

Abstract
Visceral myopathies (VMs) encompass a spectrum of disorders characterized by chronic disruption of gastrointestinal function, with or without urinary system involvement. Pathogenic missense variation in smooth muscle γ-actin gene (ACTG2) is associated with autosomal dominant VM. Whole-genome sequencing of an infant presenting with chronic intestinal pseudo-obstruction revealed a homozygous 187 bp (c.589_613 + 163del188) deletion spanning the exon 6–intron 6 boundary within ACTG2. The patient's clinical course was marked by prolonged hospitalizations, multiple surgeries, and intermittent total parenteral nutrition dependence. This case supports the emerging understanding of allelic heterogeneity in ACTG2-related VM, in which both biallelic and monoallelic variants in ACTG2 are associated with gastrointestinal dysfunction of similar severity and overlapped clinical presentation. Moreover, it illustrates the clinical utility of rapid whole-genome sequencing, which can comprehensively and precisely detect different types of genomic variants including small deletions, leading to guidance of clinical care decisions.
Keywords: chronic constipation, hypoperistalsis, ileus, intestinal pseudo-obstruction
INTRODUCTION
Visceral myopathies (VMs), or congenital defects of the smooth muscle, can lead to several clinically overlapping disorders of the gastrointestinal (GI) system, including chronic intestinal pseudo-obstruction (CIPO) and megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS). The main features of these disorders are abdominal pain and distension, constipation, and emesis. In MMIHS, bladder distension may also be present. An estimated 100 new CIPO cases are diagnosed each year in the United States, whereas a total of 227 MMIHS cases were reported in the literature between 1976 and 2011 (De Giorgio et al. 2011; Gosemann and Puri 2011).
Enteric smooth muscle γ-actin 2 (ACTG2) encodes an actin isoform that is selectively expressed in the smooth muscle layers of the GI tract (including the muscularis mucosa and muscularis propria) and is absent from skeletal muscle, heart, lung, and brain (Miwa et al. 1991; Halim et al. 2016). The muscularis propria is composed of two smooth muscle layers, the outer longitudinal layer and inner circular layer, which are innervated by the enteric nervous system and contract in an alternating pattern termed peristalsis to propel the contents GI tract onward (Lehtonen et al. 2012). Like other actin isoforms, ACTG2 forms multimers within muscle cells to control cellular contractility in conjunction with myosin. Pathogenic variation in ACTG2 disrupts this crucial function and has been estimated to account for ∼60% of VM cases (Assia Batzir et al. 2019). Several other genes have been implicated in VM, including myosin subunits MYH11 and MYL9, and proteins involved in the actin–myosin interaction such as LMOD1 and MYLK (Ambartsumyan 1993; Ravenscroft et al. 2018; Kandler et al. 2020). Enteric neuropathies such as Hirschsprung disease can also lead to CIPO (Ambartsumyan 1993). The disease progression for ACTG2-related visceral myopathy appears to be more severe than that of ACTG2-negative visceral myopathy, with high rates of parenteral nutrition dependence, visceral transplantation, and disease-related mortality (Assia Batzir et al. 2019).
In more than 50 individuals reported in the literature, heterozygous missense variants in ACTG2 have been associated with autosomal dominant (AD) visceral myopathy, manifesting variably as CIPO or MMIHS (Lehtonen et al. 2012; Holla et al. 2014; Thorson et al. 2014; Wangler et al. 2014; Klar et al. 2015; Tuzovic et al. 2015; Lu et al. 2016; Milunsky et al. 2017; Ravenscroft et al. 2018; Assia Batzir et al. 2019). Functional studies have demonstrated that the ACTG2 variants carried by these individuals lead to defects in actin polymerization and cellular contractility (Lehtonen et al. 2012; Thorson et al. 2014; Halim et al. 2016). Two cases of apparent autosomal recessive (AR) ACTG2-associated visceral myopathy (ACTG2-VM) have also been reported—one involving a homozygous loss-of-function (LOF) variant and the other involving a homozygous missense variant (Monies et al. 2017; Matera et al. 2021).
