

Summary
The Golgi apparatus is the central hub for protein trafficking and glycosylation in the secretory pathway. However, how the Golgi responds to glucose deprivation is so far unknown. Here, we report that GRASP55, the Golgi stacking protein located in medial- and trans-Golgi cisternae, is O-GlcNAcylated by the O-GlcNAc transferase OGT under growth conditions. Glucose deprivation reduces GRASP55 O-GlcNAcylation. De-O-GlcNAcylated GRASP55 forms puncta outside of the Golgi area, which colocalize with autophagosomes and late endosomes/lysosomes. GRASP55 depletion reduces autophagic flux and results in autophagosome accumulation, while expression of an O-GlcNAcylation-deficient mutant of GRASP55 accelerates autophagic flux. Biochemically, GRASP55 interacts with LC3-II on the autophagosomes and LAMP2 on late endosomes/lysosomes and functions as a bridge between LC3-II and LAMP2 for autophagosome and lysosome fusion; this function is negatively regulated by GRASP55 O-GlcNAcylation. Therefore, GRASP55 senses glucose levels through O-GlcNAcylation and acts as a tether to facilitate autophagosome maturation.
Keywords: GRASP55, LC3, LAMP2, O-GlcNAcylation, glucose starvation, tethering, autophagosome-lysosome fusion
Graphical Abstract

In brief
Zhang et al. provide insight into how the Golgi response to glucose deprivation and participates in autophagosome maturation. The Golgi stacking protein GRASP55 is de-O-GlcNAcylated upon glucose starvation and targeted to the autophagosome-lysosome interface through LC3-II and LAMP2, where it functions as a membrane tether to facilitate autophagosome-lysosome fusion.
Introduction
The Golgi apparatus is characterized by stacks of flattened cisternae, which are often laterally linked into a ribbon-like structure in mammalian cells. This unique structure is required for its proper functioning in intracellular trafficking and modifications of proteins and lipids in the secretory pathway. The exact mechanism for Golgi stack formation is unclear, but the Golgi ReAssembly Stacking Proteins, GRASP65 and GRASP55, are the only proteins shown to function in Golgi stacking so far (Xiang and Wang, 2010; Xiang et al., 2013). Their conserved N-terminal GRASP domain contains two PDZ domains that form homodimers, which subsequently oligomerize in trans between the adjacent cisternae and function as the “glue” to stick cisternae into a stack (Xiang and Wang, 2010) and link Golgi stacks into a ribbon (Puthenveedu et al., 2006). Their C-terminus Serine-Proline Rich (SPR) domains are more divergent, but both are phosphorylated in mitosis to dissociate the trans-oligomers and disassemble the Golgi (Zhang and Wang, 2015). Therefore, GRASPs are essential tethering proteins for maintaining cisternal membranes at close proximity, and their function is highly regulated. However, whether and how the Golgi responds to extracellular stimuli or stresses is largely unknown.
Autophagy, most often referred to as macroautophagy, is an evolutionarily conserved intracellular degradation process, which is crucial for clearance of protein aggregates and turnover of damaged organelles for cellular homeostasis; it is also important for survival of eukaryotic cells under stress conditions, such as nutrient and energy deprivation (Feng et al., 2014; Galluzzi et al., 2014; Nakatogawa et al., 2009; Saxton and Sabatini, 2017). Different sets of genes are involved in distinct steps of autophagy. The Atg1/Atg13 kinase complex and the Vps34/Atg14 PI(3)P kinase complex are required for the induction and initiation of the crescent-shaped isolation membrane (Chan and Tooze, 2009; Feng et al., 2014; Nakatogawa et al., 2009). The Atg8 (LC3 in mammalian cells) and Atg12 ubiquitin-like conjugation systems are involved in the expansion and closure of isolation membranes into autophagosomes (Feng et al., 2014; Nakatogawa et al., 2009). The autophagosomes are then delivered to and fused with lysosomes for degradation of the sequestrated materials (Lamb et al., 2013). Concerted action of Rab7 GTPase (Gutierrez et al., 2004; Jager et al., 2004), Rab7 effectors such as PLEKHM1 (McEwan et al., 2015), RILP (Cantalupo et al., 2001; Jordens et al., 2001), EPG5 (Wang et al., 2016), and the SNARE complex consisting of Syntaxin 17, SNASP29, and VAMP7/8 (Itakura et al., 2012), are required for autophagosome-lysosome fusion. TECPR1 (Chen et al., 2012), the HOPS complex (Jiang et al., 2014), and the Beclin 1/VPS34/VPS15/UVRAG complex, are also involved in this process (Levine et al., 2015; Liang et al., 2006). It is noteworthy that the autophagosome protein LC3 and lysosome protein LAMP2 (Saftig et al., 2008) have also been reported to participate in autophagosome maturation. LC3 binds Rab7 effectors, PLEKHM1 (McEwan et al., 2015) and EPG5 (Wang et al., 2016), which may account for its function in autophagosome and lysosome fusion; whereas the molecular mechanism for LAMP2 in this process is so far unknown.
In an effort to study how the Golgi responds to energy deprivation, we found that the Golgi stacking protein, GRASP55, is O-GlcNAcylated under growth conditions. Upon glucose starvation, GRASP55 is de-O-GlcNAcylated and targeted to the autophagosome-lysosome interface, where it interacts with LC3-II and LAMP2 to function as a bridge between autophagosomes and lysosomes. Our study reveals an unexpected role for GRASP55 as an energy sensor by O-GlcNAcylation and a membrane tether to promote autophagosome-lysosome fusion.
Results
GRASP55 is O-GlcNAcylated
The Golgi apparatus is the central subcellular membrane organelle that transports and processes a large number of secretory and transmembrane proteins in the endoplasmic reticulum (ER)-Golgi-plasma membrane trafficking axis; proper functioning of the Golgi is essential for a variety of cellular activities (Zhang and Wang, 2016). However, whether the Golgi adjusts its structure and function in response to extracellular stimuli or stresses, such as energy deprivation, is so far unknown. The basic energy source of cells is glucose. In the presence of glucose, many intracellular proteins are O-GlcNAcylated. Therefore, O-GlcNAcylation has been thought to function as a nutrient sensor in cells to regulate many cellular processes (Hardiville and Hart, 2014). So far, it is not known whether Golgi structural proteins are O-GlcNAcylated. Since O-GlcNAcylation occurs in the cytosol, we tested several Golgi peripheral membrane proteins for O-GlcNAcylation. GFP- or myc-tagged Golgi proteins were expressed in HeLa cells, immunoprecipitated, and probed with two O-GlcNAc antibodies, CTD110.6 and RL2. Among the proteins tested, including GRASP55 and GRASP65 (Fig. 1A), Golgin 45, Golgin 84, and Golgin 160 (Fig. S1A–C), only GRASP55 was O-GlcNAcylated. This result was confirmed with endogenous GRASP55; immunoprecipitated GRASP55 from normal rat kidney (NRK) cells, but not GRASP65, was recognized by the O-GlcNAc antibody CTD110.6 (Fig. 1B).
Figure 1. GRASP55 is O-GlcNAcylated.
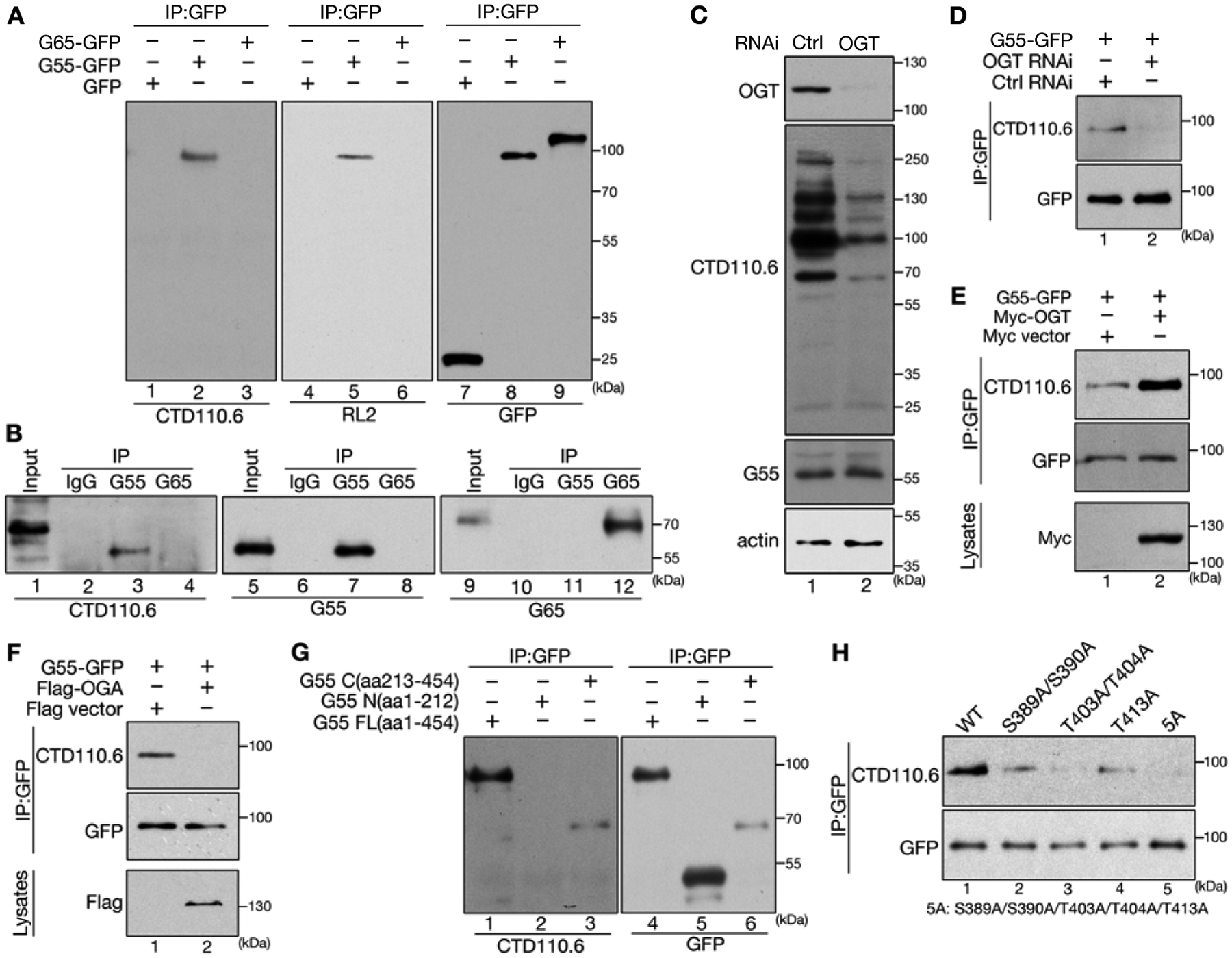
(A) GRASP55 (G55 in figures), but not GRASP65 (G65), is O-GlcNAcylated. HeLa cells transfected with GFP (as control), or GFP-tagged GRASP55 or GRASP65, were immunoprecipitated with a GFP antibody followed by Western blot for GFP and O-GlcNAc using two antibodies, CTD110.6 and RL2.
(B) Endogenous GRASP55 is O-GlcNAcylated. NRK cells were immunoprecipitated with control IgG, or GRASP55 or GRASP65 antibodies, and blotted for indicated proteins.
(C) OGT depletion decreases overall protein O-GlcNAcylation. HeLa cells were transfected with either control (ctrl) or OGT RNAi and analyzed by Western blot.
(D) OGT depletion reduces GRASP55 O-GlcNAcylation. HeLa cells were first transfected with control or OGT RNAi for 48 h and then with GRASP55-GFP for another 24 h. Cell lysates were immunoprecipitated with a GFP antibody followed by Western blot.
(E) Overexpression of OGT increases GRASP55 O-GlcNAcylation. HeLa cells co-transfected with GRASP55-GFP and a myc vector or myc-OGT were immunoprecipitated with a GFP antibody followed by Western blot. Cell lysates were probed for myc-OGT expression.
(F) Overexpression of OGA abolishes GRASP55 O-GlcNAcylation. HeLa cells were co-transfected with GRASP55-GFP and a Flag vector or Flag-OGA, lysed, immunoprecipitated with a GFP antibody, and blotted for GFP and O-GlcNAc. Cell lysates were probed for Flag-OGA expression.
(G) GRASP55 O-GlcNAcylation occurs at the C-terminus of GRASP55. HeLa cells expressing GFP-tagged GRASP55 full length (G55 FL, aa1-454), N-terminus (aa1-212), or C-terminus (aa213-454) were immunoprecipitated with a GFP antibody and blotted for GFP and O-GlcNAc.
