

Abstract
Hematological malignancies are broadly divided into myeloid and lymphoid neoplasms, reflecting the two major cellular lineages of the hematopoietic system. It is generally rare for hematological malignancies to spontaneously progress with a switch from myeloid to lymphoid lineage. We describe the exceptional case of a patient who sequentially developed myelodysplastic syndrome (MDS), chronic myelomonocytic leukemia (CMML), and B-cell acute lymphoblastic leukemia (B-ALL), as well as our investigation into the underlying pathogenesis. Using whole-exome sequencing (WES) performed on sorted CMML and B-ALL cell fractions, we identified both common and unique potential driver mutations, suggesting a branching clonal evolution giving rise to both diseases. Interestingly, we also identified a germline variant in the cancer susceptibility gene CHEK2. We validated that this variant (c.475T > C; p.Y159H), located in the forkhead-associated (FHA) domain, impairs its capacity to bind BRCA1 in cellulo. This unique case provides novel insight into the genetics of complex hematological diseases and highlights the possibility that such patients may carry inherited predispositions.
Keywords: chronic myelomonocytic leukemia, multiple lineage myelodysplasia, pre-B-cell acute lymphoblastic leukemia
INTRODUCTION
Myelodysplastic syndromes (MDSs) are a group of hematological malignancies characterized by ineffective hematopoiesis, chronic cytopenias, and dysplastic morphological changes in bone marrow cells (Cazzola 2020). MDS is associated with a wide variety of chromosomal and genetic abnormalities affecting processes such as epigenetic regulation, RNA splicing, transcription, cell cycle control, and DNA repair (Ogawa 2019). Although the severity and rate of progression of MDS is highly variable among individuals, transformation to acute myeloid leukemia (AML) is a well-described component of the natural history of the disease and occurs in ∼30% of MDS patients (Porwit and Saft 2011). Evolution of MDS to other leukemias, such as chronic myelomonocytic leukemia (CMML) and B-cell acute lymphoblastic leukemia (B-ALL), has been described in case reports but is considered exceedingly rare (Disperati et al. 2006; Niscola et al. 2013; Martins et al. 2021).
Although the mechanisms underlying MDS to ALL transformation are still poorly understood, it has been proposed that this results from the stepwise acquisition of genetic or epigenetic alterations within a self-renewing multipotent hematopoietic stem or early progenitor cell (Disperati et al. 2006; Martins et al. 2021). MDS-specific cytogenetic changes can be identified within the lymphoid compartment of some MDS patients, providing evidence for clonal lymphoid output in at least a subset of MDS cases (van Lom et al. 1995; Nilsson et al. 2000). Additionally, blood cancer development may originate within a preleukemic hematopoietic stem cell (HSC) pool, because cancer-associated mutations can be found in the normal blood cells of healthy individuals (Zink et al. 2017) and patients with AML (Jan et al. 2012) or lymphoma (Quivoron et al. 2011). From this premalignant reservoir, it is possible that branching evolution may occur. Overall, characterizing the genetic evolution of patients who develop multiple hematological malignancies may provide additional insight into their pathogenesis and guide therapy.
In this study, we leveraged fluorescence-activated cell sorting (FACS) and whole-exome sequencing (WES) to elucidate the genetic underpinnings of an exceptionally rare case of a patient who underwent transformation from MDS to CMML, followed by a terminal B-ALL blast phase. We identified suspected driver mutations shared between the myeloid and lymphoid populations, including NRAS, TET2, EZH2, and ZRSR2, supporting a common clonal origin. Other mutations were unique to the CMML (ASXL1) or B-ALL (BRD2) clones. In addition, we found a germline variant of unknown significance (VUS) in the checkpoint kinase 2 (CHEK2) tumor-suppressor gene affecting a critical protein domain. CHEK2 is a cancer susceptibility gene that has been associated with MDS (Janiszewska et al. 2018). We performed functional validation on this mutant CHK2 protein and demonstrated impaired binding to downstream BRCA1, suggesting this variant may be cancer-predisposing.
RESULTS
Case Presentation and Family History
A 56-year-old male was referred to the hematology clinic for an isolated finding of severe but asymptomatic neutropenia. He did not have any personal history of cancer or prior hematologic disease. However, his mother had been diagnosed with essential thrombocytosis (ET) and his father with colorectal cancer. He also had a brother with an unspecified malignancy. A bone marrow biopsy was consistent with a diagnosis of MDS with multilineage dysplasia (MDS-MLD) by World Health Organization (WHO) 2016 criteria (Swerdlow et al. 2017). His International Prognostic Scoring System (IPSS) risk score was low and the karyotype showed -Y in all metaphases. He was managed with observation alone for 10 years until he eventually developed worsening anemia, which was treated with recombinant erythropoietin (EPO). Following an initial response, the anemia became refractory to EPO, and he developed progressive neutrophilia and monocytosis. Repeat bone marrow examination was consistent with transformation to CMML without blast count elevation (WHO 2016 criteria, intermediate-1 IPSS score) (Swerdlow et al. 2017). The karyotype remained unchanged (-Y). The patient was started on lenalidomide 10 mg daily. This resulted in control of white blood cell counts, although he remained dependent on red blood cell transfusions. Following 2 years of stability, he experienced abrupt onset leukocytosis. Examination of the blood smear revealed abundant blasts. Bone marrow examination (morphology and flow cytometry) now showed a B-ALL clone in addition to the background CMML. The karyotype was unchanged. He was admitted to the hospital and treated with a five-drug induction consisting of prednisone, daunorubicin, vincristine, cyclophosphamide, and L-asparaginase. This was complicated by severe invasive fungal infection (fusarium species), liver toxicity, and prolonged hospital admission. He was not deemed fit for further intensive combination chemotherapy following these events. Roughly 8 months after the induction regimen, he experienced florid relapse of both the CMML and B-ALL clones. It was at this time point that a sample was collected for WES. Attempts were made to control the disease with several lines of therapy, including vincristine monotherapy, mitoxantrone monotherapy, and inotuzumab ozogamicin. Responses were brief. Eventually, both leukemias became refractory to therapy and multiple organ failure ensued. Comfort care was instituted. The patient died 14 years after the initial MDS diagnosis.