Here we present the case of a young child with severe chronic intestinal pseudo-obstruction, constipation, and bilious emesis. Whole-genome sequencing revealed a 187-bp homozygous deletion within ACTG2 that is predicted to result in a complete loss of functional γ-actin protein. A family history negative for severe GI abnormalities suggests that ACTG2 may display allelic heterogeneity, with disease caused by either monoallelic missense variation or biallelic LOF or missense variation.
RESULTS
Clinical Presentation and Family History
The patient, a former full-term infant born in Mexico, was reportedly healthy at birth but began outpatient treatment for constipation at 3 mo of age and presented with worsening constipation, abdominal distension, and bilious emesis at 8 mo of age. During the next 5 mo, the patient was hospitalized multiple times in Mexico and underwent multiple operations, including an ileostomy and lysis of adhesions. His symptoms failed to improve, and he was brought to San Diego, California for further evaluation.
Upon admission to Rady Children's Hospital, the patient was 13 mo of age and weighed 6.8 kg (<1%ile, −3.6 SD, per Centers for Disease Control [CDC] U.S. growth charts). An abdominal radiograph revealed severely dilated bowel loops throughout the abdomen (Fig. 1A). Initial treatment included rehydration and nutritional optimization via nasogastric (NG) tube and peripherally inserted central catheter (PICC) line. He was later advanced to regular feeds with total parenteral nutrition (TPN) titrated to help the patient meet nutritional goals. Small bowel follow-through imaging showed normal transit time at 75 min, and retrograde contrast enema via rectum did not display any obstruction. Biopsies of the rectum and small intestine (ileostomy site) revealed abundant ganglion cells in both tissues, ruling out Hirschsprung disease. The muscular layers of the ileostomy site were unremarkable in thickness and organization (Fig. 1B).
Figure 1.
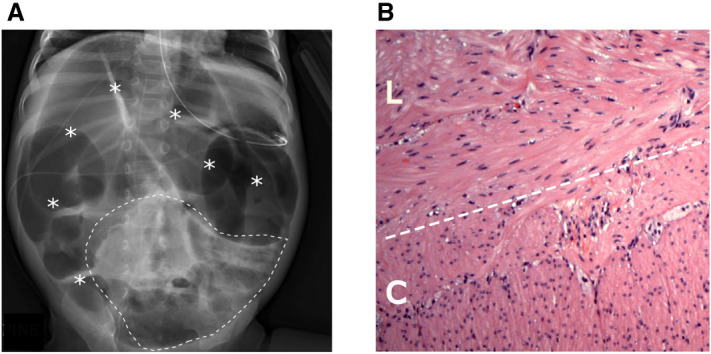
(A) Abdominal radiograph shows severe diffuse gaseous distension of bowel throughout the abdomen (asterisks). Scattered bubbly lucencies (within dashed line) may represent stool contents. Enteric tube and peripherally inserted central catheter (PICC) tip are also visible. (B) Hematoxylin and eosin (H&E) staining of the patient's small bowel (ileostomy) specimen reveals no abnormalities in smooth muscle band thickness or organization. (L) Longitudinal layer, (C) circular layer.
Based on these findings, the decision was made to proceed with ileostomy takedown. Postoperative recovery was complicated by worsening abdominal distension, fevers, and sepsis-like symptoms. A computed tomograph (CT) of the abdomen demonstrated dilated loops of bowel with a possible closed loop obstruction. The patient underwent an urgent exploratory laparotomy on postoperative day 7, during which viable bowel was visualized and adhesions were lysed.
Genomic Analyses
After 6 wk of hospitalization and in the absence of a clear diagnosis, the patient was nominated for clinical rapid whole-genome sequencing. A family history provided by the patient's mother at the time of genome sequencing revealed a history of recent intermittent constipation in a maternal half-sister (II-5) and fourth-degree consanguinity of the parents (Fig. 2A). Whole-genome sequencing of blood-derived DNA from the patient and his mother showed that the patient carried 56 regions of homozygosity (ROH) larger than 1 Mb, totaling 129.1 Mb (Supplemental Table 1). A sample from the father was not available. This total ROH is consistent with the distant parental consanguinity noted in the family history (coefficient of inbreeding F ≤ 1/32) (Fan et al. 2013).