(H) Identification of the O-GlcNAcylation sites on GRASP55. Five C-terminal serine and threonine residues were mutated individually or in combination, expressed in cells, and assessed by immunoprecipitation and Western blot.
See also Figure S1.
Protein O-GlcNAcylation is modulated by two enzymes; while the O-GlcNAc transferase (OGT) catalyzes the transfer of GlcNAc to serine or threonine residues on proteins, the O-GlcNAcase (OGA) removes GlcNAc from targeted proteins. Therefore, we manipulated OGT or OGA levels and determined the effects on GRASP55 O-GlcNAcylation. Depletion of OGT by RNA interference (RNAi) reduced the overall and GRASP55 O-GlcNAcylation, a similar effect was observed when OGA was overexpressed, while overexpression of OGT increased GRASP55 O-GlcNAcylation (Fig. 1C–F). These results demonstrate that GRASP55 is O-GlcNAcylated by OGT under normal growth conditions.
To map the O-GlcNAcylation sites on GRASP55, we expressed GRASP55 N-terminus (aa1-212), C-terminus (aa213-454) (Fig. 1G), and multiple truncation mutants (Fig. S1D) in HeLa cells and determined their O-GlcNAcylation by immunoprecipitation and Western blot for O-GlcNAc. Only the extreme C-terminus of GRASP55 (aa361-454) is required for O-GlcNAcylation (Fig. 1G; Fig. S1D). We then mutated 11 Ser/Thr residues to alanines in this region, single or in combination, and tested the effects on GRASP55 O-GlcNAcylation. Among the mutants tested, the S389A, S390A, T403A, T404A and T413A mutations reduced GRASP55 O-GlcNAcylation (Fig. S1E–F); and simultaneous mutation of all 5 sites completely abolished GRASP55 O-GlcNAcylation (Fig. 1H, Fig. S1G). This O-GlcNAcylation deficient mutant, referred to as 5A hereafter, was used to determine the function of GRASP55 O-GlcNAcylation in the following studies. Interestingly, the S380A mutation increased GRASP55 O-GlcNAcylation (Fig. S1F, lane 12); but when S380A was combined with the 5A mutant, the generated 6A mutant was not O-GlcNAcylated (Fig. S1G, lane 6). In addition, wild type (WT) GRASP55 was heavily O-GlcNAcylated when OGT was coexpressed, while the 5A mutant was not (Fig. S1H), even though they both interacted with OGT at a similar affinity (Fig. S1I). These results confirmed the five sites on which GRASP55 is O-GlcNAcylated.
Protein phosphorylation and O-GlcNAcylation are two similar post-translational modifications (PTMs) on serine and threonine residues, and mass spectrometric studies showed extensive crosstalk between these two PTMs (Hart et al., 2011). In GRASP55, sites for these two modifications are clustered in two different regions of the C-terminus (Fig. S1J). To determine whether mitotic phosphorylation of GRASP55 affects its O-GlcNAcylation, we expressed non-phosphorylatable and phosphomimetic GRASP55 mutants and tested their O-GlcNAcylation. The results showed, however, phosphorylation of GRASP55 had no effect on its O-GlcNAcylation (Fig. S1K). Consistently, GRASP55 O-GlcNAcylation remained unchanged in mitotic Golgi membranes compared to interphase Golgi membranes (Tang et al., 2010a), despite being heavily phosphorylated during mitosis as indicated by the band-shift (Fig. S1L). Therefore, GRASP55 O-GlcNAcylation is neither regulated by phosphorylation nor a cell cycle event.
GRASP55 depletion results in autophagosome accumulation
As protein O-GlcNAcylation is highly dependent on the level of glucose, we incubated cells in glucose starvation medium. GRASP55 O-GlcNAcylation was significantly reduced after 4 h and diminished after 8 h treatment (Fig. 2A). As previously reported (Kim et al., 2011), glucose deprivation induced autophagy over time, as indicated by the increased level of lipidated LC3B (LC3-II for simplicity in this study) and the decreased level of the cargo protein p62 (Fig. 2B; Fig. S1M–N). Glucose starvation, and/or treatment of cells with Bafilomycin A1 (BafA1, an inhibitor for autophagosome-lysosome fusion and autophagy mediated protein degradation) had no effect on the overall level of GRASP55 (Fig. 2B), suggesting that GRASP55 is not a cargo for autophagy-mediated protein degradation during glucose starvation. In addition, BafA1 treatment alone did not affect GRASP55 O-GlcNAcylation (Fig. 2C). These results revealed a positive correlation between increased autophagic flux and decreased GRASP55 O-GlcNAcylation upon glucose starvation, indicating a possible role for GRASP55 in autophagy.
Figure 2. GRASP55 depletion results in autophagosome accumulation.

(A) Glucose starvation significantly reduces GRASP55 O-GlcNAcylation. HeLa cells transfected with GFP-tagged GRASP55 were incubated in control (Con) or glucose starvation medium (GS) for 4 or 8 h as indicated. GRASP55 O-GlcNAcylation was assessed by immunoprecipitation and Western blot.
(B) Glucose starvation increases autophagic flux. HeLa cells were incubated for 4 h in control medium (Con), glucose starvation medium (GS), control medium with Bafilomycin A1 (BafA1) (Con+B), or glucose starvation medium with BafA1 (GS+B). Cell lysates were blotted with indicated antibodies.
(C) BafA1 treatment has no effect on GRASP55 O-GlcNAcylation. HeLa cells transfected with GRASP55-GFP were treated as in (B) followed by GFP immunoprecipitation and Western blot.
(D) GRASP55 depletion results in accumulation of autophagic cargo proteins. HeLa cells were transfected with control (ctrl), GRASP55 or GRASP65 RNAi for 72 h, lysed and analyzed by Western blot.
(E) Quantification of LC3-II and p62 levels in (D) from three independent experiments. Values are shown as mean ± SEM; statistical significance of the results was assessed by Student’s t-test in this and all following experiments. *, p<0.05; **, p<0.01; ***, p<0.001.
(F) GRASP55 depletion results in autophagosome accumulation. mRFP-GFP-LC3 cells were transfected with control (ctrl), GRASP55 or GRASP65 RNAi for 72 h, and immunostained for GARSP55 and/or GRASP65. Scale bar, 20 μm.
(G) Quantification of the number of autophagosomes (LC3 GFP+/RFP+)/cell and autolysosomes (LC3 GFP−/RFP+)/cell from (F).
See also Figure S1.
To dissect the role of GRASP55 in autophagy, we knocked down GRASP55 in cells and found that GRASP55 depletion increased the protein levels of both LC3-II and p62; this effect was not seen in control and GRASP65 depleted cells (Fig. 2D–E). As p62 and LC3-II accumulation indicates a defect in autophagosome-mediated degradation, we determined the effect of GRASP55 depletion on the autophagic flux using a HeLa cell line that stably expresses an mRFP-GFP-LC3 reporter (Vicinanza et al., 2015). In autophagosomes, which have neutral pH, both RFP (Red Fluorescent Protein) and GFP (Green Fluorescent Protein) are fluorescent and emit red and green signals, respectively. Upon autophagosome fusion with the lysosomes, the GFP signal is quenched by the acidic pH in the autolysosomes, while RFP signal remains. Therefore, the differentiation of the two-color signals faithfully represents the autophagic flux. GRASP55 knockdown resulted in an increased number of LC3 puncta that are positive for both red and green signals (autophagosomes), but decreased the red-only LC3 signals (autolysosomes) when compared with control and GRASP65 knockdown cells (Fig. 2F–G). These results demonstrate that GRASP55 depletion reduces autophagosome-lysosome fusion, suggesting an essential role for GRASP55 in autophagosome maturation.
GRASP55 is targeted to autophagosomes and late endosome/lysosomes upon glucose starvation
Since GRASP55 is a Golgi protein, it is not clear how it functions in autophagy upon energy deprivation. Therefore, we determined the subcellular localization of GRASP55 after glucose starvation. Surprisingly, in glucose starved mRFP-GFP-LC3 expressing cells, GRASP55 formed puncta outside of the Golgi area, which colocalized with autophagosomes as indicated by the dual-color GFP and RFP signals of LC3. This effect was most dramatic when glucose starvation medium contained BafA1 (Fig. 3A–B).
Figure 3. GRASP55 is targeted to autophagosomes and late endosomes/lysosomes upon glucose starvation.
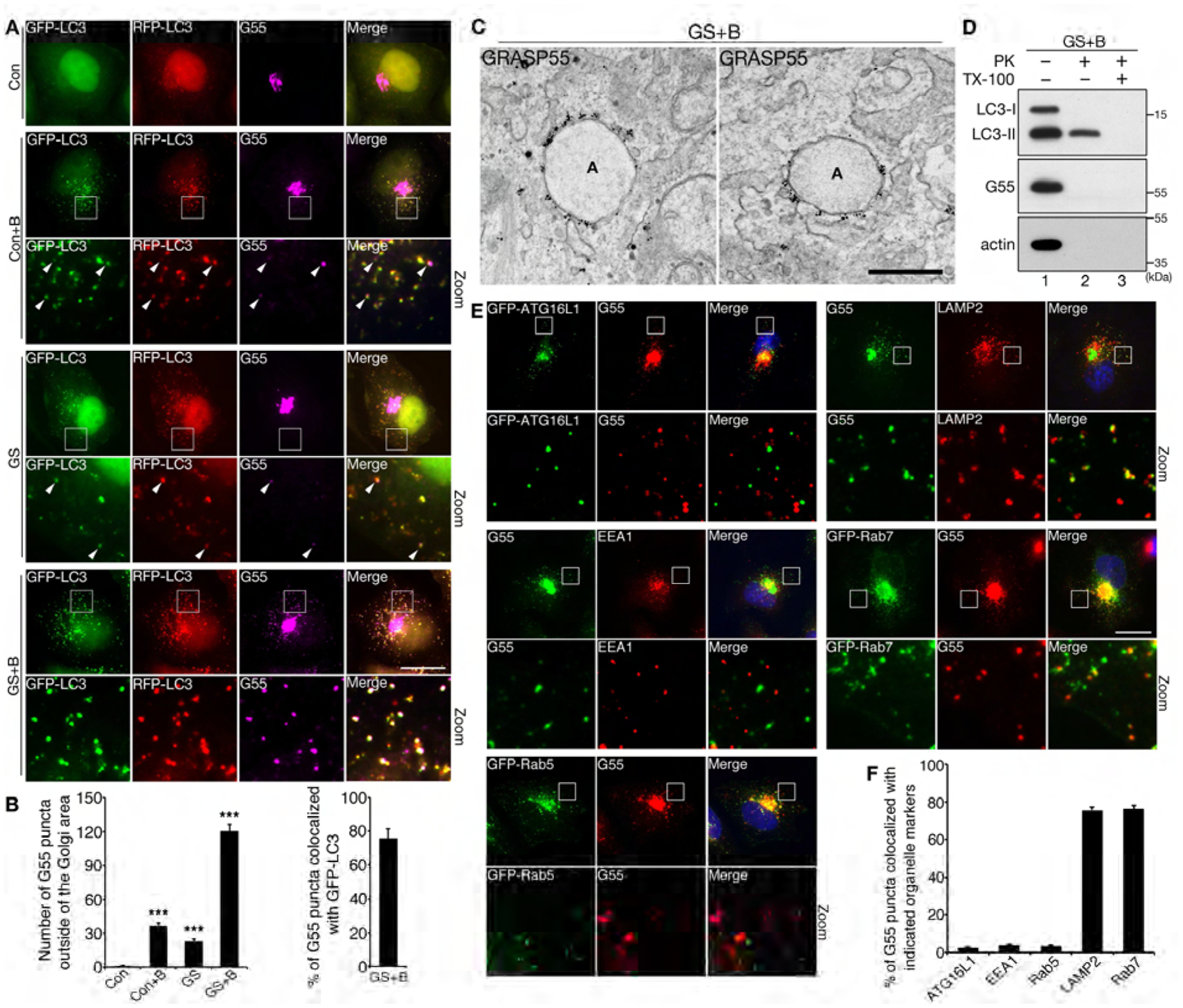
(A) GRASP55 colocalizes with LC3 upon glucose starvation. mRFP-GFP-LC3 cells were treated with control medium (Con), glucose starvation medium (GS), control medium with BafA1 (Con+B), glucose starvation medium with BafA1 (GS+B), and stained for GRASP55. Zoom in images of the indicated frames are shown underneath. Arrowheads indicate GRASP55 puncta that colocalize with mRFP-GFP-LC3. Scale bar, 20 μm.
