

Abstract
The ETS2 repressor factor (ERF) is a transcription factor in the RAS-MEK-ERK signal transduction cascade that regulates cell proliferation and differentiation, and pathogenic sequence variants in the ERF gene cause variable craniosynostosis inherited in an autosomal dominant pattern. The reported ERF variants are largely loss-of-function, implying haploinsufficiency as a primary disease mechanism; however, ERF gene deletions have not been reported previously. Here we describe three probands with macrocephaly, craniofacial dysmorphology, and global developmental delay. Clinical genetic testing for fragile X and other relevant sequencing panels were negative; however, chromosomal microarray identified heterozygous deletions (63.7–583.2 kb) on Chromosome 19q13.2 in each proband that together included five genes associated with Mendelian diseases (ATP1A3, ERF, CIC, MEGF8, and LIPE). Parental testing indicated that the aberrations were apparently de novo in two of the probands and were inherited in the one proband with the smallest deletion. Deletion of ERF is consistent with the reported loss-of-function ERF variants, prompting clinical copy-number-variant classifications of likely pathogenic. Moreover, the recent characterization of heterozygous loss-of-function CIC sequence variants as a cause of intellectual disability and neurodevelopmental disorders inherited in an autosomal dominant pattern is also consistent with the developmental delays and intellectual disabilities identified among the two probands with CIC deletions. Taken together, this case series adds to the previously reported patients with ERF and/or CIC sequence variants and supports haploinsufficiency of both genes as a mechanism for a variable syndromic cranial phenotype with developmental delays and intellectual disability inherited in an autosomal dominant pattern.
Keywords: congenital strabismus, craniosynostosis, intellectual disability, macrocephaly at birth, global developmental delay, widely patent coronal suture
INTRODUCTION
Craniosynostosis is the premature fusion of one or more cranial sutures that typically remain open to allow for postnatal brain growth, which can result in subsequent skull deformation and intracranial pressure. Craniosynostosis affects ∼1 in 2000 children (Heuze et al. 2014; Wilkie et al. 2017) and can present with other features in a syndromic form; however, the majority (∼85%) are nonsyndromic (Heuze et al. 2014). There is significant clinical variation in the number and type of sutures involved, which is influenced in part by both genetic and environmental factors (Heuze et al. 2014; Wilkie et al. 2017). Although more than 90 genes with pathogenic variants have been identified to be associated with craniosynostosis, the majority of affected individuals (70%–75%) remain without a genetic diagnosis, which is critical to guide clinical management and inform on prognosis and reproductive risk (Heuze et al. 2014; Timberlake et al. 2017; Wilkie et al. 2017; Lee et al. 2018). Several cellular pathways have emerged in the pathogenesis of craniosynostosis, including the RAS-MEK-ERK signaling pathway, which involves the FGFR1, FGFR2, FGFR3, TWIST1, and EFNB1 genes (Kim et al. 2003; Heuze et al. 2014; Timberlake et al. 2017; Wilkie et al. 2017). Additional genes that have more recently been implicated in craniosynostosis include TCF12 and ERF (Sharma et al. 2013; Twigg et al. 2013; Wilkie et al. 2017; Glass et al. 2019).
The ERF (ETS2 repressor factor) gene is located on Chromosome 19q13.2 and encodes a transcription factor that is bound directly by ERK1/2 to regulate the RAS-MEK-ERK signal transduction cascade (le Gallic et al. 1999; Twigg et al. 2013). Heterozygous loss-of-function variants in ERF (start-loss, missense, nonsense, frameshift, and splice-site) have been reported among individuals with craniosynostosis, which ranged from mild nonsyndromic craniosynostosis to a syndromic form with multisuture synostosis, craniofacial dysmorphism, central nervous system abnormalities, and speech delay (Twigg et al. 2013; Chaudhry et al. 2015; Timberlake et al. 2017; Clarke et al. 2018; Lee et al. 2018; Glass et al. 2019; Korberg et al. 2020). Importantly, although the mutational spectrum of reported sequence variants implies haploinsufficiency as the disease mechanism (Glass et al. 2019), ERF gene deletions have not been previously reported.
Herein, we report a series of three unrelated probands with heterozygous deletions of Chromosome 19q13.2 that include ERF. Proband 1 was initially identified and two additional individuals with overlapping deletions were subsequently incorporated into the series through DECIPHER (https://www.deciphergenomics.org/) (Firth et al. 2009). Moreover, heterozygous sequence variants in the neighboring CIC (capicua) gene have recently been implicated as a cause of intellectual disability and neurodevelopmental disorders inherited in an autosomal dominant pattern (Lu et al. 2017; Kim et al. 2019), which is also consistent with the identified deletions and proband phenotypes in this series.
