

Abstract
Recent developments in scanning probe block copolymer lithography (SPBCL) enable the confinement of multiple metal precursors in a polymer nanoreactor and their subsequent transformation into a single multimetallic heterostructured nanoparticle through thermal annealing. However, the process by which multimetallic nanoparticles form in SPBCL-patterned nanoreactors remains unclear. Here, we utilize the combination of PEO-b-P2VP and Au, Ag, and Cu salts as a model three-component system to investigate this process. The data suggest that the formation of single-component Au, Ag, or Cu nanoparticles within polymer nanoreactors consists of two stages: I) nucleation, growth, and coarsening of the particles to yield a single particle in each reactor; II) the particle continues to grow by depleting the remaining precursor in the reactor until it reaches a stable size. Also, different aggregation rates are observed for single-component particle formation (Au>Ag>Cu). This behavior is also observed for two-component systems, where nucleation sites have greater Au contents than the other metals. This observation can be used to trap nanoparticles with kinetic structures. High temperature treatment ultimately facilitates the structural evolution of the kinetic particle into a particle with a fixed structure. Therefore, with multicomponent systems, a third stage that involves elemental redistribution within the particle must be used to describe the synthetic process. Thus, this work not only provides a glimpse at the mechanism underlying multicomponent nanoparticle formation in SPBCL-generated nanoreactors but also illustrates, for the first time, the utility of SPBCL as a platform for controlling the architectural evolution of multimetallic nanoparticles in general.
Graphical Abstract
INTRODUCTION
Multimetallic heterostructured nanoparticles that contain well-defined interfaces between inorganic domains have recently attracted extensive interest due to their utility in catalysis,1,2 plasmonics,3,4 magnetics,5 and electronics.6 The chemical, electronic, and magnetic interactions that occur across the interfaces inside heterostructured nanoparticles can be engineered by changing the structure and elemental distribution of each inorganic domain; this allows the chemical and physical properties of nanoparticles to be finely tuned.2–4,7–9 To achieve this composition-structure-function tuning, wet chemistry approaches, especially stepwise seed-mediated growth processes in solution, have been successfully developed to prepare a wide variety of hetero-nanostructures.10–17 However, slight changes in synthetic parameters can greatly influence the nucleation and particle growth processes as well as the resulting nanostructures, making targeted synthesis of a specific particle type a complicated and challenging process.10,18 Consequently, there is a need to develop a fundamental understanding of the growth pathways for multimetallic nanoparticles.19–27 In this regard, these studies are usually performed either in an in-situ manner utilizing TEM19–23 or X-ray scattering techniques4,24 with liquid cells, or in an ex-situ manner by characterizing the particles in the reaction solution at different time points.25–27 In addition to informing synthetic protocols, mechanistic understanding provides ways of trapping reactive intermediate nanostructures, which can be useful in their own right.21,25,26–28
Recently, we reported how scanning probe block copolymer lithography (SPBCL), a technique that delivers attoliter-scale volumes of metal precursor-coordinated polymers to a desired location to form a polymer nanoreactor, could be used to synthesize well-defined multimetallic nanoparticles in terms of size and composition.29–34 This method takes the advantage of polymer nanoreactors to confine metal precursors at the nanoscale.29,35,36 Each nanoreactor, when thermally treated, yields an individual nanoparticle with a composition that mirrors the stoichiometry defined by the precursors. The method has been utilized to construct a combinatorial library of multimetallic nanoparticles consisting of all elemental combinations of Au, Ag, Cu, Co, and Ni at a fixed equal elemental stoichiometry, which includes sets of novel hetero-nanostructures that are composed of miscible and immiscible metals.32 Since multimetallic nanoparticles have not been synthesized with such precision and complexity before, the process by which the various metals in a polymer reactor aggregate to yield a single nanoparticle and then evolve into a final architecture is unclear. Therefore, this study is important for understanding how to control and design nanostructures for catalysis, plasmonics, magnetics, and electronics.
Herein, we report an in-depth study on how AuAgCu nanoparticles are formed in polymer nanoreactors. AuAgCu nanoparticles were chosen as a model system because both miscible (Au/Ag and Au/Cu) and immiscible metal pairs (Ag/Cu) are present in this system. The aggregation of different combinations of Au, Ag, and Cu in SPBCL-generated polymer reactors has been studied and characterized by XPS, STEM, and EDS. The study shows that different metal components aggregate at markedly different rates within the polymer nanoreactors, which leads to kinetically trapped states that evolve into either alloy or well-defined, phase-segregated heterostructures. By confining the reagents at a fixed stoichiometry within the polymer nanoreactors, we can systematically study how different metals migrate through a single particle to create alloys or heterostructures. This technique not only is useful for understanding particle formation but can also be used to identify conditions that trap structures in kinetic states, some of which may be useful in their own right.
RESULTS AND DISCUSSION
To investigate the nucleation and growth of metal precursors in polymers, we initially studied the simplest structures, i.e. polymer features that contain only HAuCl4. Previous studies have shown that Au salts, at elevated temperatures, can be reduced by PEO, which results in the subsequent aggregation of the metal atoms and nanoparticle nuclei in the polymer reactor.30,37 Herein, we examined the aggregation of monometallic particles in a quantitative manner. Experimentally, we find that the aggregation process can be divided into two stages (Figure 1A and 1B, reactor diameters ~1.5 μm, annealed at 100 °C). In the first stage (< 45 min), many Au particles form in a single polymer nanoreactor with the maximum average number appearing after an annealing time of ~15 min (Figure 1C). Next, the coarsening (ripening and coalescence) of the particles becomes the dominant process, overwhelming the rate of new nucleation, resulting in a decrease in the average number of particles per reactor (50 reactors monitored, Figure 1C). At the end of Stage I (~ 45 min, Figure 1C), only one nanoparticle is observed in each reactor, suggesting that the Au concentration in each polymer feature is below the threshold necessary to induce nucleation of new particles (Figure S1). During Stage I, the average particle diameter slowly increases until it reaches 46 nm due to the growth and coarsening processes (Figure 1D). In Stage II (45 min-360 min), individual particles in each reactor (Figure 1C) continue to grow by depleting the remaining Au in the polymer reactor, and ultimately reaching a final size of about 75 nm after 6 h (Figure 1D). This two-stage growth phenomenon was also observed for Au particles formed in smaller reactors (e.g. 550 and 800 nm; Figure 1C, 1D, and S2–S4). Impurity salt particles (NaCl/KCl) were observed in some polymer reactors (Figure 1B and S5A), which are a consequence of salt impurities in the commercially available polymer matrix. It is important to note that while both types of particles are present, the Au particles primarily nucleate on the substrate as opposed to salt crystals. In addition, the impurity salts can also be removed from the polymer by dialysis (Figure S5B). Since the mobilities of both Au atoms and particles are accelerated by thermal annealing, temperature plays a key role in tuning the nucleation, growth, and coarsening rates of Au particles. As shown in Figure S6, annealing the polymer feature at 150 °C led to a faster aggregation of Au, which requires only 0.5 h to generate nanoparticles that have the same size (75 nm) as those formed in polymer features annealed at 100°C for 6 h.
Figure 1.