(B) Quantification of (A) for the number of GRASP55 puncta per cell in all four conditions (left panel), and for the percentage of GRASP55 puncta that colocalized with LC3 after glucose starvation with BafA1 (right panel).
(C) Immuno-gold labeling of endogenous GRASP55 on autophagosome membranes after glucose starvation and BafA1 treatment. Two representative images are shown. Scale bar, 500 nm.
(D) GRASP55 is located on the surface of membrane compartments. HeLa cells were first incubated in glucose starvation medium in the presence of BafA1 for 4 h. Cells were homogenized and post-nuclear supernatant (PNS) was prepared. PNS was subsequently treated with proteinase K (PK) in the presence or absence of TX-100 and analyzed by Western blot.
(E) GRASP55 puncta colocalize with late endosome/lysosome markers, LAMP2 and Rab7, but not with early endosome markers, EEA1 and Rab5, or the early autophagy marker, ATG16L1. HeLa cells were untransfected or transfected with GFP-tagged ATG16L1, Rab5 or Rab7 as indicated, treated with glucose starvation medium with BafA1 for 4 h, and stained for indicated proteins. Scale bar, 20 μm.
(F) Quantification of (E) for the colocalization of GRASP55 puncta and indicated markers.
See also Figure S2.
The localization of GRASP55 on autophagosomes can be explained by autophagy-mediated degradation of Golgi elements. It is possible that some Golgi membranes or stacks are engulfed by autophagosomes for degradation upon glucose starvation. If this is the case, we expect that other Golgi proteins may also be found in autophagosomes. However, among all Golgi structural proteins we analyzed, including GRASP65 and GM130 (cis Golgi), mannosidase II (ManII, medial-Golgi transmembrane enzyme), Golgin 45 (medial and trans-Golgi), Golgin 97 (trans-Golgi), and TGN46 (TGN), GRASP55 was the only Golgi protein found in autophagosomes upon glucose starvation (Fig. S2A–E). As GRASP55 and GRASP65 are close homologues with similar functions, we directly compared their localizations in the same cell. Upon glucose starvation, GRASP55 formed puncta outside of the Golgi area, while GRASP65 remained in the Golgi (Fig. S2D). To confirm GRASP55 localization on autophagosomes, we performed Immuno-Electron Microscopy (Immuno-EM). Under growth conditions, GRASP55 specifically localized in the medial- and trans-Golgi (Fig. S2F); under glucose starvation and BafA1, GRASP55 was also concentrated on the surface of autophagosomes (Fig. 3C, Fig. S2G). Consistent with the notion that it functions on the surface of autophagosomes rather than a target for autophagy-mediated protein degradation, GRASP55 in the post-nuclear supernatant (PNS) of HeLa cells was easily accessible by added proteinase K (PK), while LC3-II was protected from PK digestion due to its partial localization in the lumen of autophagosomes (Fig. 3D). These results demonstrate that GRASP55 is a unique Golgi structural protein that is targeted to autophagosome membranes upon glucose starvation.
Autophagy is a multistep process. Therefore, we examined the colocalization of GRASP55 puncta with other markers for autophagosomes as well as endosomes and lysosomes in glucose starvation and BafA1 treated cells. GRASP55 puncta did not colocalize with the early autophagy marker ATG16L1, and are distinct from early endosome markers, EEA1 and Rab5, but largely overlapped with LAMP2- and Rab7-labeled late endosome/lysosomes (Fig. 3E–F). These observations originally made in HeLa cells were also confirmed in U2OS cells (Fig. S2H–I). Considering that GRASP55 knockdown resulted in autophagosome accumulation and p62 stabilization (Fig. 2D–G), we speculated that GRASP55 may function in a later stage of autophagy, such as autophagosome-lysosome fusion.
GRASP55 de-O-GlcNAcylation is required for autophagosome targeting
As glucose starvation decreased GRASP55 O-GlcNAcylation but increased its autophagosome localization, we asked whether there is a direct relationship between GRASP55 O-GlcNAcylation and autophagosome maturation by expressing rat WT GRASP55 or the O-GlcNAcylation deficient 5A mutant (Fig. 1H) in GRASP55-depleted HeLa cells. GRASP55 depletion reduced autophagic flux, as indicated by the increased level of the autophagy substrate p62 (Fig. 4A–B, Fig. S3A–B). Expression of WT GRASP55 rescued the autophagic flux by reducing p62 to a similar level as in control RNAi treated cells (Fig. 4A–B, lane 3 vs. 1), while expression of the 5A mutant further decreased p62 level compared to WT GRASP55 (Fig. 4A–B, lane 4 vs. 3). Expression of 5A also increased total LC3 level, indicating that the overall autophagy activity was enhanced. Consistently, when WT or 5A GRASP55 was expressed in GRASP55-depleted cells, the 5A mutant formed more puncta than WT GRASP55 under control conditions, which colocalized with LC3 (Fig. 4C–D). Furthermore, expression of WT GRASP55 in cells increased autophagy activity, as indicated by the elevated LC3 and reduced p62 levels, but again a stronger effect was observed when 5A was expressed (Fig. S3C–D). Additionally, depletion of the O-GlcNAc transferase OGT reduced GRASP55 O-GlcNAcylation (Fig. 1D), increased autophagic activity (Fig. S3E–F), and facilitated GRASP55 targeting to late endosomes/lysosomes (Fig. S3G–H). These results demonstrate that de-O-GlcNAcylated GRASP55 is more inclined to reside on autophagosomes to facilitate autophagosome-lysosome fusion.
Figure 4. De-O-GlcNAcylation is required for GRASP55 targeting to autophagosomes.
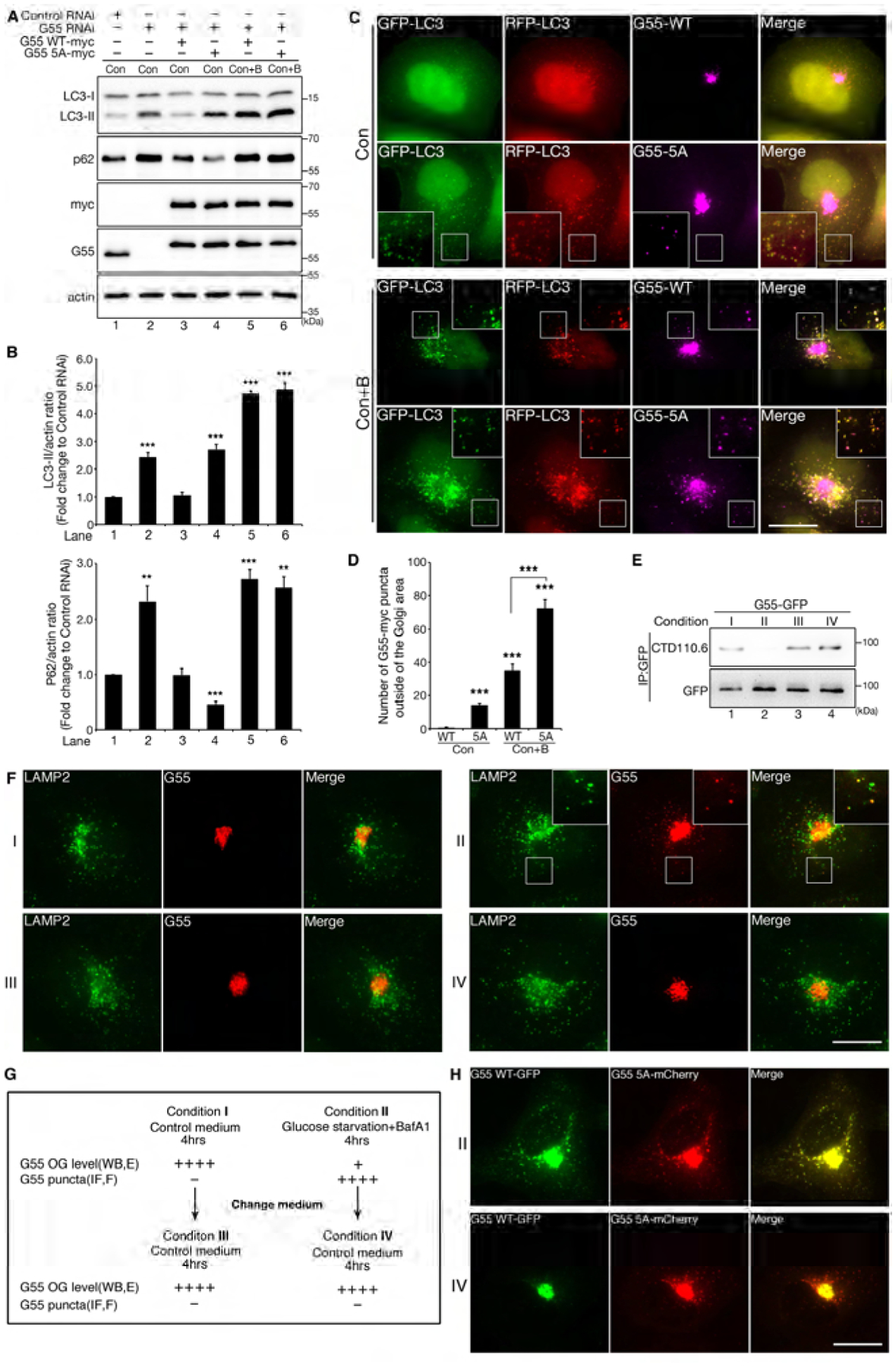
(A) Expression of the GRASP55 O-GlcNAcylation deficient mutant 5A increases autophagic flux. HeLa cells were transfected first with control RNAi or GRASP55 RNAi and then with myc-tagged WT GRASP55 or 5A in GRASP55-depleted cells. Cells were treated with control medium (Con) or control medium with BafA1 (Con+B) for 4 h, lysed and blotted with indicated antibodies.
(B) Quantification of LC3-II and p62 levels in (A) from three independent experiments.
(C) Expression of GRASP55 5A increases the number of GRASP55 puncta that colocalize with autophagosomes. mRFP-GFP-LC3 cells were first transfected with GRSAP55 RNAi and then with myc-tagged WT GRASP55 or 5A. Cells were treated with control medium without or with BafA1 and stained with a myc antibody. Scale bar, 20 μm.
(D) Quantification of the number of GRASP55 puncta per cell in (C) from three independent experiments. Statistical significance was assessed by comparing different treatments to WT GRASP55 transfected cells in control medium.
(E) Replenishment of glucose after glucose starvation restores GRASP55 O-GlcNAcylation. HeLa cells transfected with GRASP55-GFP were treated with either control medium (condition I) or glucose starvation medium with BafA1 (condition II) for 4 h followed by further incubation in control medium for another 4 h (conditions III and IV, respectively). Cell lysates were then immunoprecipitated with a GFP antibody followed by Western blot. See also panel (G) for the workflow.
(F) Replenishment of glucose after glucose starvation reduces GRASP55 puncta on autophagosomes. HeLa cells treated as in (E) were stained for LAMP2 and GRASP55. Scale bar, 20 μm. Images were enlarged in the insert to show the colocalization of GRASP55 and LAMP2 in condition II.
(G) Summary of the experimental procedure and results shown in (E–F). OG, O-GlcNAcylation; WB, Western blot; IF, immunofluorescence; E, results in panel E; F, panel F.
(H) When coexpresssed, the 5A mutant preferably remains as puncta than WT GRASP55 upon glucose replenishment. HeLa cells co-transfected with GFP-tagged WT GRASP55 and mCherry-tagged 5A were sequentially treated with conditions II and IV. Scale bar, 20 μm.
See also Figure S3.
To better understand how GRASP55 senses and dynamically responds to the glucose level in cells, we first incubated GRASP55-GFP expressing cells in growth medium (condition I) or glucose starvation medium with BafA1 (condition II) for 4 h, and then shifted to control medium for another 4 h (conditions III and IV, respectively). Western blot of immunoprecipitated GRASP55-GFP demonstrated that GRASP55 O-GlcNAcylation was high when cells were cultured in growth medium (conditions I, III & IV) but was low in starvation medium (condition II) (Fig. 4E). Fluorescence microscopy showed that GRASP55 formed puncta only when cells were incubated in glucose starvation medium (Fig. 4F). As summarized in Fig. 4G, these results revealed a dynamic negative relationship between GRASP55 O-GlcNAcylation and autophagosome localization.
To gain more information on the relationship between GRASP55 O-GlcNAcylation and autophagosome localization, we coexpressed GRASP55 WT-GFP and 5A-mCherry in HeLa cells. After glucose starvation and BafA1 treatment (condition II), both WT and 5A GRASP55 proteins formed and colocalized in puncta outside of the Golgi area, presumably autophagosomes. Upon glucose replenishment (condition IV), WT GRASP55 diminished from the puncta, while 5A persisted (Fig. 4H).
