

Abstract
Introduction:
Treatment of patients with EGFR mutant NSCLC with vascular endothelial growth factor (VEGF) inhibitors in combination with EGFR inhibitors provides greater benefit than EGFR inhibition alone, suggesting that EGFR mutation status may define a patient subgroup with greater benefit from VEGF blockade. The mechanisms driving this potentially enhanced VEGF dependence are unknown.
Methods:
We analyzed the effect of EGFR inhibition on VEGF and HIF-1α in NSCLC models in vitro and in vivo. We determined the efficacy of VEGF inhibition in xenografts and analyzed the impact of acquired EGFR inhibitor resistance on VEGF and HIF-1α.
Results:
NSCLC cells with EGFR activating mutations exhibited altered regulation of VEGF compared to EGFR wild-type cells. In EGFR mutant cells, EGFR, not hypoxia, was the dominant regulator or HIF-1α and VEGF. NSCLC tumor models bearing classical or exon 20 EGFR mutations were more sensitive to VEGF inhibition than EGFR wild-type tumors, and combination of VEGF and EGFR inhibition delayed tumor progression. In models of acquired EGFR inhibitor resistance, while VEGF remained overexpressed, the hypoxia-independent expression of HIF-1α was delinked from EGFR signaling, and EGFR inhibition no longer diminished HIF-1α or VEGF expression.
Conclusions:
In EGFR mutant NSCLC, EGFR signaling is the dominant regulator of HIF-1α and VEGF in a hypoxia-independent manner, hijacking an important cellular response regulating tumor aggressiveness. Cells with acquired EGFR inhibitor resistance retained elevated expression of HIF-1α/VEGF, and the pathways was no longer EGFR-regulated. This supports VEGF targeting in EGFR mutant tumors in the EGFR inhibitor naïve and refractory settings.
Keywords: Non-small cell lung cancer, epidermal growth factor receptor, hypoxia-inducible factor, vascular endothelial growth factor
Introduction
Hypoxia-inducible factor-1 (HIF-1) is a master regulator of the hypoxic transcriptional response and is composed of α and β-subunits. The β-subunit is constitutively expressed, while the α-subunit is regulated primarily by oxygen-dependent proteasome degradation, which is hindered under hypoxic conditions1. Several oxygen-independent mechanisms have also been shown to regulate HIF expression1. For instance, HIF-1α is transcriptionally upregulated in a cell type–specific manner by various growth factors and receptor tyrosine kinases (RTKs), including EGF, VEGFR and RET1, 2. EGFR has been implicated as a hypoxia-independent driver of HIF expression as inhibition of EGFR activation has been shown to reduce HIF protein translation in some cells3. We previously reported that expression of EGFR mutations is associated with increased HIF-1α levels in NSCLC and NIH-3T3 cells even under normoxic conditions, which implies that cells harboring EGFR activating mutations may have distinct regulation of HIF-1α expression4.
HIF-1α drives the transcription of genes involved in glycolysis and angiogenesis. VEGF, a HIF-1α target gene, is modulated by activation of several RTKs, and EGFR inhibition has been shown to decrease VEGF expression in many tumor types5–7. The molecular mechanisms underlying the link between EGFR signaling and VEGF expression include both HIF-1–dependent and –independent mechanisms3, 8. The effects of EGFR-activating mutations leading to altered regulation of HIF may play a critical role in the cross-talk between EGFR downstream signaling and effects on angiogenesis and tumor growth. Therefore, we hypothesized that regulation of VEGF and HIF-1α may be distinct in EGFR mutant NSCLC tumor cells, and VEGF blockade may be particularly effective in EGFR mutant tumors.
We examined the relationship between EGFR activating mutations and HIF-1α in NSCLC cells. Our data revealed that EGFR mutant tumors are highly dependent on VEGF, and in tumor cells with EGFR mutations, EGFR is the predominant regulator of HIF expression and generates a hypoxic gene signature in normoxia. Moreover, in EGFR mutant NSCLC cells with acquired resistance to EGFR TKIs, HIF-1α expression becomes disassociated from EGFR signaling. These findings offer insight into the mechanism by which EGFR activating mutations promote tumor angiogenesis and aggressiveness in NSCLC and provide a mechanism for the clinical observations indicating that VEGF blockade may enhance the efficacy of EGFR TKIs.
Materials and Methods
Cell lines and reagents.
H3255, H1975, H1993, and HCC827 cells were obtained from Drs. Minna and Gazdar (UT Southwestern Medical School, Dallas, TX, USA). A549, H1299, H1650, H23, and Calu-6 cells were obtained from ATCC. Ba/F3 cells were obtained from Creative-Biogene. YUL-0019 cells were obtained from Dr. Politi (Yale Medical School)9. VEGF and HIF-1α ELISAs were obtained from R&D systems. Plasmids used for for promoter assays included pGL2-VEGF-luciferase, pRL-TK (Promega), empty vector, a WT EGFR, or EGFR E746_A750del (a gift from Dr. Kurie, MD Anderson Cancer Center)10. Detailed methods for generation of resistant cell lines, real-time PCR, measurement of HIF-1α half-life,Western blotting, immunohistochemistry (IHC), promoter assays and ELISA assays are listed in Supplementary Material and Methods.
Microarray analysis of NSCLC cell lines.
Two microarray gene expression datasets were used.We analyzed gene expression microarray data from 53 NSCLC cell lines using the gene chip HG-U133A (Affymetrix, Santa Clara, CA) as previously described11. CEL type data files were downloaded from the publicly available NCBI-GEO datasets (www.ncbi.nlm.nih.gov/gds, GSE 4824). The dCHIP software package was used to analyze HIF-1α gene expression levels in cell lines expressing wild-type (n = 45) or mutant EGFR (n = 8). We also analyzed illumina microarray gene expression data from NSCLC cell lines (n=110) expressing wild-type EGFR or classical EGFR activating mutations (Illumina WG v2 and v3 platform) generated and processed by Dr. Minna at the University of Texas Southwestern Medical Center (Dallas, TX).
Gene set enrichment analysis.
Gene set enrichment analysis (GSEA) was performed using GSEA software v2.0.4 (www.broad.mit.edu/gsea/msigdb/msigdb_index.html)12 using gene expression data publicly available in the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/gds, GSE 4824)and a hypoxia-related gene set containing 125 probesets from 69 individual genes1. From the downloaded CEL files from the 53 NSCLC cell lines already described, including eight with EGFR mutations, raw microarray data were processed using quartile normalization and the RMA algorithm13.
Reverse phase protein array (RPPA).
RPPA slides were printed from NSCLC cell line lysates, and RPPA studies were performed and analyzed as previously described14.
Mouse xenograft studies.
Male athymic nude mice (NCI-nu) were obtained from the National Cancer Institute (Frederick Cancer Center, Frederick, MD). Animal studies were conducted under an institutionally approved protocol in compliance with NIH guidelines. Details on xenograft experiments can be found in Supplementary Material and Methods.
Statistical analysis.
Two-tailed paired Student t-tests were used to detect differences in data generated from real-time PCR, ELISA assays, HIF protein half-life assays, luciferase assays, and mouse xenograft studies. P-values less than 0.05 were considered significant. Mantel-Cox Log-rank test was used to determine statistical differences and hazard ratios in PFS for PDX studies.
Results
NSCLC cells with EGFR activating mutations express higher levels of VEGF than EGFR wild-type cells
We evaluated VEGF gene expression in a panel of 110 NSCLC cell lines with or without EGFR activating mutations. VEGF RNA levels were significantly elevated in EGFR mutant NSCLC cell lines compared to EGFR wild-type cell lines (p = 0.02; Fig. 1A). There was no significant difference in the expression of angiogenic factors not regulated by HIF-1α including FGF2, IL6, IL8 and PDGFB (Fig. 1 B–E) demonstrating that the elevated VEGF expression did not reflect a general upregulation of cytokines and angiogenic factors. Among EGFR wild-type cell lines, VEGF expression was not not significantly different between cells with or without EGFR copy number gains (Supplementary Fig. 1A). Together these data suggest that mutant but not wild-type EGFR increase VEGF expression.
Fig 1. In NSCLC cells with EGFR activating mutations, VEGF is elevated and regulated by EGFR.