Figure 2.
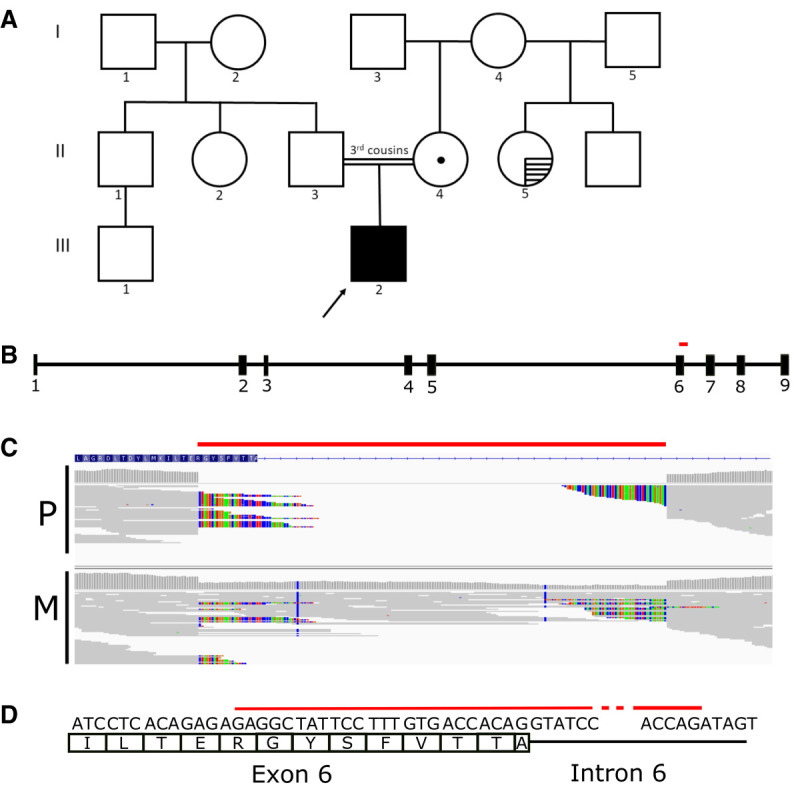
(A) Pedigree of the patient (III-2), indicated with the arrow. The dot (II-4) indicates heterozygous carrier status; horizontal hatching (II-5) indicates a recent history of intermittent constipation. (B) Gene schema of ACTG2 with deletion indicated (red bar). The deletion harbored by the patient corresponds to part of exon 6 of 9, and part of intron 6. (C) Representation of whole-genome sequencing reads overlapping the intragenic ACTG2 deletion (red bar) for the proband (P) and mother (M). Colored nucleotides indicate bases that are mismatched with respect to the reference genome, because of the deletion. (D) Schema of the deletion (red bar) at the single base level, with the 24 deleted base pairs of exon 6 shown.
Within a 4.8-Mb region of homozygosity on Chromosome 2, a homozygous 187-bp intragenic deletion within the ACTG2 gene was detected in the patient (Chr 2:74140750-74140936 × 0) (Tables 1 and 2). This variant was detected by one of the two copy-number variant (CNV) callers used in the genomic analysis pipeline: Manta but not CNVnator, consistent with Manta's higher sensitivity for CNVs 100 bp to 1 kb (Kosugi et al. 2019). It was not detected through DRAGEN-based single-nucleotide variant (SNV)/indel calling. The deletion spanned 24 bp of the 3′ end of exon 6 (out of 9) of ACTG2, and the following 163 bp of intron 6, denoted as c.589_613 + 163del188 (NM_001615.3) (Fig. 2B–D). The deletion was present in the heterozygous state in the mother and was presumed heterozygous in the father, who was not available for testing, given the homozygous status in proband and known parental consanguinity (Fig. 2C; Supplemental Table 1). Multiplex ligation-dependent probe amplification was used to orthogonally confirm these results in the patient's and mother's samples. This deletion is absent from population databases gnomAD SV and DGV. The deletion creates a frameshift ending in a premature stop codon at position 80, which is predicted to lead to nonsense-mediated decay of aberrant ACTG2 transcripts from both alleles in the patient.