To determine whether autophagosome localized GRASP55 is newly synthesized or retargeted from an existing pool, we included the protein synthesis inhibitor cycloheximide (CHX) in the glucose starvation medium. In the presence of CHX, GRASP55 still formed puncta outside of the Golgi area, but the number is slightly reduced (Fig. S3I–J). These results indicate that preexisting GRASP55 is recruited to autophagosomes upon glucose starvation, but also suggest that newly synthesized GRASP55 may also be directly targeted to autophagosomes. However, given that CHX treatment itself inhibits autophagy (Watanabe-Asano et al., 2014), a more likely explanation for the decreased localization of GRASP55 on autophagosomes is the reduction of autophagosome formation by CHX. Taken together, de-O-GlcNAcylation is required for GRASP55 targeting to autophagosomes.
De-O-GlcNAcylated GRASP55 interacts with LC3-II and LAMP2
GRASP55 has been shown to link Golgi cisternal membranes into stacks by forming trans-oligomers (Xiang and Wang, 2010). The localization of GRASP55 to autophagosomes and lysosomes described above provided a possibility that GRASP55 oligomers may also tether autophagosomes and lysosomes to promote membrane fusion. As GRASP55 colocalized with LC3 upon glucose starvation, we first determined whether GRASP55 interacts with LC3 by co-immunoprecipitation. mRFP-GFP-LC3 weakly interacted with GRASP55 under growth conditions, but the interaction was significantly stronger after glucose starvation with BafA1 (Fig. S4A). As a control, no interaction was detected between GFP and GRASP55 regardless of the glucose level. Treatment of cells with glucose starvation or BafA1 alone significantly increased LC3-GRASP55 interaction, a combination of both yielded a stronger effect (Fig. 5A–B); this trend is consistent with GRASP55 puncta formation and autophagosome targeting under different conditions (Fig. 3A–B).
Figure 5. De-O-GlcNAcylated GRASP55 interacts with LC3-II and LAMP2.
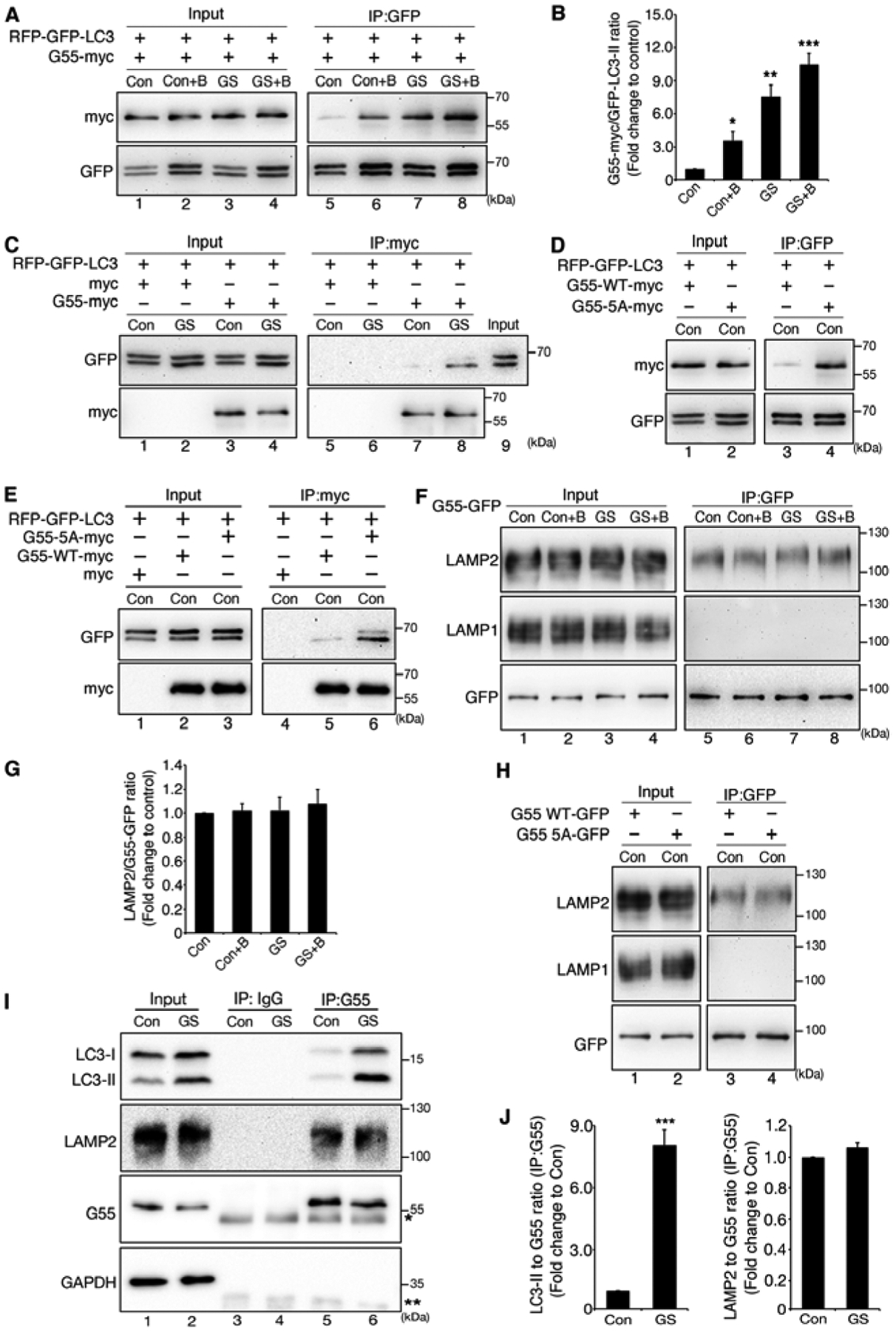
(A) GRASP55 interacts with LC3 upon glucose starvation. mRFP-GFP-LC3 cells transfected with GRASP55-myc were treated with control medium (Con), glucose starvation medium (GS), control medium with BafA1 (Con+B), or glucose starvation medium with BafA1 (GS+B), immunoprecipitated with a GFP antibody, and blotted for GFP and myc (right panel). Whole cell lysates were also blotted to show RFP-GFP-LC3 and GRASP55-myc expression.
(B) Quantification of the interaction between GRASP55-myc and RFP-GFP-LC3 in (A) from three independent experiments.
(C) GRASP55 preferably interacts with LC3-II. mRFP-GFP-LC3 cells transfected with a myc vector or GRASP55-myc were treated with control medium (Con) or glucose starvation medium (GS). Cells were then immunoprecipitated with a myc antibody and blotted for GFP and myc. Control cell lysate was loaded in lane 9 to indicate the positions of LC3-I and LC-II on the gel.
(D) Mutation of the O-GlcNAcylation sites on GRASP55 increases its interaction with LC3 under growth condition. mRFP-GFP-LC3 cells transfected with myc tagged WT GRASP55 or 5A were immunoprecipitated with a GFP antibody followed by Western blot.
(E) GRASP55 5A binds LC3-II under growth conditions at a higher affinity than WT GRASP55. mRFP-GFP-LC3 cells transfected with a myc vector or myc tagged WT GRASP55 or the 5A mutant were cultured in control medium (Con). Cells were then immunoprecipitated with a myc antibody and blotted for GFP and myc.
(F) GRASP55 interacts with LAMP2 but not LAMP1. HeLa cells transfected with GRASP55-GFP were treated as in (A) and immunoprecipitated with a GFP antibody and blotted for GFP, LAMP1 and LAMP2.
(G) Quantification of (F) from three independent experiments.
(H) GRASP55-LAMP2 interaction is independent of GRASP55 O-GlcNAcylation. HeLa cells transfected with GFP-tagged WT GRASP55 or 5A were immunoprecipitated with a GFP antibody and blotted for GFP, LAMP1 and LAMP2.
(I) Endogenous GRASP55 preferably interacts with LC3-II upon glucose starvation. HeLa cells were treated with control medium (Con) or glucose starvation medium (GS), immunoprecipitated with rabbit IgG or GRASP55 antibodies, and blotted for indicated proteins. *, IgG heavy chain; **, IgG light chain.
(J) Quantification of (I) from three independent experiments.
See also Figure S4.
LC3 belongs to the Atg8 protein family that has 6 members in mammalian: GABARAP, GABARAPL1, GABARAPL2 (GATE-16), MAP1LC3A, MAP1LC3B and MAP1LC3C; and so far, only LC3B has been tested for GRASP55 interaction (Fig. 5A–B; Fig. S4A). Therefore, we determined the interaction between GRASP55 and all 6 proteins by in vitro pull-down assays. Among these homologues, GRASP55 interacted with LC3B (LC3 in this study), LC3C and GABARAP, but at the highest affinity with LC3B (Fig. S4B). This validated a direct interaction between GRASP55 and LC3B, and justified the use of LC3B for further studies.
LC3 has two forms, the soluble form LC3-I in the cytosol and the lipidated form LC3-II on autophagosomes. To determine whether GRASP55 preferably interacts with LC3-I or LC3-II, we immunoprecipitated GRASP55 and blotted for LC3 in the bound fraction. Interestingly, although LC3-I and LC3-II were expressed at a comparable level, GRASP55 preferably bound to LC3-II under glucose starvation (Fig. 5C, lane 8 vs. 9). To determine whether GRASP55 O-GlcNAcylation directly affects GRASP55-LC3 interaction, we compared WT GRASP55 and the 5A mutant for LC3 interaction. Under growth conditions, WT GRASP55 had little if any interaction with LC3, while this interaction was much stronger between 5A and LC3 (Fig. 5D). Consistent with the results shown above, 5A preferably interacted with LC3-II than LC3-I (Fig. 5E). Taken together, glucose starvation reduces GRASP55 O-GlcNAcylation and increases GRASP55 interaction with LC3-II, indicating that de-O-GlcNAcylation is required for GRASP55-LC3 interaction.
GRASP55 interacts with LC3 on autophagosomes. To function as a membrane linker for autophagosome-lysosome fusion, it also needs a membrane anchor on the lysosomes. Consistent with this notion, GRASP55 colocalized with the late endosome/lysosome marker LAMP2 (Fig. 3E–F, Fig. S2H–I). The lysosome membrane proteins LAMP1 and LAMP2 are estimated to contribute to about 50% of all proteins on lysosome membranes (Eskelinen, 2006). Interestingly, GRASP55 co-immunoprecipitated with LAMP2 but not LAMP1, and the GRASP55-LAMP2 interaction was independent of the glucose level (Fig. 5F–G; Fig. S4C) and GRASP55 O-GlcNAcylation (Fig. 5H). Therefore, GRASP55 specifically interacts with LAMP2 regardless of the glucose level.
LAMP2 has three isoforms, A, B and C, by alternative splicing; the antibody we used for LAMP2 recognizes all three isoforms. The major difference between these isoforms lies in their transmembrane and cytoplasmic tails. Both LAMP2A and LAMP2B are ubiquitously expressed, while LAMP2C is tissue specific (Perez et al., 2016). Among these isoforms, LAMP2A is well characterized for its function in chaperone mediated autophagy (CMA) (Cuervo and Wong, 2014), and LAMP2B has been indicated in macroautophagy (Nishino et al., 2000; Tanaka et al., 2000). To specify which LAMP2 isoform interacts with GRASP55, we expressed GFP-tagged LAMP2A and LAMP2B and determined their interaction with GRASP55 by co-immunoprecipitation. Both LAMP2A and LAMP2B interacted with GRASP55, while LAMP2B exhibited a stronger signal (Fig. S4D), consistent with the role of LAMP2B in macroautophagy.
Since most of these results were obtained using exogenously expressed proteins, we determined the interaction between endogenous GRASP55, LC3 and LAMP2 by co-immunoprecipitation in both HeLa and U2OS cells. Our results confirmed that endogenous GRASP55 interacts with LC3 and LAMP2, and that the GRASP55-LC3-II interaction was significantly enhanced by glucose starvation (Fig. 5I–J, Fig. S4E–F). Taken together, GRASP55 interacts with LC3-II on autophagosomes and LAMP2 on lysosomes.
GRASP55 links LC3 and LAMP2 for autophagosome-lysosome fusion
The finding that GRASP55 binds LC3-II on autophagosomes and LAMP2 on lysosomes (Fig. 5) under glucose starvation prompted us to consider GRASP55 as a linker between autophagosomes and lysosomes through the interactions with LC3 and LAMP2. To test this hypothesis, we determined whether LC3 interacts with LAMP2 in dependence of GRASP55 by co-immunoprecipitation. A weak, but specific, interaction between LC3 and LAMP2 was detected under glucose starvation condition (Fig. S5A). Addition of purified recombinant GRASP55 into the cell lysate significantly increased LC3-LAMP2 interaction in a dose-dependent manner (Fig. 6A, lanes 4&5 vs. 1), while addition of bovine serum albumin (BSA) had no effect. In this experiment, GRASP55, but not BSA, co-immuoprecipitated with LC3 and LAMP2 (Fig. 6A–B).