Genomic Analyses
We used FACS to separate the peripheral blood sample into B lymphoid (CD19+/CD33−) and myelomonocytic (CD19−/CD33+) populations. We extracted genomic DNA from these two fractions and a buccal swab. We performed WES on these three samples. Following the bioinformatics analysis (see Methods), we identified 11 potential driver mutations meeting retention criteria. These variants (Table 1) included six nonsynonymous single-nucleotide variants (SNVs), four indels, and one stop-gain. Some variants were shared across all three samples (buccal swab, B lymphoid, myelomonocytic), suggesting a germline origin. Others were unique to one or both sorted cellular fractions, indicating somatic mutation events. The buccal swab yielded lower DNA quantity, and WES performed on this sample had 12.72× mean coverage. Therefore, we confirmed the presence or absence of each variant reported in Table 1 for all samples using Sanger sequencing as an orthogonal validation method.
Table 1.
Variants identified in the buccal swab sample and sorted cell populations using whole-exome sequencing
| Gene | Chromosome and GRCh37 position | HGVS DNA reference | HGVS protein reference | Variant type | In silico pathogenicity assessment (see Methods) | ClinVar status (if available) | dbSNP/dbVar ID | Buccal swab | Myeloid | Lymphoid |
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Variant allele frequency | |||||||||
| NRAS | Chr 1:115258747 | NM_002524:c.35G > A | p.G12D | Nonsynonymous SNV | VUSa | Pathogenic | rs121913237 | - | 0.47 | 0.39 |
| CLOCK | Chr 4:56322165 | NM_001267843:c.881C > T | p.P294L | Nonsynonymous SNV | Likely pathogenic | Not reported | rs1362924961 | - | 0.41 | 0.38 |
| TET2 | Chr 4:106158398_ 106158399 | NM_001127208: c.3299_3300del2 | p.V1100Afs*2 | Frameshift deletion | Pathogenic | Not reported | None | - | 1.00 | 1.00 |
| BRD2 | Chr 6:32943865_32943865 | NM_001113182: c.529_530insTGA | p.V177_A178insI | Nonframeshift insertion | Unknown | Not reported | None | - | - | 0.41 |
| CDKN1A | Chr 6:36652068 | NM_001291549:c.292G > A | p.A98T | Nonsynonymous SNV | VUS | Not reported | rs780909704 | Heterozygous | 0.47 | 0.47 |
| EZH2 | Chr 7: 148507467_148507468 | NM_004456:c.1986_1987del2 | p.Y663fsa0 | Frameshift deletion | Pathogenic | Not reported | None | - | 0.54 | 0.39 |
| BMPR1A | Chr 10:88683261 | NM_004329:c.1471G > A | p.E491K | Nonsynonymous SNV | VUS | VUS | rs1329735599 | Heterozygous | 0.39 | 0.64 |
| FOXA2 | Chr 20:22562771 | NM_021784:c.1109T > G | p.L370R | Nonsynonymous SNV | Pathogenic | Not reported | None | Heterozygous | 0.50 | 0.58 |
| ASXL1 | Chr 20: 31022922_31022922 | NM_015338:c.2407delC | p.Q803Kfs*14 | Frameshift deletion | Pathogenic | Not reported | None | - | 0.41 | - |
| CHEK2 | Chr 22:29121082 | NM_007194:c.475T > C | p.Y159H | Nonsynonymous SNV | Pathogenic | VUS | rs781254437 | Heterozygous | 0.40 | 0.38 |
| ZRSR2 | Chr X:15809088 | NM_005089:c.73G > T | p.E25X | Stopgain | Pathogenic | Not reported | None | - | 1.00 | 0.75 |
(SNV) Single-nucleotide variant, (VUS) variant of unknown significance.
aAlthough this variant was classified as a VUS using the combination of in silico tools, it is a well-validated pathogenic variant in ClinVar and we considered it as such.
To evaluate our samples for allelic imbalance (AI), we used the web-based application ExomeAI (Nadaf et al. 2015). This analysis revealed a large AI in the long arm of Chromosome 4 (Chr 4q) in both the CMML and B-ALL samples. This finding is consistent with the identification of a frameshift deletion in TET2 (located on Chr 4q) with a variant allele frequency (VAF) of 1.0 in both tumor samples. In addition, we also identified a large AI on Chromosome 16q unique to the B-ALL sample.