Table 1.
Variant table
| Gene | Chromosomal variant coordinate (build 37.1) | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP ID | Genotype | ClinVar ID |
|---|---|---|---|---|---|---|---|---|
| ACTG2 | Chr 2:74140749-74140936 | c.589_613 + 163del188 | p.? | Deletion | Frameshift | n/a | Homozygous | 872896 |
Table 2.
Proband genome sequencing metrics
| Metric | Value |
|---|---|
| Read length | 100 |
| Total reads | 1,392,792,700 |
| Total mapped reads | 1,372,429,438 |
| Nucleotide variants identified | 4,913,732 |
| Variants with quality scores >40 | 94.36% |
| Coding nucleotide variants identified | 25,501 |
| Homozygous: heterozygous ratio of coding nucleotide variants | 0.62 |
| Transition: transition ratio of coding nucleotide variants | 2.86 |
| Mean coverage | 39.5% |
| Coverage at location of diagnostic variant | 36 |
In previous studies, intestinal biopsies from patients with AD ACTG2-VM have shown thinning of the longitudinal muscle layer and a disordered arrangement of smooth muscle fibers in the circular and longitudinal muscle bands of the muscularis propria (Tuzovic et al. 2015; Lu et al. 2016). However, hematoxylin and eosin (H&E) staining of this patient's ileostomy resection revealed neither longitudinal muscle thinning nor smooth muscle fiber disorganization (Fig. 1B).
Treatment Outcomes
Following molecular diagnosis, the patient was started on augmentin (10 mg/kg/dose BID) and erythromycin ethylsuccinate (EES) (3 mg/kg/dose four times daily [QID]), with EES replaced by cisapride (0.1mg/kg/dose QID) following insurance approval. Gut function returned postileostomy takedown and exploratory laparotomy. Per os (PO) feeds were trialed and up-titrated as tolerated, along with TPN. Eventually the patient progressed to PO formula (Pediasure), purees, and solids, and TPN was discontinued. Because of advancement of oral feeds and stable weight gain, the patient was discharged after a 56-d hospital stay at a discharge weight of 8.18 kg (<1%ile, −2.3 SD), per CDC U.S. growth charts.
He was readmitted 1.5 mo later with increased abdominal distension and nonbilious emesis despite regular stooling. Decompression was achieved with an NG tube and rectal tube along with Pedialax and fleet enemas. The GI motility specialist recommended increasing the cisapride dosage to 0.2 mg/kg/dose QID. Despite these interventions, the patient continued to have worsening abdominal distension and bilious emesis. The decision was made to proceed with an exploratory laparotomy, lysis of adhesions, redo end ileostomy with mucus fistula creation, and gastrostomy tube placement. The patient remained hospitalized for 6.5 mo, as a result of difficulty advancing enteral feeds to goal rate, cycling between vomiting and distension and increased ostomy output. He continues to be followed clinically by several specialties and is noted to experience moderate urinary retention not requiring catheterization.
DISCUSSION
In contrast to numerous reported cases of AD ACTG2-VM, we report here a case of severe visceral myopathy with apparent AR inheritance. Whereas nearly all reported patients with AD ACTG2-VM carry heterozygous missense variants in the ACTG2 gene, this patient carries a homozygous deletion predicted to result in complete loss of functional ACTG2 protein. Two other kindreds with suspected AR ACTG2-associated visceral myopathy have been reported to date, one carrying homozygous LOF variant p.(Arg168*) and the other carrying a homozygous missense variant p.(Arg336Gln). (Monies et al. 2017; Matera et al. 2021). In addition, there is one reported ACTG2-VM case with a deletion of exons 8–9 for which zygosity was not stated (Lee et al. 2019).