Figure 6. De-O-GlcNAcylated GRASP55 links LC3 and LAMP2.
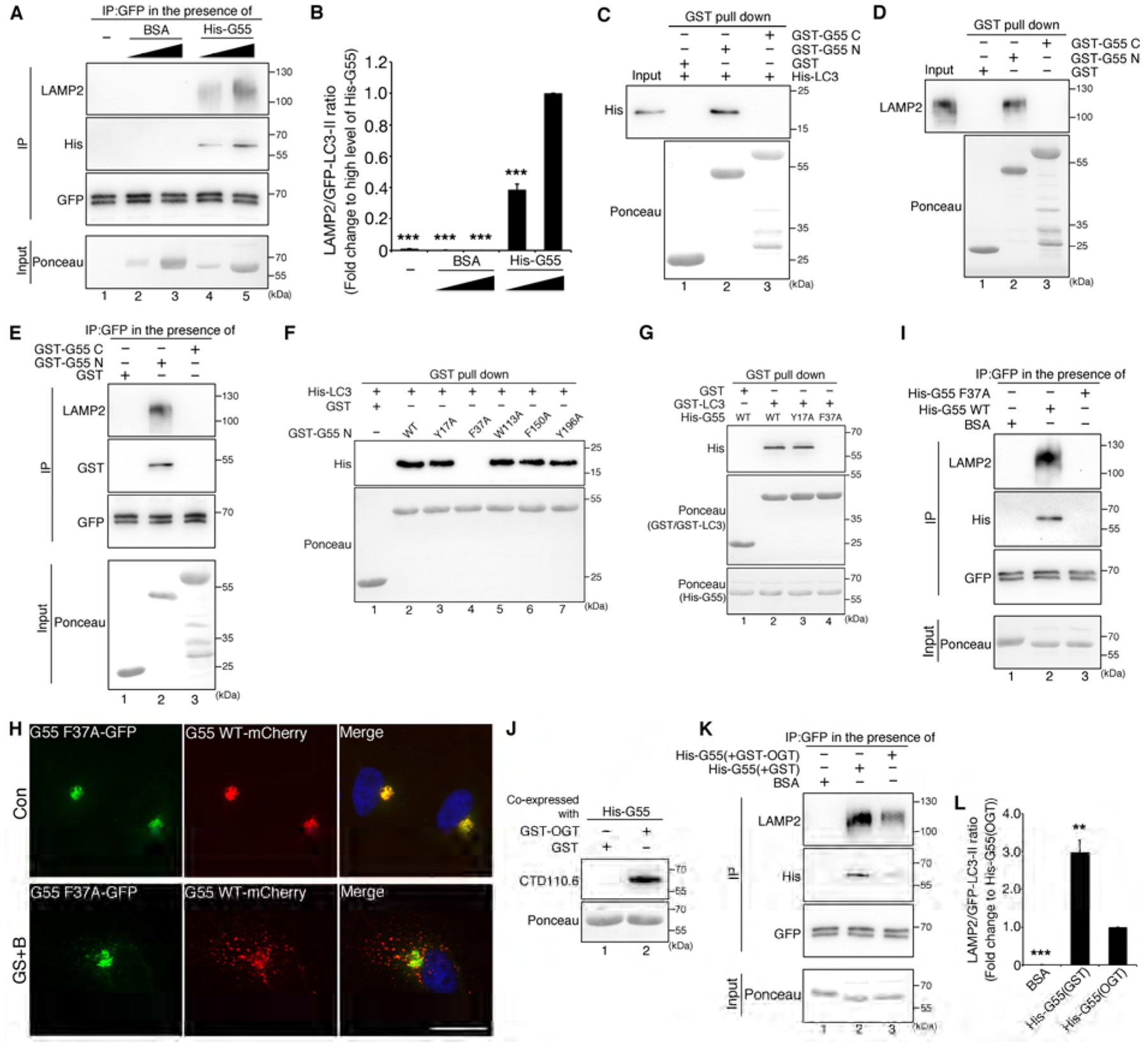
(A) GRASP55 meditates LC3-LAMP2 interaction. mRFP-GFP-LC3 cells treated with BafA1 were immunoprecipitated with a GFP antibody in the presence of nothing (lane 1), or increasing amount of BSA (as control, lanes 2&3) or His-tagged GRASP55 (lanes 4&5), followed by Western blot for LAMP2, His and GFP. Ponceau stain shows the input.
(B) Quantification of LAMP2 and LC3-II interaction in (A) from three independent experiments.
(C) The GRASP domain (N-terminus) of GRASP55 interacts with LC3. His-tagged LC3 was pulled down by GST-tagged GRASP55 N-terminus, but not by GST alone (as control) or GRASP55 C-terminus.
(D) The GRASP domain of GRASP55 interacts with LAMP2. HeLa cells were lysed and subjected to GST pull-down by indicated GST proteins followed by Western blot of LAMP2.
(E) The GRASP domain of GRASP55 mediates LC3-LAMP2 interaction. mRFP-GFP-LC3 cells were treated with BafA1, immunoprecipitated with a GFP antibody in the presence of indicated GST-tagged proteins, and blotted for LAMP2, GST and GFP.
(F) Identification of the F37 LIR motif on GRASP55 as the LC3 binding site. Five potential LIR motifs in the GRASP domain were mutated as indicated. The mutants were expressed and purified as GST-fusion proteins, incubated with recombinant His-tagged LC3, pulled down with glutathione beads, and blotted for His.
(G) F37A mutation abolishes GRASP55 and LC3 interaction. His-tagged full length WT GRASP55, Y17A, and F37A recombinant proteins were incubated with GST or GST-tagged LC3, pulled down with glutathione beads, and blotted for His.
(H) The F37 LIR motif on GRASP55 is required for GRASP55 targeting to autophagosomes. HeLa cells co-transfected with GRASP55 F37A-GFP and GRASP55 WT-mCherry were incubated in control medium (Con) or glucose starvation medium with BafA1 (GS+B) and analyzed by fluorescence microscopy. Scale bar, 20 μm.
(I) The F37 LIR motif on GRASP55 is required for GRASP55 to link LC3 and LAMP2. mRFP-GFP-LC3 cells were treated with BafA1, immunoprecipitated with a GFP antibody in the presence of BSA (as control, lane 1), His-tagged WT GRASP55 (lane 2), or the F37A mutant (lane 3) followed by Western blot for LAMP2, His and GFP.
(J) Preparation of O-GlcNAcylated GRASP55 by coexpression of OGT and His-tagged GRASP55 in bacteria. Purified proteins were probed with CTD110.6.
(K) O-GlcNAcylation of GRASP55 reduces its capability of linking LC3 and LAMP2. mRFP-GFP-LC3 cells were treated with BafA1, immunoprecipitated with a GFP antibody in the presence of indicated proteins, and blotted for LAMP2, His and GFP. Note that O-GlcNAcylated His-GRASP55 (coexpressed with GST-OGT) had lower activity in linking LC3 and LAMP2 compared with non-O-GlcNAcylated His-GRASP55 (coexpressed with GST).
(L) Quantification of LAMP2 and RFP-GFP-LC3-II interaction in (K) from three independent experiments.
See also Figure S5.
GRASP55 contains two major domains, a GRASP domain at the N-terminus (aa1-212) that forms dimers and oligomers and physically “glue” Golgi cisternal membranes into stacks, and a C-terminal SPR domain that contains multiple phosphorylation sites for cell cycle regulation of the Golgi structure (Fig. S1J) (Xiang and Wang, 2010). As shown using an in vitro GST pull-down assay, GRASP55 N-terminus, but not C-terminus, interacted with both LC3 and LAMP2 (Fig. 6C–D). Consistently, GRASP55 N-terminus but not C-terminus, or GST, enhanced LC3 and LAMP2 association (Fig. 6E), indicating that the GRASP domain could bridge autophagosomes and lysosomes in a similar way as it glues Golgi cisternae into a stack.
Within the N-terminal GRASP domain of GRASP55, there are 5 potential LC3-interacting region (LIR) motifs that fit in the W/F/Y-X/(D,E,S,T)-X-L/I/V consensus sequence (Deretic et al., 2013). Therefore, we mutated the first W/F/Y residue of each motif into alanine to determine the effect on GRASP55-LC3 interaction. Among these five mutants, only the F37A mutation abolished GRASP55-LC3 interaction (Fig. 6F–G). Since the effect is complete, it is likely that the F37 LIR motif is the only one on GRASP55 for LC3 interaction. When GRASP55 F37A-GFP and GRASP55 WT-mCherry were coexpressed, WT and F37A colocalized well in the Golgi area in control medium; however, after glucose starvation and BafA1 treatment, WT GRASP55 formed puncta outside of the Golgi area, while F37A mutant largely stayed in the Golgi region (Fig. 6H). In addition, F37A failed to enhance the interaction between LC3 and LAMP2 (Fig. 6I). These results identified the F37 LIR motif as the binding site of LC3 on GRASP55 and further confirmed that the GRASP55-LC3 interaction is essential for targeting GRASP55 onto autophagosomes.
It is noteworthy that the recombinant GRASP55 used in the above experiments was prepared from bacteria and was not O-GlcNAcylated. The obtained results support that LC3 preferentially interacts with de-O-GlcNAcylated GRASP55 (Fig. 5). To further determine the effect of O-GlcNAcylation on GRASP55 in bridging LC3 and LAMP2, we prepared O-GlcNAcylated GRASP55 by coexpressing OGT and GRASP55 in bacteria (Fig. 6J). Compared to non-O-GlcNAcylated GRASP55, O-GlcNAcylated GRASP55 had much lower activity in linking LC3 and LAMP2 (Fig. 6K–L). These results demonstrate that de-O-GlcNAcylated GRASP55 directly interacts with LC3 and LAMP2 through its oligomeric GRASP domain and that GRASP55 O-GlcNAcylation at the C-terminus inhibits GRASP55-LC3 interaction.
Consistent with the biochemical data described above (Fig. 6), we observed that GRASP55 concentrated at the autophagosome-lysosome interface under glucose starvation conditions on the immuno-EM images (Fig. S7A). To directly test whether GRASP55 links autophagosomes and lysosomes, we determined the effect of GRASP55 depletion on LC3 and LAMP2 colocalization after glucose starvation and BafA1 treatment. In the presence of GRASP55, a vast majority of autophagosomes (indicated by the dual color LC3 signals) colocalized with lysosomes (indicated by LAMP2) where GRASP55 was also detected. When GRASP55 was depleted, colocalization between autophagosomes and lysosomes was significantly reduced (Fig. 7A–C). Expression of WT GRASP55 and the 5A mutant, but not the F37A mutant, rescued LC3-LAMP2 colocalization in GRASP55-depleted cells (Fig. 7A–C).
Figure 7. GRASP55 facilitates autophagosome-lysosome fusion upon glucose starvation.