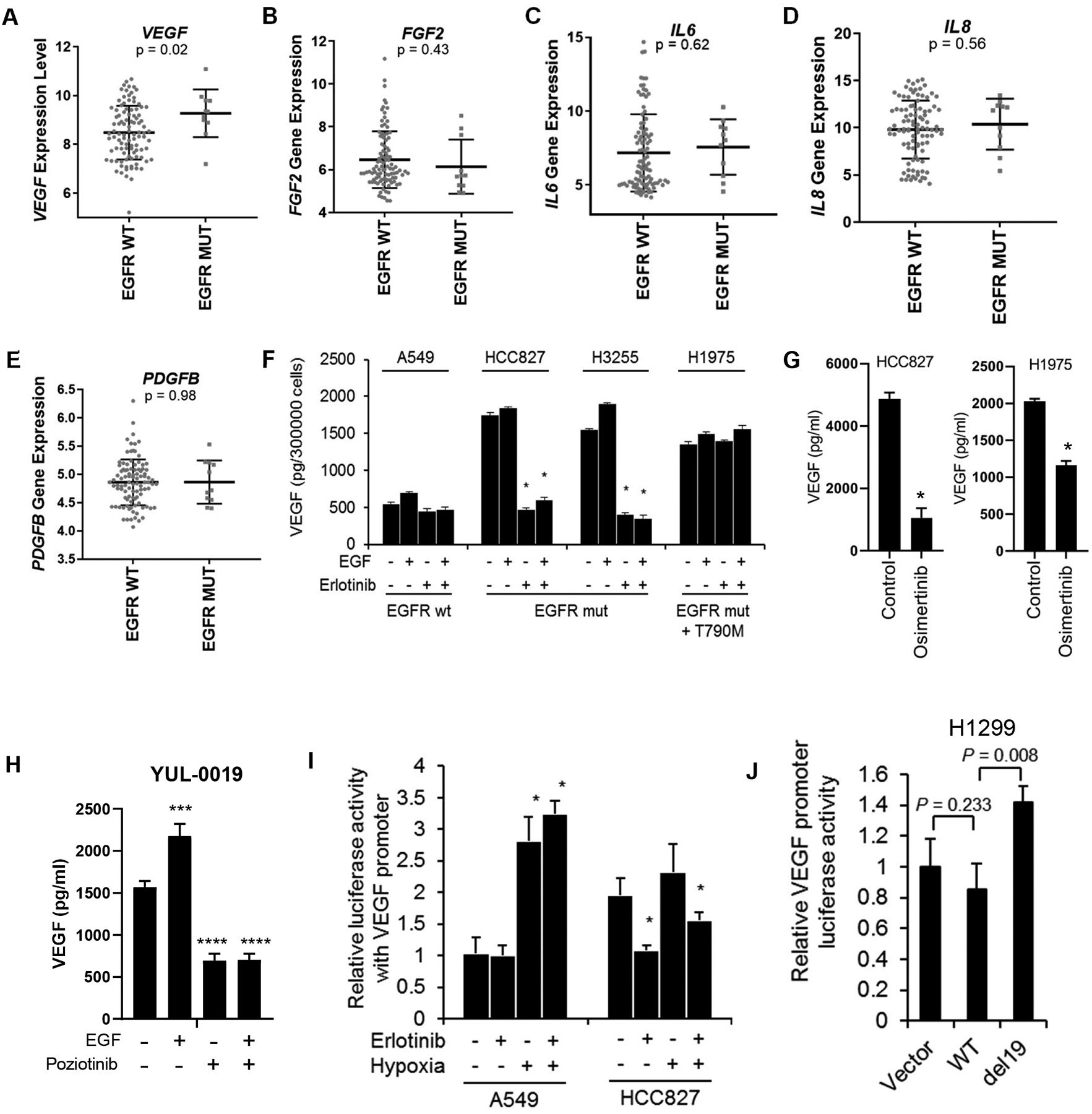
(A-E) Gene expression of VEGF (A), FGF2 (B), IL6 (C), IL8 (D), and PDGFB (E) in a panel of 110 NSCLC cell lines with or without EGFR activating mutations. (F) VEGF secretion as determined by ELISA in EGFR mutant (mut) or wild-type (wt) NSCLC cells treated with EGF or erlotinib. Bars are mean ± SEM; *p<0.05. (G) VEGF production following osimertinib treatment as determined by ELISA. *p<0.01. (H) VEGF secretion as determined by ELISA in YUL-0019 EGFR exon 20 mutant NSCLC cells treated with EGF or poziotinib. ***p = 0.0002; ****p <0.0001. (I) VEGF promoter activity was inhibited by erlotinib in cells with mutant EGFR (HCC827) but not in cells with wild-type EGFR (A549) in normoxic conditions. Cells transfected with a VEGF-luciferase construct were treated with or without erlotinib in normoxic or hypoxic conditions (1% O2). *p <0.05. (J) Expression of mutant EGFR (del19) increased VEGF promoter activity in H1299 cells. WT: wild-type EGFR; del19: del19 mutant EGFR. Data in panels A–J are shown as mean ± SD.
EGFR regulates VEGF expression in EGFR mutant NSCLC cells
NSCLC cell lines were cultured in serum-free media with or without EGF, the EGFR TKI erlotinib, or EGF plus erlotinib, and VEGF was measured in the conditioned media by ELISA. NSCLC cells harboring EGFR activating mutations (HCC827, H3255, and H1975 cells) expressed higher baseline levels of VEGF than A549 cells (EGFR wild-type) (Fig. 1F). Treatment of erlotinib-sensitive HCC827 or H3255 cells with erlotinib for 24 hours significantly reduced VEGF to levels similar to A549 cells. Demonstrating pharmacologic specificity, we found erlotinib did not alter VEGF secretion in erlotinib-resistant H1975 cells which express both L858R and T790M mutations15 (Fig. 1F). A549 cells, which express wild-type EGFR, exhibited the lowest level of VEGF secretion, and VEGF production was not altered by erlotinib. In all NSCLC lines tested, EGF treatment resulted in a slight increase in secreted VEGF, and the effects of EGF were blocked by erlotinib. Treatment of cells with the EGFR TKI osimertinib which has activity against EGFR T790M mutations significantly inhibited VEGF secretion by HCC827 and H1975 cells (Fig. 1G). Likewise, poziotinib-sensitive YUL-0019 cells9, which harbor an EGFR exon 20 activating mutation (N771del insFH) were treated with EGF with or without poziotinib. Poziotinib significantly reduced VEGF secretion when used alone or in combination with EGF (Fig. 1H). To confirm that the observed effects of EGFR TKIs on VEGF secretion was not due to changes in cell viability, we evaluated cell viability after 24 hour treatment with erlotinib, osimertinib or poziotinib. No change in cell viability was observed (Supplementary Fig. 1B–E). Collectively, these data demonstrate that in NSCLC cells with different activating EGFR mutations, VEGF secretion is EGFR-dependent.
To further investigate the role of EGFR activating mutations on VEGF expression, we analyzed transcriptional activation of the VEGF promoter. In HCC827 cells, erlotinib significantly reduced VEGF promoter activity (Fig. 1I), while VEGF promoter activity was unchanged in hypoxic conditions in HCC827 cells, indicating that VEGF promoter activity was predominantly regulated by EGFR signaling rather than hypoxia. In A549 cells, erlotinib had no effect on VEGF promoter activity, while hypoxia induced a 2.7-fold increase in VEGF promoter activity (Fig. 1I). We next transfected H1299 cells (EGFR wild-type) with wild-type or mutant EGFR (E746_A750del; del19) expression constructs, and found expression of mutant, but not wild-type, EGFR increased VEGF promoter activity (Fig. 1J). Likewise, expression of EGFR exon 19 deletion (Ex19del) in H23 cells (EGFR wild-type) and expression of EGFR Ex19del/T790M or the EGFR exon 20 insertion D770insSVD in Ba/F3 cells (EGFR negative) increased VEGF secretion (Supplementary Fig. S1F&G). Together these data indicate that NSCLC cells expressing mutant EGFR have diminished hypoxic regulation of VEGF and that mutant EGFR promotes VEGF expression.