Functional Analyses
The tumor-suppressor CHEK2 c.475T > C variant identified using WES was of interest to us given its possible role as a cancer-predisposing mutation. We confirmed the presence of this mutation by Sanger sequencing of the buccal swab and myelomonocytic and B-lymphoid samples. We also found this mutation in nonmalignant T cells sorted from this patient (Supplemental Fig. S1). This strongly suggested a germline origin for this variant. The resulting amino acid change (CHK2 Y159H) is only two positions away from the known pathogenic variant I157T (Fig. 1), which affects the forkhead-associated (FHA) domain and impairs binding of downstream effectors such as BRCA1, p53, and CDC25 family proteins (Li et al. 2002). We hypothesized that the Y159H variant could have a similar deleterious effect on the FHA domain as it was predicted as pathogenic by all six in silico prediction tools that we used (see Methods). Currently, ClinVar classifies Y159H as being of uncertain significance, in part due to lack of growth perturbation in a yeast-based functional assay (Delimitsou et al. 2019). Similarly, the variant is considered a VUS on VarSome based on the American College of Medical Genetics and Genomics (ACMG) classification (Richards et al. 2015; Kopanos et al. 2019).
Figure 1.

Schematic representation of the CHK2 protein and its major domains. A selection of five pathogenic mutations described in the literature is depicted in red. The position of the CHK2 Y159H variant described in this report is shown in black, highlighting its proximity to the pathogenic I157T variant. The SCD region is the site of phosphorylation and activation by ATM. The FHA domain is critical for the binding of downstream proteins such as BRCA1, p53, and CDC25A (Li et al. 2002). Figure redrawn using data from Cai et al. (2009) and Stolarova et al. (2020). (ATM) Ataxia-telangiectasia mutated, (BRCA1) breast cancer type 1 susceptibility protein, (CDC25A) M-phase inducer phosphatase 1, (FHA) forkhead-associated domain, (KD) kinase domain, (p53) cellular tumor antigen p53, (SCD) serine-glutamine/threonine-glutamine cluster domain.
To test our hypothesis, we took advantage of a single-cell assay in which the association of two proteins can be assessed at an integrated LacO array by tethering an mCherry-LacR-tagged version of any prey of interest (Fig. 2). This LacO/LacR system has been successfully used to characterize the association between BRCA1 and PALB2 (Orthwein et al. 2015) and to determine the impact of a pathogenic RAD51D variant on its association to XRCC2 (Rivera et al. 2017). Using this approach, we first confirmed that an mCherry-LacR-tagged version of CHK2 can recruit endogenous BRCA1 to the LacO array (Fig. 2). We then evaluated the impact of the Y159H variant on the CHK2–BRCA1 association. As comparators, we used wild-type (WT) CHK2 and two previously described loss-of-function variants (I157T and H143Y). The I157T variant disrupts the FHA domain, and the H143Y variant produces a highly unstable protein (Bell et al. 2007, Li et al. 2002). Our results showed that the Y159H mutation drastically impairs CHK2 association to BRCA1 similarly to the other two loss-of-function variants, suggesting that it significantly disrupts the FHA domain (Fig. 2). These data support the categorization of CHK2 Y159H as likely pathogenic.
Figure 2.

CHK2–BRCA1 colocalization assay. (A) Schematic representation of the experimental assay (redrawn from Orthwein et al. 2015). (B) Representative confocal microscopy images. The location of the LacO array (white square) is identified by the accumulation of mCherry signal. Colocalization of BRCA1 and CHK2 is assessed at the mCherry focus by measuring the AF-647 signal, representing endogenous BRCA1. (C) Relative AF-647 signal (representing BRCA1) at the mCherry focus for the various CHK2 variants studied. The control (Ctrl) is an mCherry-LacR construct lacking CHK2. Statistical significance was assessed using Dunnett's multiple comparisons test using wild-type CHK2 as the comparator. (*) P ≤ 0.05, (**) P ≤ 0.01. (AF-647) Alexa Fluor 647, (BRCA1) breast cancer type 1 susceptibility protein, (CHK2) checkpoint kinase 2, (WT) wild type.
DISCUSSION
In this study, we leveraged FACS and WES to elucidate the genetic underpinnings of a highly unusual case of transformation from a chronic myeloid malignancy (MDS, CMML) to an aggressive lymphoid malignancy (B-ALL). We identified 11 putative driver mutations with various distributions between the buccal swab and malignant samples that allowed us to propose a model of branching clonal evolution and identify a possible inherited predisposition.
Four variants (CDKN1A, BMPR1A, FOXA2, CHEK2) were shared across the buccal swab, CMML clone, and B-ALL clone, suggesting these were germline mutations. Although CDKN1A, BMPR1A, and FOXA2 have all been implicated in human malignancies to various degrees (Halmos et al. 2004; Calva-Cerqueira et al. 2009; Kreis et al. 2019), there is little evidence to suggest these were pathogenic in this specific case beyond in silico predictions. CHEK2 demonstrates a broader spectrum of associated malignancies and is considered a cancer susceptibility gene with a low-to-moderate penetrance (Stolarova et al. 2020). It encodes the protein CHK2, which regulates the DNA damage response by phosphorylating and activating BRCA1, promoting the homologous recombination repair pathway. CHK2 also appears to be implicated in the control of apoptosis, senescence, and p53-mediated cell cycle arrest, although it does not appear to be indispensable for the latter (Antoni et al. 2007). We suspected a possible pathogenic role for CHEK2 c.475T > C (Y159H) because of in silico predictions and proximity to the known I157T FHA variant, which impairs BRCA1, p53, and CDC25 binding, thus potentially promoting tumorigenesis (Li et al. 2002). Notably, CHEK2 I157T has been identified as a risk factor for the development of MDS and ET in Polish populations (Janiszewska et al. 2012; Janiszewska et al. 2018). Recently, it was also shown to increase the risk of myeloproliferative neoplasms by promoting self-renewal of HSCs (Bao et al. 2020). Using a LacO–LacR system, we demonstrated that the Y159H variant behaves similarly to I157T in terms of impaired binding to BRCA1. This suggests that the variant we identified in this patient may possibly be cancer-predisposing and warrant further study. Prior to this new data, we considered the Y159H variant as meeting the ACMG criteria PM1 (located in a critical functional domain), PM2 (absent in healthy controls), and PP3 (in silico predictions), giving an overall classification of VUS (2 moderate, 1 supporting criteria) (Richards et al. 2015). Our new functional data would qualify as an additional supporting criterion (PS3_supporting) as per recent Clinical Genome Resource (ClinGen) recommendations (Brnich et al. 2019). If our findings can be reproduced by other laboratories, this has the potential to reclassify Y159H as likely pathogenic as per ACMG criteria (two moderate, two supporting) (Richards et al. 2015).
