

Summary
Interferon regulatory factor 3 (IRF3) is a critical transcription factor for inducing production of type I interferons (IFN‐I) and regulating host antiviral response. Although IRF3 activation during viral infection has been extensively studied, the inhibitory regulation of IRF3 remains largely unexplored. Here, we revealed that Midline‐1 (MID1) is a ubiquitin E3 ligase of IRF3 that plays essential roles in regulating the production of IFN‐I. We found that MID1 physically interacts with IRF3 and downregulates IRF3 protein levels. Next, we demonstrated that MID1 can induce K48‐linked polyubiquitination of IRF3, thus lowing the protein stability of IRF3. Our further studies identified Lys313 as a major ubiquitin acceptor lysine of IRF3 induced by MID1. Finally, MID1‐mediated ubiquitination and degradation of IRF3 restrict IFN‐I production and cellular antiviral response. This study uncovers a role of MID1 in regulating innate antiviral immunity and may provide a potential target for enhancing host antiviral activity.
Keywords: antiviral response, innate immunity, interferon, IRF3, MID1, ubiquitination
The ubiquitin E3 ligase MID1 interacts with and ubiquitinates IRF3, thus regulating IRF3 protein stability and type I interferon antiviral response.

Abbreviations
- CHX
cycloheximide
- H1N1
influenza A virus
- HSV
herpes simplex virus
- IFNAR1
type I interferon receptor 1
- IFN‐I
type I interferons
- IRF3
interferon regulatory factor 3
- ISGs
interferon‐stimulated genes
- MA
methylamine hydrochloride
- MG132
proteasome inhibitor
- MID1
E3 ubiquitin‐protein ligase Midline‐1
- MOI
multiplicity of infection
- PRRs
pattern‐recognition receptors
- RLRs
RIG‐I‐like receptors
- SeV
Sendai virus
- SLE
systemic lupus erythematosus
- TCID50
50% tissue culture infective dose
- TLRs
Toll‐like receptors
- Ub
ubiquitin
- VSV‐GFP
vesicular stomatitis virus with a green fluorescent protein gene
Introduction
The innate immune system is essential for host cells to fight against viral infection. When host cells suffer from viruses, the pattern‐recognition receptors (PRRs), such as RIG‐I‐like receptors (RLRs) and Toll‐like receptors (TLRs), can rapidly recognize viral nucleic acids. 1 , 2 , 3 , 4 Then, PRRs trigger antiviral signalling to activate the production of IFN‐I and proinflammatory cytokines, which eventually restricts viral infection. 3 , 5 Exploring the regulation of PRRs signalling pathways could provide new strategies for enhancing host antiviral defence.
In the RLR signalling pathway, activated RLRs interact with the mitochondrial antiviral signalling protein (MAVS), which in turn recruits the tumour necrosis factor receptor‐associated factor (TRAF) family proteins to form a MAVS/TRAF3/TRAF6 signalosome. 6 , 7 Subsequently, the signalosome induces activation of serine/threonine‐protein kinase (TBK1) and inhibitor of nuclear factor kappa‐B kinase subunit epsilon (IKKε), which finally activates interferon regulatory factor 3 (IRF3). Activated IRF3 translocates to the nucleus to induce transcriptional expression of the antiviral cytokine IFN‐I. 8 , 9 , 10 Although IRF3 activation has been extensively explored, the negative regulation of IRF3 signals remains to be illuminated.
It has been reported that excessive production of IFNs could be associated with multiple autoimmune diseases, such as systemic lupus erythematosus (SLE). 11 Therefore, one can understand that the control of IRF3 protein levels or activation is important for balancing cellular IFN response. For example, recent studies have shown that SUMOylation of IRF3 at the lysine (K) 152 site represses IRF3 activation and therefore inhibits IFN‐I production. 12 In addition, the regulation of ubiquitination of IRF3 also significantly affects activation of IRF3. The ubiquitin E3 ligase RAUL can downregulate proteins levels of IRF3 and limit the production of IFN‐I by catalysing K48‐linked polyubiquitination of IRF3· 13 Tripartite motif‐containing protein 26 (TRIM26) inhibits cellular antiviral response by degrading nuclear IRF3 protein levels in the nucleus. 14 Interestingly, our preliminary results showed that MID1 could be a potential ubiquitin E3 ligase of IRF3.
MID1 belongs to a member of the RING domain ubiquitin E3 ligases. Mutation of the MID1 gene could lead to X‐linked Opitz BBB/G syndrome (OS). 15 MID1 has been revealed to be associated with cell migration and adhesion, epithelial–mesenchymal differentiation and some other biological functions. MID1 consists of seven conserved domains. Among them, the major E3 active domains are the RING domain and B‐boxes domain, which are rich in cysteine residues. 16 , 17 So far, the reported substrates of MID1 are very limited. PP2A is the first reported substrate of MID1· 18 MID1 could promote degradation of the protein kinase Fu, which is a natural activator of the hedgehog pathway. 19 In addition, MID1 could target Pax6 to regulate the development of the eyes, brain and pancreas. 20 However, whether MID1 targets any substrates to regulate innate antiviral immunity remains largely unknown.
Here, we demonstrated that MID1 as a ubiquitin E3 ligase regulates innate antiviral immunity by targeting IRF3. MID1 interacts with IRF3, induces both K48‐linked and K63‐linked polyubiquitination of IRF3 and negatively regulates IRF3 proteins levels, thus inhibiting IFN‐I production and attenuating cellular antiviral activity. These effects of MID1 are dependent on its ubiquitin E3 ligase activity. Furthermore, we identified Lys313 as a major residue of IRF3 for MID1‐induced polyubiquitination. These findings uncovered that MID1 plays crucial roles in regulating IFN antiviral immunity.
Materials and methods
Mice
C57BL/6 mice were purchased from the Laboratory Animal Center of Soochow University. Mice were bred and housed in the animal facility of the Soochow University under specific pathogen‐free (SPF) conditions. All protocols and procedures for mice study have been approved by the Commission of the Scientific Investigation Board of Soochow University.
Cell culture and transfection
HEK293T, HeLa and Vero cell lines were purchased from ATCC. 2fTGH cell line was a gift from S. Y. Fuchs (University of Pennsylvania). All cells were cultured at 37 °C under 5% CO2. All cells were cultured in DMEM (HyClone) supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. All transient transfections were performed using either LongTrans (UcallM, TF/07) or GenePORTER® 2 Transfection Reagent (Genlantis) according to the manufacturer’s instructions.
Cell isolation from mice
Ifnar1+/+ and Ifnar1−/− embryos were obtained from the pregnant (13·5 days) mice. The mouse embryonic tissues were first cut into small pieces and then digested with collagenases. After centrifugation, MEFs were collected and cultured in DMEM.
Liver tissue from mice
Mouse tissues were prepared from the 6‐ to 8‐week‐old adult wild‐type mice. Briefly, mouse liver tissues were cut into pieces and grinded to cell suspension. Mouse primary liver cells were collected and prepared for further experiments.
Expression constructs and reagents
Human FH‐MID1 was purchased from Vigene Biosciences (CH820124). Human HA‐IRF3 was a gift from Dr. F. Zhou (Soochow University). IFN‐β (P125)‐Luc was provided by Dr. J. Dai (Soochow University). Renilla, HA‐ubiquitin (HA‐Ub), HA‐R6K, HA‐R11K, HA‐R27K, HA‐R29K, HA‐R33K, HA‐R48K and HA‐R63K (all lysines on the ubiquitin gene are mutated to arginines except the corresponding lysine) were as described previously. 21 Flag‐ubiquitin (Flag‐Ub) was a gift from Dr. Feng Shao (National Institute of Biological Sciences, Beijing).