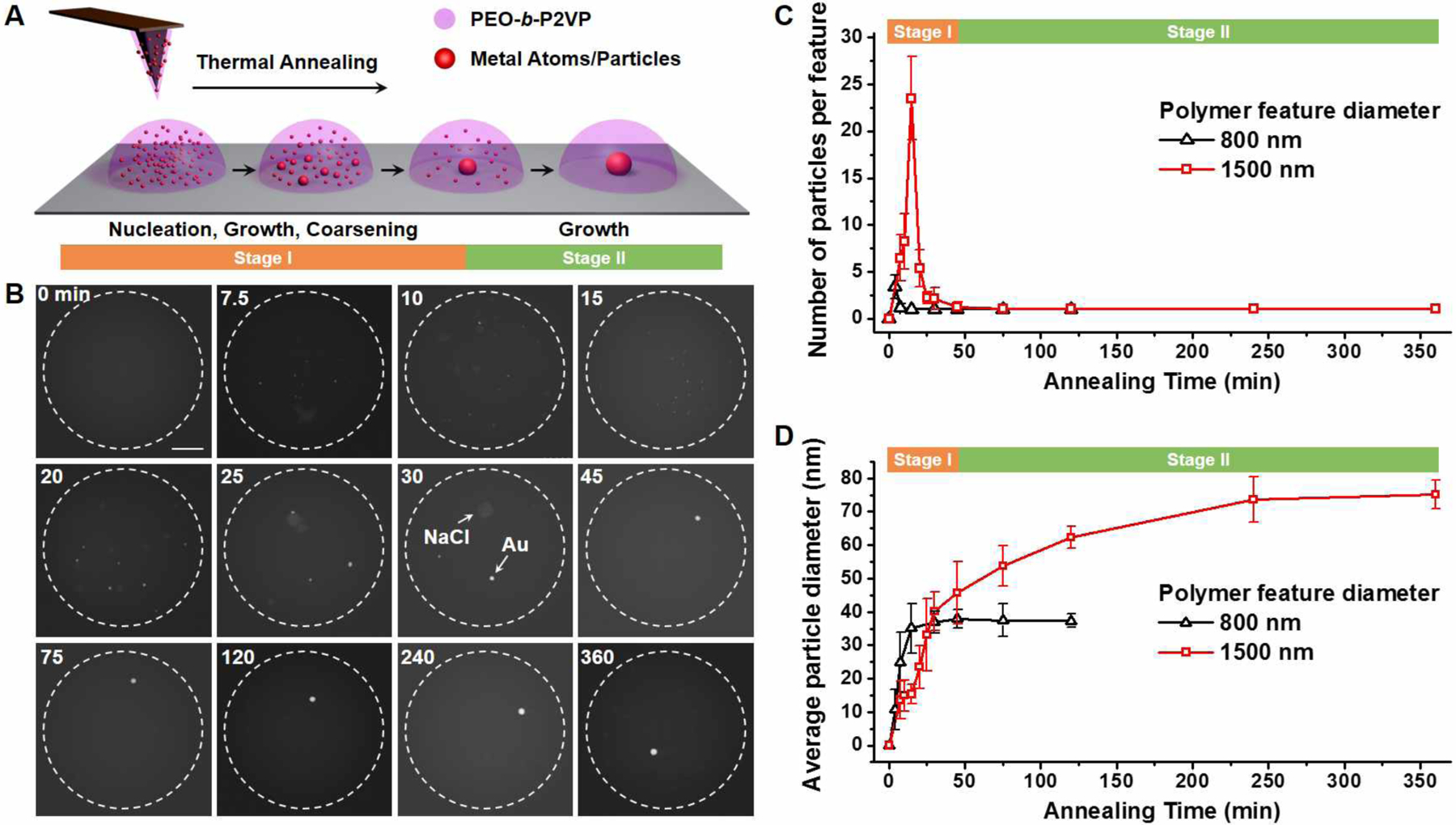
Nucleation and growth of monometallic nanoparticles in polymer nanoreactors. (A) A scheme of the nucleation and growth process. (B) Ex-situ ADF-STEM images of the aggregation and growth of Au nanoparticles in polymer features annealed at 100 °C. Dashed circles indicate the edge of polymer features. Scale bar: 300 nm. (C-D) The number (C) and average diameter (D) of the nanoparticles formed in polymer features annealed at 100 °C. The assignment of Stage I and II in (C) and (D) applies to the polymer features with diameters of 1500 nm.
To evaluate the generality of the aggregation processes, we next investigated polymer features that contain AgNO3 or Cu(NO3)2. Unlike Au, which precipitates directly in the form of Au nanoparticles, Ag initially aggregates by forming a mixture of Ag and AgCl particles (annealed at 120 °C for 0.5 h, Figure S7 and S8). This is likely due to the presence of Cl− ions in the reactor that come from the starting polymer matrix. After being annealed at 120 °C for 2 h, all AgCl particles were completely transformed into Ag particles through either thermal decomposition or reduction by PEO,30 resulting in multiple Ag nanoparticles in each reactor. In general, Ag aggregates more slowly than Au when being annealed under identical conditions (Figure S7). This rate difference suggests a higher mobility of Au atoms/particles in polymer reactors than that of Ag atoms/particles. In addition, the higher reduction potential of AuCl4−/Au (E° = 1.002 V) than that of Ag+/Ag (E° = 0.7996 V) also contributes to faster precipitation of Au. The last element, Cu, requires a longer annealing time or higher annealing temperature for nucleation to occur. As shown in Figure S9, particles that contain Cu are only observable in reactors that are annealed at 160 °C for more than 18 h or at temperatures above 200 °C for 4 h. These Cu-containing particles likely consist of CuII species such as CuO, a decomposition product from thermally annealing the Cu(NO3)2 precursor;38 the oxidation state of Cu at this stage was confirmed by XPS (Figure S9G). The slower nucleation of CuII species can be attributed to the higher thermal stability of the Cu precursor (CuNO3), which decomposes at temperature above 180 °C.38 In contrast, Au3+ and Ag+ are reduced to the metallic state by PEO at 120 °C for nucleation (Figure S7). In order to construct a systematic comparison of Au, Ag, and Cu, we annealed polymer reactors that contain each precursor at 240 °C for a predetermined time (Figure 2). At this temperature, the Au and Ag salts are reduced quickly and form nanoparticles (Figure 2D and 2E). Interestingly, while Au undergoes Stage I and II processes in 1 h and forms a single particle per reactor, Ag takes 2 h to finish Stage I and it is not until after 16 h of annealing that Ag reaches the end of Stage II (Figure 1A and S10). By contrast, Cu requires 8 h of annealing to finish Stage I, and it is not until 48 h that Stage II is completed (Figure S10 and S11). The slower aggregation of the CuII species is presumably due to the higher binding strength between CuII and the substrate (Si3N4 substrate with a native oxide top layer) than that between the other elements and the substrate. It should be noted that the presence of impurity salts has little effect on the aggregation of Au, Ag, or Cu at 240 °C, since Na, K, and Cl signals are not observed in the EDS spectra of the initially nucleated particles (Figure S12). The data for the monometallic systems clearly show that there are markedly different rates of particle formation, an observation that will be used to help understand the multimetallic systems.
Figure 2.