Cells with EGFR activating mutations have increased HIF-1α expression
Cell lines expressing mutant EGFR including L858R and del19 mutations (n = 8) expressed 1.5 fold higher levels of HIF-1α mRNA than cell lines expressing wild-type EGFR (n = 45) (Fig. 2A). Furthermore, HIF-1α protein levels were higher in cell lines expressing mutant EGFR than in cell lines with wild-type EGFR (Fig. 2B). HIF-1α mRNA levels were not significantly different between cell lines expressing EGFR L858R mutation (n = 2) and cell lines expressing EGFR del19 mutation (n = 6).
Fig 2. HIF-1α expression is elevated in EGFR mutant NSCLC cells and is dependent on EGFR.
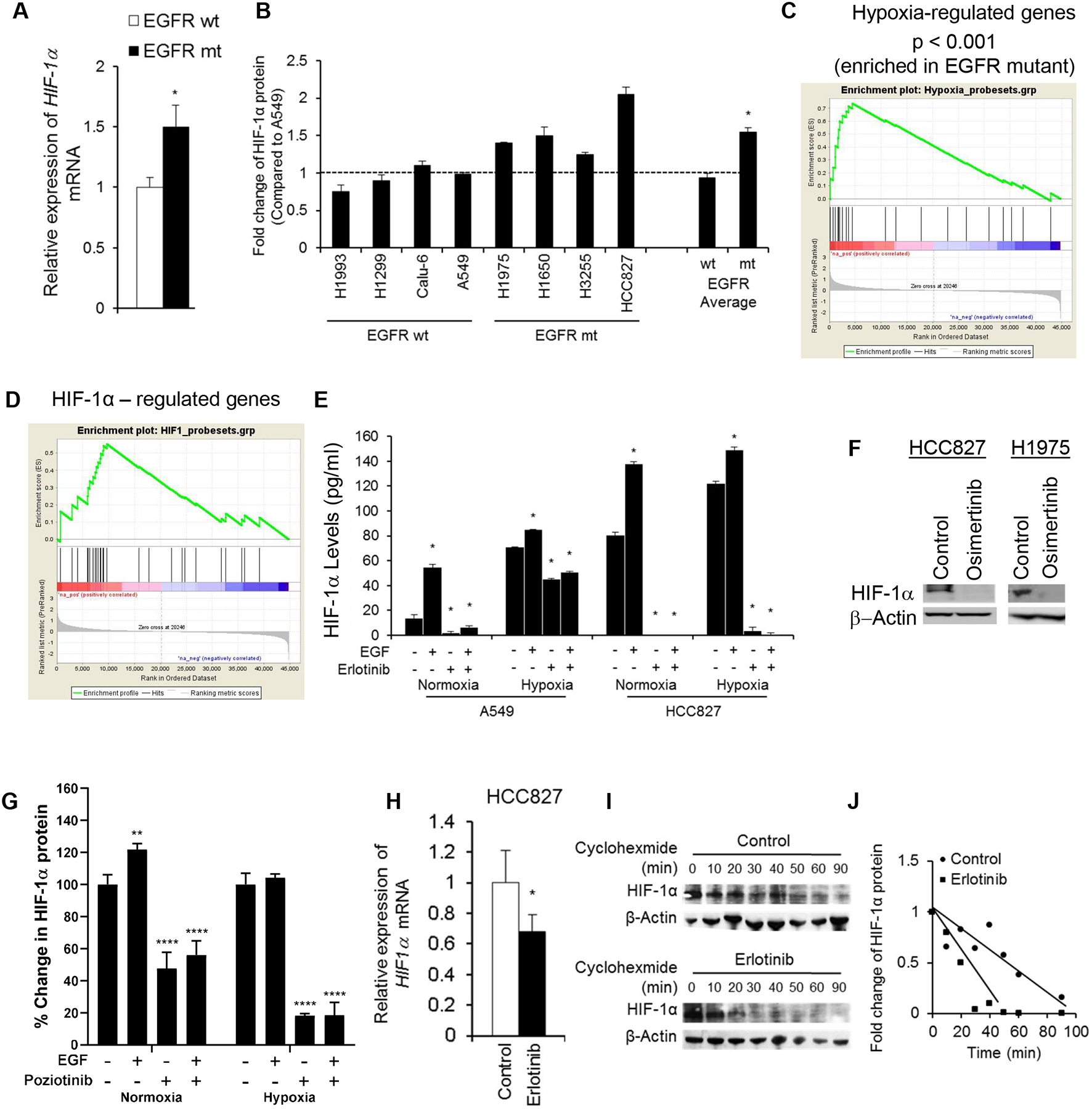
(A) Fifty-three EGFR wild-type (wt) or mutant (mt) NSCLC cell lines were analyzed by Affymetrix genechip analysis. Mean expression levels of HIF-1α mRNA are shown. * p <0.05. (B) HIF-1α expression as determined by ELISA in whole cell lysates from NSCLC cell lines grown under normoxic conditions. *p < 0.05. (C & D) GSEA analysis revealed that NSCLC cells with EGFR activating mutations displayed enriched expression of hypoxia- responsive genes and HIF-1α-regulated genes. (E) HIF-1α protein expression as measured by ELISA in A549 and HCC827 cells treated with erlotinib (1 μmol/L) and/or EGF (60 ng/mL) for 24 hours in normoxic or hypoxic conditions. *p <0.05 vs untreated control. (F) HIF-1α expression as determined by Western blotting after treatment with 200 nM osimertinib. (G) HIF-1α protein expression as measured by ELISA in YUL-0019 cells treated with EGF (60 ng/ml) or poziotinib (0.1 μmol/L). **p <0.0098, ***p <0.0001 vs untreated control. (H) HIF-1α mRNA expression was measured by real-time PCR in HCC827 cells treated with or without 1 μmol/L erlotinib in normoxic conditions. * p <0.05. (I) HIF-1α protein expression was analyzed by Western blotting in HCC827 cells treated with cycloheximide. One representative blot of three is shown. (J) Densitometry of HIF-1α expression in (I) was plotted to determine its half-life. Data in panels B, E, G, H are shown as mean ± SD.
NSCLC cells with mutant EGFR express increased levels of hypoxia-regulated genes
The elevated HIF-1α expression in EGFR mutant cells suggested that these cells might exhibit a hypoxic gene signature even in normoxic conditions. We performed GSEA on 53 NSCLC cell lines to determine whether cells with EGFR activating mutations were enriched for expression of hypoxia-regulated genes12. The probesets of hypoxia-inducible genes revealed a significant enrichment in cell lines expressing EGFR activating mutations (P <0.001, FDR q <0.001; Fig. 2C). We next examined genes specifically upregulated by HIF-1α16. EGFR mutant cell lines were significantly enriched for expression of HIF-1α-regulated genes (P = 0.037, FDR q < 0.06; Fig. 2D).
HIF-1α is predominantly regulated by EGFR, and not hypoxia, in EGFR mutant NSCLC cells
We next investigated whether the regulation of HIF-1α by EGFR impacted the normal hypoxia-induced regulation of HIF-1α. HCC827 cells or A549 cells were treated with erlotinib, EGF, or erlotinib plus EGF under normoxic or hypoxic conditions. HCC827 cells expressed high basal level of HIF-1α, which was only minimally increased by hypoxia (1.5-fold) or EGF (1.7-fold; Fig. 2E). Erlotinib completely diminished HIF-1α expression in HCC827 cells even in the presence of EGF and hypoxia, demonstrating a critical role for EGFR signaling in the regulation of HIF-1α in EGFR mutant NSCLC cells. By contrast, A549 cells, expressed markedly lower basal levels of HIF-1α compared to HCC827 cells, and HIF-1α expression significantly increased with hypoxia (>5-fold) or EGF stimulation (4-fold; Fig. 2E). When A549 cells were cultured in hypoxia, erlotinib only partially reduced HIF-1α (Fig. 2E). The EGFR TKI osimertinib (200 nM) similarly diminished HIF-1α expression in HCC827 and H1975 cells (Fig. 2F). EGFR exon 20 mutant YUL-0019 cells expressed HIF-1α in normoxia, and the addition of poziotinib significantly decreased HIF-1α levels (Fig. 2G), and EGF stimulated an increase in HIF-1α. Hypoxia treatment did not increase HIF-1α levels, but even in hypoxia, poziotinib diminished HIF-1α expression (Fig. 2G). To further confirm these findings, we evaluated protein levels of two hypoxia-regulated genes (hexokinase II and SLC2A1) included in the hypoxia-regulated geneset. Erlotinib decreased EGFR phosphorylation, hexokinase II and SLC2A1 expression in HCC827 cells (Supplementary Fig. S2A). These data indicate that in NSCLC cells harboring EGFR activating mutations, HIF-1α expression is predominantly regulated by EGFR signaling and the response to hypoxia is blunted, whereas in NSCLC cells expressing wild-type EGFR, HIF-1α expression is predominantly regulated by hypoxia.