The clinical presentation of this patient is comparable to the severe end of the spectrum of AD ACTG2-VM, in which there is evidence of incomplete penetrance and variable intrafamilial expressivity, as well as some concordance between specific recurrent variants and particularly poor clinical outcomes (Lehtonen et al. 2012; Wangler et al. 2014; Klar et al. 2015; Matera et al. 2016; Milunsky et al. 2017; Assia Batzir et al. 2019). In AD ACTG2-VM, many patients are symptomatic at birth; 58% of patients undergo abdominal surgery within the first few weeks of life and 44% of patients have a poor outcome defined as reliance on TPN, visceral transplantation, or death in early childhood (Assia Batzir et al. 2019). In contrast, this patient was reportedly healthy until 3 mo of age but has since undergone numerous hospitalizations and several surgeries and relied on TPN sporadically. Ninety-three percent of patients with AD ACTG2-VM experience urine retention and megacystis requiring catheterization (Assia Batzir et al. 2019). This patient has a history of moderate urinary retention, which has not required catheterization. The family history for this patient was notable only for intermittent constipation in one maternal half-sibling who has not had ACTG2 genetic testing to our knowledge. The mother denies any personal history of GI issues including diarrhea, constipation, vomiting, bloating, or recurrent abdominal pain for herself and the patient's father.
In one reported case of AR ACTG2-VM, two affected siblings carrying a homozygous p.(Arg336Gln) variant presented with severe CIPO and megacystis, whereas heterozygote parents were unaffected (Matera et al. 2021). In another reported case of AR ACTG2-VM, the severely affected patient carrying a homozygous p.(Arg168*) variant presented with CIPO but no urinary symptoms (Monies et al. 2017). This patient had one presumed homozygote sibling who died of intestinal obstruction and perforation, one mildly symptomatic homozygote sibling, and several heterozygote family members reported chronic intermittent constipation (Monies et al. 2017). Taken together, these cases suggest that AR ACTG2-VM is associated with variable expression, and that heterozygote relatives may also exhibit visceral myopathy with variable expression and incomplete penetrance.
The allelic heterogeneity of ACTG2 resembles the pattern reported for ACTA1, which encodes skeletal muscle α-actin 1 and is associated with a spectrum of myopathies. AD ACTA1-related myopathies are overwhelmingly caused by missense variants, many of which have been shown to act in a dominant negative fashion to impair overall assembly, stability, or contraction of actin polymers, whereas AR ACTA1-related myopathies are caused by loss-of function variants (Feng and Marston 2009; Laing et al. 2009). Functional testing of several recurrent heterozygous missense ACTG2 variants has shown that they impair actin multimer assembly and contraction, supporting a dominant negative mechanism (Lehtonen et al. 2012; Thorson et al. 2014). For ACTG2-VM, the report of an AR case associated with homozygous missense variants, and the presence of symptomatic heterozygote carrier relatives in at least one AR case complicates this picture. Ascertainment of additional AR ACTG2-VM cases and functional testing of more pathogenic ACTG2 variants is required for a complete understanding of the spectrum of ACTG2-VM and its molecular pathogenesis.
The pathogenic deletion reported in this patient was discovered through whole-genome sequencing, which is unparalleled for scope of variant discovery (Belkadi et al. 2015). Exome sequencing can generate uneven coverage across and within exons, complicating CNV discovery, and low or absent intronic coverage may prevent detection of intronic breakpoints such as the 3′ breakpoint reported here. A 187-bp deletion is beyond the lower limit of detection by even the highest coverage single-nucleotide polymorphism (SNP) arrays. In this case, a rapid molecular diagnosis provided guidance for drug trialing, obviated the need for further diagnostic workup such as tissue biopsies or radiologic studies, and informed genetic counseling. Furthermore, this result supports the allelic heterogeneity of ACTG2-related VM, which may now merit attention as a potential diagnosis in cases in which parental consanguinity is reported.