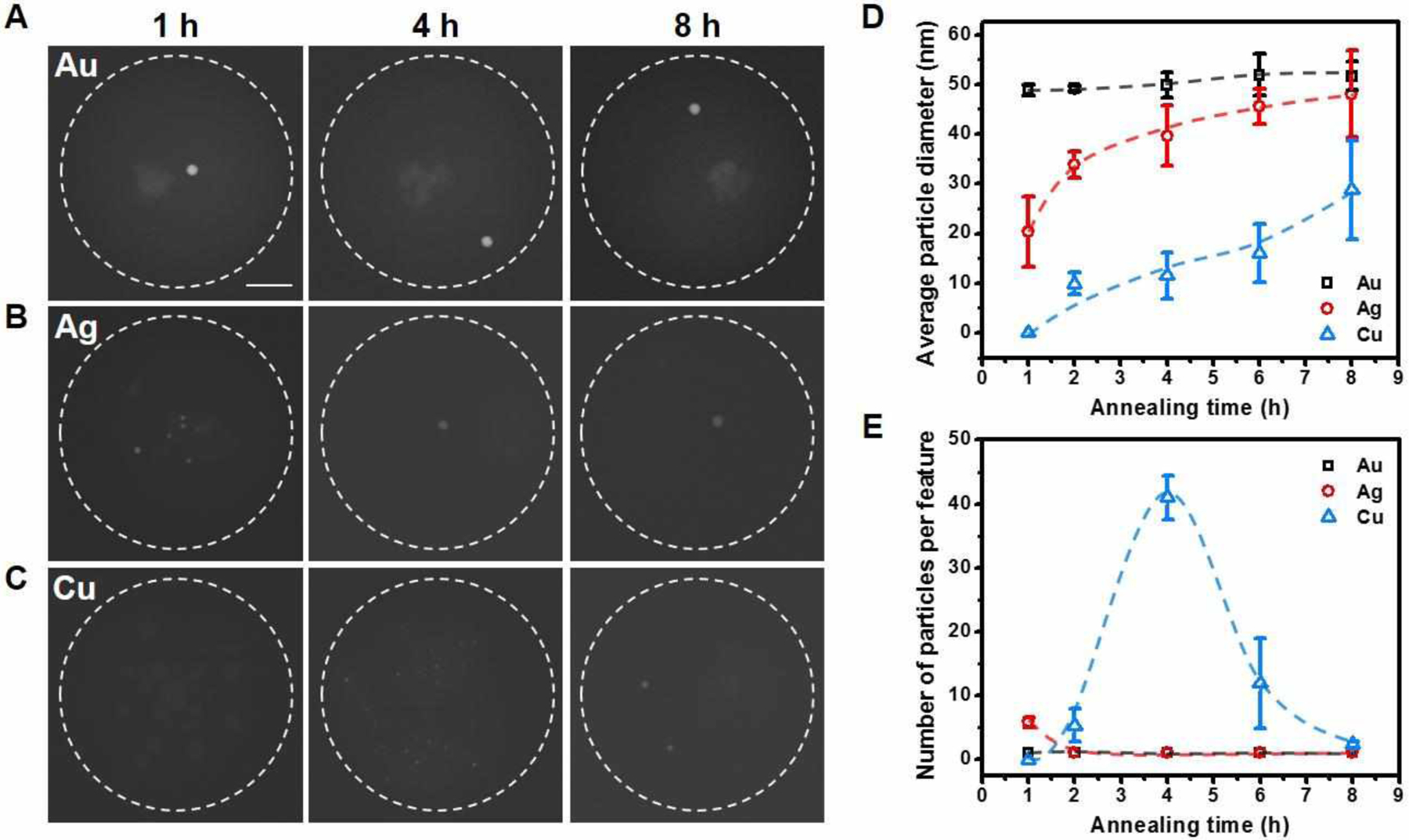
Comparison of the aggregation rates during the formation of different monometallic nanoparticles. (A-C) ADF-STEM images showing the aggregation of Au, Ag, or CuII species in polymer features annealed at 240 °C. Dashed circles indicate the edge of polymer features. Scale bar: 200 nm. (D-E) The average diameter (D) and number (E) of the nanoparticles formed in polymer features annealed at 240 °C. Dashed curves are guide to the eye.
With the knowledge gained in the monometallic study, we next examined the co-nucleation/growth process of all bimetallic combinations of Au, Ag, and Cu at fixed 1:1 stoichiometries. For the first binary combination (HAuCl4/AgNO3; reduced metals are miscible), Au, Ag, and AgCl particles initially form and are observable at early time points within the polymer reactors (after being annealed at 120 °C for 30 min, Figure S13). Continuous additional thermal treatment at 120 °C for another 1.5 h completely decomposes the AgCl and results in the formation of multiple irregular-shaped particles in each reactor (Figure 3). The irregular-shaped particles are composed of Au and Ag and are structurally inhomogeneous, as evidenced by the Au-enriched and Ag-enriched domains in the EDS elemental mapping images of the particles (Figure 3A and 3B). The inhomogeneous structure is caused by the coalescence of the Au and Ag particles early in the particle formation process. It should be noted that generally, the aggregation of Au is slower for the two-component Au/Ag system when compared to the monometallic system, pure Au system. Indeed, when polymer reactors containing only HAuCl4 are heated at 120 °C for 2 h (identical conditions used for the binary system), single well-formed Au nanoparticles form (Figure 3A). When one compares the three types of reactors at the same time point (Figures 3A), it is clear that the presence of Ag significantly depresses the mobility of Au. In addition, while Au@Ag core-shell nanostructures are observed for some particles, Ag@Au core-shell nanostructures were never found (Figure 3B and S13). This is consistent with the conclusion that Au still aggregates at a faster rate than Ag, and can act as a seed for heterogeneous nucleation of an Ag shell. Finally, for the two component system, when it is annealed at 160 °C for 4h, the vast majority of the precursor goes into forming a single primary two-component particle (~50 nm) within each reactor (Figure 3C), which is surrounded by much smaller, primarily silver particles (< 3 nm) that continue to contribute to coarsening as the reactor is heated. EDS elemental mapping of the primary particle shows that Au and Ag do not fully alloy even after being heated at 160 °C for 4 h (Figure 3D, here an Au@Ag particle is shown). To further explore the temperature-induced structural evolution of the particle, the polymer reactor shown in Figure 3D was annealed stepwise at 160 °C for 18 h and at 260 °C for 4 h with EDS mapping carried out after each step. Ag partly diffused into the Au core after being annealed at 160 °C for 18 h, while Au and Ag are fully mixed after being annealed at 260 °C for just 4 h (Figure 3D). As such, high temperature and short annealing times exhibit higher efficacy than low temperature and long annealing periods in facilitating the structural evolution of SPBCL nanoparticles into thermodynamic structures. The entire aggregation process of Au/Ag is summarized in Figure 3E. When two metals are present in one reactor, co-nucleation and growth of the two metals result in both monometallic and bimetallic particles in the reactor. Upon particle growth and coarsening of the particles, an individual structurally inhomogeneous particle is obtained. Ultimately, thermal annealing at a relatively high temperature allows the constituent metals to be transformed into a stable configuration.
Figure 3.