RESULTS
Clinical Presentation and Family History
Proband 1 (Icahn School of Medicine at Mount Sinai, ISMMS)
The proband was a 3.4-yr-old male, who presented for evaluation of global developmental delay, macrocephaly, and dysmorphic features (Table 1; Fig. 1). He was the second child of nonconsanguineous parents. His older brother was 10 yr old and had speech delay during preschool that subsequently resolved. His brother's height was in the 75th centile. The mother's family originated from the Philippines, and she was healthy, with no relevant family history. Her height was 160 cm (31st centile) and head circumference was 54.8 cm (74th centile). The father was of European descent and healthy. His height was 188 cm (95th centile), head circumference was 59 cm (>99th centile), and he had big thumbs and toes. Among his eight siblings, some were also tall and/or had big thumbs or toes. The paternal grandfather and his five siblings died with colon cancer. There was no history of other birth defects, developmental delay, psychiatric disease, or hydrocephalus in the family.
Table 1.
Clinical and molecular features of the probands with heterozygous deletions of ERF and CIC
| Proband 1 (ISMMS) | Proband 2 (KEMH) | Proband 3 (Rigshospitalet) | Decipher—249925a | Decipher—381692a | |
|---|---|---|---|---|---|
| Sex | Male | Female | Male | Female | Male |
| DECIPHER ID | N/A | 421530 | 251589 | 249925 | 381692 |
| MOLECULAR DIAGNOSIS | |||||
| Detection | CMA | CMA | CMA | CMA | Unknown |
| Deletion coordinates (hg19)—minimum | Chr 19: 42632535_42947378 | Chr 19: 42433934_43017156 | Chr 19: 42696659_42760365 | Chr 19: 42462460_42894328 | Chr 19: 42702762_42754032 |
| Deletion size (bp)— minimum | 314,844 | 583,223 | 63,707 | 431,869 | 51,271 |
| Deletion size (bp)— maximum | 420,515 | 592,717 | 75,512 | Unknown | Unknown |
| Inheritance | Apparently de novo | Apparently de novo | Inherited (from a parent with learning disabilities) | De novo | Unknown |
| Deleted OMIM genes | ERF, CIC, MEGF8, LIPE | ATP1A3, ERF, CIC, MEGF8, LIPE | ERF | ATP1A3, ERF, CIC, MEGF8 | ERF |
| Classification | Likely pathogenic | Likely pathogenic | VOUS | Unknown | Unknown |
| CLINICAL ASSESSMENT | |||||
| Age at last assessment | 3 yr and 5 mo | 17 yr | 15 yr | 8 yr | Unknown |
| Maternal and paternal ages at birth (years) | 35, 43 | 38, 36 | 21, 23 | Unknown | Unknown |
| Craniofacial features | |||||
| Macrocephaly | + (prominent coronal suture) | + | + | Unknown | Unknown |
| Craniofacial dysmorphology | + (low-set ears, asymmetry) | + (long face, everted lower lip, narrow jaw) | + (square face, tall forehead, full lower lip, absent Cupid´s bow) | Unknown | Unknown |
| Prominent forehead | + | + | - | Unknown | Unknown |
| Prominent orbits | - | - | - | Unknown | Unknown |
| Widely spaced eyes | - | + | - | Unknown | Unknown |
| Strabismus (esotropia) | + | + | - | Unknown | Unknown |
| Midface retrusion | - | - | - | Unknown | Unknown |
| Limb features | |||||
| Femoral torsion | - | + | - | Unknown | Unknown |
| Tibial torsion and pronation | + | - | - | Unknown | Unknown |
| Digit abnormalities | + (broad thumbs and toes) | - | - | Unknown | Unknown |
| Neurological Features | |||||
| global developmental delay | + | + | + | Unknown | Unknown |
| Behavioral problems | - | - | + | Unknown | Unknown |
| Seizure | + (febrile) | + (tonic clonic, absence) | - | Unknown | Unknown |
| Chiari type 1 malformation | - | - | - | Unknown | Unknown |
| Increased intracranial pressure | - | - | - | Unknown | Unknown |
| Hypotonia | + | - | - | Unknown | Unknown |
| Other clinical features (previously noted in individuals with variants—epicanthus, depressed nasal bridge, long philtrum, micrognathia, high arched palate, dental malocclusion, dysplastic ears, clinodactyly, radio-ulnar synostosis, scoliosis, inverted nipples, inguinal hernia, sacral dimples, ectopic posterior pituitary, speech delay, atrial septal defect, ventricular septal defect, platelet disorder, adenoidectomy) | Overweight, speech delay | Hypermobile joints | Overweight, short stature, short neck, incontinence, grommets, adenoidectomy, speech delay | Unknown | Unknown |
| Imaging studies | |||||
| CT scan | + (poor quality) | - | - | Unknown | Unknown |
| MRI scan | - | + (normal) | - | Unknown | Unknown |
| Surgery (type of surgery, age of surgery, number of surgeries) | - | - | - | Unknown | Unknown |
| DECIPHER phenotypes | N/A | Ataxia, esotropia, intellectual disability, seizure | None listed | None listed | Language impairment, psychomotor retardation |
(N/A) Not available, (CMA) chromosomal microarray, (OMIM) Online Mendelian Inheritance in Man, (VOUS) variant of uncertain significance, (+) present, (-) absent, (CT) computed tomography, (MRI) magnetic resonance imaging.