Mutant EGFR regulates HIF-1α transcription and protein stability in NSCLC cells
To determine whether mutant EGFR modulates HIF-1α at the mRNA level, HIF-1α transcripts were analyzed by real-time PCR. In HCC827 cells, erlotinib treatment resulted in a 32% (P = 0.019) reduction in HIF-1α mRNA compared to control-treated cells (Fig. 2H). To determine whether EGFR regulates the protein stability of HIF-1α in NSCLC cells with EGFR activating mutations, we treated HCC827 cells with or without erlotinib in the presence of the protein biosynthesis inhibitor, cycloheximide, and evaluated the effect on the half-life of HIF-1α protein. In the absence of erlotinib, HIF-1α protein demonstrated a half-life of greater than 55 minutes (Fig. 2I&J). In the presence of erlotinib, the half-life of HIF-1α was reduced to approximately 20 minutes (Fig. 2I&J). The erlotinib-induced decrease in HIF-1α protein levels was blocked by the proteosome inhibitor MG132 (Supplementary Fig. S2B). Thus, EGFR inhibition increased loss of expression of HIF-1α protein in a proteosome-dependent manner. These data demonstrate that mutant EGFR regulates HIF-1α expression both at the mRNA level and through increased protein stability.
Cells expressing EGFR activating mutations have elevated expression of CA9 in vivo
Carbonic anhydrase 9 (CA9, CA IX) is a HIF-1α–regulated protein used as a marker of hypoxia in clinical and xenograft specimens17. We evaluated whether xenografts harboring classical or atypical mutant EGFR express higher levels of CA9 protein than xenografts with wild-type EGFR. We found that intra-tumoral levels of hypoxia, as quantitated by pimonidazole staining, were similar between xenografts expressing wild-type EGFR or mutant EGFR (Fig. 3A). Despite the similar levels of hypoxia, both EGFR mutant xenografts expressed higher levels of CA9 than EGFR wild-type xenografts (20–80% vs. 0–20%, Fig. 3A&B), indicating increased expression of HIF-1α target genes independent of intra-tumoral hypoxia. These data confirmed our in vitro findings and further indicate that NSCLC tumors expressing EGFR activating mutations upregulate HIF-1α target genes including VEGF and CA9 and that regulation of these genes is uncoupled from the hypoxia, the canonical regulator of this pathway.
Fig. 3. EGFR mutant xenografts have elevated expression of HIF-1α target genes and are sensitive to VEGF inhibition.
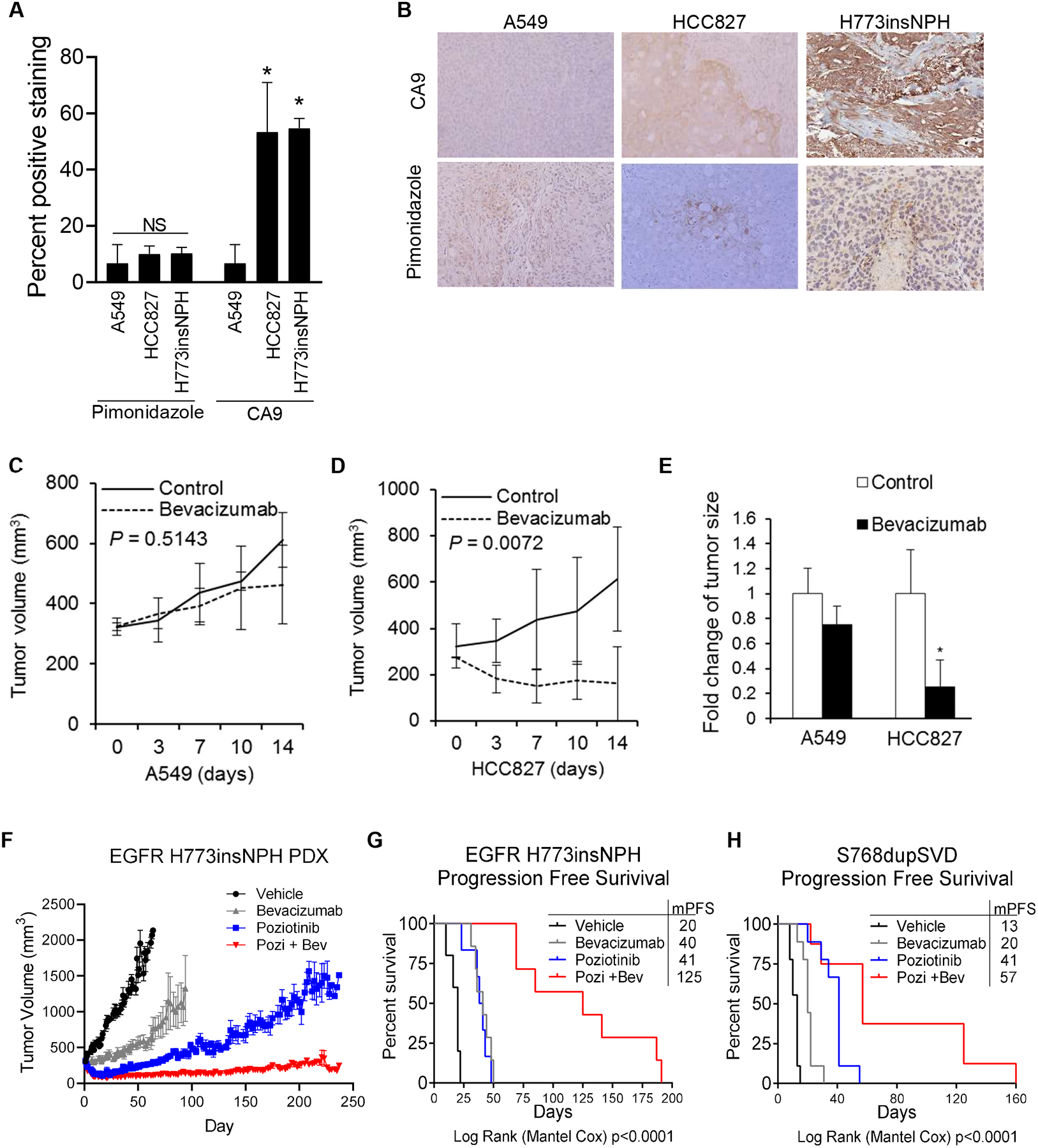
(A) HCC827 xenografts and H773insNPH EGFR exon 20 PDX tumors display elevated CA9 expression compared to A549 xenografts, although pimonidazole staining was similar. *p <0.05. (B) Representative CA9 immunohistochemical staining at 100× magnification. (C&D) Tumor growth curves for A549 (C) or HCC827 (D) tumor-bearing animals treated with bevacizumab (10 mg/kg) or vehicle for 14 days. (E) Fold-changes in size of A549 and HCC827-derived xenografts, compared to controls, at the end of bevacizumab treatment. *p <0.05. (F) Tumor growth curve of NSCLC PDX harboring EGFR H773insNPH mutation treated with vehicle (black), bevacizumab (5mg/kg twice per week, grey), poziotinib (2 mg/kg five times per week, blue) or combination of bevacizumab and poziotinib (red) for 225 days. Tumor size was measured three to five times per week. (G-H) Kaplan-Meier curves of PFS for PDX models expressing (G) EGFR H773insNPH or (H) EGFR S768dupSVD treated with vehicle, bevacizumab (5 mg/kg twice per week), poziotinib (2 mg/kg five times per week) or combination of bevacizumab and poziotinib. Progression was defined as time until tumor doubling from best response. Log-rank (Mantel Cox) was used to determine statistical differences between groups. In panels A, C, D, E, F data are shown as mean ± SEM.