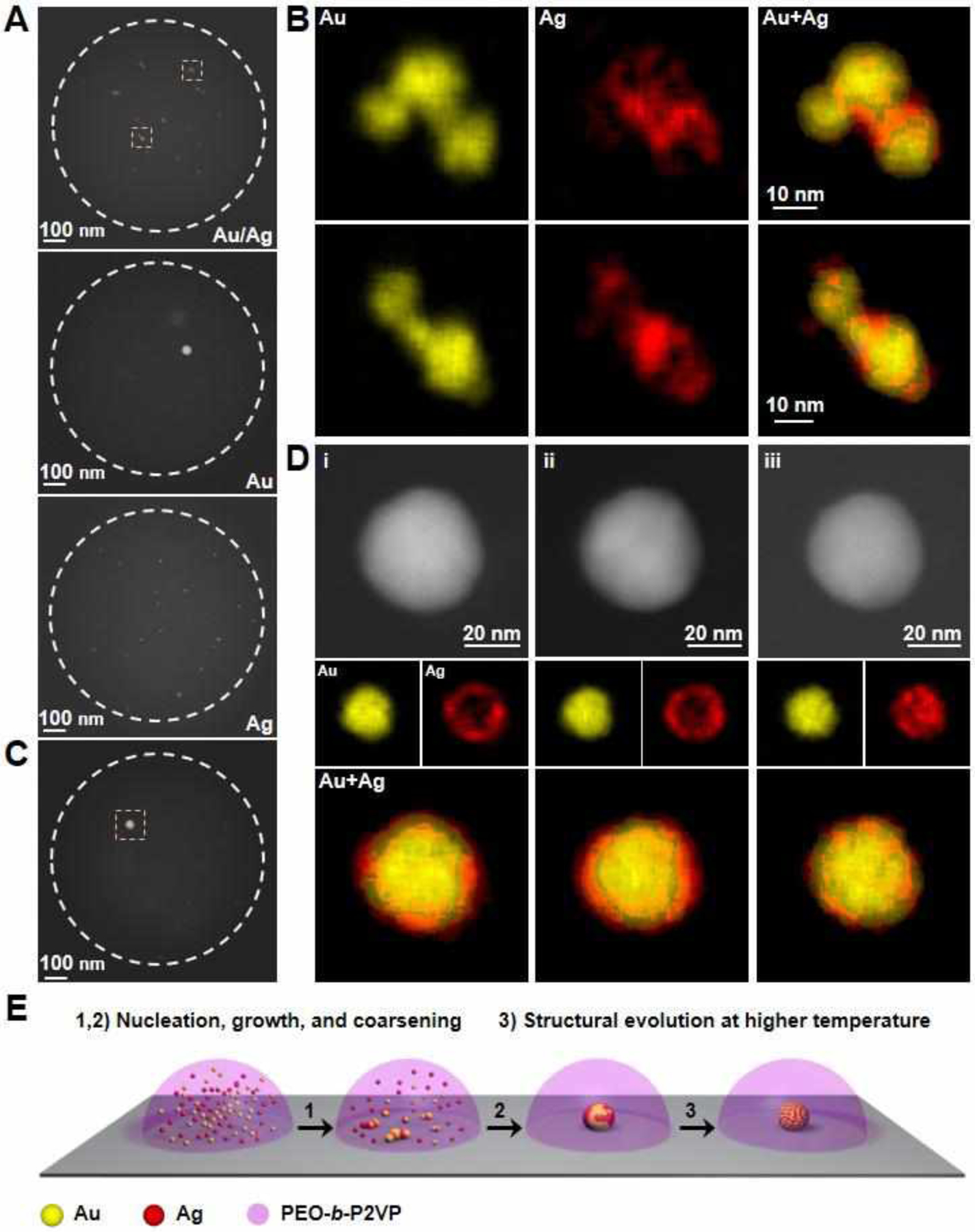
Formation of AuAg nanoparticles in polymer nanoreactors. (A) ADF-STEM images of representative polymer features that contain either equal amounts of HAuCl4 and AgNO3, only HAuCl4, or only AgNO3 metal salt precursors. The features were annealed at 120 °C for 2h. White dotted circles indicate the edge of polymer features. (B) EDS elemental maps of the AuAg particles located inside the yellow dashed squares in (A). (C) An ADF-STEM image of a polymer feature that contains equal amounts of the metal salt precursors, HAuCl4 and AgNO3, annealed at 160 °C for 4h. (D) ADF-STEM images and EDS elemental maps of the AuAg particle (51% Au, 49% Ag, atomic composition) located inside a yellow dashed square in (C). The particle was stepwise annealed at (i) 160 °C for 4h, (ii) 160 °C for 18h, and (iii) 260 °C for 4h. (E) Schematic of the structural evolution of AuAg nanoparticles in polymer nanoreactors deposited with SPBCL.
For the second binary combination (Au/Cu, miscible metals), primary nucleated particles are also formed by a combination of nucleation, growth, and coarsening (Figure S14 and S15), which is similar to the process observed in the AuAg system. The structure of the primary nucleated particles obtained at low annealing temperature (160 °C for 4 h) is inhomogeneous, as evidenced by the Au-enriched and Cu-enriched domains observed by EDS elemental mapping of the particles (Figure 4A). Either longer annealing times (18 h, Figure S15) or higher annealing temperatures (240–260 °C, Figure 4B–4E) increase the alloying of the Au and Cu. The oxidation states of Au and Cu that comprise the nucleated particles were examined by XPS using samples prepared by drop-casting (Figure 4F). When Au is present in the reactor, the Cu component is in the 0 valence state after being annealed at 260 °C for 18h, as compared to the formation of particles consisting of CuII species in reactors containing only Cu(NO3)2. HR-TEM characterization of the nucleated particles corroborates the formation of AuCu alloy particles in AuCu reactors (Figure 4G). Another distinct difference between the pure Cu and AuCu alloy system is the faster precipitation of Cu observed for the reactors containing Au. The majority of the available Cu accumulates into an alloy particle after being annealed at 160 °C for 4 h (Figure 4A), while for the pure Cu system, a higher annealing temperature (240 °C) and a longer annealing time (48 h) are required (Figure 2). Clearly, Au facilitates the reduction and aggregation of Cu. It has been reported that the free electron cloud on the surface of noble metal nanoparticles (e.g. Au) can induce the reduction of non-noble metal ions (e.g. Co2+, Cu2+) that are adsorbed on particle surface, i.e., the noble-metal-induced reduction (NMIR) process.39,40 In AuCu polymer reactors, the CuII species presumably adsorbed on the primarily nucleated Au nanoparticles and are then reduced to form AuCu alloy particles. The high mobility of Au atoms/particles expedites the aggregation of AuCu particles, as compared to the slow aggregation of a pure Cu system.
Figure 4.