METHODS
Consent and Enrollment
Parental consent was obtained for this study through Rady Children's Hospital, San Diego.
Genetic Sequencing and Analysis
Following DNA isolation from whole blood, sequencing libraries were generated using TruSeq kits (Illumina). Whole-genome sequencing was performed on NovaSeq 6000 instruments (Illumina), generating paired 101-nt reads with an average coverage of 39.5× for the proband and 52.9× for the mother. Alignment and SNV calling were performed using the DRAGEN hardware and software platform, yielding 4,913,732 calls for the proband and 5,041,148 for the mother.
CNV calling was performed with CNVnator and Manta (Abyzov et al. 2011; Chen et al. 2016). Variant call format (VCF) files incorporating SNV and CNV calls were annotated and analyzed using Fabric Enterprise version 6.5.0 (Fabric Genomics) according to standard guidelines (Coonrod et al. 2013; Richards et al. 2015).
Multiplex ligation-dependent probe amplification (MLPA) was performed on the patient and parental samples and three unrelated control samples, using two custom-designed probes falling within the deleted region. The MLPA products were separated using a 3500 Genetic Analyzer (ThermoFisher), and the results were analyzed using Coffalyser software (MRC-Holland). For each probe, patient to averaged control peak intensity ratios of 0.40–0.65 were considered indicative of a heterozygous deletion, and ratios of 0 were considered indicative of a homozygous deletion.
Histopathology
Full-thickness specimens obtained through small intestinal (ileostomy site) biopsies were formalin fixed, and H&E staining was performed.
ADDITIONAL INFORMATION
Data Deposition and Access
Neither sharing of sequencing data for this patient nor its upload to public repositories is possible, because of parental opt-out during consenting. The ClinVar ID for the ACTG2 deletion reported in this patient is VCV000872896.1.
Ethics Statement
Parental consent for duo whole-genome sequencing was obtained in the course of clinical care. Written consent for publication of clinical details was obtained via a Rady Children's Hospital San Diego (RCHSD) release form, reviewed by the RCHSD Privacy Board, in accordance with RCHSD rules for case reports.
Acknowledgments
The authors thank the family for participation in the study.
Author Contributions
K.N.J., M.L., and K.A.E. conceived the study design; K.S., J.L., R.M., R.I., J.H., L.C., and S.K. acquired and analyzed the clinical data; K.N.J. and M.L. drafted the manuscript; and K.N.J., M.L., R.I, S.K., and K.A.E. revised the manuscript for intellectual content.
Funding
Funding for this study was provided by Rady Children's Institute for Genomic Medicine.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Caleb Bupp
M.M. Alves
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Abyzov A, Urban AE, Snyder M, Gerstein M. 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21: 974–984. 10.1101/gr.114876.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambartsumyan L. 1993. Megacystis-microcolon-intestinal hypoperistalsis syndrome overview. In Gene Reviews® (ed. Adam MP, Ardinger HH, Pagon RA, et al. ). University of Washington, Seattle. [Google Scholar]
- Assia Batzir N, Kishor Bhagwat P, Larson A, Coban Akdemir Z, Bagłaj M, Bofferding L, Bosanko KB, Bouassida S, Callewaert B, Cannon A, et al. 2019. Recurrent arginine substitutions in the ACTG2 gene are the primary driver of disease burden and severity in visceral myopathy. Hum Mutat 41: 641–654. 10.1002/humu.23960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, Shang L, Boisson B, Casanova J-L, Abel L. 2015. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci 112: 5473–5478. 10.1073/pnas.1418631112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, Saunders CT. 2016. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32: 1220–1222. 10.1093/bioinformatics/btv710 [DOI] [PubMed] [Google Scholar]