The shMID1 targeting sequences were as follows:
shMID1‐1,5′‐gatccacgtcaccctacagaacatttcaagagaatgttctgtagggtgacgtttttttacgcgtg‐3′ and 5′‐aattcacgcgtaaaaaaacgtcaccctacagaacattctcttgaaatgttctgtagggtgacgtg‐3′; shMID1‐2,5′‐gatccaccgcatcctagtatcacattcaagagatgtgatactaggatgcggtttttttacgcgtg‐3′ and 5′‐aattcacgcgtaaaaaaaccgcatcctagtatcacatctcttgaatgtgatactaggatgcggtg‐3′.
Myc‐MID1 was made using PCR amplification from the FH‐MID1 plasmids and cloned into the Myc‐N1 vector. MID1‐ΔRING (Leu65‐Pro667) and MID1‐Δ(RING + Bbox1) (Ile165‐Pro667) were made using PCR amplification from the FH‐MID1 plasmids. MH‐IRF3 was made using PCR amplification from the HA‐IRF3 plasmids and cloned into the Myc‐His vector. MH‐IRF3‐ΔSR (Met1‐Ala384) and MH‐IRF3‐Δ(SR + IAD) (Met1‐Asn190) were made using PCR amplification from the FH‐MID1 plasmid and cloned into the Myc‐His vector. HA‐IRF3‐K193R, HA‐IRF3‐K313R, HA‐IRF3‐K315R, HA‐IRF3‐K360R and HA‐IRF3‐K366R were generated by the QuikChange site‐directed mutagenesis kit (Stratagene, Cat. no. 210518). All of the plasmids were confirmed by DNA sequencing. Human IFN‐α was purchased from PBL Interferon Source. CHX and MG132, and other chemicals were purchased from Sigma.
CRISPR/Cas9‐mediated genome editing
The lentiCRISPRv2 vector was a gift from F. Zhou (Soochow University). Small guide Cas9‐MID1 RNAs were cloned into the lentiCRISPRv2 vector and transfected into HEK293T cells. The supernatant was collected 48 h after transfection. The mouse peritoneal macrophage cells were obtained from C57BL/6 mice and then infected with the supernatant. Cells were cultured under puromycin (1·5 μg/ml) selection until further experiments. The guide RNA sequence of mouse Mid1 is 5′‐GCTGACCTGTCCTATTTGTC‐3′.
Immunoblotting and immunoprecipitation
The cells were harvested using lysis buffer containing 150 mm NaCl, 1% Nonidet P‐40, 0·5 mm EDTA, 20 mm Tris–HCl (pH 7·4), PMSF (50 μg/ml) and protease inhibitor mixtures (Sigma). N‐ethylmaleimide (10 mm) was added into the above lysis buffer when protein ubiquitination was analysed. Immunoprecipitation and immunoblotting were carried out as described previously. 22 The following antibodies were used: MID1 (1:750, Abcam, ab70770), HA (1:3000, Abcam, ab9110), VSV‐G (1:5000, Abcam, ab1874), GAPDH (1:1000, Goodhere Biological Technology, AB‐M‐M001), IRF3 (1:3000, Proteintech, 11312‐1‐AP), IRF3 (1:100, Santa Cruz, sc‐9082), Myc (1:5000, Abmart, m2002), Flag (1:5000, Sigma, F7425), Ub (1:750, Santa Cruz, sc‐8017), H1N1 (1:3000, Sino Biological, 11684‐T56) and Tubulin (1:5000, Proteintech, 66031‐1‐Ig).
Virus and viral infection
The SeV was a gift from Dr. C. Wang (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences). H1N1 (PR/8/34) was a gift from Dr. J. Dai (Soochow University). VSV–GFP was a gift from Dr. C. Dong (Soochow University). HSV‐1 was a gift from Dr. C. Zheng (Fujian Medical University). Briefly, cells were transfected with FH/Myc‐MID1 or shMID1 for 48 or 72 h. Then, cells were infected with viruses for 2 h. Next, the medium was moved, and cells were cultured in fresh medium containing 10% FBS for continuous 20 h.
TCID50 assay
Cells were first transfected with different constructs. Then, cells were infected with either VSV or HSV‐1 for 20 h. Culture supernatants containing VSV or HSV‐1 viruses were serially diluted with DMEM and then placed on the monolayer of Vero cells in 96‐well plates. The TCID50 was calculated using the Spearman–Karber algorithm.
Immunofluorescence and confocal microscopy assay
Cells were infected with VSV–GFP for 20 h. Then, cells were subjected to analysis by immunofluorescence microscopy. The images were magnified at 200×. Confocal assay was as described previously. Briefly, cells were fixed in 4% paraformaldehyde at room temperature. Then, cells were permeabilized with 0·5% Triton X‐100 and blocked with 5% BSA. The cells were incubated with either an anti‐MID1 antibody (1:100, Abcam, ab70770) or an anti‐IRF3 antibody (1:50, Santa Cruz, sc‐33641) overnight, followed by staining with either 488 goat anti‐rabbit IgG (Alexa Fluor, A11008) or 633 goat anti‐mouse IgG (Alexa Fluor, A21053). Cell nuclei were stained with DAPI, and the fluorescent images were captured with the Nikon A1 confocal microscope.
Cycloheximide chase assay
In order to analyse the half‐life of IRF3, HEK293T cells were transfected with FH‐MID1, together with HA‐IRF3. Forty‐eight hours after transfection, cells were treated with DMSO or CHX (50 mg/ml) for different times. Next, cells were collected and analysed by Western blot.
In vivo ubiquitination assay
Cells were transfected with the corresponding plasmids. After forty‐eight or seventy‐two hours, cells were collected in the strong RIPA lysis buffer. In order to explore the role of MID1 in regulating ubiquitination of IRF3, IRF3 proteins were immunoprecipitated by a specific anti‐IRF3 Ab. The immunoprecipitates were extensively washed by a high salt (500 mm) washing buffer for three times and then subjected to Western blot analyses using the anti‐HA, anti‐Flag or anti‐ubiquitin Ab.
Reporter gene assay
In order to analyse IFN‐β production, cells were transfected with the IFN‐β–luciferase plasmid (P125), together with Renilla plasmid and FH‐MID1 plasmid. Forty‐eight hours later, SeV viruses were used to infect cells. Next, the medium was moved, and cells were cultured in fresh medium containing 10% FBS for different times. The luciferase activity was measured by using the Dual‐Luciferase Reporter Assay System (Promega, E1910).
ELISA
The concentrations of human IFN‐α (E‐EL‐H2532c; Elabscience) and mouse IFN‐β (E‐EL‐M0033c; Elabscience) were measured by ELISA kits according to the manufacturer’s instructions.
RNA isolation and RT‐qPCR assay
Total RNAs were extracted from cells using TRIzol reagent (Invitrogen) in accordance with the manufacturer’s instructions. All cDNA was synthesized from 600 ng of total RNA using the 5X All‐In‐One RT MasterMix (Abcam, cat. no. G490). Then, the mRNA levels were analysed by RT‐qPCR using H1N1, SeV, VSV, β‐actin, IFN‐β and IRF3 primers using 2× SYBR Green qPCR Master Mix (Selleck, cat. no. B21202). The primer sequences were as follows:
IRF3: 5′‐gatgcacagcaggaggattt‐3′ and 5′‐gtcctctgctaaacgcaacc‐3′;
VSV: 5′‐acggcgtacttccagatgg‐3′ and 5′‐ctcggttcaagatccaggt‐3′;
SeV: 5′‐gatgacgatgccgcagcagtag‐3′ and 5′‐cctccgatgtcagttggttcactc‐3′;
H1N1: 5′‐ttctaaccgaggtcgaaacg‐3′ and 5′‐acaaagcgtctacgctgcag‐3′;
β‐actin: 5′‐accaactgggacgacatggagaaa‐3′ and 5′‐atagcacagcctggatagcaacg‐3′;
Human IFN‐β: 5′‐cattacctgaaggccaagga‐3′ and 5′‐cagcatctgctggttgaaga‐3′.