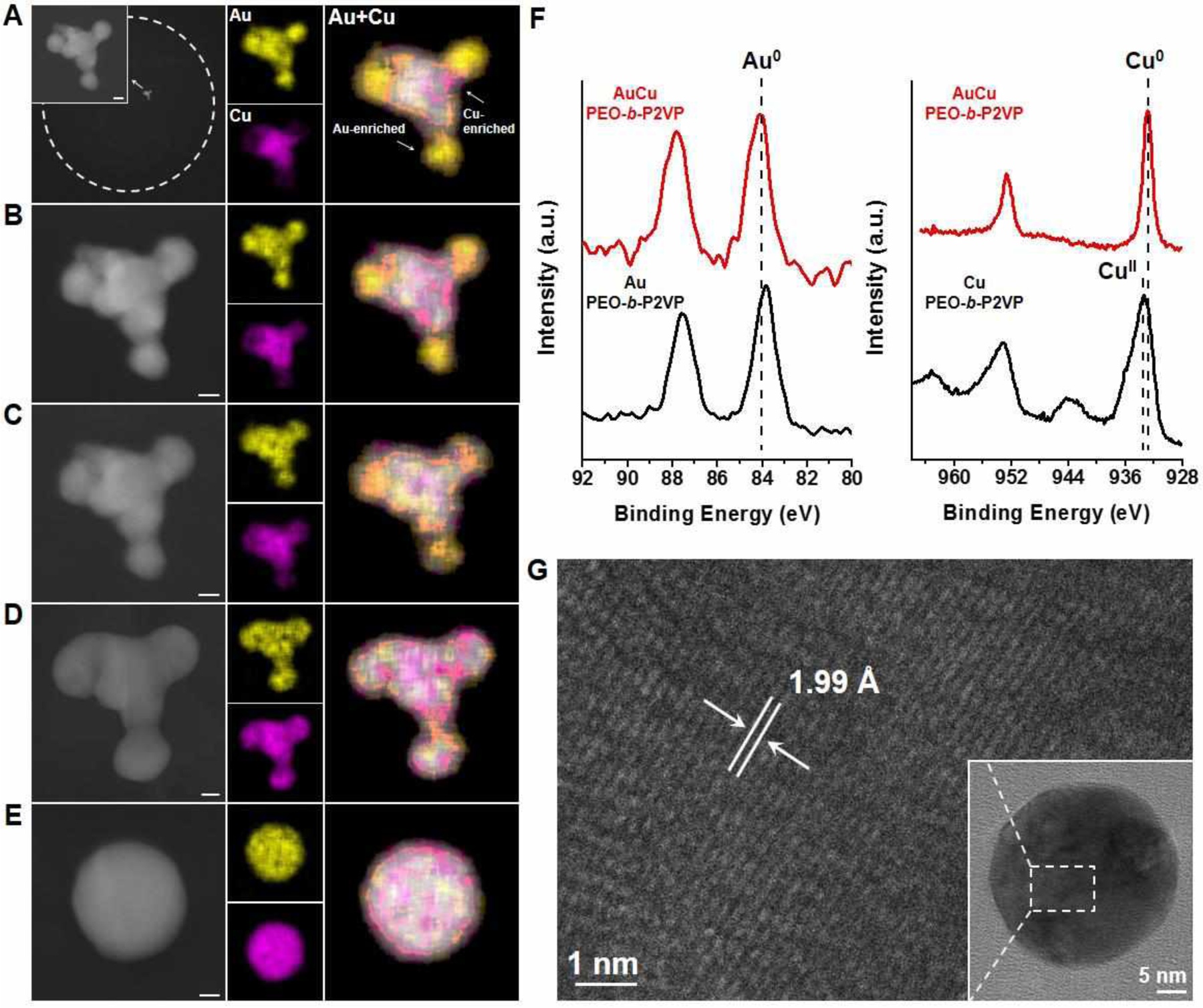
Formation of AuCu nanoparticles in polymer nanoreactors. (A-E) ADF-STEM images and EDS elemental maps of an AuCu particle (44% Au, 56% Cu, atomic composition) formed in a representative polymer feature that contains equal amounts of the metal salt precursors, HAuCl4 and Cu(NO3)2. White dashed circles indicate the edge of polymer features. The polymer feature was stepwise annealed at (A) 160 °C under Ar for 4 h, (B) 240 °C under Ar for 2 h, (C) 240 °C under Ar for 2 h, (D) 260 °C under Ar for 2 h, and (E) 500 °C under H2 for 12 h. Scale bars: 10 nm. (F) Au 4f and Cu 2p XPS spectra of particles synthesized by thermally annealing a drop-cast polymer ink solution at 260 °C for 18 h. Red lines correspond to polymer ink containing both Au and Cu precursors. Black lines correspond to polymer inks containing only one type of precursor. (G) HR-TEM image of an AuCu nanoparticle in a polymer feature that is formed by being annealed at 260 °C for 18h. The observed lattice spacing is 1.99 Å (extracted from fast Fourier transform), which closely matches the AuCu alloy (200) plane.
For the final binary combination considered for this study, Ag and Cu are immiscible metals, which results in phase-segregated Ag-Cu heterodimers after high temperature treatment (500 °C).32 To get a glimpse at how this process works, we initially annealed polymer reactors containing AgNO3 and Cu(NO3)2 at 160 °C for 4 h, the conditions that yielded the AuCu particles in the aforementioned experiment (Figure 4A). As evidenced by ADF-STEM images and EDS measurements, only Ag nanoparticles were observed in the reactors (Figures 5A and 5B). Continuous annealing at 160 °C for 18 h results in heterogeneous nucleation or attachment of CuII clusters onto the Ag cores, as observed by EDS elemental mapping of the particles (Figure 5D). The precipitating sequence of Ag followed by Cu is consistent with the monometallic cases (Figure 2) because Ag cannot alloy with Cu. XPS characterization validates that Ag cannot facilitate the reduction of CuII into metallic Cu. The clusters formed around the Ag core particle are composed of CuII species (Figure 5C). Thermal treatment at a higher temperature (260 °C) is effective in triggering the coarsening (ripening and coalescence) of the CuII clusters on the surface of the Ag core particle (Figure 5E and 5F). This intraparticle coarsening process decreases the total surface energy of the CuII domains as multiple small CuII clusters are transformed into a single large CuII cluster on the particle. The resulting Ag-CuII heterodimers can be further transformed into Ag-Cu heterodimers upon subsequent thermal annealing at 500 °C under H2 (Figure 5G, 5H, and S16). Similar intraparticle coalescence phenomena have been reported in the solution phase synthesis of heterostructured nanoparticles.41–43 It should be noted that the similarity between nanoreactors made from the same polymer solution ensures that each nanoreactor generates a multimetallic particle through the same process. Due to the different aggregation rates of each element and the random nature of the merging of particles/atoms, kinetic particles made from the same conditions have similar inhomogeneous structures but may vary in morphology (Figure S15 and S17).
Figure 5.
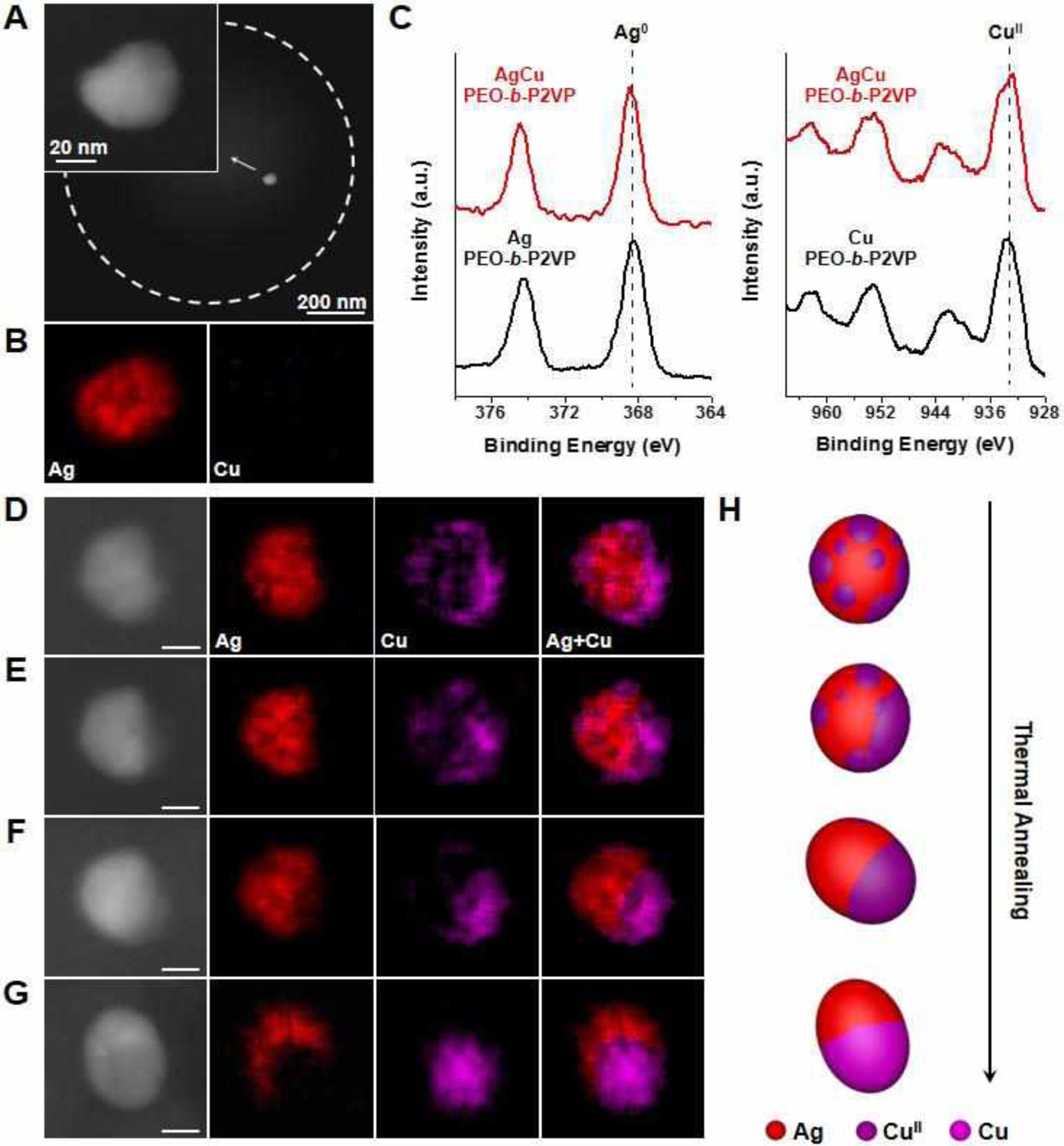
Formation of AgCu nanoparticles in polymer nanoreactors. (A) ADF-STEM images and (B) EDS elemental maps of a particle formed in a representative polymer feature annealed at 160 °C for 4 h that contains equal amounts of the metal salt precursors, AgNO3 and Cu(NO3)2. White dashed circles indicate the edge of the polymer feature. (C) Ag 3d and Cu 2p XPS spectra of particles synthesized by thermally annealing a drop-cast polymer ink solution at 260 °C for 18h. Red lines correspond to polymer ink containing both Ag and Cu precursors. Black lines correspond to polymer inks that contain only one type of precursor. (D-G) ADF-STEM images and EDS elemental maps of an AgCu particle (55% Ag, 45% Cu, atomic composition) formed in a representative polymer feature that was stepwise annealed at (D) 160 °C under Ar for 18 h, (E) 260 °C under Ar for 2 h, (F) 260 °C under Ar for 4 h, and (G) 500 °C under H2 for 12 h. Scale bars: 15 nm. (H) Schematic depicting the structural evolution of AgCu nanoparticles in polymer features during thermal treatment.