- Coonrod EM, Margraf RL, Russell A, Voelkerding KV, Reese MG. 2013. Clinical analysis of genome next-generation sequencing data using the Omicia platform. Expert Rev Mol Diagn 13: 529–540. 10.1586/14737159.2013.811907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Giorgio R, Cogliandro RF, Barbara G, Corinaldesi R, Stanghellini V. 2011. Chronic intestinal pseudo-obstruction: clinical features, diagnosis, and therapy. Gastroenterol Clin North Am 40: 787–807. 10.1016/j.gtc.2011.09.005 [DOI] [PubMed] [Google Scholar]
- Fan Y-S, Ouyang X, Peng J, Sacharow S, Tekin M, Barbouth D, Bodamer O, Yusupov R, Navarrete C, Heller AH, et al. 2013. Frequent detection of parental consanguinity in children with developmental disorders by a combined CGH and SNP microarray. Mol Cytogenet 6: 38. 10.1186/1755-8166-6-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J-J, Marston S. 2009. Genotype–phenotype correlations in ACTA1 mutations that cause congenital myopathies. Neuromuscul Disord 19: 6–16. 10.1016/j.nmd.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Gosemann J-H, Puri P. 2011. Megacystis microcolon intestinal hypoperistalsis syndrome: systematic review of outcome. Pediatr Surg Int 27: 1041–1046. 10.1007/s00383-011-2954-9 [DOI] [PubMed] [Google Scholar]
- Halim D, Hofstra RMW, Signorile L, Verdijk RM, van der Werf CS, Sribudiani Y, Brouwer RWW, van IJcken WFJ, Dahl N, Verheij JBGM, et al. 2016. ACTG2 variants impair actin polymerization in sporadic megacystis microcolon intestinal hypoperistalsis syndrome. Hum Mol Genet 25: 571–583. 10.1093/hmg/ddv497 [DOI] [PubMed] [Google Scholar]
- Holla OL, Bock G, Busk OL, Isfoss BL. 2014. Familial visceral myopathy diagnosed by exome sequencing of a patient with chronic intestinal pseudo-obstruction. Endoscopy 46: 533–537. 10.1055/s-0034-1365142 [DOI] [PubMed] [Google Scholar]
- Kandler JL, Sklirou E, Woerner A, Walsh L, Cox E, Xue Y. 2020. Compound heterozygous loss of function variants in MYL9 in a child with megacystis–microcolon–intestinal hypoperistalsis syndrome. Mol Genet Genomic Med 8: e1516. 10.1002/mgg3.1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar J, Raykova D, Gustafson E, Tóthová I, Ameur A, Wanders A, Dahl N. 2015. Phenotypic expansion of visceral myopathy associated with ACTG2 tandem base substitution. Eur J Hum Genet 23: 1679–1683. 10.1038/ejhg.2015.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, Kamatani Y. 2019. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol 20: 117. 10.1186/s13059-019-1720-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing NG, Dye DE, Wallgren-Pettersson C, Richard G, Monnier N, Lillis S, Winder TL, Lochmüller H, Graziano C, Mitrani-Rosenbaum S, et al. 2009. Mutations and polymorphisms of the skeletal muscle α-actin gene (ACTA1). Hum Mutat 30: 1267–1277. 10.1002/humu.21059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Park S, Oh J-T, Kim HM, Kim S, Lee J-S. 2019. Oral pyridostigmine-responsive visceral myopathy with ACTG2 mutations: a case series. J Pediatr Gastroenterol Nutr 68: e16. 10.1097/MPG.0000000000002183 [DOI] [PubMed] [Google Scholar]