IFIT1: 5′‐cacaagccattttctttgct‐3′ and 5′‐acttggctgcatatcgaaag‐3′;
ISG54: 5′‐cacctctggactggcaatagc‐3′ and 5′‐gtcaggattcagccgaatgg‐3′;
GAPDH: 5′‐ggccttccgtgttcctacc‐3′ and 5′‐agcccaagatgcccttcagt‐3′;
Mouse IFN‐β: 5’‐cttcgtgtttggtagtgatggt‐3’ and 5’‐ggggatgatttccagccga‐3’;
Statistical analysis
Different groups were compared by a two‐tailed Student t‐test. All differences were regarded as statistically significant when P < 0·05.
Results
MID1 promotes viral infection
To explore the role of MID1 in regulating the innate antiviral response, we first used an RNA virus, vesicular stomatitis virus (VSV), which has been used as a sensitive viral model to assess the activity of cellular antiviral response. By knocking down endogenous MID1 by two specific shRNAs, we noticed that MID1 deficiency inhibited viral infection (Fig. 1a). Furthermore, we employed another RNA virus influenza A virus H1N1 (PR/8/34) and a DNA virus herpes simplex virus (HSV). The results showed that knockdown of MID1 suppressed infection by both H1N1 (Fig. 1b) and HSV (Fig. 1c). In contrast, overexpression of MID1 promoted cellular infection by viruses, including SeV, H1N1 and Sendai viruses (SeV) (Fig. 1d). Additionally, by Western blot we confirmed the effect of MID1 knockdown on virus infection (Fig. 1e,f). Moreover, MID1 overexpression increased viral titres in cells infected with both VSV and HSV (Fig. 1g). To directly observe the effect of MID1 on viral infection, an immunofluorescence assay was carried out in HEK293T cells infected with VSV‐GFP, a VSV virus with a GFP gene. The results demonstrated that MID1 overexpression promoted viral infection, as shown by increased GFP signals (Fig. 1h). Importantly, MID1 knockout in mouse primary macrophages significantly restricted viral infection (Fig. 1i). Together, these findings suggested that MID1 could promote viral infection.
Figure 1.
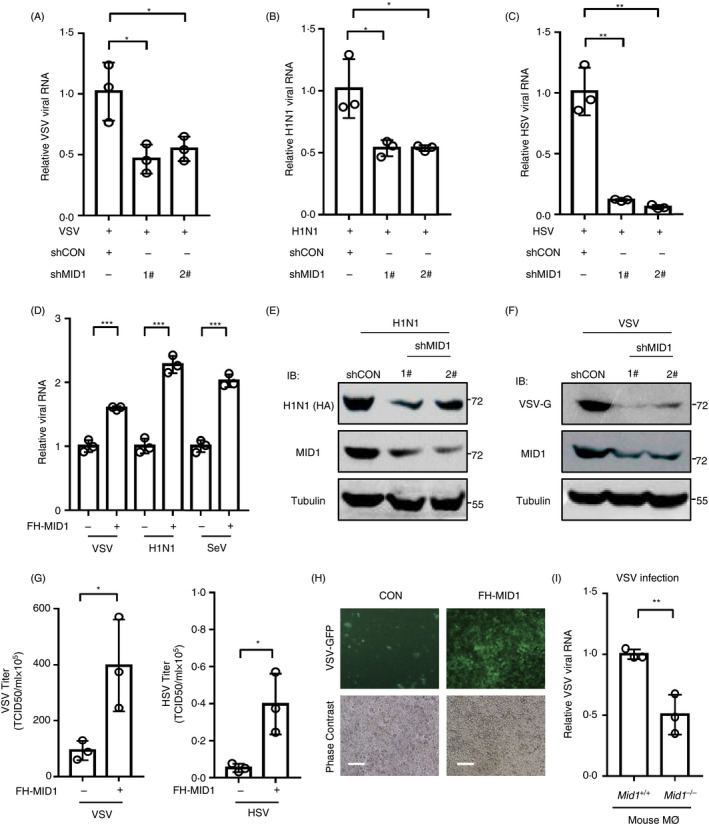
MID1 promotes virus replication. (a–c) HEK293T cells were transfected with control shRNAs (shCON) or two shRNAs against MID1 (shMID1, 1# and 2#). Then, cells were infected with VSV (a) (MOI = 1·0), H1N1 (b) (MOI = 1·0) or HSV (c) (MOI = 1·0) for 20 h. (d) 2fTGH cells were transfected with or without Flag‐His‐tagged MID1 (FH‐MID1). Then, 2fTGH cells were infected with VSV H1N1 or SeV (MOI = 1·0) for 20 h. (e, f) HEK293T cells were transfected with control shRNAs (shCON) or two shMID1. 72 h after transfection, cells were infected with H1N1 (e) or VSV (f) (MOI = 1·0) for 20 h. Whole‐cell extracts were analysed by Western blot as indicated. (g) HEK293T cells were transfected with or without FH‐MID1 for 48 h. Then, cells were infected with VSV or HSV (MOI = 1·0) for 20 h. VSV and HSV viral titres in culture supernatants were determined by a TCID50 assay. (h) HEK293T cells were transfected with FH‐MID1. 48 h after transfection, cells were infected with VSV‐GFP (MOI = 1·0) for 20 h. The VSV‐GFP signal was detected by fluorescence microscopy. Scale bars: 100 µm. (i) RT‐qPCR analysis of VSV RNA levels in Mid1 +/+ and Mid1 −/− mouse peritoneal macrophages (Mø) infected with VSV (MOI = 0·1).
MID1 inhibits virus‐induced production of type I IFNs
We next sought to explore the mechanism by which MID1 promotes viral infection. To this end, we first utilized type I interferon receptor 1 (IFNAR1)‐deficient MEF cells. Ifnar1+/+ and Ifnar1−/− MEF cells were transfected with Myc‐MID1 and then infected with VSV. As shown above, MID1 overexpression promoted viral infection in Ifnar1+/+ cells (Fig. 2a). However, overexpression of MID1 did not noticeably affect viral infection in Ifnar1−/− MEF cells (Fig. 2a). Furthermore, in Vero cells, which are deficient to produce IFN‐I during viral infection, overexpression of MID1 cannot promote viral infection any longer (Fig. 2b). These findings suggested that MID1‐mediated regulation of viral infection could be associated with type I IFN. Therefore, we next determined whether MID1 could influence IFN‐I production. Using two specific shRNAs, we observed that knockdown of endogenous MID1 markedly promoted SeV‐induced IFN‐β production (Fig. 2c). Conversely, overexpression of MID1 substantially restricted IFN‐I production during SeV infection (Fig. 2d). To confirm the effect of MID1 on IFN‐I transcriptional expression, we determined the IFN‐β promoter activity using the luciferase constructs with an IFN‐β promoter. Our data showed that overexpression of MID1 strongly attenuated IFN‐β–luciferase activity induced by viral infection (Fig. 2e). Importantly, knockout of MID1 promotes SeV‐induced IFN‐β production (Fig. 2f). Consistently, MID1 overexpression reduced intracellular IFN‐I protein levels (Fig. 2g) and the concentration of secreted IFN‐I proteins (Fig. 2h) induced by SeV. MID1 knockout upregulated the levels of virus‐induced IFN‐I proteins (Fig. 2i). In line with the increased IFN‐I proteins, knockdown of MID1 upregulated the expression of IFN‐stimulated genes (ISGs) (Fig. 2j). Collectively, we demonstrated that MID1 inhibits IFN‐I production during viral infection and therefore negatively regulates IFN‐I antiviral response.