Xenografts expressing mutant EGFR display increased sensitivity to anti-VEGF therapy
We next assessed whether EGFR mutant tumors display a greater sensitivity to the VEGF inhibitor, bevacizumab, than EGFR wild-type tumors. HCC827 or A549 cells were implanted in nude mice. Bevacizumab treatment began when tumors reached 300 mm3. Growth of A549 tumors was modestly inhibited by bevacizumab (Fig. 3C). However, bevacizumab completely suppressed the growth of HCC827 tumors (Fig. 3D). At day 14, wild-type EGFR tumor growth was reduced by 30%, while EGFR mutant tumor growth was reduced by 70% (Fig. 3E). Next, we evaluated the anti-tumor activity of bevacizumab in NSCLC EGFR exon 20 mutant PDX models. In PDXs harboring EGFR H773insNPH, bevacizumab or poziotinib significantly impaired the growth of EGFR H773insNPH tumors (Log Rank (Mantel-Cox) p = 0.0004 and 0.0008; Fig. 3F–G). The combination of bevacizumab plus poziotinib resulted in near complete tumor regression (Fig. 3F) and significantly prolonged progression free survival (PFS) compared to vehicle or either agent alone (PFS; p = <0.0001; Fig. 3G). Similar results were obtained using an additional EGFR exon 20 mutant PDX model. Bevacizumab alone induced a modest but significant improvement in PFS (Log Rank (Mantel-Cox) p = 0.0001) and the combination of bevacizumab plus poziotinib significantly improved the efficacy of poziotinib treatment (p = 0.0107; Fig. 3H). We analyzed RNA from tumors harvested at the end of the experiment (resistance) by qRT-PCTR. In EGFR H773insNPH expressing tumors, treatment with bevacizumab caused a modest increase in CA9 and VEGF (Supplementary Fig. 3A&B). Poziotinib, alone or in combination with bevacizumab, caused a significant decrease in CA9 and VEGF RNA levels. In the EGFR S768dupSVD model, the combination of poziotinib and bevacizumab resulted in reduced CA9 RNA levels (Supplementary Fig. 3C&D). In light of our in vivo findings, we assessed whether hypoxia increases tumor cell sensitivity to poziotinib. Poziotinib sensitivity was similar between YUL-0019 cells grown in normoxia or hypoxia (Supplementary Fig. 3E). Collectively, these data suggest that tumor cells expressing mutant EGFR are highly sensitive to VEGF inhibition and VEGF blockade may improve the efficacy of EGFR TKIs in this patient population
HIF-1α and VEGF are uncoupled from EGFR in cells with acquired EGFR TKI resistance
While patients with EGFR activating mutations are initially sensitive to EGFR inhibition, resistance inevitably occurs and often times is EGFR-independent15, 18, 19. We generated EGFR TKI resistant cells by culturing HCC827 and HCC4006 cells in erlotinib or YUL-0019 cells in poziotinib until erlotinib-resistant (ER) and poziotinib resistant (PR) clones developed. HCC827-ER and HCC4006-ER cells were negative for secondary T790M EGFR mutations20. EGFR inhibitor treatment reduced EGFR phosphorylation in HCC827, HCC4006, and YUL-0019 parental cells and ER and PR cell lines (Fig. 4A–C). Under normoxic conditions, HCC827-ER HCC4006-ER, and YUL-0019-PR cells expressed high levels of HIF-1α similar to that observed in parental cells (Fig. 4D–E). While EGFR inhibitor treatment significantly reduced HIF-1α levels in HCC827, HCC4006, and YUL-0019 parental cells, EGFR inhibition did not alter HIF-1α levels in HCC827-ER, HCC4006-ER, or YUL-0019-PR cell lines (Fig. 4D–F). In HCC827, HCC4006, and YUL-0019 parental cells EGFR-TKI treatment resulted in a significant reduction in VEGF production (Fig. 4G–I). In contrast, EGFR-TKI treatment did not decreased VEGF levels in any of the ER or PR variants and in fact a trend towards increased VEGF was observed after erlotinib treatment in the HCC4006-ER lines. In the majority of EGFR TKI cell lines, exposure to hypoxia did not significantly increase HIF-1α protein levels (Supplementary Fig. 4). These data indicate that in NSCLC with acquired EGFR-independent EGFR TKI resistance, HIF-1α expression remains independent of oxygen concentration but de-linked from EGFR signaling.
Fig 4. HIF-1α expression is uncoupled from EGFR in NSCLC cells with acquired resistance to EGFR TKIs.
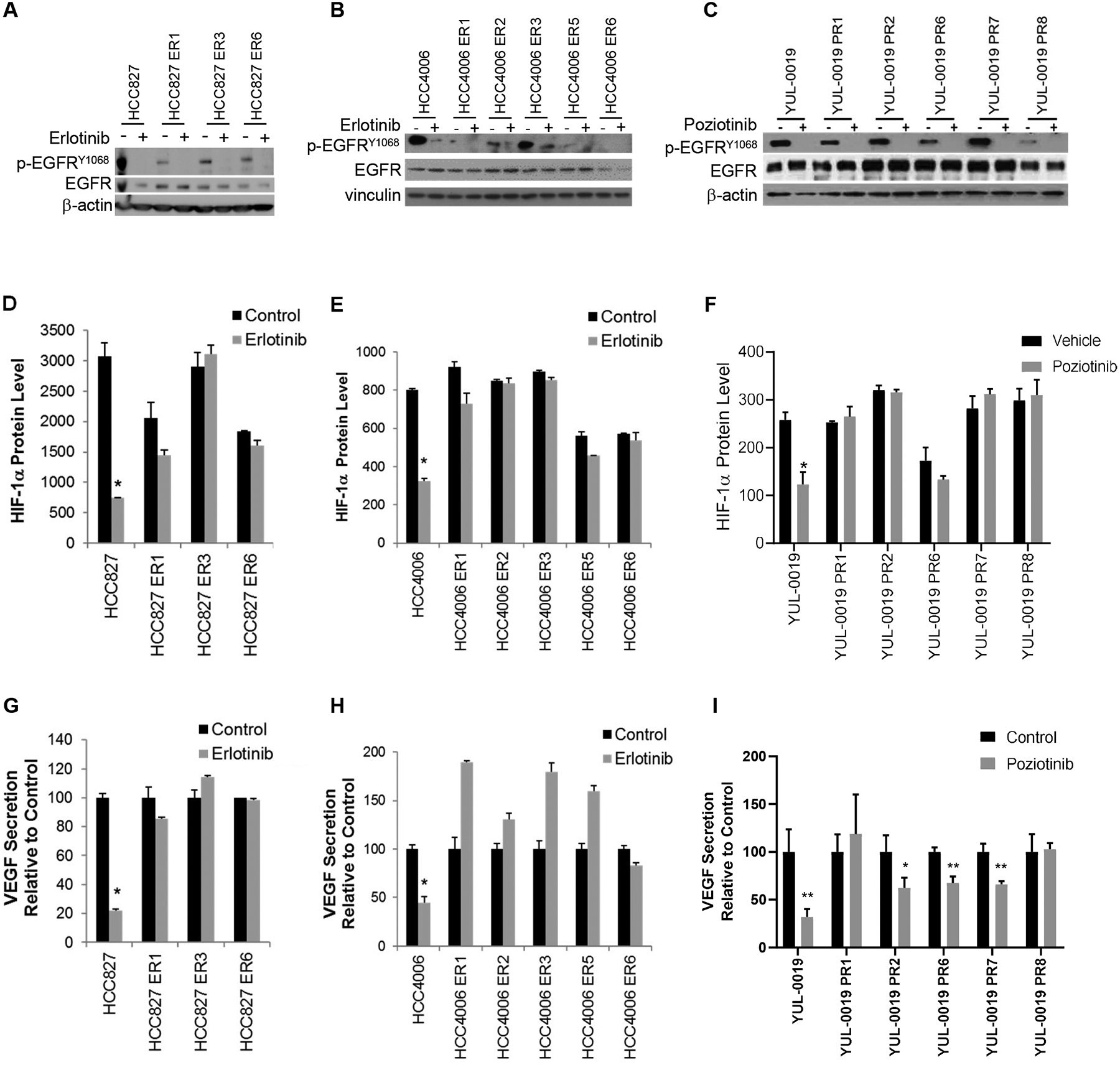
(A&B) Erlotinib (1 μM) decreased protein levels of pEGFR in HCC827 and HCC827 ER cells and HCC4006 and HCC4006 ER cells as determined by Western blotting. (C) Poziotinib (1 μM) decreased protein levels of pEGFR in YUL-0019 and YUL-0019 PR cells as determined by Western blotting. (D&E) Effect of erlotinib (1μM) on HIF-1α protein levels in HCC827 and HCC4006 parental and ER cells as determined by ELISA. *p < 0.05 versus control. (F) Effect of poziotinib (1μM) on HIF-1α protein levels as determined by ELISA. *p = 0.002 versus control. (G&H), Effect of erlotinib (1 μM) on VEGF secretion by HCC827 and HCC4006 ER cells as determined by ELISA. *p < 0.05 versus control. (I) Effect of poziotinib (1 μM) on VEGF secretion by YUL-0019 and YUL-0019 PR cells as determined by ELISA. *p = 0.03, **p < 0.01 versus control. Data in panels D-I are shown as mean ± SD.