We identified other variants that were absent from the buccal swab but shared between the CMML and B-ALL clones. This suggests that these mutations were somatic and likely arose earlier in the disease course, probably at the MDS or CMML stage, prior to the emergence of the B-ALL clone. Of these variants, three consisted of highly typical MDS mutations (TET2, EZH2, ZRSR2). These genes regulate DNA methylation, histone modification, and RNA splicing, respectively, and are implicated in the pathophysiology of MDS (Ogawa 2019). TET2 mutations are also common in clonal hematopoiesis (CH), which may precede MDS (Jaiswal et al. 2014). An activating NRAS mutation was identified in both leukemic clones. This variant affects the G12 hotspot of the NRAS protein and leads to aberrant cellular proliferation (Prior et al. 2012). We hypothesize that this mutation may have been acquired at the MDS to CMML transition when the disease began to behave more aggressively. As we did not have access to samples earlier in the disease course, the attribution of a mutation to the MDS or CMML stage is based on the knowledge of common driver mutations in these entities. The identification of a somatic mutation in the circadian rhythm regulator CLOCK was intriguing. Circadian disruption linked to night-shift work has been associated with increased risk of a wide range of human malignancies (Fu and Kettner 2013). The circadian clock is thought to act as a tumor suppressor by modulating cellular proliferation, apoptosis, and the immune system (Fu and Lee 2003). In microsatellite unstable colorectal cancers, somatic loss-of-function CLOCK mutations appear to be very common and may promote tumorigenesis via dysregulation of DNA damage response genes such as CDKN1A and RAD50, as suggested by a chromatin immunoprecipitation sequencing (ChIP-seq) approach (Alhopuro et al. 2010). The role of CLOCK mutations in MDS and other hematologic malignancies remains to be defined.
Two variants were exclusively identified in a single clone. These included a frameshift deletion in ASXL1 for the CMML clone and a nonframeshift insertion in BRD2 in the B-ALL clone. Truncating exon 12 mutations in ASXL1, such as the one identified in this patient, are commonly described in myeloid malignancies but are particularly frequent in CMML (∼45% of cases) (Gelsi-Boyer et al. 2012). These mutations cause loss of ASXL1 protein and aberrant chromatin remodeling, leading to overexpression of oncogenes such as HOXA genes and activation of the PI3K/AKT pathway via suppression of PTEN (Abdel-Wahab et al. 2012; Cao et al. 2020). Interestingly, ASXL1 mutations seem to cooperate with NRAS G12D in CMML (Abdel-Wahab et al. 2012), as was observed in our patient. ASXL1 mutations are generally regarded as early events in leukemogenesis (Fujino and Kitamura 2020). However, in our case, the ASXL1 mutation was only seen in the CMML clone and not the B-ALL clone. It is possible that the subclone responsible for the B-ALL transformation simply did not carry the ASXL1 variant or that the variant was lost in the B-ALL clone as a result of lack of selective advantage. The B-ALL clone could either have arisen from the CMML clone or from a shared progenitor. The only retained mutation found to be unique to the B-ALL clone in our analysis was a 3-bp insertion in the gene BRD2 resulting in the in-frame addition of a single amino acid. This gene encodes BRD2, a BET protein family member involved in regulating transcription (French 2016). Little is known on the role of BRD2 in human cancers. However, overexpression of BRD2 induces B-cell lymphoma/leukemia in murine models (Greenwald et al. 2004). The insertion that we identified falls within the functionally important first bromodomain (BD1); however, it is unclear how this could lead to gain of function of the protein (Yu et al. 2015). Overall, we believe that this mutation is unlikely to be solely responsible for the B-ALL transformation in this patient. Because we did not obtain excellent coverage with WES, it is possible that other driver mutations were missed in the B-ALL clone. In addition, our study design could not evaluate epigenetic changes, which are highly relevant to cancer biology (Sharma et al. 2010). It is also possible additional karyotypic abnormalities beyond -Y may have been missed in the B-ALL clone given the imperfect sensitivity of G-band karyotyping.
Overall, our study identified a common clonal origin and provided evidence for the sequential acquisition of driver mutations in a patient with complex hematological disease progressing through three stages (MDS, CMML, B-ALL). This allowed us to construct a putative sequence of mutation acquisition (Fig. 3). We also validated the likely pathogenic effects of a germline CHEK2 variant (CHK2 Y159H), which possibly represented a cancer-predisposing condition in this case.
Figure 3.