Figure 2.
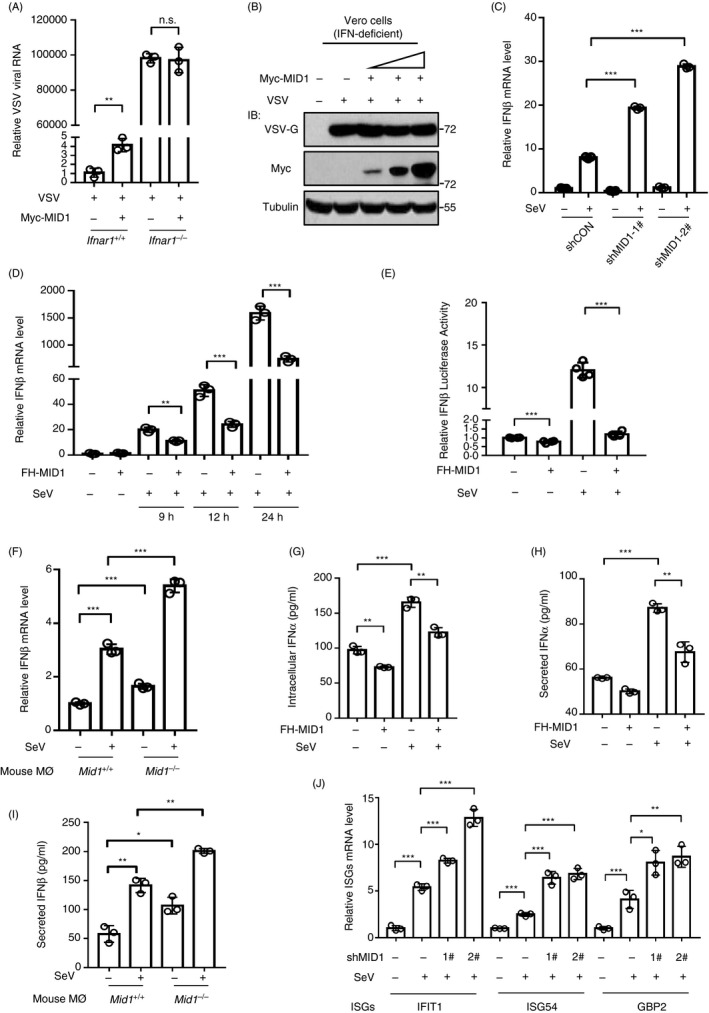
MID1 inhibits virus‐induced production of type I IFNs. (a) RT‐qPCR analysis of VSV RNA levels in Ifnar1+/+ and Ifnar1−/− MEF cells transfected with Myc‐MID1 and then infected with VSV (MOI = 1·0) for 20 h. (b) Vero cells were transfected with Myc‐MID1. 48 h after transfection, cells were infected with VSV (MOI = 1·0) for 20 h. Whole‐cell extracts were analysed by Western blot using the indicated antibodies. (c) RT‐qPCR analysis of IFN‐β mRNA levels in HEK293T cells transfected with shCON or two shMID1, followed by SeV infection (MOI = 1·0) for 12 h. (d) RT‐qPCR analysis of IFN‐β mRNA levels in HEK293T cells transfected with FH‐MID1, followed by SeV infection (MOI = 1·0) for indicated times. (e) HEK293T cells were transfected with FH‐MID1, together with IFN‐β–luciferase (IFN‐β‐Luc) and Renilla. The luciferase activity was measured after cells were infected with SeV (MOI = 1·0) for 20 h. (f) RT‐qPCR analysis of IFN‐β mRNA levels in Mid1 +/+ and Mid1 −/− mouse peritoneal macrophages followed by SeV infection (MOI = 1·0) for 20 h. (g, h) HEK293T cells transfected with or without FH‐MID1 were infected with SeV for 20 h. Intracellular IFN‐α proteins (g) and secreted IFN‐α proteins in culture supernatants (h) were analysed by ELISA. (i) Mid1 +/+ and Mid1 −/− mouse macrophages were infected with SeV (MOI = 1·0) for 20 h. Levels of secreted IFN‐β proteins in culture supernatants were analysed by ELISA. (j) HEK293T cells transfected with or without shMID1 were infected with SeV (MOI = 1·0) for 12 h. The levels of IFIT1, ISG54 and GBP2 mRNA were analysed by RT‐qPCR.
MID1 interacts with and downregulates IRF3
We further explored the detailed mechanisms of MID1‐mediated inhibition of IFN‐I production. By using an overexpression model, which is widely used to activate IFN‐I production, we noticed that MID1 can regulate IFN‐I production induced by overexpression of each component of the RIG‐MAVS signalling pathway, including RIG‐I, MAVS, TBK1 and IRF3 (Fig. 3a). The results suggested that MID1 could be able to target IRF3 to affect IFN‐I production. MID1 has been reported to be a ubiquitin E3 ligase. Given that our above data demonstrated that MID1 inhibits IFN‐I production, we speculated that MID1 could downregulate IRF3 protein levels. Consistent with our speculation, knockdown of endogenous MID1 by two specific shRNAs strongly upregulated cellular IRF3 protein levels (Fig. 3b). In contrast, overexpression of MID1 remarkably downregulated IRF3 protein levels (Fig. 3c). Importantly, MID1 knockout robustly upregulated IRF3 protein levels in mouse primary macrophages (Fig. 3d). Furthermore, we demonstrated that FH‐MID1 was capable of interacting with exogenously expressed HA‐IRF3 (Fig. 3e), and endogenous MID1 constitutively interacts with endogenous IRF3 in not only HEK293T cells (Fig. 3f) but also mouse primary liver cells (Fig. 3g). A confocal microscopy assay confirmed the interaction between endogenous MID1 and IRF3 (Fig. 3h). Interestingly, viral infection promoted MID1 and IRF3 interaction (Fig. 3i), which could facilitate viral immune evasion. However, we did not observe the upregulation of MID1 mRNA expression within six hours of infection with viruses, including H1N1, VSV and SeV (Fig. 3j). Taken together, these findings suggest that MID1 interacts with IRF3 and negatively regulates IRF3 protein levels.
Figure 3.