VEGFR inhibitors do not have direct activity against EGFR TKI resistant cells
We next assessed whether VEGFR inhibitors have direct anti-tumor cell activity on EGFR TKI refractory cell lines. VEGFR2 levels were not altered between parental and EGFR TKI resisitant cells (Supplementary Fig. 5A). We screened parental and resistant variants against bevacizumab and the VEGFR TKIs cederinib, pazopanib, and sorafenib in vitro and observed that both parental and resistant cells were resistant to VEGFR2 inhibition (Supplementary Fig. 5B–E).
Discussion
These data offer insight into the mechanism by which EGFR activating mutations promote tumor angiogenesis and aggressiveness in NSCLC. Our findings indicate that EGFR mutant NSCLC cells display a VEGF-dependent phenotype and that in EGFR-mutant tumor cells, EGFR, but not hypoxia, is the predominant regulator of HIF-1α expression, whereas in EGFR wild-type NSCLC hypoxia is the primary regulator of HIF-1α. Moreover, in cells with acquired EGFR-independent EGFR TKI resistance, regulation of HIF-1α becomes re-wired and dissociated from EGFR signaling. These findings indicate that patients with EGFR mutations may represent a highly VEGF-dependent subgroup that may benefit from combined EGFR and VEGF blockade.
Uncoupling of HIF expression and hypoxia in cells expressing mutant EGFR demonstrates an important mechanism by which cells can upregulate HIF and its target genes in the absence of hypoxia. This aberrant upregulation can trigger an overall hypoxic gene signature within the tumor microenvironment creating pseudohypoxia, which has been proposed as a mechanism of EGFR inhibitor resistance21. Hypoxia has been shown to lead to more aggressive tumors through a number of biological mechanisms and is a principal regulator of tumor angiogenesis. Hypoxia has also been shown to contribute to therapeutic resistance22–24 and enhanced invasion and metastases24, 25. We have previously demonstrated a role for HIF-1α in the upregulation of c-Met4, which when activated leads to increased invasiveness in EGFR mutant NSCLC26, 27. Therefore, the presence of EGFR activating mutations would give cells a “head start” in hypoxia-induced tumor aggressiveness and therapeutic resistance.
While ligand-induced activation of EGFR has been shown to modulate HIF-1α levels1, our finding that EGFR activating mutations regulate HIF-1α in a manner that is dominant over hypoxia-controlled HIF expression has important biological implications. In tumors driven primarily by the EGFR pathway, targeting HIF or key HIF-regulated genes (i.e. VEGF) may further inhibit tumor growth and invasiveness beyond the effect of EGFR inhibition alone, and delay drug tolerance. Moreover, VEGFR and EGFR activate common downstream signaling pathways including the PI3K/Akt and MAPK pathways in endothelial and tumor cells expressing VEGF receptors (VEGFR). Therefore, VEGF overexpression may further drive signal bypass-mediated resistance to EGFR TKIs, which is supported by preclinical findings that VEGF expression is further elevated in vivo following EGFR TKI resistance28 and that blockade of both VEGFR2 and EGFR has greater anti-tumor activity than the respective monotherapies29. While the data presented here using preclinical models indicates that EGFR activating mutations promote increased levels of tumor-derived VEGF, the study is limited by the fact that it does not compare tumor VEGF levels in EGFR wild-type and mutant NSCLC clinical specimens. The comparison of VEGF levels is challenging as publicly available datasets are not controlled for tumor size which would impact tumor hypoxia and VEGF levels.
To date, several clinical studies have provided evidence supporting the dual targeting of EGFR and VEGFR pathways. In an exploratory analysis from the double-blinded, placebo-controlled Phase 3 BeTa trial, it was observed that that patients with EGFR mutant, but not EGFR wild-type, tumors demonstrated a trend towards benefit from the addition of erlotinib to bevacizumab (overall survival hazard ratio 0.44 for EGFR mutant vs 1.11 for EGFR wild-type)30. Further clinical evidence is provided in clinical study J025567 where the addition of bevacizumab with erlotinib significantly improved progression free survival (PFS) in NSCLC patients harboring EGFR mutations31. A subgroup analysis included in the phase 3 maintenance study, ATLAS, revealed that patients with EGFR mutations had an improved PFS and overall survival (OS) when treated with bevacizumab plus erlotinib32. Moreover, in the RELAY study (NCT02411448) the VEGFR2 antagonist, ramucirumab, plus erlotinib yielded a significantly improved PFS compared to erlotinib treatment alone in metastatic EGFR mutant NSCLC33. Our preclinical data indicates that EGFR mutant tumors have a VEGF-dependent phenotype and that VEGF is regulated by EGFR in EGFR-mutant NSCLC, providing a mechanism for the clinical observations that EGFR mutant patients benefit from VEGF blockade.
While studies of combination EGFR and VEGF inhibition have yielded encouraging results in patients with classical mutations, no clinical studies of combination EGFR and VEGF inhibition have been completed with patients harboring exon 20 mutations due to a lack of an effective EGFR TKI for this subpopulation. Our data indicate that not only would patients with classical EGFR mutations receive clinical benefit from the addition of VEGF inhibition, but that also patients with EGFR exon 20 insertion may benefit with the addition of up-front VEGF inhibition in combination with an EGFR TKI.
In models of acquired EGFR TKI resistance, we observed that the hypoxia-independent expression of HIF-1α was re-wired and no longer EGFR-dependent. The finding that all resistant cells tested retained elevated expression of HIF-1α and VEGF supports the notion that the HIF-1α/VEGF axis is an important pathway in EGFR mutant NSCLC and further suggests that targeting VEGF may be clinically useful in delaying the emergence of EGFR TKI resistance. It is worth noting, however, that the specific role that VEGF plays in these tumors is not definitively addressed in this study. We do not see evidence that the survival or growth of EGFR mutant tumor cells are directly impacted by VEGF pathway inhibition in a cell autonmomous manner (Supplementary Fig. 5B–E), and we previously observed that tumor expression of VEGFR-2 (KDR) was not a predictor of benefit from vandetinib34, providing evidence that the primary effect of VEGF is unlikely to be directly on the tumor cells. We and others have reported that bevacizumab28, 35 and other VEGFR TKIs36 do cause a marked reduction in tumor microvessel density and alterations in tumor vessel tortuosity in human EGFR mutant xenografts in vivo, suggesting that the effects are likely due, at least in part, to effects on tumor vasculature. These preclinical models conducted in immunodeficient models, and it is possible that VEGF may exert additional immunomodulatory effects not captured in these preclinical models.
Collectively, our results indicate that in EGFR mutant NSCLC, EGFR is the predominant regulator of VEGF and HIF-1α independent of hypoxia and EGF ligand binding, likely contributing to the enhanced sensitivity to VEGF inhibitors observed clinically, while hypoxia is the primary driver of HIF-1 α in the EGFR wild-type NSCLC models studied. Mutant EGFR–mediated HIF-1α upregulation is initially completely attenuated by inhibition of EGFR, raising the possibility that the high efficacy of EGFR TKIs in EGFR mutant NSCLC may be due not only to the blockade of EGFR-driven survival pathways, but also to their effects on abolishing HIF-1α and HIF-1α-mediated VEGF expression. These data suggest that EGFR mutant NSCLC patients may receive greater benefit for VEGF blockade than patients with wild-type EGFR and supports the clinical testing and use of VEGF inhibitors in combination with EGFR TKIs.