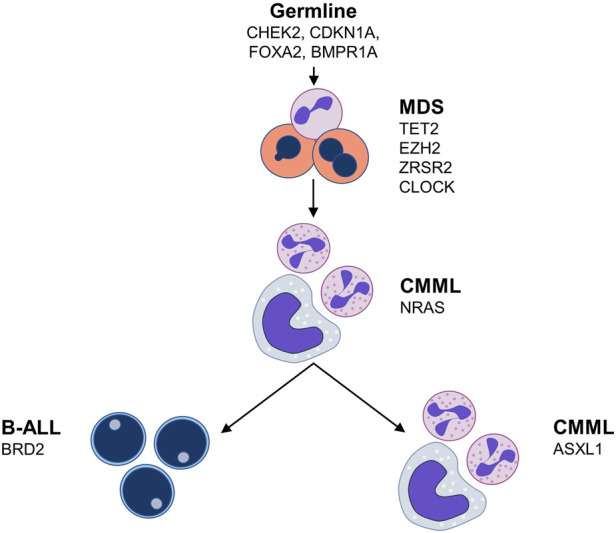
Putative sequence of clonal evolution in the presented case. (B-ALL) B-cell acute lymphoblastic leukemia, (CMML) chronic myelomonocytic leukemia, (MDS) myelodysplastic syndrome.
METHODS
Flow Cytometry and Cell Sorting
We processed the patient blood sample using Ficoll density gradient centrifugation to obtain peripheral blood mononuclear cells (PBMCs). The PBMCs were cryopreserved at −80°C until recovery for processing. After thawing, we stained the PBMCs using CD19-APC (eBioscience, clone HIB19, 17-0199-41) and CD33-FITC (eBioscience, clone WM53, 11-0338-41). These cell surface markers were chosen based on the clinical flow cytometry panels performed on this patient that showed the B-ALL clone was CD19+/CD33− and the CMML clone was CD19−/CD33+. We used a BD FACSAria Fusion cell sorter to isolate the B-lymphoid fraction (CD19+/CD33−) and the myelomonocytic fraction (CD19−/CD33+). To isolate T cells for CHEK2 variant detection, we added CD3-BV786 (BD Biosciences, clone SK7, 563800) and sorted the CD19−/CD33−/CD3+ fraction. The cell sorting strategy is outlined in Supplemental Figure S2. A postsorting purity check revealed >99% purity for the target populations.
Whole-Exome Sequencing
We extracted genomic DNA from the sorted cell populations as well as a concomitantly collected buccal swab using the DNeasy Blood and Tissue Kit (QIAGEN 69506). WES was performed on the B-lymphoid, myelomonocytic, and buccal swab DNA samples by Novogene Co. The Agilent SureSelect Human All Exon kit (Agilent Technologies) was used to generate the sequencing libraries. Sequencing was performed on an Illumina platform (150-bp paired-end reads). Clean reads were aligned to the human reference genome GRCh37/hg19 using BWA-MEM (Li and Durbin 2009). Sequencing metrics are presented in Table 2. For the buccal swab sample, germline variants were called using HaplotypeCaller (GATK, Broad Institute) and hard-filtered using the following parameters: QD < 2.0, FS > 60.0, MQ < 40.0, HaplotypeScore > 13.0, MappingQualityRankSum < −12.5, ReadPosRankSum < −8.0. We used Mutect2 with a panel of normals (PON) to call somatic variants in the B-lymphoid and myelomonocytic fractions, followed by filtering using FilterMutectCalls (GATK, Broad Institute). Annotation was performed using ANNOVAR (Wang et al. 2010). We further refined the list of variants by excluding synonymous SNVs, noncoding region variants, variants identified at a rate >0.01% in population databases (1000G, ExAC, ESP6500si, gnomAD), and known benign variants on ClinVar. Finally, we identified likely driver mutations using Cancer Genome Interpreter (Tamborero et al. 2018) to obtain the final list presented in Table 1. In parallel, we performed a pathogenicity assessment as previously described (de Kock et al. 2019). Briefly, frameshift and nonsense variants were classified as pathogenic. Otherwise, nonsynonymous SNVs were classified as either pathogenic, likely pathogenic, VUS, or benign using a score derived from a combination of six in silico tools (SIFT, PolyPhen-2, M-CAP, FATHMM, PROVEAN, CADD). We did not retain any predicted benign variants in the curated list. We retained only variants with at least 10× coverage in at least one of the samples. We used Sanger sequencing to validate all variants reported in Table 1. The ExomeAI analysis for AI assessment was performed using the web-based application (http://genomequebec.mcgill.ca/exomeai) with the default settings (Nadaf et al. 2015).
Table 2.
Whole-exome sequencing metrics
| Sample | Reads | % Duplicate | % Mapped | Mean coverage on target | % of target covered with at least 10× |
|---|---|---|---|---|---|
| B lymphoid | 47,361,768 | 25.10 | 99.88 | 79.17× | 97.0 |
| Myeloid | 63,717,732 | 25.45 | 99.92 | 97.19× | 97.8 |
| Buccal swab | 6,849,050 | 23.71 | 99.89 | 12.72× | 49.8 |
Functional Validation of the CHK2 Y159H Variant
To evaluate the effect of the CHK2 Y159H variant on BRCA1 binding, we used the previously described LacO–LacR system (Tang et al. 2013; Orthwein et al. 2015). We obtained the CHEK2 open reading frame (ORF) vector from the ORFeome Collaboration provided by the McGill Platform for Cellular Perturbation of the Goodman Cancer Research Centre and Biochemistry at McGill University. We generated the Y159H mutant variant using the QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies). We also generated I157T and H143Y mutants (known dysfunctional variants) for comparison (Bell et al. 2007). The oligonucleotides used in the QuikChange protocol are detailed in Supplemental Table S1. Gateway cloning technology (Invitrogen) was used to insert the CHEK2 mutant and WT vectors into a pDEST-mCherry-LacR vector. We confirmed the expected sequence and mutations in the final constructs using Sanger sequencing.