- Lehtonen HJ, Sipponen T, Tojkander S, Karikoski R, Järvinen H, Laing NG, Lappalainen P, Aaltonen LA, Tuupanen S. 2012. Segregation of a missense variant in enteric smooth muscle actin γ-2 with autosomal dominant familial visceral myopathy. Gastroenterology 143: 1482–1491.e3. 10.1053/j.gastro.2012.08.045 [DOI] [PubMed] [Google Scholar]
- Lu W, Xiao Y, Huang J, Tao Y, Yan W, Lu L, Cao Y, Cai W. 2016. Mutation in actin γ-2 responsible for megacystis microcolon intestinal hypoperistalsis syndrome in 4 Chinese patients. J Pediatr Gastroenterol Nutr 63: 624–626. 10.1097/MPG.0000000000001204 [DOI] [PubMed] [Google Scholar]
- Matera I, Rusmini M, Guo Y, Lerone M, Li J, Zhang J, Di Duca M, Nozza P, Mosconi M, Pini Prato A, et al. 2016. Variants of the ACTG2 gene correlate with degree of severity and presence of megacystis in chronic intestinal pseudo-obstruction. Eur J Hum Genet 24: 1211–1215. 10.1038/ejhg.2015.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera I, Bordo D, Di Duca M, Lerone M, Santamaria G, Pongiglione M, Lezo A, Diamanti A, Spagnuolo MI, Pini Prato A, et al. 2021. Novel ACTG2 variants disclose allelic heterogeneity and bi-allelic inheritance in pediatric chronic intestinal pseudo-obstruction. Clin Genet 99: 430–436. 10.1111/cge.13895 [DOI] [PubMed] [Google Scholar]
- Milunsky A, Baldwin C, Zhang X, Primack D, Curnow A, Milunsky J. 2017. Diagnosis of chronic intestinal pseudo-obstruction and megacystis by sequencing the ACTG2 gene. J Pediatr Gastroenterol Nutr 65: 384–387. 10.1097/MPG.0000000000001608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa T, Manabe Y, Kurokawa K, Kamada S, Kanda N, Bruns G, Ueyama H, Kakunaga T. 1991. Structure, chromosome location, and expression of the human smooth muscle (enteric type) γ-actin gene: evolution of six human actin genes. Mol Cell Biol 11: 3296–3306. 10.1128/mcb.11.6.3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monies D, Maddirevula S, Kurdi W, Alanazy MH, Alkhalidi H, Al-Owain M, Sulaiman RA, Faqeih E, Goljan E, Ibrahim N, et al. 2017. Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation. Genet Med 19: 1144–1150. 10.1038/gim.2017.22 [DOI] [PubMed] [Google Scholar]
- Ravenscroft G, Pannell S, O'Grady G, Ong R, Ee HC, Faiz F, Marns L, Goel H, Kumarasinghe P, Sollis E, et al. 2018. Variants in ACTG2 underlie a substantial number of Australasian patients with primary chronic intestinal pseudo-obstruction. Neurogastroenterol Motil 30: e13371. 10.1111/nmo.13371 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorson W, Diaz-Horta O, Foster J, Spiliopoulos M, Quintero R, Farooq A, Blanton S, Tekin M. 2014. De novo ACTG2 mutations cause congenital distended bladder, microcolon, and intestinal hypoperistalsis. Hum Genet 133: 737–742. 10.1007/s00439-013-1406-0 [DOI] [PubMed] [Google Scholar]
- Tuzovic L, Tang S, Miller RS, Rohena L, Shahmirzadi L, Gonzalez K, Li X, LeDuc CA, Guo J, Wilson A, et al. 2015. New insights into the genetics of fetal megacystis: ACTG2 mutations, encoding γ-2 smooth muscle actin in megacystis microcolon intestinal hypoperistalsis syndrome (Berdon syndrome). Fetal Diagn Ther 38: 296–306. 10.1159/000381638 [DOI] [PubMed] [Google Scholar]
- Wangler MF, Gonzaga-Jauregui C, Gambin T, Penney S, Moss T, Chopra A, Probst FJ, Xia F, Yang Y, Werlin S, et al. 2014. Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis–microcolon–intestinal hypoperistalsis syndrome. PLoS Genet 10: e1004258. 10.1371/journal.pgen.1004258 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Neither sharing of sequencing data for this patient nor its upload to public repositories is possible, because of parental opt-out during consenting. The ClinVar ID for the ACTG2 deletion reported in this patient is VCV000872896.1.