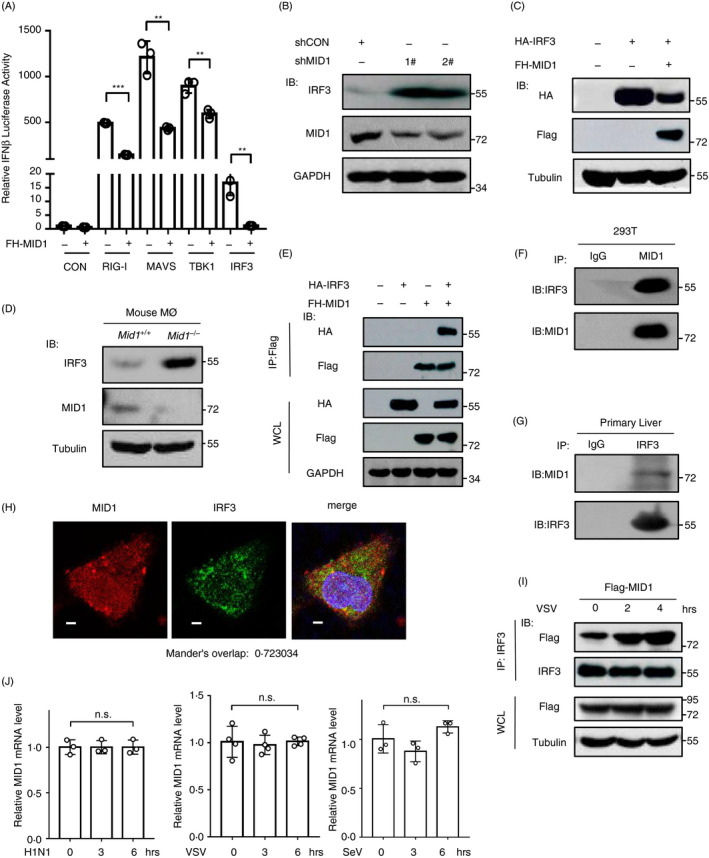
MID1 interacts with and downregulates IRF3. (a) HEK293T cells were transfected with empty vectors (CON) or RIG‐I/MAVS/TBK1/IRF3, together with FH‐MID1, IFN‐β–luciferase (IFN‐β‐Luc) and Renilla. 48 h after transfection, IFN‐β–luciferase activity was analysed. (b) HEK293T cells were transfected with shCON or two shMID1 for 72 h. Protein levels of endogenous IRF3 were detected by Western blot using an anti‐IRF3 Ab. (c) HEK293T cells were transfected with HA‐IRF3 together with FH‐MID1. Protein levels of HA‐IRF3 were detected by Western blot using an anti‐HA Ab. (d) Western blot analysis of endogenous IRF3 levels in Mid1 +/+ and Mid1 −/− mouse macrophages. (e) HEK293T cells were transfected with HA‐IRF3 together with FH‐MID1. Immunoprecipitation was performed using an anti‐Flag (M2) beads, followed by immunoblotting as indicated. (f) Endogenous MID1 proteins from HEK293T cells were immunoprecipitated, and then, immunoblotting was performed using an anti‐IRF3 Ab. (g) Endogenous IRF3 proteins in mouse primary liver cells were immunoprecipitated. Then immunoblotting was performed using an anti‐MID1 Ab. (h) HeLa cells were stained by IRF3 and MID1 antibodies. Cell nuclei were stained by DAPI. The fluorescent images were captured with the Nikon A1 confocal microscope. Scale bars: 2·5 µm. (i) HEK293T cells transfected with FH‐MID1 were infected with VSV (MOI = 1·0) for 0, 2 or 4 h. Whole‐cell lysates were subjected to immunoprecipitation with an anti‐IRF3 Ab, followed by immunoblotting using the indicated Abs. (j) Raw264·7 cells were infected with either H1N1, or VSV, or SeV (MOI = 1·0) for 0, 3 and 6 h. The levels of MID1 mRNA were analysed by RT‐qPCR.
MID1 regulates ubiquitination and protein stability of IRF3
We further found that either knockdown (Fig. 4a) or overexpression (Fig. 4b) of MID1 did not affect IRF3 mRNA levels. In conjugation with the above results showing that MID1 downregulates the level of IRF3 proteins, we think that MID1 regulates IRF3 at protein level as a ubiquitin E3 ligase. Consistent with this idea, by carrying out a CHX pulse‐chase assay, we found that overexpression of MID1 substantially promoted the degradation of HA‐IRF3 proteins (Fig. 4c), suggesting that MID1 is a regulator of IRF3 protein stability. Next, we used a proteasome inhibitor MG132 to observe whether MID1‐mediated degradation of IRF3 proteins is through the proteasome. The results showed that MG132 treatment largely blocked MID1‐induced degradation of IRF3 proteins (Fig. 4d). In contrast, methylamine hydrochloride (MA), a widely used lysosomal inhibitor, did not inhibit MID1‐induced IRF3 degradation (Fig. 4d). Collectively, these findings demonstrated that MID1 induces proteasome‐dependent degradation of IRF3 proteins.
Figure 4.
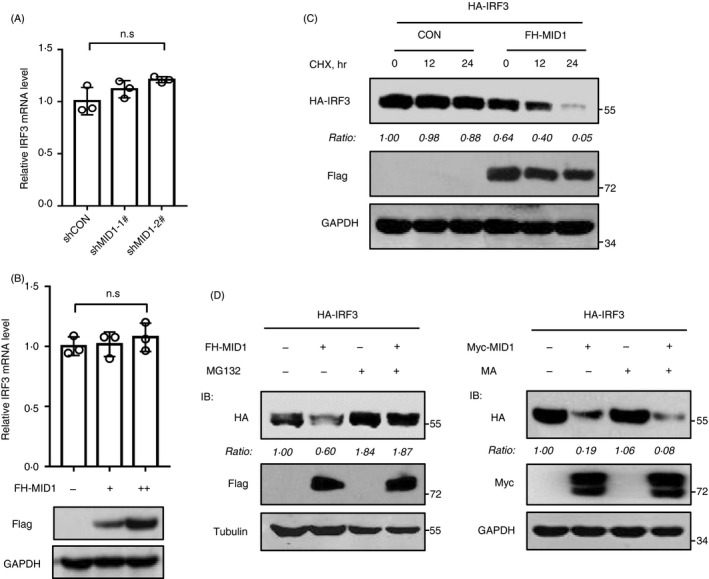
MID1 regulates protein stability of IRF3. (a) RT‐qPCR analysis of endogenous IRF3 mRNA in HEK293T cells transfected with shCON or two shMID1. (b) RT‐qPCR analysis of endogenous IRF3 mRNA in HEK293T cells transfected with increased amounts of FH‐MID1. Flag‐MID1 protein levels were detected by immunoblotting using an anti‐Flag antibody. (c) HEK293T cells were transfected with HA‐IRF3, together with or without FH‐MID1. Then, cells were treated with CHX (50 mg/ml) for different times. Whole‐cell extracts were analysed by immunoblotting using the indicated Abs. (d) HEK293T cells were transfected with HA‐IRF3, together with or without FH‐MID1. Then, cells were treated with MG132 (10 mm) for 12 h. Whole‐cell extracts were analysed by immunoblotting using the indicated Abs (left). HEK293T cells transfected with HA‐IRF3 and (or) Myc‐MID1 were treated with methylamine hydrochloride (MA; 20 mm) for 12 h. Whole‐cell lysates were analysed by immunoblotting using the indicated Abs (right).
Based on the above findings, we speculated that MID1 could regulate ubiquitination of IRF3 proteins. Our results showed that knockdown of MID1 dramatically downregulated polyubiquitination levels of exogenously expressed IRF3 (Fig. 5a). Further studies demonstrated that knockdown of endogenous MID1 significantly attenuated endogenous IRF3 ubiquitination (Fig. 5b). Conversely, overexpression of MID1 enhanced IRF3 ubiquitination modifications (Fig. 5c). Furthermore, we analysed the type(s) of IRF3 polyubiquitination mediated by MID1. The results showed that overexpression of MID1 largely upregulated K48‐linked polyubiquitination of IRF3 and also promoted K63‐linked polyubiquitination of IRF3 slightly, but did not significantly affect other types of IRF3 polyubiquitination (Fig. 5d). Thus, these findings suggested that MID1 is able to induce K48‐linked polyubiquitination and degradation of IRF3.
Figure 5.