U2OS cells containing a stable integration of a Lac operator repeat sequence (U2OS-LacO) were plated onto #1.5 coverslips in a 12-well plate. We transfected the U2OS-LacO cells at 60% confluency with 1 µg of each mCherry-LacR-CHEK2 construct. The coverslips were then rinsed twice with PBS, fixed with PBS + 2% paraformaldehyde (Thermofisher) for 20 min at room temperature (RT), washed twice again with PBS, and then followed by a 20-min permeabilization in PBS + 0.3% Triton-100 solution at RT. The coverslips were washed twice with PBS and blocked for 1 h in PBS + 5% BSA + 0.1% Triton-100 (PBSA-T) at RT. These were then incubated for 1 h with a 1:400 dilution of rabbit anti-BRCA1 primary antibody (EMD Millipore 07-434) at RT, washed twice with PBS, and incubated with secondary antibody (goat anti-rabbit Alexa Fluor 647, Thermofisher A-21244) in PBSA-T for 1 h at RT. The coverslips were washed twice in PBS, washed once in ddH2O, and then mounted with Fluoromount-G (Thermofisher). Images were acquired with a LSM 800 confocal microscope (Carl Zeiss AG) and analyzed as previously described (Findlay et al. 2018). The colocalization of BRCA1 (AF647 signal) with WT and variant CHK2 was measured at the LacO repeat array (Fig. 2B) and quantified by calculating the mean fluorescence intensity (MFI) of AF647 at the mCherry focus divided by the MFI of AF647 in the background region of the nucleus (relative fluorescence). Two biological replicates were performed for each variant, and a minimum of 25 cells were evaluated per replicate. Statistical significance was assessed using ordinary one-way ANOVA with Dunnett's multiple comparisons test in GraphPad Prism software (version 9.0.0) using the WT CHK2 as a control (Fig. 2C).
ADDITIONAL INFORMATION
Data Deposition and Access
The raw WES data is available on the European Genome-Phenome Archive (EGA) under study accession ID #EGAS00001005117. The CHEK2 variant was submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under the accession number SCV001593269.
Ethics Statement
The patient provided written informed consent for the collection of blood samples for research according to our Research Ethics Board (REB) protocol 11-047. Use of these samples for genetic testing was approved by REB protocol 12-052. The consent obtained from the patient allows for the controlled-access deposition of anonymized genetic data.
Acknowledgments
The authors thank Dr. Gabriela Galicia Vazquez, Yoon Kow Young, Maja Jankovic, Wai Lam Poon, and Katharine Fooks for technical assistance and fruitful discussion. We thank the patient and his family for agreeing to participate in this research project. We also thank the Jewish General Hospital Foundation for supporting in part the biobank Banque de cellules leucémiques du Québec (BCLQ)—axe lymphome. The authors A.B. and F.E.M. thank the Fonds de la recherche en santé du Québec (FRQS) for salary support and the Cole Leukemia Foundation and Richard and Edith Strauss Foundation for funding for this project.
Author's Contributions
A.B., F.E.M., and A.O. designed the study. A.B., S.W., and J.H. performed the experiments. S.C. provided the clinical data. A.B. and J.H. analyzed the data. A.-S.C., B.R.P., and W.D.F. helped design and guide the analysis of the sequencing data. A.O., E-R.S.-C., and J.H. helped design and guide the analysis of the functional validation data. N.A.J. established the biobanking framework for sample collection and processing. A.O. and F.E.M. supervised the experimental work. A.B. wrote the manuscript. All authors reviewed and provided input for the manuscript.
Funding
A.B. and F.E.M. received salary support from the FRQS. A.O. is the Canada Research Chair (Tier 2) in Genome Stability and Hematological Malignancies. Work in the A.O. laboratory was supported by a Canadian Institutes of Health Research (CIHR) Project Grant (#376245), a Transition Grant from the Cole Foundation, and an internal Operating Fund from the Sir Mortimer B. Davis Foundation of the Jewish General Hospital. Work in the F.E.M. laboratory was supported by a Transition Grant from the Cole Foundation, an Operating Grant from the Richard and Edith Strauss Foundation, and an internal Operating Fund from the Sir Mortimer B. Davis Foundation of the Jewish General Hospital.