The synthesis of multimetallic nanoparticles containing more than two components usually requires delicate control of metal reduction kinetics in order to obtain a desired stoichiometric ratio between constituent metals. By contrast, the composition of SPBCL nanoparticle composition is mainly determined by the nanoreactor composition due to the forced aggregation of the metals in the reactor to yield an individual particle.31,32 This advantage is particularly useful when studying the formation of trimetallic nanoparticles with target compositions. For example, to reveal the formation mechanism of AuAgCu trimetallic nanoparticles, we annealed polymer reactors containing equal amounts of HAuCl4, AgNO3, and Cu(NO3)2 at 260 °C for 0.5h. Through these studies, it is clear that AuAg initially precipitates to form primary particles composed of predominately AuAg alloy and a small amount of AuCu alloy (Figure S18). The formation of AuAg primary particles is due to the faster aggregation of Au and Ag than Cu (Figure 2). In addition, Ag is not miscible with Cu, which makes the AuAg particle less effective in facilitating the reduction of Cu precursors. With prolonged thermal treatment at 260 °C (1.5 h), CuII clusters are observed on the fringe of the primary particle through heterogeneous nucleation or attachment of CuII clusters onto the primary particle (Figure 6A, S19, and S20). After that, the CuII clusters were reduced and diffuse into the AuAg particle to form a metastable AuAgCu particle, which contains discrete AuAg grains and AuCu grains, as indicated by EDS measurements (Figure 6B, 6C, and S21). The AuAg grains and AuCu grains are mobile when annealed at 260 °C (Figure 6B–6D and S22), which tend to segregate into a single AuAg domain and a single AuCu domain. Ultimately, when the reactor is annealed at 500 °C under H2, the polymer is decomposed and the phase segregation between AuAg and AuCu is completed, generating an AuAgCu heterodimer with one domain being an AuAg alloy and the other one being an AuCu alloy (Figure 6D).
Figure 6.
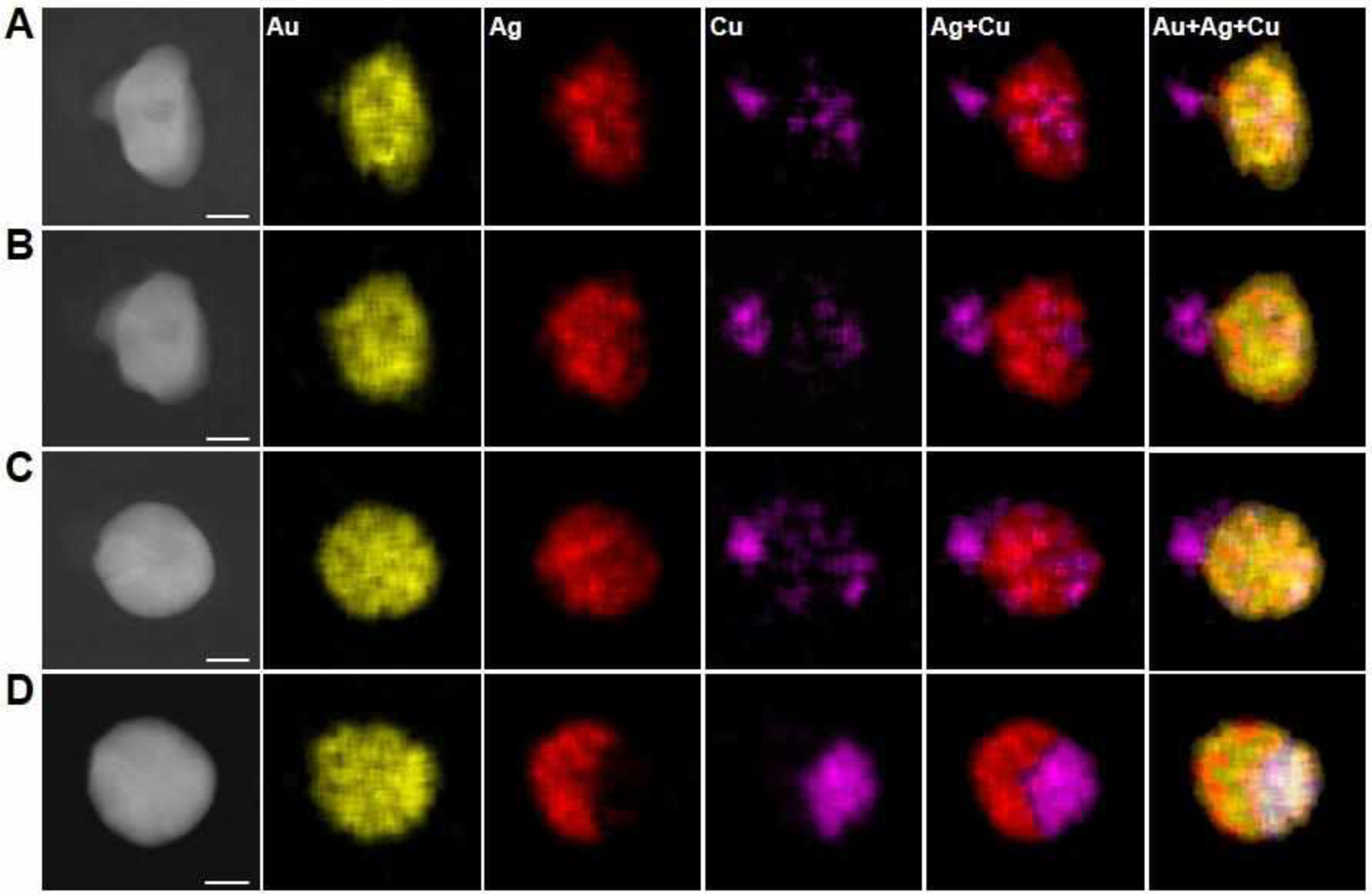
Formation of AuAgCu nanoparticles in polymer nanoreactors. (A-D) ADF-STEM images and EDS elemental maps of an AuAgCu particle (33% Au, 39% Ag, 28% Cu, atomic composition) formed in a representative polymer feature that contains equal amounts of the metal salt precursors, HAuCl4, AgNO3, and Cu(NO3)2. The polymer nanoreactor was stepwise annealed at (A) 260 °C under Ar for 1.5 h, (B) 260 °C under Ar for 2 h, (C) 260 °C under Ar for 3 h, and (D) 500 °C under H2 for 12 h. Scale bars: 20 nm.