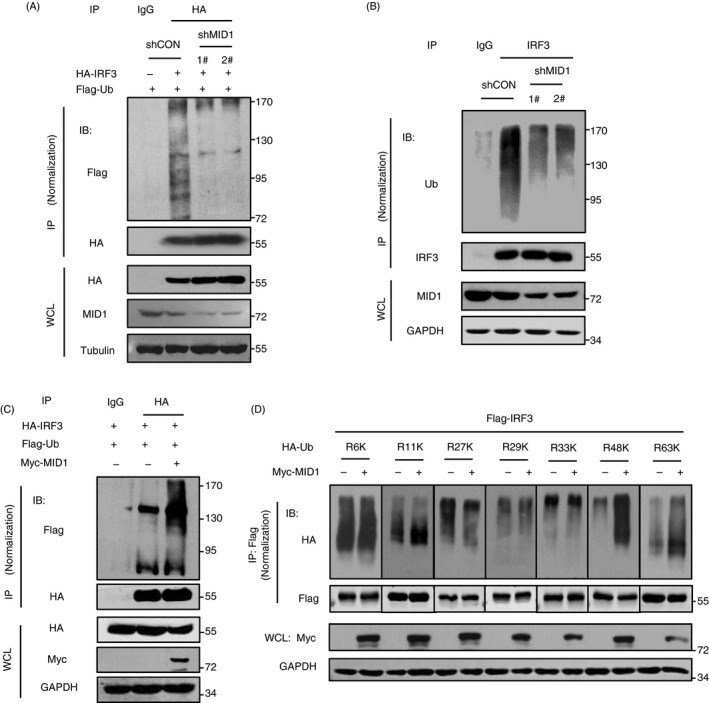
MID1 regulates ubiquitination of IRF3. (a) HEK293T cells were transfected with Flag‐Ub, together with HA‐IRF3 and two shMID1. HA‐IRF3 was immunoprecipitated using anti‐HA antibodies, and then, immunoblotting was performed as indicated. (b) HEK293T cells were transfected with two shMID1. Endogenous IRF3 was immunoprecipitated, and immunoblotting was performed as indicated. (c) HEK293T cells were transfected with Flag‐Ub, together with HA‐IRF3 and Myc‐MID1. HA‐IRF3 was immunoprecipitated using anti‐HA antibodies, and then, immunoblotting was performed as indicated. (d) HEK293T cells were transfected with Flag‐IRF3, Myc‐MID1 and different HA‐Ub mutants, including HA‐Ub‐K6 (R6K), HA‐Ub‐K11 (R11K), HA‐Ub‐K27 (R27K), HA‐Ub‐K29 (R29K), HA‐Ub‐K33 (R33K), HA‐Ub‐K48 (R48K) or HA‐Ub‐K63 (R63K). Flag‐IRF3 was immunoprecipitated using anti‐Flag antibodies, and then, Flag‐IRF3 ubiquitination levels were analysed by immunoblotting as indicated.
Ubiquitin E3 ligase activity of MID1 is required for MID1‐mediated regulation of IRF3, IFN‐I production and cellular antiviral response
As described above, we have demonstrated that MID1 can interact with, ubiquitinate and downregulate IRF3. Next, we determined whether these effects are dependent on the ubiquitin E3 ligase activity of MID1. MID1 consists of seven conserved domains (Fig. 6a). It has been reported that mutation of the RING domain of MID1 attenuated the ubiquitin E3 ligase activity of MID1· 16 Recent studies have revealed that both the RING and B‐box1 domain contribute to the ubiquitin E3 ligase activity of MID1· 17 Thus, according to the literature, we constructed two catalytically inactive mutants, MID1‐ΔRING (ΔR) and MID1‐Δ(RING + B‐box1) (ΔRB1) (Fig. 6a). We found that overexpression of MID1 wild type (WT) induced strong ubiquitination of IRF3, while overexpression of any of the inactive mutants did not noticeably upregulate ubiquitination levels of IRF3 (Fig. 6b). Furthermore, we determined the effect of MID1‐ΔRB1 on IRF3 protein levels. The results showed that MID1‐ΔRB1 lost the ability to decrease IRF3 proteins levels (Fig. 6c). These findings suggested that MID1‐mediated regulation of IRF3 ubiquitination and degradation is dependent on the ubiquitin E3 ligase activity of MID1. We further found that the B‐box1 domain of MID1 is required for its interaction with IRF3 (Fig. 6d). And MID1‐ΔRB1 cannot inhibit virus‐induced production of IFN‐I (Fig. 6e). Consistently, overexpression of the catalytically inactive MID1‐ΔRB1 lost the ability to promote viral infection (Fig. 6f). In addition, by fluorescence microscopy, we further confirmed that MID1‐mediated regulation of cellular antiviral response is dependent on its ubiquitin E3 ligase activity (Fig. 6g). In conclusion, these findings suggested that MID1 is a ubiquitin E3 ligase of IRF3, which regulates IRF3 ubiquitination and protein levels, as well as IFN‐I antiviral response.
Figure 6.
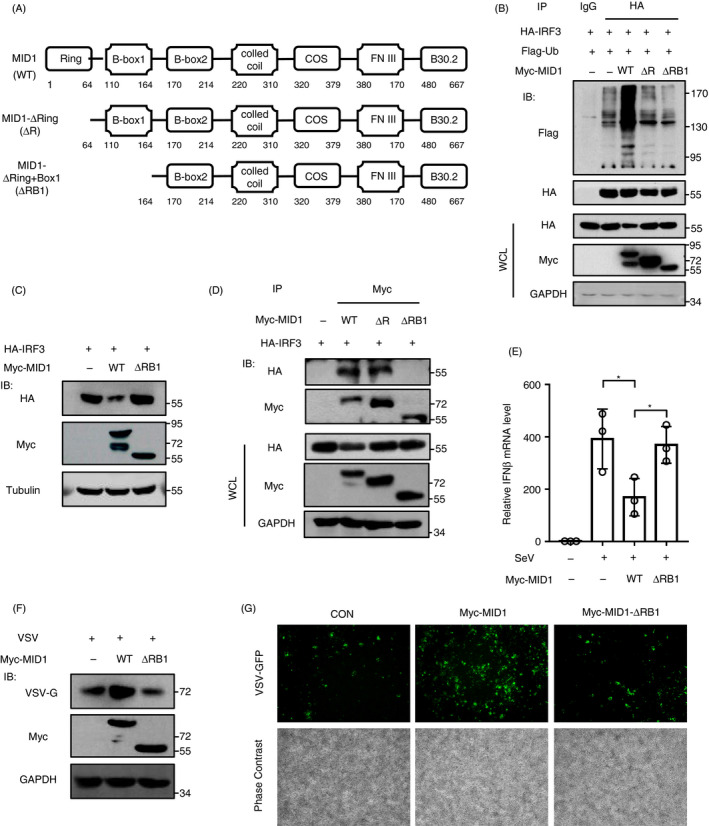
Ubiquitin E3 ligase activity of MID1 is required for MID1‐mediated regulation of IRF3, IFN‐I production and cellular antiviral response. (a) The domain of MID1 protein and MID1 deletion mutants. (b) HEK293T cells were transfected with HA‐IRF3 and Flag‐Ub, together with Myc‐MID1 (WT, ΔRING or Δ(RING + Bbox1)). HA‐IRF3 was immunoprecipitated, and immunoblotting was performed as indicated. (c) HEK293T cells were transfected with HA‐IRF3, together with Myc‐MID1(WT) or Myc‐MID1‐Δ(RING + Bbox1) (ΔRB1). The level of HA‐IRF3 was analysed by Western blot as indicated. (d) HEK293T cells were transfected with HA‐IRF3 and Myc‐MID1 (WT or its mutants). Myc‐MID1 or its mutants were immunoprecipitated, and immunoblotting was performed as indicated. (e) RT‐qPCR analysis of IFN‐β mRNA in HEK293T cells transfected with Myc‐MID1 (WT) or Myc‐MID‐ΔRB1, followed by SeV infection (MOI = 1·0) for 12 h. (f) HEK293T cells were transfected with Myc‐MID1 (WT) or Myc‐MID‐ΔRB1. Then, cells were infected with VSV (MOI = 1·0) for 20 h. Whole‐cell extracts were analysed by Western blot using the indicated Abs. (g) HEK293T cells were transfected with Myc‐MID1 (WT) or Myc‐MID‐ΔRB1. Then, cells were infected with VSV‐GFP (MOI = 1·0) for 20 h. The VSV‐GFP signal was detected by fluorescence microscopy. Scale bars: 500 µm.