Supplementary Material
Supplementary Fig 1. (A) VEGFA gene expression in EGFR wild-type NSCLC cell lines with or without EGFR copy number gains (CNG). (B - E) Cell viability of EGFR mutant cell lines after 24-hour treatment with EGFR TKIs erlotinib (ERL), osimertinib (OSI) or (pozi) poziotinib. Bars are mean ± SD. (F & G) VEGF secretion as determined by ELISA in H23 (F) or Ba/F3 (G) cells with or without expression of mutant EGFR. Bars represent mean ± SD. *** p < 0.001.
Supplementary Fig. 2. (A) Erlotinib inhibited expression of HIF-1α target genes hexokinase II and SLC2A1 in HCC827 cells, analyzed by Western blotting. (B) MG132 blocked the degradation of HIF-1α in HCC827 cells. HIF-1α levels were measured by ELISA. Bars represent mean ± SD. *p <0.05 vs without MG132.
Supplementary Fig. 3. (A-D) RNA expression of CA9 and VEGF in EGFR H773insNPH (A & B) and S768dupSVD (C & D) tumors as determined by qRT-PCR. * p ≤ 0.05. (E) Dose response curve of YUL-0019 cells treated with poziotinib in normoxic or hypoxic conditions. In panels A–E bars represent mean ± SD.
Supplementary Fig. 4. (A - C) HIF-1α protein expression as determined by ELISA in EGFR TKI sensitive and resistant cells cultured in normoxia or hypoxia. Bars represent mean + SD. * p ≤ 0.05; ** p ≤ 0.005; *** p = 0.0007.
Supplementary Fig. 5. (A) Expression of VEGFR2 in EGFR TKI sensitive and resistant cell lines as determined by RPPA. (B - E) Dose response curve of EGFR TKI sensitive and resistant cells treated with bevacizumab or VEGFR TKIs cediranib, pazopanib, and sorafenib.
Fig. 5.
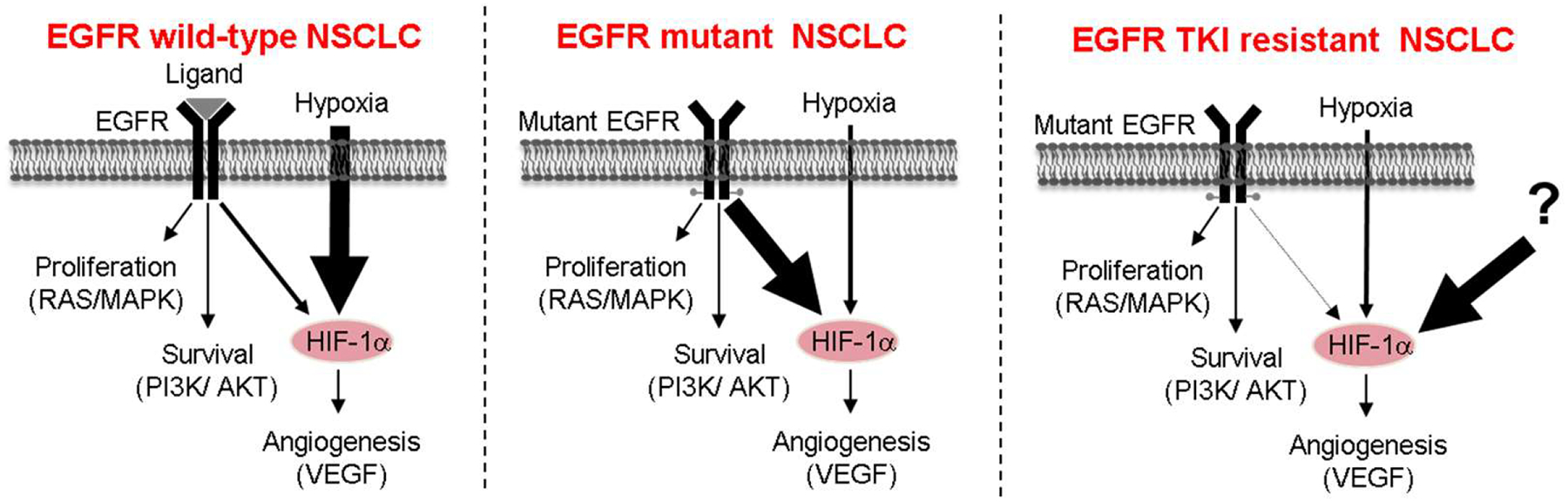
In NSCLC cells with EGFR activating mutations, mutant EGFR is the predominant regulator of HIF-1α, while hypoxia is the major regulator of HIF-1α in cells expressing wild-type EGFR. NCLC cells with acquired resistance to EGFR TKIs become rewired and HIF-1α expression is no longer coupled to EGFR signaling.
Funding
This work was supported by a Joan’s Legacy award and the Flight Attendant’s Medical Research Institute, the Exon 20 group, the David Bruton, Jr. Endowment, the Rexanna Foundation for Fighting Lung Cancer, LUNGevity Foundation, NIH CCSG (CA016672), 1R01 CA190628, 1R01 CA234183-01A1, Lung SPORE grant 5 P50 CA070907, Lung Cancer Moon Shot Program, the Rexanna Foundation for Fighting Lung Cancer, Bruton Endowed Chair in Tumor Biology, Stading Fund for EGFR inhibitor resistance, the Fox Lung EGFR Inhibitor Fund, the Hanlon Fund, the Richardson fund, the Kopelman Foundation and the Margot Johnson Cancer Research Fund. T.C. is partially supported by the Conquer Cancer Foundation of the American Society of Clinical Oncology (ASCO) Career Development Award 2018, Khalifa Bin Zayed Al Nahyan Foundation, the Physician Scientist Program, the T.J. Martell Foundation, and the Bob Mayberry Foundation.
Conflict of Interests/Disclosures:
Dr. Heymach reports grants, personal fees and other from Spectrum, personal fees from Genentech, during the conduct of the study; grants and personal fees from AstraZeneca, and GlaxoSmithKline, personal fees from Boehringer Ingelheim, Bristol Myers Squibb, Merck, Catalyst, Guardant Health, Foundation Medicine, Hengrui, Lilly, Novartis, EMD Serono, Sanofi, Biotree, Takeda, outside the submitted work; In addition, Dr. Heymach has a patent PCT/US2019/022067 pending, and a patent PCT/US2017/062326 and U.S. Provisional Patent Application Nos. 62/423,732; 62/427,692 and 62/572,716, with royalties paid to The University of Texas System Board of Regents. Dr. Nilsson reports personal fees from Spectrum, during the conduct of the study; In addition, Dr. Nilsson has a patent PCT/US2019/022067 pending, and a patent PCT/US2017/062326 and U.S. Provisional Patent Application Nos. 62/423,732; 62/427,692 and 62/572,716, with royalties paid to The University of Texas System Board of Regents. Dr. Robichaux reports grants and licensing fees from Spectrum, and is an inventor on patents related to treatment of EGFR and HER2 mutant tumors. Dr. Cascone reports personal fees and other from Bristol-Myers Squibb, personal fees and other from MedImmune/AstraZeneca, other from Boehringer Ingelheim, personal fees and other from EMD Serono, outside the submitted work. Dr. Le reports personal fees from EMD Serono, grants and personal fees from Eli Lilly , personal fees from AstraZeneca , Boehringer Ingraham, outside the submitted work. Dr. Saintigny reports grants from Astra-Zeneca, Bristol-Myers-Squibb, Roche, HTG Molecular Diagnostics, Illumina, OSE Immunotherapeutics, HITACHI, outside the submitted work. Dr. Minna reports grants from National Cancer Institute and Margot Johnson Cancer Research Fund , during the conduct of the study; personal fees from National Institutes of Health, personal fees from University of Texas Southwestern Medical Center, outside the submitted work. Dr. Wistuba reports grants and personal fees from Genentech/Roche, Bayer, Bristol-Myers Squibb, AstraZeneca/Medimmune, Pfizer, HTG Molecular, Merck, personal fees from GlaxoSmithKline, grants and personal fees from Guardant Health, personal fees from MSD, grants from Oncoplex, grants from DepArray, Adaptive, Adaptimmune, EMD Serono, Takeda, Amgen, Karus, Johnson & Johnson, Iovance, 4D, Novartis, Oncocyte, Akoya, personal fees from Flame, outside the submitted work.