CONCLUSIONS
In summary, the formation processes for multimetallic nanoparticles within PEO-b-P2VP polymer nanoreactors were studied with Au, Ag, and Cu salts as model systems. The ability to confine multiple precursors to one reactor and follow the transformation of such precursors into a single particle sets this work apart from previous attempts to understand alloy and phase-segregation in nanoparticle structures.32 Moreover, this research contributes to our understanding of nanoparticle formation in two important ways. First, it provides insight into the kinetics of nanoparticle formation for different metals, and second, it reveals that, for two-component systems in which the metals are miscible (e.g. Au and Cu), the metal that exhibits a faster aggregation rate facilitates the aggregation and reduction of the slower one to form alloy particles; in contrast, immiscible metals (e.g. Ag and Cu) aggregate at rates comparable to those for corresponding single-component systems. Significantly, this understanding of nanoparticle formation enables the kinetic trapping of particles with distinct architectures that may otherwise be missed, and such structures may be extremely useful in areas that rely on the emergent properties of nanoparticles, for example, in catalysis, magnetics, plasmonics, and electronics. As this field continues to move towards combinatorial nanoscience,32 this study illustrates that thermal treatment conditions, in addition to particle composition and size, will be an important library parameter.
EXPERIMENTAL SECTION
Chemicals.
Poly(ethylene oxide)-b-poly(2-vinyl pyridine) (PEO-b-P2VP, Mn = 2.8-b-1.5 kg/mol, polydispersity index = 1.11) was purchased from Polymer Source, Inc. and used as received. Metal compounds, HAuCl4·3H2O, AgNO3, and Cu(NO3)2·xH2O were purchased from Sigma-Aldrich, Inc. and used without further purification. DPN 1D pen arrays (type M, no gold coating) were purchased from Nanoink, Inc. Hexamethyldisilazane (HMDS) was purchased from Sigma-Aldrich. TEM grids with silicon nitride support films (Si3N4 film thickness = 15 or 50 nm) were purchased from Ted Pella, Inc.
Preparation of block copolymer inks.
Polymer inks were prepared by dissolving PEO-b-P2VP and different metal compounds in de-ionized water in predetermined molar ratios. The ink solution had a polymer concentration of 5 mg/mL and the molar ratio of pyridyl group to total metal precursors was 64:1. The use of an excess of pyridyl group facilitate a complete complexation of metal ions from the metal precursor salt onto PEO-b-P2VP. The pH of the ink solution was adjusted to between 3 and 4 by the addition of HNO3. The ink solution was stirred for 24 h at room temperature prior to use.
SPBCL patterning process.
Hydrophobic TEM grids were obtained by vapor coating the grids with HMDS for 24 h in a desiccator that contained one vial of a HMDS and hexane mixture (1:1, v/v). DPN pen arrays were dip-coated with inks and dried under ambient conditions. Subsequently, the pen arrays were mounted onto an AFM (XE-150, Park Systems) and brought into contact with the hydrophobic TEM grid to deposit arrays of polymer nanoreactors. The patterning process was performed in a chamber at a controlled temperature of 25 °C and relative humidity of 90%. In order to convert polymer features into nanoparticles, the TEM grids were put into a tube furnace and thermally annealed with programmed conditions.
Characterization.
Scanning transmission electron microscopy (STEM) characterization of nanoparticles synthesized on TEM grids with 50 nm Si3N4 support films was performed on an in-house designed dual-energy dispersive X-ray spectroscopy (EDS) detector equipped Hitachi HD-2300 dedicated STEM. The dark-field images were taken with an annular dark-field (ADF) detector at an electron acceleration voltage of 200 kV. The detection limit of the size of nanoparticles is around 1 nm. Nanoparticle composition was studied using the equipped dual EDS detectors (Thermo Scientific) on the HD-2300 STEM with a 200 kV acceleration voltage. The Lα peaks of Au and Ag, and the Kα peaks of Cu in the EDS spectra were used for elemental mapping and for composition quantification with standardless Cliff-Lorimer correction method. The atomic composition measured by EDS has an inherent error of less than 5% due to X-ray absorption and fluorescence. Each EDS map is built based on 30 frames with pixel dimensions of 256×192 and pixel dwell time of 203 μs. Thermo Scientific NSS software was used for EDS data processing. High-resolution TEM (HR-TEM) images were taken on a JEOL 3200FS transmission electron microscope at an acceleration voltage of 300 kV with nanoparticles prepared on TEM grids with 15 nm Si3N4 support films. X-ray photoelectron spectroscopy (XPS) spectra were recorded on a Thermo Scientific ESCALAB 250Xi with samples made by drop-casting polymer ink solutions, followed by predetermined thermal annealing process.
Supplementary Material
ACKNOWLEDGEMENT
This material is based upon work supported by GlaxoSmithKline LLC; the Air Force Office of Scientific Research awards FA9550-12-1-0141, FA9550-12-1-0280, and FA9550-16-1-0150; National Science Foundation award DBI-1353682; and the Vannevar Bush Faculty Fellowship program sponsored by the Basic Research Office of the Assistant Secretary of Defense for Research and Engineering and funded by the Office of Naval Research through grant N00014-15-1-0043. P.-C.C. and J.L.H. gratefully acknowledge support from the Ryan Fellowship and the Northwestern University International Institute for Nanotechnology. J.S.D. acknowledges support from the Hierarchical Materials Cluster Program Fellowship from Northwestern University. J.L.H. acknowledges support from the Department of Defense (DoD) through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program. This work made use of the EPIC facility of Northwestern University’s NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205), the MRSEC program (NSF DMR-1121262) at the Materials Research Center, the International Institute for Nanotechnology (IIN), the Keck Foundation, and the State of Illinois through the IIN. This work made use of the Structural Biology Facility supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, and the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust.
Footnotes
The authors declare no competing financial interest.
Supporting Information
Additional experimental data, Figures S1–S22.
