition as a new treatment strategy for patients with COVID‐19. Int Arch Allergy Immunol. 2020;181:467‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bagheri M, Niavarani A. Molecular dynamics analysis predicts ritonavir and naloxegol strongly block the SARS‐CoV‐2 spike protein‐hACE2 binding. J Biomol Struct Dyn. 2020;8:1‐10. [DOI] [PubMed] [Google Scholar]
- 6. Rothan HA, Stone S, Natekar J, Kumari P, Arora K, Kumar M. The FDA‐approved gold drug auranofin inhibits novel coronavirus (SARS‐COV‐2) replication and attenuates inflammation in human cells. Virology. 2020;547:7‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ryser HJ, Levy EM, Mandel R, DiSciullo GJ. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol‐disulfide interchange upon virus‐receptor interaction. Proc Natl Acad Sci U S A. 1994;91:4559‐4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gallagher TM. Murine coronavirus membrane fusion is blocked by modification of thiols buried within the spike protein. J Virol. 1996;70:4683‐4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lavillette D, Barbouche R, Yao Y, et al. Significant redox insensitivity of the functions of the SARS‐CoV spike glycoprotein: comparison with HIV envelope. J Biol Chem. 2006;281:9200‐9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hati S, Bhattacharyya S. Impact of thiol‐disulfide balance on the binding of Covid‐19 spike protein with angiotensin‐converting enzyme 2 receptor. ACS Omega. 2020;5:16292‐16298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ates B, Abraham L, Ercal N. Antioxidant and free radical scavenging properties of N‐acetylcysteine amide (NACA) and comparison with N‐acetylcysteine (NAC). Free Radical Res. 2008;42:372‐377. [DOI] [PubMed] [Google Scholar]
- 12. Ibrahim H, Perl A, Smith D, et al. Therapeutic blockade of inflammation in severe COVID‐19 infection with intravenous N‐acetylcysteine. Clin Immunol. 2020;219:108544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Colunga Biancatelli RML, Berrill M, Catravas JD, Marik PE. Quercetin and vitamin C: an experimental, synergistic therapy for the prevention and treatment of SARS‐CoV‐2 related disease (COVID‐19). Front Immunol. 2020;11:1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roder C, Thomson MJ. Auranofin: repurposing an old drug for a golden new age. Drugs R&D. 2015;15:13‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mancek‐Keber M, Gradisar H, Iñigo Pestaña M, Martinez de Tejada G, Jerala R. Free thiol group of MD‐2 as the target for inhibition of the lipopolysaccharide‐induced cell activation. J Biol Chem. 2009;284:19493‐19500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mohammadpour AH, Akhlaghi F. Future of cholesteryl ester transfer protein (CETP) inhibitors: a pharmacological perspective. Clin Pharmacokinet. 2013;52:615‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271‐280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kamiyama D, Sekine S, Barsi‐Rhyne B, et al. Versatile protein tagging in cells with split fluorescent protein. Nat Commun. 2016;7:11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plaper T, Aupič J, Dekleva P, et al. Coiled‐coil heterodimers with increased stability for cellular regulation and sensing SARS‐CoV‐2 spike 1 protein‐mediated cell fusion. Sci Rep. 2021;11:9136‐9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berger Rentsch M, Zimmer G. A vesicular stomatitis virus replicon‐based bioassay for the rapid and sensitive determination of multi‐species type I interferon. PLoS ONE. 2011;6:e25858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lainšček D, Fink T, Forstnerič V, et al. Immune response to vaccine candidates based on different types of nanoscaffolded RBD domain of the SARS‐CoV‐2 spike protein. Vaccines (Basel). 2021;9:431‐451.33925446 [Google Scholar]
- 22. Meško M, Lebar T, Dekleva P, Jerala R, Benčina M. Engineering and rewiring of a calcium‐dependent signaling pathway. ACS Synth Biol. 2020;9:2055‐2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. The SARS‐CoV S glycoprotein: expression and functional characterization. Biochem Biophys Res Comm. 2003;312:1159‐1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia S, Zhu Y, Liu M, et al. Fusion mechanism of 2019‐nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol. 2020;17:765‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome‐associated coronavirus (SARS‐CoV) spike glycoprotein‐mediated viral entry. Proc Natl Acad Sci U S A. 2004;101:4240‐4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Papa G, Mallery DL, Albecka A, et al. Furin cleavage of SARS‐CoV‐2 Spike promotes but is not essential for infection and cell‐cell fusion. PloS Pathog. 2021;17:e1009246‐1009265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. García‐Murria MJ, Expósito‐Domínguez N, Duart G, Mingarro I, Martinez‐Gil L. A bimolecular multicellular complementation system for the detection of syncytium formation: a new methodology for the identification of Nipah virus entry inhibitors. Viruses. 2019;11:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lan J, Ge J, Yu J, et al. Structure of the SARS‐CoV‐2 spike receptor‐binding domain bound to the ACE2 receptor. Nature. 2020;581:215‐220. [DOI] [PubMed] [Google Scholar]
- 29. Stukalov A, Girault V, Grass V, et al. Multi‐level proteomics reveals host‐perturbation strategies of SARS‐CoV‐2 and SARS‐CoV. Nature. 2021;1‐7. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 30. Fenouillet E, Barbouche R, Jones IM. Cell entry by enveloped viruses: redox considerations for HIV and SARS‐coronavirus. Antioxid Redox Signal. 2007;9:1009‐1034. [DOI] [PubMed] [Google Scholar]
- 31. Suhail S, Zajac J, Fossum C, et al. Role of oxidative stress on SARS‐CoV (SARS) and SARS‐CoV‐2 (COVID‐19) infection: a review. Protein J. 2020;39:644‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Markovic I, Stantchev TS, Fields KH, et al. Thiol/disulfide exchange is a prerequisite for CXCR4‐tropic HIV‐1 envelope‐mediated T‐cell fusion during viral entry. Blood. 2004;103:1586‐1594. [DOI] [PubMed] [Google Scholar]
- 33. Walls AC, Tortorici MA, Snijder J, et al. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci U S A. 2017;114:11157‐11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193‐amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin‐converting enzyme 2. J Biol Chem. 2004;279:3197‐3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meirson T, Bomze D, Markel G. Structural basis of SARS‐CoV‐2 spike protein induced by ACE2. Bioinformatics. 2020;btaa744. [Epub ahead of print]. 10.1093/bioinformatics/btaa744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gur M, Taka E, Yilmaz SZ, Kilinc C, Aktas U, Golcuk M. Conformational transition of SARS‐CoV‐2 spike glycoprotein between its closed and open states. J Chem Phys. 2020;153:075101. [DOI] [PubMed] [Google Scholar]
- 37. Cecchini R, Cecchini AL. SARS‐CoV‐2 infection pathogenesis is related to oxidative stress as a response to aggression. Med Hypotheses. 2020;143:110102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arslan B, Ergun N, Topuz S, et al. Synergistic effect of quercetin and vitamin C against COVID‐19: is a possible guard for front liners? 2020. Preprint.
- 39. Thomas S, Patel D, Bittel B, et al. Effect of high‐dose zinc and ascorbic acid supplementation vs usual care on symptom length and reduction among ambulatory patients with SARS‐CoV‐2 infection: the COVID A to Z randomized clinical trial. JAMA Netw Open. 2021;4:e210369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Andreou A, Trantza S, Filippou D, Sipsas N, Tsiodras S. COVID‐19: the potential role of copper and N‐acetylcysteine (NAC) in a combination of candidate antiviral treatments against SARS‐CoV‐2. In Vivo. 2020;34:1567‐1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5