References
- 1.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003;3:721–732. [DOI] [PubMed] [Google Scholar]
- 2.Nilsson MB, Zage PE, Zeng L, et al. Multiple receptor tyrosine kinases regulate HIF-1alpha and HIF-2alpha in normoxia and hypoxia in neuroblastoma: implications for antiangiogenic mechanisms of multikinase inhibitors. Oncogene 2010. [DOI] [PubMed] [Google Scholar]
- 3.Pore N, Jiang Z, Gupta A, et al. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer research 2006;66:3197–3204. [DOI] [PubMed] [Google Scholar]
- 4.Xu L, Nilsson MB, Saintigny P, et al. Epidermal growth factor receptor regulates MET levels and invasiveness through hypoxia-inducible factor-1alpha in non-small cell lung cancer cells. Oncogene 2010;29:2616–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciardiello F, Bianco R, Damiano V, et al. Antiangiogenic and antitumor activity of anti-epidermal growth factor receptor C225 monoclonal antibody in combination with vascular endothelial growth factor antisense oligonucleotide in human GEO colon cancer cells. Clinical cancer research : an official journal of the American Association for Cancer Research 2000;6:3739–3747. [PubMed] [Google Scholar]
- 6.Perrotte P, Matsumoto T, Inoue K, et al. Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clinical cancer research : an official journal of the American Association for Cancer Research 1999;5:257–265. [PubMed] [Google Scholar]
- 7.Petit AM, Rak J, Hung MC, et al. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Am J Pathol 1997;151:1523–1530. [PMC free article] [PubMed] [Google Scholar]
- 8.Luwor RB, Lu Y, Li X, et al. The antiepidermal growth factor receptor monoclonal antibody cetuximab/C225 reduces hypoxia-inducible factor-1 alpha, leading to transcriptional inhibition of vascular endothelial growth factor expression. Oncogene 2005;24:4433–4441. [DOI] [PubMed] [Google Scholar]
- 9.Robichaux JP, Elamin YY, Tan Z, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med 2018;24:638–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amann J, Kalyankrishna S, Massion PP, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer research 2005;65:226–235. [PubMed] [Google Scholar]
- 11.Zhou BB, Peyton M, He B, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 2006;10:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003;4:249–264. [DOI] [PubMed] [Google Scholar]
- 14.Byers LA, Sen B, Saigal B, et al. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res 2009;15:6852–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. The New England journal of medicine 2005;352:786–792. [DOI] [PubMed] [Google Scholar]
- 16.Wang V, Davis DA, Haque M, et al. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res 2005;65:3299–3306. [DOI] [PubMed] [Google Scholar]
- 17.Potter CP, Harris AL. Diagnostic, prognostic and therapeutic implications of carbonic anhydrases in cancer. British journal of cancer 2003;89:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schoenfeld AJ, Chan JM, Kubota D, et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin Cancer Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le X, Puri S, Negrao MV, et al. Landscape of EGFR -dependent and -independent resistance mechanisms to osimertinib and continuation therapy post-progression in EGFR-mutant NSCLC. Clin Cancer Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nilsson MB, Sun H, Diao L, et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with beta-blockers. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang WC, Wells JM, Chow KH, et al. miR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat Metab 2019;1:460–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rho JK, Choi YJ, Lee JK, et al. Gefitinib circumvents hypoxia-induced drug resistance by the modulation of HIF-1alpha. Oncol Rep 2009;21:801–807. [PubMed] [Google Scholar]
- 23.Unruh A, Ressel A, Mohamed HG, et al. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene 2003;22:3213–3220. [DOI] [PubMed] [Google Scholar]
- 24.Chan DA, Giaccia AJ. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev 2007;26:333–339. [DOI] [PubMed] [Google Scholar]
- 25.Gort EH, Groot AJ, van der Wall E, et al. Hypoxic regulation of metastasis via hypoxia-inducible factors. Curr Mol Med 2008;8:60–67. [DOI] [PubMed] [Google Scholar]
- 26.Ma PC, Maulik G, Christensen J, et al. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev 2003;22:309–325. [DOI] [PubMed] [Google Scholar]
- 27.Herynk MH, Stoeltzing O, Reinmuth N, et al. Down-regulation of c-Met inhibits growth in the liver of human colorectal carcinoma cells. Cancer Res 2003;63:2990–2996. [PubMed] [Google Scholar]
- 28.Naumov GN, Nilsson MB, Cascone T, et al. Combined vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) blockade inhibits tumor growth in xenograft models of EGFR inhibitor resistance. Clin Cancer Res 2009;15:3484–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kayatani H, Ohashi K, Imao T, et al. Abstract 5198: Combination effect of anti-VEGFR-2 antibody with erlotinib on EGFR mutant non-small cell lung cancer. Cancer research 2016;76:5198–5198. [Google Scholar]
- 30.Roy S Herbst HS, Amler Lukas, Otterson Gregory, Lin Ming, Paula O’Connor, Hainsworth John. Biomarker evaluation in the phase III, placebo (P)-controlled, randomized BeTa trial of bevacizumab (B) and erlotinib (E) for patients (Pts) with advanced non-small cell lung cancer (NSCLC) after failure of standard 1st-line chemotherapy: correlation with treatment outcomes. J Thorac Oncol 2009;4:S323. [Google Scholar]
- 31.Seto T, Kato T, Nishio M, et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomised, multicentre, phase 2 study. Lancet Oncol 2014;15:1236–1244. [DOI] [PubMed] [Google Scholar]
- 32.Johnson BE, Kabbinavar F, Fehrenbacher L, et al. ATLAS: randomized, double-blind, placebo-controlled, phase IIIB trial comparing bevacizumab therapy with or without erlotinib, after completion of chemotherapy, with bevacizumab for first-line treatment of advanced non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2013;31:3926–3934. [DOI] [PubMed] [Google Scholar]
- 33.Nakagawa K, Garon EB, Seto T, et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology 2019;20:1655–1669. [DOI] [PubMed] [Google Scholar]
- 34.Nilsson MB, Giri U, Gudikote J, et al. KDR Amplification Is Associated with VEGF-Induced Activation of the mTOR and Invasion Pathways but does not Predict Clinical Benefit to the VEGFR TKI Vandetanib. Clin Cancer Res 2016;22:1940–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cascone T, Herynk MH, Xu L, et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. J Clin Invest 2011;121:1313–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cascone T, Xu L, Lin HY, et al. The HGF/c-MET Pathway Is a Driver and Biomarker of VEGFR-inhibitor Resistance and Vascular Remodeling in Non-Small Cell Lung Cancer. Clin Cancer Res 2017;23:5489–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig 1. (A) VEGFA gene expression in EGFR wild-type NSCLC cell lines with or without EGFR copy number gains (CNG). (B - E) Cell viability of EGFR mutant cell lines after 24-hour treatment with EGFR TKIs erlotinib (ERL), osimertinib (OSI) or (pozi) poziotinib. Bars are mean ± SD. (F & G) VEGF secretion as determined by ELISA in H23 (F) or Ba/F3 (G) cells with or without expression of mutant EGFR. Bars represent mean ± SD. *** p < 0.001.
Supplementary Fig. 2. (A) Erlotinib inhibited expression of HIF-1α target genes hexokinase II and SLC2A1 in HCC827 cells, analyzed by Western blotting. (B) MG132 blocked the degradation of HIF-1α in HCC827 cells. HIF-1α levels were measured by ELISA. Bars represent mean ± SD. *p <0.05 vs without MG132.
Supplementary Fig. 3. (A-D) RNA expression of CA9 and VEGF in EGFR H773insNPH (A & B) and S768dupSVD (C & D) tumors as determined by qRT-PCR. * p ≤ 0.05. (E) Dose response curve of YUL-0019 cells treated with poziotinib in normoxic or hypoxic conditions. In panels A–E bars represent mean ± SD.
Supplementary Fig. 4. (A - C) HIF-1α protein expression as determined by ELISA in EGFR TKI sensitive and resistant cells cultured in normoxia or hypoxia. Bars represent mean + SD. * p ≤ 0.05; ** p ≤ 0.005; *** p = 0.0007.
Supplementary Fig. 5. (A) Expression of VEGFR2 in EGFR TKI sensitive and resistant cell lines as determined by RPPA. (B - E) Dose response curve of EGFR TKI sensitive and resistant cells treated with bevacizumab or VEGFR TKIs cediranib, pazopanib, and sorafenib.