Lys313 is a major lysine residue of IRF3 for ubiquitination induced by MID1
To clarify the effect of MID1 on IRF3 ubiquitination, we further identified the potential ubiquitination residue(s) of IRF3 induced by MID1. Human IRF3 protein has three conserved domains and consists of more than 400 amino acids with many lysine residues. Thus, we first constructed several deletion mutants of IRF3, IRF3‐ΔSR and IRF3‐Δ(SR + IAD), in which different domains of IRF3 were deleted (Fig. 7a). We noticed that both IRF3‐WT and IRF3‐ΔSR can be strongly ubiquitinated by MID1 (Fig. 7b), suggesting that the SR domain of IRF3 does not harbour the major ubiquitin acceptor residue(s) induced by MID1. However, MID1 cannot noticeably stimulate ubiquitination modifications of IRF3‐Δ(SR + IAD) (Fig. 7b), suggesting that the major ubiquitin acceptor residue(s) of IRF3 induced by MID1 are located within the IAD domain. We further mutated five lysine residues, including Lys193, Lys313, Lys315, Lys360 and Lys366, in the IAD domain of IRF3. We found that two of these IRF3 mutants, IRF3‐K193R and IRF3‐K366R, can be strongly downregulated by MID1, which is comparable to IRF3‐WT (Fig. 7c). However, mutation of either K313, or K315, or K360 of IRF3 attenuated MID1‐mediated downregulation of their protein levels to some extent (Fig. 7c). In particular, mutation of K313 largely abolished the degradation of IRF3 induced by MID1 (Fig. 7c), suggesting that Lys313 could be a major ubiquitin acceptor residue of IRF3 induced by MID1. Consistent with these findings, mutation of K313 largely inhibited MID1‐mediated ubiquitination of IRF3 (Fig. 7d). We further found that the IAD domain of IRF3 is required for its interaction with MID1 (Fig. 7e). Next, we found that mutation of K313 largely attenuated K48‐linked ubiquitination of IRF3 (Fig. 7f). Importantly, MID1 lost the ability to restrict IRF3‐K313R mutant‐induced production of IFN‐I (Fig. 7g). Finally, we demonstrated that IRF3‐K313R mutant was much stabler than IRF3‐WT in cells (Fig. 7h). Together, these findings revealed that MID1 as a ubiquitin E3 ligase can induce ubiquitination at Lys 313 of IRF3 and therefore regulated IRF3 protein stability.
Figure 7.
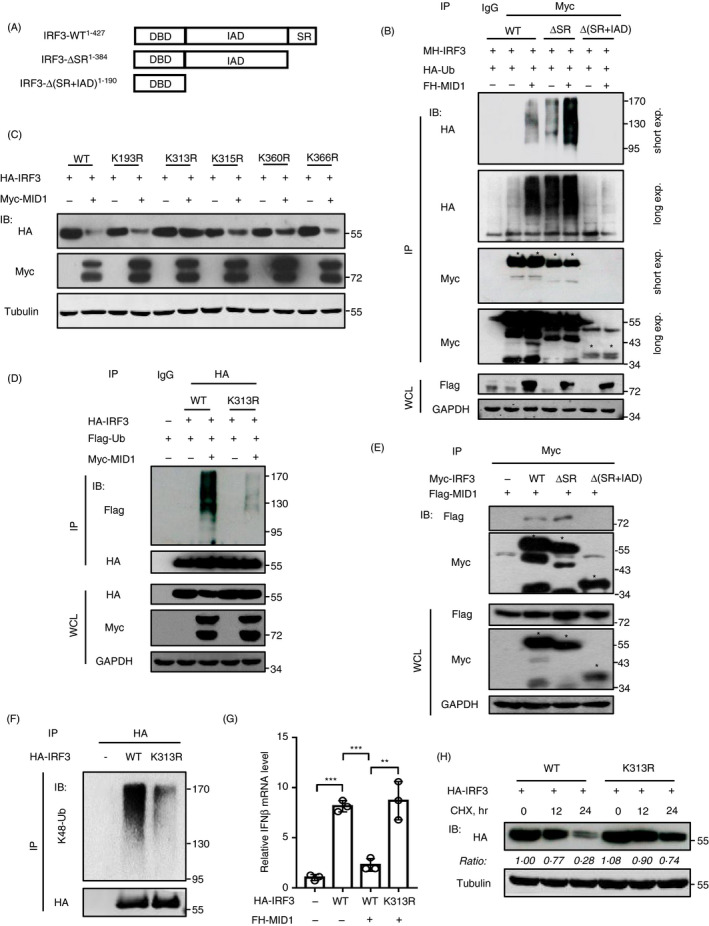
Lys313 is a major lysine residue of IRF3 for ubiquitination induced by MID1. (a) Wild‐type IRF3 and two IRF3 deletion mutants. (b) HEK293T cells were transfected with HA‐Ub, together with FH‐MID1 and MH‐IRF3 (WT, ΔSR or Δ(SR + IAD)) as indicated. Myc‐IRF3 and its mutants were immunoprecipitated, and then, immunoblotting was performed as indicated. (c) HEK293T cells were transfected with Myc‐MID1, together with HA‐IRF3 (WT) or HA‐IRF3‐K193R/K313R/K315R/K360R/366R mutants. The levels of HA‐IRF3 and its mutants were analysed as indicated. (d) HEK293T cells were transfected with Myc‐MID1, together with Flag‐Ub and HA‐IRF3 (WT or K313R). HA‐IRF3 (WT or K313R) proteins were immunoprecipitated, and then, immunoblotting was performed as indicated. (e) HEK293T cells were transfected with Flag‐MID1 and Myc‐IRF3 (WT or its mutants). Myc‐IRF3 and its mutants were immunoprecipitated, and then, immunoblotting was performed as indicated. (f) HEK293T cells were transfected with HA‐IRF3 (WT or K313R). HA‐IRF3 (WT or K313R) proteins were immunoprecipitated using anti‐HA antibodies, and immunoblotting was performed using specific anti‐K48‐linked ubiquitination antibodies. (g) RT‐qPCR analysis of IFN‐β mRNA in HEK293T cells transfected with HA‐IRF3 (WT/K313R) and FH‐MID1 as indicated. (h) HEK293T cells were transfected with HA‐IRF3 (WT or K313R). Then, cells were treated with CHX (50 mg/ml) for different times. HA‐IRF3 (WT or K313R) protein levels were detected by immunoblotting as indicated.
Discussion
MID1 was first noticed because of its association with a rare human genetic disease Opitz BBB/G syndrome (OS), which results in hypospadias, heart defects, hypertelorism, cleft lip and other symptoms. 15 Currently, most studies on MID1 focused on OS, cell cycle regulation and carcinogenesis, while the other biological functions of MID1 are poorly understood. 19 , 23 , 24 , 25 In this study, we revealed a new biological function of MID1. We demonstrated that MID1 is a crucial regulator of IFN‐mediated antiviral innate immune response. Our data suggested that MID1 is a ubiquitin E3 ligase of IRF3, which induces ubiquitination and degradation of IRF3 proteins, thus negatively regulating IFN‐I production and cellular antiviral activity.
Recent reports have demonstrated that MID1 could regulate both K48‐linked and K63‐linked polyubiquitination of its substrates. For example, MID1 catalysed both K48‐linked and K63‐linked polyubiquitination of Fu proteins. Consistent with our findings, we showed that MID1 induces both K48‐linked and K63‐linked polyubiquitination of IRF3. Although the biological functions of K63‐linked polyubiquitination of IRF3 induced by MID1 remain unknown, MID1‐induced K48‐linked polyubiquitination of IRF3 could promote IRF3 protein degradation. As shown in our results, MID1 lowed the protein stability of IRF3 and resulted in proteasome‐dependent degradation of IRF3. We think that it would be interesting to explore the significance of K63‐linked ubiquitination of IRF3 in the future.