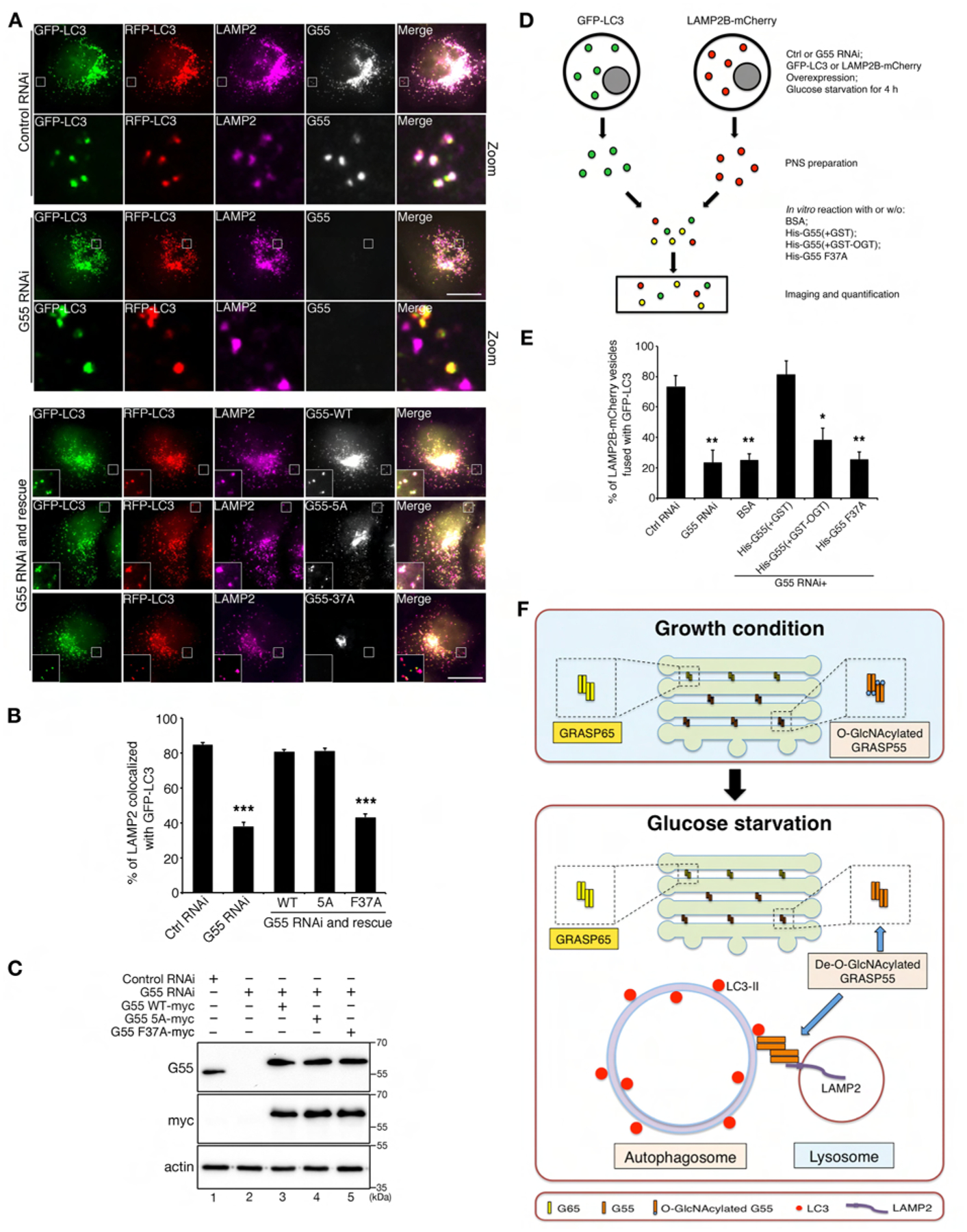
(A) GRASP55 is required for LC3-II and LAMP2 colocalization. mRFP-GFP-LC3 cells were first transfected with control or GRASP55 RNAi and then with myc-tagged WT GRASP55, 5A or F37A in GRASP55 depleted cells, treated with glucose starvation medium with BafA1, and stained for LAMP2 and GRASP55 or myc. Scale bar, 20 μm.
(B) Quantitation of LAMP2-LC3 colocalization in (A). Statistical significance was assessed by comparing different treatments to control RNAi.
(C) Cells as in (A) were analyzed by Western blot to show the knockdown efficiency of endogenous GRASP55 and the expression level of myc-tagged GRASP55.
(D) Schematic paradigm of in vitro fusion of GFP-LC3 and LAMP2B-mCherry vesicles. Control and GRASP55 RNAi treated cells were transfected with GFP-LC3 or LAMP2B-mCherry and incubated with glucose starvation medium. Post-nuclear supernatants (PNS) was prepared from GFP-LC3 and LAMP2B-mCherry expressing cells, mixed and incubated in the absence and presence of GRASP55 recombinant proteins. Samples were centrifuged onto coverslips and analyzed for colocalization of GFP-LC3 and LAMP2B-mCherry positive vesicles.
(E) GRASP55 facilitates autophagosome-lysosome fusion in vitro. Color-labeled autophagosomes and lysosomes prepared from GFP-LC3 and LAMP2B-mCherry expressing cells were mixed and incubated with or without recombinant GRASP55 as in (D), and analyzed by microscopy. Shown are the quantitation results of the percentage of LAMP2B vesicles that fused with GFP-LC3 under the indicated six conditions. Example images are shown in Fig. S6.
(F) Schematic model of GRASP55 function in Golgi stacking and autophagosomes-lysosomes tethering. During growth conditions, GRASP55 is O-GlcNAcylated and localized in medial- and trans- Golgi to glue adjacent cisternae together. Upon glucose deprivation, de-O-GlcNAcylated GRASP55 is targeted to the autophagosome and lysosome interface, where it binds LC3-II on autophagosomes and LAMP2B on lysosomes, and thereby links autophagosomes and lysosomes to facilitate fusion.
To better understand how GRASP55 mutation affects autophagic flux, we expressed these mutants in GRASP55-depleted mRFP-GFP-LC3 cells and incubated the cells in control medium. GRASP55 knockdown increased the number of autophagosomes but decreased that of autolysosomes, which was rescued by the expression of WT GRASP55 (Fig. S5B–C). Compared to WT GRASP55, expression of GRASP55 5A further increased the number of both autophagosomes and autolysosomes, suggesting that GRASP55 5A enhanced the autophagic flux. In contrast, GRASP55 F37A failed to rescue the effects caused by GRASP55 depletion, with a high number of autophagosomes and lower level of autolysosomes in the cell (Fig. S5B–C). These results demonstrated that de-O-GlcNAcylation and interaction with LC3 are both required for GRASP55 to promote autophagosome maturation.
To further confirm that GRASP55 links autophagosomes and lysosomes under glucose starvation, we prepared post-nuclear supernatant (PNS) from GFP-LC3 and LAMP2B-mCherry expressing cells, which contained color-labeled autophagosomes and lysosomes, respectively (Fig. 7D). When the two membranes were mixed and incubated, LAMP2B-mCherry positive lysosomes (red) fused with GFP-LC3 containing autophagosomes (green) and generated autolysosomes that are positive for both colors and thus shown as yellow puncta under a fluorescence microscope (Fig. 7D–E, Fig. S6). When GRASP55 was knocked down before PNS was prepared, the fusion activity was significantly reduced. This effect was rescued by supplementing recombinant non-O-GlcNAcylated His-GRASP55 proteins into the in vitro fusion assay, but not O-GlcNAcylated His-GRASP55, the F37A mutant, or BSA (Fig. 7E, Fig. S6). These results further demonstrated that autophagososome-lysosome fusion is enhanced by de-O-GlcNAcylated GRASP55 and GRASP55-LC3 interaction is required for this process.
It is worth mentioning that GRASP55 de-O-GlcNAcylation occurs not only under glucose starvation, but also under amino acid starvation (Fig. S7B) and upon inhibition of mTOR by Torin 1 (Fig. S7C). Torin 1 treatment decreases GRASP55 O-GlcNAcylation by reducing OGT expression (Fig. S7C). Like glucose starvation, Torin 1 treatment also triggers the formation of GRASP55 puncta that are positive for LC3 (Fig. S7D–E). Significantly, depletion of GRASP55 reduced p62 degradation in glucose starvation, amino acid starvation, and mTOR inhibition induced autophagy (Fig. S7F–K, lane 4 vs. 3), suggesting a common mechanism of de-O-GlcNAcylated GRASP55 in autophagy.
Taken together, our study has made an unexpected finding that the Golgi stacking protein GRASP55 functions as a membrane linker in autophagosome and lysosome fusion; this function is regulated by O-GlcNAcylation. Under growth conditions, GRASP55 is O-GlcNAcylated and localized in the Golgi to perform its role in Golgi structure formation and function. Upon glucose deprivation, some GRASP55 is de-O-GlcNAcylated and targeted to the autophagosome and lysosome interface, where it binds LC3-II on autophagosomes and LAMP2B on lysosomes, both through the GRASP domain that is known to form oligomers, and thereby links autophagosomes and lysosomes to facilitate fusion (Fig. 7F). These results indicate that GRASP55 senses glucose deprivation through de-O-GlcNAcylation to promote the autophagic flux.
Discussion
In this study, we have discovered an essential role for GRASP55 in autophagy, where it acts as a bridge to link LC3-II on autophagosomes and LAMP2B on lysosomes, and facilitates autophagy mediated protein degradation. GRASP55, originally identified as a Golgi stacking factor (Shorter et al., 1999), possesses two major molecular properties that assign its function in holding adjacent membranes together in a regulated manner. First, the N-terminal conserved GRASP domain, which is attached to the membrane by myristoylation, contains two tandem PDZ domains that allow it to form homodimers and trans-oligomers that function as a glue to bring two adjacent membranes together (Tang et al., 2010b; Wang et al., 2003). The fact that the GRASP domain binds both LC3 and LAMP2 indicates that GRASP55 may function as a membrane tether between autophagosomes and lysosomes in a similar way as it stacks Golgi cisternal membranes.
Second, GRASP55 contains a more divergent C-terminal SPR domain, which is phosphorylated in mitosis (Xiang and Wang, 2010) or O-GlcNAcylated in nutrient-rich conditions, making its function highly regulated. How exactly phosphorylation regulates GRASP oligomerization is so far unknown, but deletion of the SPR domain or mutation of the phosphorylation sites into alanines increased the efficiency of GRASP65 oligomerization (Wang et al., 2005). Therefore, it is hypothesized that phosphorylation in the C-terminus affects the oligomerization of the protein (Xiang and Wang, 2010), although no structural basis has been provided so far. Likewise, O-GlcNAcylation at its C-terminus may restrict the GRASP domain from interacting with LC3 via steric hindrance, while glucose starvation-induced de-O-GlcNAcylation may allow GRASP55-LC3 interaction. As GRASP55 constitutively interacts with the lysosome membrane protein LAMP2, the LC3-GRASP55-LAMP2 ternary complex formed upon glucose starvation may function as a linker between autophagosomes and lysosomes to promote fusion.
In addition to GRASP55, the SNARE protein, SNAP29, is also O-GlcNAcylated under growth conditions (Guo et al., 2014). Upon glucose starvation, de-O-GlcNAcylated SNAP29 forms a more stable complex with Syntaxin 17 and VAMP8 to mediate autophagosome-lysosome fusion. Therefore, protein O-GlcNAcylation by OGT is a common pathway that modulates multiple steps of the autophagy process. In addition, the protein level of OGT is regulated by autophagy (Park et al., 2014); inhibition of mTOR by Torin 1 reduced OGT protein level, decreased GRASP55 O-GlcNAcylation, but increased GRASP55 localization to autophagosomes (Fig. S7C–E), indicating an extensive role of GRASP55 in autophagy.
The major components of lysosome membranes are the homologous transmembrane proteins, LAMP1 and LAMP2. Mice deficient in either LAMP1 or LAMP2 are viable, while mice deficient in both LAMPs are lethal, suggesting that these two proteins share redundant functions in vivo (Eskelinen, 2006). However, compared to the mild phenotype in LAMP1 deficient mice, LAMP2 single deficiency is characterized by an increased postnatal lethality due to a massive accumulation of autophagosomes in various tissues (Saftig et al., 2008; Tanaka et al., 2000). Therefore, LAMP2 is thought to play a more important role in autophagy, but the molecular mechanism is missing. In this study, we identified GRASP55 as a binding partner for LAMP2, but not LAMP1, and thus revealed the molecular mechanism of LAMP2, in particular LAMP2B, in autophagosome maturation.
Membrane trafficking and fusion depends on a concerted action of different Rab GTPases, tethers, and SNARE complexes in distinct membrane organelles for vesicle docking and fusion (Yu and Hughson, 2010). In autophagy, Rab7, the HOPS tethering complex, and the STX17-SNAP29-VAMP7/8 SNARE complex are well documented for their roles in autophagosome-lysosome tethering and fusion. Here, we discovered that GRASP55 also bridges autophagosomes and lysosomes. The relative relationship between the Rab7/HOPS machinery and GRASP55 is so far unknown, but depletion of GRASP55 significantly reduced autophagosome-lysosome colocalization, suggesting that GRASP55 plays an indispensable role in autophagosome-lysosome tethering. It is possible that GRASP55 and Rab7/HOPS function as a cascade, or coordinate with each other. In the Golgi, it has been proposed that GRASPs help build the framework of the stacked structure, while Rabs, Golgins, and multisubunit COG tethers, as well as SNAREs, help locate the Golgi enzymes and cargo proteins inside this framework by controlling membrane fusion (Fisher and Ungar, 2016; Willett et al., 2013; Zhang and Wang, 2015, 2016). Extending from this hypothesis, we propose that GRASP55 holds autophagosomes and lysosomes at a relatively close proximity, while Rab7, HOPS, and subsequent SNARE complexes catalyze the membrane fusion process. This will be a future direction of this study.