REFERENCES
- (1).George C; Genovese A; Casu A; Prato M; Povia M; Manna L; Montanari T Nano Lett. 2013, 13, 752. [DOI] [PubMed] [Google Scholar]
- (2).Weng L; Zhang H; Govorov AO; Ouyang M Nat. Commun 2014, 5, 4792. [DOI] [PubMed] [Google Scholar]
- (3).Ye XC; Hickey DR; Fei JY; Diroll BT; Paik T; Chen J; Murray CB J. Am. Chem. Soc 2014, 136, 5106. [DOI] [PubMed] [Google Scholar]
- (4).Demortiere A; Schaller RD; Li T; Chattopadhyay S; Krylova G; Shibata T; Claro PCD; Rowland CE; Miller JT; Cook R; Lee B; Shevchenko EV J. Am. Chem. Soc 2014, 136, 2342. [DOI] [PubMed] [Google Scholar]
- (5).Yu H; Chen M; Rice PM; Wang SX; White RL; Sun SH Nano Lett. 2005, 5, 379. [DOI] [PubMed] [Google Scholar]
- (6).Mokari T; Rothenberg E; Popov I; Costi R; Banin U Science 2004, 304, 1787. [DOI] [PubMed] [Google Scholar]
- (7).Zhang HY; Pokhrel S; Ji ZX; Meng H; Wang X; Lin SJ; Chang CH; Li LJ; Li RB; Sun BB; Wang MY; Liao YP; Liu R; Xia T; Madler L; Nel AE J. Am. Chem. Soc 2014, 136, 6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yu XL; Shavel A; An XQ; Luo ZS; Ibanez M; Cabot A J. Am. Chem. Soc 2014, 136, 9236. [DOI] [PubMed] [Google Scholar]
- (9).Acharya KP; Khnayzer RS; O’Connor T; Diederich G; Kirsanova M; Klinkova A; Roth D; Kinder E; Imboden M; Zamkov M Nano Lett. 2011, 11, 2919. [DOI] [PubMed] [Google Scholar]
- (10).Buck MR; Schaak RE Angew. Chem. Int. Ed 2013, 52, 6154. [DOI] [PubMed] [Google Scholar]
- (11).Buck MR; Bondi JF; Schaak RE Nat. Chem 2012, 4, 37. [DOI] [PubMed] [Google Scholar]
- (12).Mazumder V; Chi MF; More KL; Sun SH Angew. Chem. Int. Ed 2010, 49, 9368. [DOI] [PubMed] [Google Scholar]
- (13).Li LL; Chen XB; Wu YE; Wang DS; Peng Q; Zhou G; Li YD Angew. Chem. Int. Ed 2013, 52, 11049. [DOI] [PubMed] [Google Scholar]
- (14).Hodges JM; Kletetschka K; Fenton JL; Read CG; Schaak RE Angew. Chem. Int. Ed 2015, 54, 8669. [DOI] [PubMed] [Google Scholar]
- (15).Sneed BT; Brodsky CN; Kuo CH; Lamontagne LK; Jiang Y; Wang Y; Tao F; Huang WX; Tsung CK J. Am. Chem. Soc 2013, 135, 14691. [DOI] [PubMed] [Google Scholar]
- (16).Read CG; Gordon TR; Hodges JM; Schaak RE J. Am. Chem. Soc 2015, 137, 12514. [DOI] [PubMed] [Google Scholar]
- (17).Liu MH; Zeng HC Langmuir 2014, 30, 9838. [DOI] [PubMed] [Google Scholar]
- (18).Min Y; Kwak J; Soon A; Jeong U Acc. Chem. Res 2014, 47, 2887. [DOI] [PubMed] [Google Scholar]
- (19).Liao HG; Cui LK; Whitelam S; Zheng HM Science 2012, 336, 1011. [DOI] [PubMed] [Google Scholar]
- (20).Jungjohann KL; Bliznakov S; Sutter PW; Stach EA; Sutter EA Nano Lett. 2013, 13, 2964. [DOI] [PubMed] [Google Scholar]
- (21).Tan SF; Chee SW; Lin GH; Bosman M; Lin M; Mirsaidov U; Nijhuis CA J. Am. Chem. Soc 2016, 138, 5190. [DOI] [PubMed] [Google Scholar]
- (22).De Clercq A; Dachraoui W; Margeat O; Pelzer K; Henry CR; Giorgio S J. Phys. Chem. Lett 2014, 5, 2126. [DOI] [PubMed] [Google Scholar]
- (23).Loh ND; Sen S; Bosman M; Tan SF; Zhong J; Nijhuis CA; Král P; Matsudaira P; Mirsaidov U Nat. Chem 2017, 9, 77. [DOI] [PubMed] [Google Scholar]
- (24).Kwon SG; Krylova G; Phillips PJ; Klie RF; Chattopadhyay S; Shibata T; Bunel EE; Liu YZ; Prakapenka VB; Lee B; Shevchenko EV Nat. Mater 2015, 14, 215. [DOI] [PubMed] [Google Scholar]
- (25).Hodges JM; Morse JR; Williams ME; Schaak RE J. Am. Chem. Soc 2015, 137, 15493. [DOI] [PubMed] [Google Scholar]
- (26).Gan L; Cui CH; Heggen M; Dionigi F; Rudi S; Strasser P Science 2014, 346, 1502. [DOI] [PubMed] [Google Scholar]
- (27).Niu ZQ; Becknell N; Yu Y; Kim D; Chen C; Kornienko N; Somorjai GA; Yang PD Nat. Mater 2016, 15, 1188. [DOI] [PubMed] [Google Scholar]
- (28).Liao HG; Zheng HM J. Am. Chem. Soc 2013, 135, 5038. [DOI] [PubMed] [Google Scholar]
- (29).Chai JA; Huo FW; Zheng ZJ; Giam LR; Shim W; Mirkin CA Proc. Natl. Acad. Sci. U. S. A 2010, 107, 20202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Liu GL; Eichelsdoerfer DJ; Rasin B; Zhou Y; Brown KA; Liao X; Mirkin CA Proc. Natl. Acad. Sci. U. S. A 2013, 110, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chen P-C; Liu G; Zhou Y; Brown KA; Chernyak N; Hedrick JL; He S; Xie Z; Lin Q-Y; Dravid VP; O’Neill-Slawecki SA; Mirkin CA J. Am. Chem. Soc 2015, 137, 9167. [DOI] [PubMed] [Google Scholar]
- (32).Chen P-C; Liu XL; Hedrick JL; Xie Z; Wang SZ; Lin Q-Y; Hersam MC; Dravid VP; Mirkin CA Science 2016, 352, 1565. [DOI] [PubMed] [Google Scholar]
- (33).Piner RD; Zhu J; Xu F; Hong SH; Mirkin CA Science 1999, 283, 661. [DOI] [PubMed] [Google Scholar]
- (34).Huo FW; Zheng ZJ; Zheng GF; Giam LR; Zhang H; Mirkin CA Science 2008, 321, 1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Scott RWJ; Datye AK; Crooks RM J. Am. Chem. Soc 2003, 125, 3708. [DOI] [PubMed] [Google Scholar]
- (36).Iyyamperumal R; Zhang L; Henkelman G; Crooks RM J. Am. Chem. Soc 2013, 135, 5521. [DOI] [PubMed] [Google Scholar]
- (37).Chai J; Liao X; Giam LR; Mirkin CA J. Am. Chem. Soc 2012, 134, 158. [DOI] [PubMed] [Google Scholar]
- (38).Morozov IV; Znamenkov KO; Korenev YM; Shlyakhtin OA Thermochim. Acta 2003, 403, 173. [Google Scholar]
- (39).Wang DS; Li YD J. Am. Chem. Soc 2010, 132, 6280. [DOI] [PubMed] [Google Scholar]
- (40).Wang DS; Li YD Inorg. Chem 2011, 50, 5196. [DOI] [PubMed] [Google Scholar]
- (41).De Yoreo JJ; Gilbert P; Sommerdijk N; Penn RL; Whitelam S; Joester D; Zhang HZ; Rimer JD; Navrotsky A; Banfield JF; Wallace AF; Michel FM; Meldrum FC; Colfen H; Dove PM Science 2015, 349, aaa6760. [DOI] [PubMed] [Google Scholar]
- (42).Peng S; Lei CH; Ren Y; Cook RE; Sun YG Angew. Chem. Int. Ed 2011, 50, 3158. [DOI] [PubMed] [Google Scholar]
- (43).Wu HM; Chen O; Zhuang JQ; Lynch J; LaMontagne D; Nagaoka Y; Cao YC J. Am. Chem. Soc 2011, 133, 14327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.