Previous studies reported that linear ubiquitination of IRF3 at Lys193 and Lys313 could activate the RLR‐induced IRF3‐mediated pathway of apoptosis (RIPA), which could play an antiviral role by inducing cell apoptosis. 26 In our study, we found that MID1 induces K48‐linked polyubiquitination at Lys313 residue of IRF3. Based on these findings, we speculated that different ubiquitination types at Lys313 of IRF3 could compete each other to contribute to the switch between antiviral response and cell apoptosis. Further extensive studies in the future could promote the understanding of IRF3‐mediated biological functions. In addition, we noticed that the ability of MID1 to upregulate ubiquitination of IRF3 was significantly inhibited, but was not completely blocked after mutation of the Lys313 residue of IRF3. These results suggested that the Lys313 residue of IRF3 could be not the only site of IRF3 ubiquitination induced by MID1.
Virus infection could strongly promote IFN production and antiviral response. However, excessive production of IFNs has been recognized to be associated with some physiological disorders. Here, we found an important mechanism to restrict excessive production of IFNs. This mechanism may potentially protect cells from excessive inflammation. We revealed that MID1 physically interacts with IRF3 and regulates cellular IRF3 protein levels. And during viral infection, MID1 inhibits virus‐induced production of IFN‐I by degrading IRF3. These results implied that MID1 could act as a brake to limit IFN production under both physiological conditions and viral infection. Thus, we think that MID1‐mediated regulation of IRF3 has an important significance to maintain host homeostasis.
In summary, our study revealed that MID1 is a negative regulator of IRF3 protein levels, IFN‐I production and cellular antiviral activity. MID1 constitutively interacts with IRF3 and regulates IRF3 protein stability through mediating polyubiquitination modifications of IRF3 at Lys313 residue. This study could promote our understanding of MID1‐related biological functions and IRF3‐mediated antiviral response.
Disclosures
The authors declare no competing interests.
Date availability statement
All data are either presented in the manuscript or available from the corresponding author (Hui Zheng, Soochow University, huizheng@suda.edu.cn) upon request.
Author contributions
HZ and JW conceived and designed the projects. XC, YX, WT and FH performed most of the experiments and data analysis. YZ, HGZ and LJ contributed to cellular experiments and plasmid purification. QF, TR, JH, YM, YY, QZ, JL and RZ provided technical support. LZ, FQ and CZ helped data analysis and discussions. HZ and XC wrote the manuscript.
Acknowledgements
We thank Dr. Serge Y. Fuchs from University of Pennsylvania, Dr. Fangfang Zhou from Soochow University, Dr. Jianfeng Dai from Soochow University, Dr. Chunfu Zheng from Fujian Medical University and Dr. Feng Shao from National Institute of Biological Sciences of Beijing in China for important reagents. This work was supported by the National Key R&D Program of China (2018YFC1705500): 2018YFC1705505, the National Natural Science Foundation of China (31770177, 31970846), the Key Project of University Natural Science Foundation of Jiangsu Province (18KJA180010), the Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (2020KY368), the Gusu Medical Talent (Jun Wang) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
These authors Xiangjie Chen, Ying Xu, Wenhui Tu, and Fan Huang contributed equally.
Senior author: Chuanwu Zhu, Email: zhuchw@126.com.
Contributor Information
Hui Zheng, Email: huizheng@suda.edu.cn.
Jun Wang, Email: wjsdfyy@163.com.
References
- 1. Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol 2011; 29:185–214. [DOI] [PubMed] [Google Scholar]
- 2. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity 2013; 38:870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rehwinkel J, Reis e Sousa C. RIGorous detection: exposing virus through RNA sensing. Science 2010; 327:284–6. [DOI] [PubMed] [Google Scholar]
- 4. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M et al. The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 2004; 5:730–7. [DOI] [PubMed] [Google Scholar]
- 5. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol 2010; 11:373–84. [DOI] [PubMed] [Google Scholar]
- 6. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H et al. IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol 2005; 6:981–8. [DOI] [PubMed] [Google Scholar]
- 7. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell 2005; 122:669–82. [DOI] [PubMed] [Google Scholar]
- 8. Belgnaoui SM, Paz S, Hiscott J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr Opin Immunol. 2011; 23:564–72. [DOI] [PubMed] [Google Scholar]
- 9. Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010; 141:668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sadler AJ, Williams BR. Interferon‐inducible antiviral effectors. Nat Rev Immunol 2008; 8:559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shrivastav M, Niewold TB. Nucleic acid sensors and type I interferon production in systemic lupus erythematosus. Front Immunol 2013; 4:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kubota T, Matsuoka M, Chang TH, Tailor P, Sasaki T, Tashiro M et al. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J Biol Chem. 2008; 283:25660–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu Y, Hayward GS. The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010; 33:863–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang P, Zhao W, Zhao K, Zhang L, Gao C. TRIM26 negatively regulates interferon‐β production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog 2015; 11:e1004726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W et al. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet 1997; 17:285–91. [DOI] [PubMed] [Google Scholar]
- 16. Han X, Du H, Massiah MA. Detection and characterization of the in vitro e3 ligase activity of the human MID1 protein. J Mol Biol 2011; 407:505–20. [DOI] [PubMed] [Google Scholar]
- 17. Du H, Huang Y, Zaghlula M, Walters E, Cox TC, Massiah MA. The MID1 E3 ligase catalyzes the polyubiquitination of Alpha4 (α4), a regulatory subunit of protein phosphatase 2A (PP2A): novel insights into MID1‐mediated regulation of PP2A. J Biol Chem. 2013; 288:21341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S, Ropers HH et al. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat Genet 2001; 29:287–94. [DOI] [PubMed] [Google Scholar]
- 19. Schweiger S, Dorn S, Fuchs M, Köhler A, Matthes F, Müller EC et al. The E3 ubiquitin ligase MID1 catalyzes ubiquitination and cleavage of Fu. J Biol Chem 2014; 289:31805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pfirrmann T, Jandt E, Ranft S, Lokapally A, Neuhaus H, Perron M et al. Hedgehog‐dependent E3‐ligase Midline1 regulates ubiquitin‐mediated proteasomal degradation of Pax6 during visual system development. Proc Natl Acad Sci USA 2016; 113:10103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo T, Zuo Y, Qian L, Liu J, Yuan Y, Xu K et al. ADP‐ribosyltransferase PARP11 modulates the interferon antiviral response by mono‐ADP‐ribosylating the ubiquitin E3 ligase β‐TrCP. Nat Microbiol 2019; 4:1872–84. [DOI] [PubMed] [Google Scholar]
- 22. Ren Y, Zhao P, Liu J, Yuan Y, Cheng Q, Zuo Y et al. Deubiquitinase USP2a sustains interferons antiviral activity by restricting ubiquitination of activated STAT1 in the nucleus. PLoS Pathog 2016; 12:e1005764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winter J, Basilicata MF, Stemmler MP, Krauss S. The MID1 protein is a central player during development and in disease. Front Biosci (Landmark Ed) 2016; 21:664–82. [DOI] [PubMed] [Google Scholar]
- 24. Zanchetta ME, Meroni G. Emerging roles of the TRIM E3 ubiquitin ligases MID1 and MID2 in cytokinesis. Front Physiol 2019; 10:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Köhler A, Demir U, Kickstein E, Krauss S, Aigner J, Aranda‐Orgillés B et al. A hormone‐dependent feedback‐loop controls androgen receptor levels by limiting MID1, a novel translation enhancer and promoter of oncogenic signaling. Mol Cancer 2014; 13:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel JL, Sen GC. Ubiquitination of the transcription factor IRF‐3 activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity 2016; 44:1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]