Given the roles of GRASP55 in Golgi structure formation, it would be interesting to test whether glucose starvation and GRASP55 de-O-GlcNAcylation cause Golgi morphological changes. In this study, we stained glucose-starved cells with different Golgi makers, the results showed that the Golgi was still intact (Fig. S2A–E). This is not surprising, as our previous studies have demonstrated that knockdown of GRASP55 had only minor effect on the Golgi morphology under a fluorescence microscope (Xiang and Wang, 2010). Furthermore, the treatment of cells is relatively mild; most of our experiments were performed after 4 h incubation in glucose starvation medium, with serum included at all times. Under these conditions, only a relatively small portion of GRASP55 molecules function in autophagy, and most of the GRASP55 molecules still localize on the Golgi, and thus the Golgi is still intact.
How is GRASP55 targeted to autophagosomes? The answer is missing, but we can speculate on two possibilities. One is that upon glucose starvation, Golgi-derived GRASP55-containing membrane domains are targeted to autophagosomes. This possibility is relatively low, as we do not detect other Golgi markers on autophagosomes than GRASP55 (Fig. S2A–E). The other possibility is that a small cytosolic pool of GRASP55 dynamically exchanges with Golgi-anchored GRASP55. When cytosolic GRASP55 is de-O-GlcNAcylated, it is recruited to autophagosomes by the increased interaction with LC3. This speculation is supported by the results that interaction with LC3 is necessary for GRASP55 targeting to autophagosomes (Fig. 6H) and that only de-O-GlcNAcylated GRASP55 is targeted to autophagosomes (Fig. 4H). Since OGT overexpression increases the O-GlcNAc signal on GRASP55 (Fig. 1E), it is possible that a portion of GRASP55 is not O-GlcNAcylated under growth condition, which may contribute to basal levels autophagy. As GRASP55 interacts constitutively to LAMP2, we estimate that there is a small fraction of GRASP55 that is on the surface of late endosomes/lysosomes, possibly by recruiting GRASP55 from the cytosolic pool through GRASP55-LAMP2 interaction.
GRASP55 has also been shown to be required for unconventional secretion, an autophagy-dependent pathway that enables cytosolic proteins to be secreted (Dupont et al., 2011; Duran et al., 2010; Manjithaya et al., 2010; Pfeffer, 2010), although the underlying molecular mechanism is still not fully understood (Jiang et al., 2013; Rabouille, 2017). As the cytosolic proteins sequestered in autophagosomes are often secreted by fusion with the plasma membrane, the identification of GRASP55 as an autophagosome-lysosome tether provided a possibility that GRASP55 may be required for the release of cytosolic proteins to the extracellular space by facilitating autophagosome and plasma membrane fusion. This hypothesis will be tested in future studies.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yanzhuang Wang (yzwang@umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETALS
HeLa cells (female), mRFP-GFP-LC3 HeLa cells (female), U2OS cells (female), and NRK cells were cultured in Dubelcco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 units/ml penicillin-streptomycin at 37°C with 5% CO2.
METHODS DETAILS
Plasmids and antibodies
Constructs for GRASP55 truncation mutants, aa1-212, aa213-454, aa1-250, aa1-300, aa1-310, aa1-320, aa1-360, aa1-410, aa1-420, and GRASP55 O-GlcNAcylation deficient mutants, S350A, S379A, S380A, S379A/S380A, S389A, S390A, S389A/S390A, T403A, T404A, T403A/T404A, T413A, T425A/T426A, S389A/S390A/T403A/T404A/T413A and S380A/S389A/S390A/T403A/T404A/T413A, and GRASP55 potential LIR motif mutants, aa1-212 Y17A, aa1-212 F37A, aa1-212 W113A, aa1-212 F150A, aa1-212 Y196A, full length Y17A, and full length F37A were constructed in pEGFP-N1, pmCherry-N1, pcDNA3.1/Myc-His(-)A, pET-30a(+) or pGEX-6P-1 vectors. Constructs for GFP-tagged full length GRASP65 and GRASP55, GFP-tagged phosphorylation deficient mutants T222A/T225A, T222A/T225A/S245A, and phosphomimetic mutant T222E/T225E of GRASP55, were previously described (Xiang and Wang, 2010).
pEGFP-N1-mGolgin 45, pEGFP-C2-hGolgin 84, pEGFP-C1-ATG16L1, pET-30a(+)-rLC3B, pGEX-6P-1-OGT, pEGFP-N1-LAMP2A, pEGFP-N1-LAMP2B, pmCherry-N1-LAMP2B were constructed in this study by DNA cloning. pGEX4T1-hGABARAP(ΔG), pGEX4T1-hGABARAPL1(ΔG), pGEX4T1-hGABARAPL2 (GATE-16)(ΔG), pGEX4T1-hMAP1LC3A(ΔG), pGEX4T1-hMAP1LC3B(ΔG) and pGEX4T1-hMAP1LC3C(ΔG) were kindly provided by Dr. Ivan Dikic. Myc-tagged OGT and Flag-tagged OGA were kindly provided by Dr. Xiaochun Yu. Myc-tagged Golgin 160, GFP-tagged Rab7 and Rab5 were kindly provided by Drs. Carolyn Machamer, Haoxing Xu, and David Sheff, respectively. All cDNAs generated in this study were confirmed by DNA sequencing.
CTD110.6, LC3 and pS6K antibodies were purchased from Cell signaling; RL2 and GAPDH antibodies were from Abcam; Mouse GFP antibody, human GRASP55, OGT, actin, His, and Golgin 97 antibodies were from Proteintech; Flag and p62 antibodies were from Sigma; EEA1 and GST antibodies were from Santa Cruz; GM130 antibody was from BD Biosciences; LAMP1 and LAMP2 antibodies were from Developmental Studies Hybridoma Bank (DSHB); TGN46 antibody was from Bio-Rad. Antibodies for rat GRAP55, rat GRASP55, human GRASP65, ManII were previously described (Xiang and Wang, 2010). Myc antibody was from the Hybridoma Core Facility in University of Michigan.
Cell transfection
Lipofectamine 2000 (Invitrogen) was used for transient transfection of plasmids. In brief, cells were seeded and grown to ~80% confluency. Plasmids and Lipofectamine 2000 reagent were diluted into DMEM and mixed at a 1:1 ratio, incubated for 5 min at room temperature and added onto the cells. For knockdown experiments, HeLa cells or mRFP-GFP-LC3 cells were transfected with Lipofectamine RNAiMAX (Invitrogen). Briefly, cells were seeded and grown to ~60% confluency; siRNA and Lipofectamine RNAiMAX reagent were diluted into Opti-MEM medium and mixed at a 1:1 ratio, the mixture were incubated for 5 min at room temperature and added onto the cells. For OGT, GRASP55 or GRASP65 depletion, cells were analyzed 72 h after specific RNAi transfection. For the rescue experiment, HeLa cells or mRFP-GFP-LC3 cells were transfected with human GRASP55 specific RNAi for 48 h and then with rat WT GRASP55, O-GlcNAcylation mutant 5A, or LIR motif deficient mutant F37A for another 24 h. Control non-specific RNAi was purchased from Ambion. RNAi for human OGT (5’- GCACATAGCAATCTGGCTTCC-3’) (Chen et al., 2013) was purchased from Invitrogen. Human GRASP55 and GRASP65 targeting sequences were previously described (Xiang and Wang, 2010; Xiang et al., 2013).
Modulation of autophagy
To compare the phenotypes of GRASP55 puncta formation between normal and glucose starvation conditions, HeLa, U2OS or mRFP-GFP-LC3 HeLa cells cultured in growth medium were extensively washed with phosphate buffered saline (PBS) and then treated with control medium (DMEM medium, 25 mM glucose), control medium with 400 nM Bafilomycin A1 (BafA1), glucose starvation medium (DMEM medium without glucose, 0 mM glucose), or glucose starvation medium with 400 nM BafA1 for 4 h in most experiments or 8 h if indicated. To induce autophagy by amino acid starvation, HeLa cells were extensively washed with PBS and then incubated in Earle’s buffered salt solution (EBSS) for 4 h. To induce autophagy in an mTOR-dependent manner, cells were treated with Torin 1 at a working concentration of 0.1 μM for 24 h. All media were purchased from Invitrogen.
Proteinase K protection assay
HeLa cells were treated with glucose starvation medium with 400 nM BafA1 for 4 h. After treatment, cells were homogenized in 20 mM Hepes, pH 7.4, 220 mM mannitol, 70 mM sucrose, 1 mM EDTA. After a centrifugation at 500 g and 4°C for 5 min, the post-nuclear supernatant (PNS) was collected and equally divided into three tubes. One sample was left untreated, the other two were incubated with 2.5 μg/mL proteinase K (PK) only or with both PK and Triton X-100 (TX-100; 1% [vol/vol]) for 10 min on ice. Proteinase K was then inhibited by the addition of 1 mM PMSF and 10 min incubation on ice. All samples were subjected to Methanol/Chloroform precipitation, and protein pellets were resuspended in the same volume of sample buffer and analyzed by Western blot.
Immuno-electron microscopy (immuno-EM)
Pre-embedding immuno-EM was done as described in (Salonen et al., 2003). Briefly, HeLa cells grown on glass coverslips were fixed with PLP-fixative (McLean and Nakane, 1974) for 2 h at room temperature, permeabilized with 0.01% saponin, and immunolabeled with anti-GRASP55 (Proteintech) and nano-gold-conjugated anti-Rabbit IgG (Nanoprobes). Cells were then postfixed with 1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, and quenched with 50 mM glycine. Nano-gold particles were intensified using the HQ SILVER Enhancement kit (Nanoprobes) followed by gold toning in subsequent incubations in 2% NaAcetate, 0.05% HAuCl4 and 0.3% Na2S2O3 .5H2O. Cells were embedded into Epon (Puhka et al., 2007), and ultrathin sections were cut using Leica UCT6 microtome. Sections were post-stained with uranyl acetate and lead citrate, and observed with a Jeol JEM-1400 microscope operated at 80 kV. Images were acquired with a Gatan Orius SC 1000B camera.
Fluorescence microscopy
HeLa, U2OS, or mRFP-GFP LC3 cells were grown on glass coverslips and fixed in 3.7% paraformaldehyde, permeabilized with 0.3% Triton X-100 and processed for immunofluorescence microscopy with indicated antibodies. Cells were observed using a 63x oil objective on a Zeiss Observer Z1 epifluorescence microscope; Axiovision Rel. 4 was used for image acquisition and analysis. For quantification, ImageJ was used to count the number of LC3 and GRASP55 puncta and for the colocalization analysis.
Immunoprecipitation
To determine the protein O-GlcNAcylation level, HeLa cells transfected with indicated plasmids, or untransfected NRK cells, were lysed in 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100 and EDTA-free protease inhibitors (Roche). Lysate was cleared by centrifugation, incubated with indicated antibodies overnight at 4°C, subsequently incubated with protein A beads for another 2 h, reisolated and blotted with O-GlcNAc antibodies, CTD110.6 or RL2. To determine whether phosphorylation of GRASP55 in mitosis affects its O-GlcNAcylation, Rat Liver Golgi (RLG) membranes (Tang and Wang, 2015) were first incubated with interphase cytosol (IC) or mitotic cytosol (MC) for 1 h at 37°C, then recovered by ultracentrifugation in a TLA55 rotor at 55,000 rpm and 4°C for 1 h. The membrane pellets were lysed in 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100 and protease inhibitors and subjected to immunoprecipitation with a rat GRASP55 antibody. Rat liver Golgi membranes, and cytosols were prepared as previously described (Rabouille et al., 1995; Tang et al., 2010a).
To determine the interaction between GRASP55 and LC3 or LAMP2, HeLa cells or mRFP-GFP-LC3 cells were lysed in 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% NP-40 and protease inhibitors. Lysate was cleared by centrifugation, immunoprecipitated with indicated antibodies, and analyzed by Western blot.
To determine the interaction between LC3 and LAMP2, BSA or purified His-tagged GRASP55 was introduced into the co-immunoprecipitation assay. Briefly, mRFP-GFP-LC3 cells were lysed and protein concentration was measured, BSA or His-GRASP55 was added into the cell lysates at a 1:2500 (lower level) or 1:500 (higher level) ratio, mixed with GFP antibodies for co-immunoprecipitation (Fig. 6A). 1:500 was used as the ratio for subsequent co-immunoprecipitation assays (Fig. 6E, I and K).