Competing Interest Statement
E.R.S.-C. is employed by Eisai Canada. N.A.J. received honoraria or consultancy fees from Roche/Genentech, Merck, Bristol Myers Squibb, and AbbVie and research funding from AbbVie. F.E.M. received consultancy fees from Sanofi Genzyme. All other authors have no competing interests to declare.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, et al. 2012. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22: 180–193. 10.1016/j.ccr.2012.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhopuro P, Bjorklund M, Sammalkorpi H, Turunen M, Tuupanen S, Bistrom M, Niittymaki I, Lehtonen HJ, Kivioja T, Launonen V, et al. 2010. Mutations in the circadian gene CLOCK in colorectal cancer. Mol Cancer Res 8: 952–960. 10.1158/1541-7786.MCR-10-0086 [DOI] [PubMed] [Google Scholar]
- Antoni L, Sodha N, Collins I, Garrett MD. 2007. CHK2 kinase: cancer susceptibility and cancer therapy—two sides of the same coin? Nat Rev Cancer 7: 925–936. 10.1038/nrc2251 [DOI] [PubMed] [Google Scholar]
- Bao EL, Nandakumar SK, Liao X, Bick AG, Karjalainen J, Tabaka M, Gan OI, Havulinna AS, Kiiskinen TTJ, Lareau CA, et al. 2020. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 586: 769–775. 10.1038/s41586-020-2786-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell DW, Kim SH, Godwin AK, Schiripo TA, Harris PL, Haserlat SM, Wahrer DC, Haiman CA, Daly MB, Niendorf KB, et al. 2007. Genetic and functional analysis of CHEK2 (CHK2) variants in multiethnic cohorts. Int J Cancer 121: 2661–2667. 10.1002/ijc.23026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, Kanavy DM, Luo X, McNulty SM, Starita LM, et al. 2019. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med 12: 3. 10.1186/s13073-019-0690-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Chehab NH, Pavletich NP. 2009. Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. Mol Cell 35: 818–829. 10.1016/j.molcel.2009.09.007 [DOI] [PubMed] [Google Scholar]
- Calva-Cerqueira D, Chinnathambi S, Pechman B, Bair J, Larsen-Haidle J, Howe JR. 2009. The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clin Genet 75: 79–85. 10.1111/j.1399-0004.2008.01091.x [DOI] [PubMed] [Google Scholar]
- Cao L, Xia X, Kong Y, Jia F, Yuan B, Li R, Li Q, Wang Y, Cui M, Dai Z, et al. 2020. Deregulation of tumor suppressive ASXL1-PTEN/AKT axis in myeloid malignancies. J Mol Cell Biol 12: 688–699. 10.1093/jmcb/mjaa011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M. 2020. Myelodysplastic syndromes. N Engl J Med 383: 1358–1374. 10.1056/NEJMra1904794 [DOI] [PubMed] [Google Scholar]
- de Kock L, Wu MK, Foulkes WD. 2019. Ten years of DICER1 mutations: provenance, distribution, and associated phenotypes. Hum Mutat 40: 1939–1953. 10.1002/humu.23877 [DOI] [PubMed] [Google Scholar]
- Delimitsou A, Fostira F, Kalfakakou D, Apostolou P, Konstantopoulou I, Kroupis C, Papavassiliou AG, Kleibl Z, Stratikos E, Voutsinas GE, et al. 2019. Functional characterization of CHEK2 variants in a Saccharomyces cerevisiae system. Hum Mutat 40: 631–648. 10.1002/humu.23728 [DOI] [PubMed] [Google Scholar]
- Disperati P, Ichim CV, Tkachuk D, Chun K, Schuh AC, Wells RA. 2006. Progression of myelodysplasia to acute lymphoblastic leukaemia: implications for disease biology. Leuk Res 30: 233–239. 10.1016/j.leukres.2005.06.011 [DOI] [PubMed] [Google Scholar]
- Findlay S, Heath J, Luo VM, Malina A, Morin T, Coulombe Y, Djerir B, Li Z, Samiei A, Simo-Cheyou E, et al. 2018. SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J 37: 1–20. 10.15252/embj.2018100158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CA. 2016. Small-molecule targeting of BET proteins in cancer. Adv Cancer Res 131: 21–58. 10.1016/bs.acr.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Fu L, Kettner NM. 2013. The circadian clock in cancer development and therapy. Prog Mol Biol Transl Sci 119: 221–282. 10.1016/B978-0-12-396971-2.00009-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Lee CC. 2003. The circadian clock: pacemaker and tumour suppressor. Nat Rev Cancer 3: 350–361. 10.1038/nrc1072 [DOI] [PubMed] [Google Scholar]
- Fujino T, Kitamura T. 2020. ASXL1 mutation in clonal hematopoiesis. Exp Hematol 83: 74–84. 10.1016/j.exphem.2020.01.002 [DOI] [PubMed] [Google Scholar]
- Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. 2012. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol 5: 12. 10.1186/1756-8722-5-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald RJ, Tumang JR, Sinha A, Currier N, Cardiff RD, Rothstein TL, Faller DV, Denis GV. 2004. E mu-BRD2 transgenic mice develop B-cell lymphoma and leukemia. Blood 103: 1475–1484. 10.1182/blood-2003-06-2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halmos B, Bassères DS, Monti S, D'Alo F, Dayaram T, Ferenczi K, Wouters BJ, Huettner CS, Golub TR, Tenen DG. 2004. A transcriptional profiling study of CCAAT/enhancer binding protein targets identifies hepatocyte nuclear factor 3 β as a novel tumor suppressor in lung cancer. Cancer Res 64: 4137–4147. 10.1158/0008-5472.CAN-03-4052 [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. 2014. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371: 2488–2498. 10.1056/NEJMoa1408617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. 2012. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med 4: 149ra18. 10.1126/scitranslmed.3004315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska H, Ba¸k A, Pilarska M, Heise M, Junkiert-Czarnecka A, Kuliszkiewicz-Janus M, Całbecka M, Jaźwiec B, Wolowiec D, Kuliczkowski K, et al. 2012. A risk of essential thrombocythemia in carriers of constitutional CHEK2 gene mutations. Haematologica 97: 366–370. 10.3324/haematol.2011.049494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janiszewska H, Ba¸k A, Skonieczka K, Jas´kowiec A, Kiełbinski M, Jachalska A, Czyzewska M, Jaźwiec B, Kuliszkiewicz-Janus M, Czyz J, et al. 2018. Constitutional mutations of the CHEK2 gene are a risk factor for MDS, but not for de novo AML. Leuk Res 70: 74–78. 10.1016/j.leukres.2018.05.013 [DOI] [PubMed] [Google Scholar]
- Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, Massouras A. 2019. VarSome: the human genomic variant search engine. Bioinformatics 35: 1978–1980. 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis NN, Louwen F, Yuan J. 2019. The multifaceted p21 (Cip1/Waf1/CDKN1A) in cell differentiation, migration and cancer therapy. Cancers (Basel) 11: 1220. 10.3390/cancers11091220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Williams BL, Haire LF, Goldberg M, Wilker E, Durocher D, Yaffe MB, Jackson SP, Smerdon SJ. 2002. Structural and functional versatility of the FHA domain in DNA-damage signaling by the tumor suppressor kinase Chk2. Mol Cell 9: 1045–1054. 10.1016/S1097-2765(02)00527-0 [DOI] [PubMed] [Google Scholar]
- Martins F, Kruszewski M, Scarpelli I, Schoumans J, Spertini O, Lubbert M, Blum S. 2021. Characterization of myelodysplastic syndromes progressing to acute lymphoblastic leukemia. Ann Hematol 100: 63–78. 10.1007/s00277-020-04114-2 [DOI] [PubMed] [Google Scholar]
- Nadaf J, Majewski J, Fahiminiya S. 2015. ExomeAI: detection of recurrent allelic imbalance in tumors using whole-exome sequencing data. Bioinformatics 31: 429–431. 10.1093/bioinformatics/btu665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson L, Astrand-Grundström I, Arvidsson I, Jacobsson B, Hellström-Lindberg E, Hast R, Jacobsen SE. 2000. Isolation and characterization of hematopoietic progenitor/stem cells in 5q-deleted myelodysplastic syndromes: evidence for involvement at the hematopoietic stem cell level. Blood 96: 2012–2021. 10.1182/blood.V96.6.2012 [DOI] [PubMed] [Google Scholar]
- Niscola P, Tendas A, Scaramucci L, Giovannini M, Fratoni S, de Fabritiis P. 2013. Evolution of chronic myelomonocytic leukemia from refractory anemia: the unusual course of a myelodysplastic syndrome. Blood Res 48: 152–153. 10.5045/br.2013.48.2.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S. 2019. Genetics of MDS. Blood 133: 1049–1059. 10.1182/blood-2018-10-844621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, et al. 2015. A mechanism for the suppression of homologous recombination in G1 cells. Nature 528: 422–426. 10.1038/nature16142 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Porwit A, Saft L. 2011. The AML–MDS interface—leukemic transformation in myelodysplastic syndromes. J Hematopathol 4: 69–79. 10.1007/s12308-011-0088-6 [DOI] [Google Scholar]
- Prior IA, Lewis PD, Mattos C. 2012. A comprehensive survey of Ras mutations in cancer. Cancer Res 72: 2457–2467. 10.1158/0008-5472.CAN-11-2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. 2011. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20: 25–38. 10.1016/j.ccr.2011.06.003 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera B, Di Iorio M, Frankum J, Nadaf J, Fahiminiya S, Arcand SL, Burk DL, Grapton D, Tomiak E, Hastings V, et al. 2017. Functionally null RAD51D missense mutation associates strongly with ovarian carcinoma. Cancer Res 77: 4517–4529. 10.1158/0008-5472.CAN-17-0190 [DOI] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA. 2010. Epigenetics in cancer. Carcinogenesis 31: 27–36. 10.1093/carcin/bgp220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolarova L, Kleiblova P, Janatova M, Soukupova J, Zemankova P, Macurek L, Kleibl Z. 2020. CHEK2 germline variants in cancer predisposition: stalemate rather than checkmate. Cells 9: 2675. 10.3390/cells9122675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow SH, Campo E, Lee Harris N, Jaffe ES, Pileri SA, Stein H, Thiele J, Arber DA, Hasserjian RP, Le Beau MM, et al. 2017. WHO classification of tumours of haematopoietic and lymphoid tissues. International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, Tusquets I, Albanell J, Rodon J, Tabernero J, et al. 2018. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med 10: 25. 10.1186/s13073-018-0531-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. 2013. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 20: 317–325. 10.1038/nsmb.2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lom K, Hagemeijer A, Smit E, Hählen K, Groeneveld K, Löwenberg B. 1995. Cytogenetic clonality analysis in myelodysplastic syndrome: monosomy 7 can be demonstrated in the myeloid and in the lymphoid lineage. Leukemia 9: 1818–1821. [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Wang Z, Zhang Z, Ren X, Lu X, Ding K. 2015. Small-molecule BET inhibitors in clinical and preclinical development and their therapeutic potential. Curr Top Med Chem 15: 776–794. 10.2174/1568026615666150302110135 [DOI] [PubMed] [Google Scholar]
- Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J, et al. 2017. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130: 742–752. 10.1182/blood-2017-02-769869 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw WES data is available on the European Genome-Phenome Archive (EGA) under study accession ID #EGAS00001005117. The CHEK2 variant was submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under the accession number SCV001593269.