Preparation of O-GlcNAcylated GRASP55
pET-30a(+)-GRASP55 plasmid was co-transformed with pGEX-6P-1-OGT or pGEX-6P-1 vector into E. coli BL21(DE3) competent cells and selected on LB-agar plates containing 12.5 μg/ml kanamycin and 25 μg/ml ampicillin. One clone with both proteins expressed was picked and bacteria were induced at 30°C for 6 h with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) after OD600 nm reached 0.6. His-tagged GRASP55 was purified with Ni-NTA agarose beads and concentrated in a buffer containing Tris-HCl (pH 7.4), 150 mM KCl, 1 mM MgCl2, 1 mM DTT and 10% glycerol. Purified His-GRASP55 that was coexpressed with GST or GST-OGT was subjected to Western blotting for O-GlcNAc using the CTD110.6 antibody.
In vitro GST pull-down assay
GST, GST-tagged GRASP55 N-terminus (aa1-212), GRASP55 C-terminus (aa213-454), hGABARAP, hGABARAPL1, hGABARAPL2, hMAP1LC3A, hMAP1LC3B, hMAP1LC3C, or His-tagged GRASP55 and LC3B proteins were expressed in BL21(DE3) bacteria and purified with glutathione Sepharose 4B beads and Ni-NTA agarose beads, respectively.
To determine GRASP55 and LC3 interaction, 5 μg of GST-tagged GRASP55 proteins were incubated with 5 μg of His-tagged LC3 in reaction buffer (20 mM Tris-HCl, pH8.0, 1M NaCl, 1% Triton X-100) at 4°C for 30 min, glutathione Sepharose 4B beads were then added into the mixture for another 30 min. After extensive washing, bound proteins were analyzed by Western blot with an anti-His antibody. The same procedure was applied to the interaction between GST-tagged LC3 homologues and His-tagged GRASP55. In Brief, 5 μg of GST-tagged six human LC3 homologues (hGABARAP, hGABARAPL1, hGABARAPL2, hMAP1LC3A, hMAP1LC3B, hMAP1LC3C) were incubated with 5 μg of His-tagged GRASP55 proteins in same reaction buffer, and glutathione Sepharose 4B beads were used to pull down the protein complex.
To determine GRASP55-LAMP2 interaction, 5 μg GST-tagged GRASP55 proteins were incubated with HeLa cell lysates in lysis buffer (20 mM Tris-HCl, pH8.0, 150 mM NaCl, 0.5% NP-40, protease inhibitor) at 4°C overnight, glutathione Sepharose 4B beads were then added into the mixture for another 30 min. After extensive washing, bound proteins were analyzed by Western blot with an anti-LAMP2 antibody.
In vitro fusion assay
The assay was performed as previously described (Barysch et al., 2010; Moreau et al., 2011). Briefly, HeLa cells were first transfected with control or GRASP55 RNAi for 48 h, then with GFP-LC3 or LAMP2B-mCherry for another 20 h. All cells were incubated with glucose starvation medium for 4 h. Post-nuclear supernatant (PNS) was prepared from HeLa cells expressing either GFP-LC3 or LAMP2B-mCherry separately, mixed and incubated for 30 min at 37°C in the presence an ATP regenerative system (10 mM creatine phosphate, 1 mM ATP, 20 μg/ml creatine kinase and 20 ng/ml cytochalasin B). To test the role of GRASP55 in autophagosome-lysosome fusion in vitro, His-tagged WT or mutant GRASP55 proteins, or BSA (as control), were added into the reactions using PNS prepared from GRASP55 RNAi treated cells. After incubation, all the samples were centrifuged and immobilized on glass coverslips at 4000 rpm at 4°C for 1 h, and single and double-labeled vesicles were visualized by fluorescence microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data represent the mean ± SEM (standard error of the mean) of at least three independent experiments unless noted. At least 20 cells were counted for colocalization analysis or puncta number measurement. A statistical analysis was conducted with two-tailed Student’s t-test. Differences in means were considered statistically significant at p < 0.05. Significance levels are: *, p<0.05; **, p<0.01; ***, p<0.001. Analyses were performed using Fiji. Figures were assembled with Photoshop CS6.
Supplementary Material
Highlights:
GRASP55 is O-GlcNAcylated under growth condition
Upon glucose starvation GRASP55 is de-O-GlcNAcylated and targeted to autophagosomes
GRASP55 interacts with LC3-II on autophagosomes and LAMP2 on lysosomes
GRASP55 bridges LC3-II and LAMP2 to facilitate autophagosome-lysosome fusion
Acknowledgements
We thank Dr. Ivan Dikic for the cDNAs of the six Atg8 human homologues, Dr. Carolyn Machamer for the Golgin 160 cDNA, Dr. David Sheff for Rab5 cDNA, Dr. Xiaochun Yu for the OGT and OGA cDNAs, Dr. Kelley Moremen for the ManII antibody, Dr. David Rubinsztein for the mRFP-GFP-LC3 cell line and Dr. Haoxing Xu for Rab7 cDNA, Drs. Michael Tiemeyer and Lance Wells for the CTD110.6 antibody and for suggestions, members of the Wang lab for helpful and constructive discussions and comments on the project, and Courtney Killeen, Erpan Ahat, Dr. Michael Bekier and Dr. Shijiao Huang for proofreading of the manuscript. This work was supported in part by the National Institutes of Health (Grant GM112786), MCubed and the Fast Forward Protein Folding Disease Initiative of the University of Michigan to Y. Wang. E.J. is funded by the Academy of Finland (project 1287975) and Biocenter Finland. B.L. is a graduate student in Integrative Life Science (ILS) PhD program, University of Helsinki, Finland.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures.
References
- Barysch SV, Jahn R, and Rizzoli SO (2010). A fluorescence-based in vitro assay for investigating early endosome dynamics. Nat Protoc 5, 1127–1137. [DOI] [PubMed] [Google Scholar]
- Cantalupo G, Alifano P, Roberti V, Bruni CB, and Bucci C (2001). Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. The EMBO journal 20, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EY, and Tooze SA (2009). Evolution of Atg1 function and regulation. Autophagy 5, 758–765. [DOI] [PubMed] [Google Scholar]
- Chen D, Fan W, Lu Y, Ding X, Chen S, and Zhong Q (2012). A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Molecular cell 45, 629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Chen Y, Bian C, Fujiki R, and Yu X (2013). TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, and Wong E (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V, Saitoh T, and Akira S (2013). Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, and Deretic V (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. The EMBO journal 30, 4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran JM, Anjard C, Stefan C, Loomis WF, and Malhotra V (2010). Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol 188, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL (2006). Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Molecular aspects of medicine 27, 495–502. [DOI] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, and Klionsky DJ (2014). The machinery of macroautophagy. Cell Res 24, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher P, and Ungar D (2016). Bridging the Gap between Glycosylation and Vesicle Traffic. Front Cell Dev Biol 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Levine B, and Kroemer G (2014). Metabolic control of autophagy. Cell 159, 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovacs AL, Zhang Z, et al. (2014). O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nature cell biology 16, 1215–1226. [DOI] [PubMed] [Google Scholar]
- Gutierrez MG, Munafo DB, Beron W, and Colombo MI (2004). Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. Journal of cell science 117, 2687–2697. [DOI] [PubMed] [Google Scholar]
- Hardiville S, and Hart GW (2014). Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell metabolism 20, 208–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Slawson C, Ramirez-Correa G, and Lagerlof O (2011). Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annual review of biochemistry 80, 825–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi-Itakura C, and Mizushima N (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269. [DOI] [PubMed] [Google Scholar]
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, and Eskelinen EL (2004). Role for Rab7 in maturation of late autophagic vacuoles. Journal of cell science 117, 4837–4848. [DOI] [PubMed] [Google Scholar]
- Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, and Mizushima N (2014). The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Molecular biology of the cell 25, 1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Dupont N, Castillo EF, and Deretic V (2013). Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. Journal of innate immunity 5, 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordens I, Fernandez-Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, Janssen H, Wubbolts R, and Neefjes J (2001). The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Current biology : CB 11, 1680–1685. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb CA, Dooley HC, and Tooze SA (2013). Endocytosis and autophagy: Shared machinery for degradation. Bioessays 35, 34–45. [DOI] [PubMed] [Google Scholar]
- Levine B, Liu R, Dong X, and Zhong Q (2015). Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends in cell biology 25, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, and Jung JU (2006). Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nature cell biology 8, 688–699. [DOI] [PubMed] [Google Scholar]
- Manjithaya R, Anjard C, Loomis WF, and Subramani S (2010). Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol 188, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, et al. (2015). PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Molecular cell 57, 39–54. [DOI] [PubMed] [Google Scholar]
- McLean IW, and Nakane PK (1974). Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem 22, 1077–1083. [DOI] [PubMed] [Google Scholar]
- Moreau K, Ravikumar B, Renna M, Puri C, and Rubinsztein DC (2011). Autophagosome precursor maturation requires homotypic fusion. Cell 146, 303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, and Ohsumi Y (2009). Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature reviews 10, 458–467. [DOI] [PubMed] [Google Scholar]
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. (2000). Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910. [DOI] [PubMed] [Google Scholar]
- Park S, Pak J, Jang I, and Cho JW (2014). Inhibition of mTOR affects protein stability of OGT. Biochem Biophys Res Commun 453, 208–212. [DOI] [PubMed] [Google Scholar]
- Perez L, McLetchie S, Gardiner GJ, Deffit SN, Zhou D, and Blum JS (2016). LAMP-2C Inhibits MHC Class II Presentation of Cytoplasmic Antigens by Disrupting Chaperone-Mediated Autophagy. Journal of immunology 196, 2457–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR (2010). Unconventional secretion by autophagosome exocytosis. J Cell Biol 188, 451–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhka M, Vihinen H, Joensuu M, and Jokitalo E (2007). Endoplasmic reticulum remains continuous and undergoes sheet-to-tubule transformation during cell division in mammalian cells. J Cell Biol 179, 895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu MA, Bachert C, Puri S, Lanni F, and Linstedt AD (2006). GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nature cell biology 8, 238–248. [DOI] [PubMed] [Google Scholar]
- Rabouille C (2017). Pathways of Unconventional Protein Secretion. Trends in cell biology 27, 230–240. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Misteli T, Watson R, and Warren G (1995). Reassembly of Golgi stacks from mitotic Golgi fragments in a cell-free system. J Cell Biol 129, 605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saftig P, Beertsen W, and Eskelinen EL (2008). LAMP-2: a control step for phagosome and autophagosome maturation. Autophagy 4, 510–512. [DOI] [PubMed] [Google Scholar]
- Salonen A, Vasiljeva L, Merits A, Magden J, Jokitalo E, and Kaariainen L (2003). Properly folded nonstructural polyprotein directs the semliki forest virus replication complex to the endosomal compartment. J Virol 77, 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- Shorter J, Watson R, Giannakou ME, Clarke M, Warren G, and Barr FA (1999). GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. The EMBO journal 18, 4949–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, and Saftig P (2000). Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906. [DOI] [PubMed] [Google Scholar]
- Tang D, and Wang Y (2015). Golgi isolation. Cold Spring Harb Protoc 2015, 562–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Xiang Y, and Wang Y (2010a). Reconstitution of the cell cycle-regulated Golgi disassembly and reassembly in a cell-free system. Nat Protoc 5, 758–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Yuan H, and Wang Y (2010b). The Role of GRASP65 in Golgi Cisternal Stacking and Cell Cycle Progression. Traffic 11, 827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicinanza M, Korolchuk VI, Ashkenazi A, Puri C, Menzies FM, Clarke JH, and Rubinsztein DC (2015). PI(5)P regulates autophagosome biogenesis. Molecular cell 57, 219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Satoh A, and Warren G (2005). Mapping the functional domains of the Golgi stacking factor GRASP65. J Biol Chem 280, 4921–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Seemann J, Pypaert M, Shorter J, and Warren G (2003). A direct role for GRASP65 as a mitotically regulated Golgi stacking factor. The EMBO journal 22, 3279–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J, et al. (2016). The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Molecular cell 63, 781–795. [DOI] [PubMed] [Google Scholar]
- Willett R, Ungar D, and Lupashin V (2013). The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochemistry and cell biology 140, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, and Wang Y (2010). GRASP55 and GRASP65 play complementary and essential roles in Golgi cisternal stacking. J Cell Biol 188, 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Zhang X, Nix DB, Katoh T, Aoki K, Tiemeyer M, and Wang Y (2013). Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat Commun 4, 1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu IM, and Hughson FM (2010). Tethering factors as organizers of intracellular vesicular traffic. Annu Rev Cell Dev Biol 26, 137–156. [DOI] [PubMed] [Google Scholar]
- Zhang X, and Wang Y (2015). GRASPs in Golgi Structure and Function. Frontiers in Cell and Developmental Biology 3, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Wang Y (2016). Glycosylation Quality Control by the Golgi Structure. J Mol Biol 428, 3183–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.