aTwo reports were deposited in DECIPHER, but no additional information was available for this report.
Figure 1.
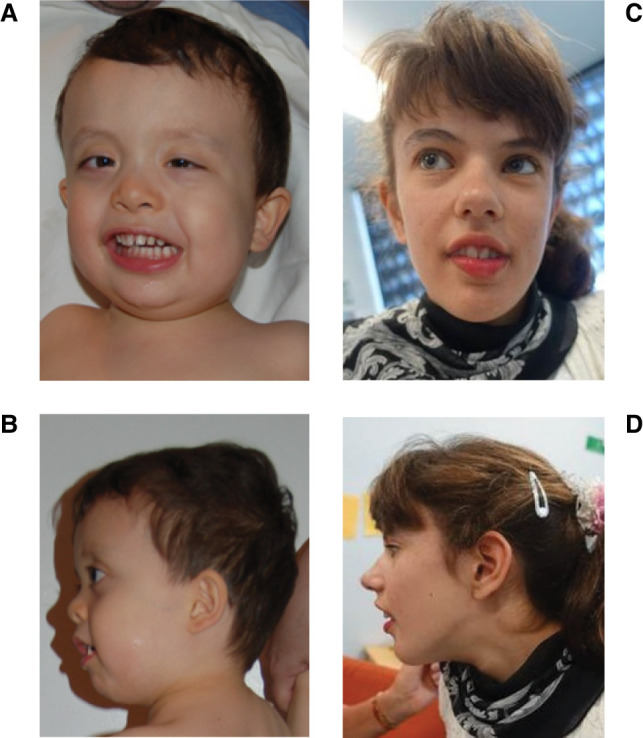
Clinical features of proband 1 at age 3.4 yr (A,B) and proband 2 at 14 yr (C,D). Notable features include macrocephaly, brachycephaly, prominent forehead, facial asymmetry, and mild exophthalmos with strabismus. Consent for publication of photos for proband 3 was not granted.
The proband was born via repeat Cesarean delivery at 38 wk gestation. A fetal ultrasound at ∼6 mo gestation indicated macrocephaly; prenatal history was otherwise unremarkable with no maternal exposures, and a normal 46, XY G-banded karyotype was reported by amniocentesis, which was carried out for advanced maternal age. Apgar scores were 9 and 9. He appeared well and had normal newborn screening, including hearing. He was discharged without complications. His birth length was 49.5 cm (50–75th centile), weight was 4.035 kg (>90th centile), and head circumference was 36 cm (>90th centile). His head circumference remained below +2 standard deviations until 5 mo, and then increased above the 97th centile. Head ultrasound at 7 mo of age showed borderline enlarged ventricles, but no evidence for external hydrocephalus. At 1 yr, 10 mo, he developed a febrile seizure due to sinusitis; cranial computed tomography (CT) was performed that was reported normal, and no electroencephalogram (EEG) was done. Global developmental delay was evident: He rolled over at 10 mo, sat without support at 12 mo, stood up at 15 mo, and talked at 1 yr, 6 mo after initiating speech therapy. A formal developmental evaluation at 2 yr, 6 mo placed his skills at the age of a 12- to 13-mo-old. He began physical therapy at 2 yr and occupational therapy at 3 yr, both three times a week, and started special education at 3 yr with speech therapy four times a week. He had no reportable behavioral issues that warranted formal assessment. Endocrinology evaluation at 3 yr and 3 mo noted steady growth with height consistently between 50th and 75th centile and body mass index (BMI) of 18.07 kg/m2 (height 0.97 m, weight 17 kg; BMI 95th centile, Z = 1.62) at the border of the overweight/obese range. His thyroid stimulating hormone (2.67 mU/L) and free thyroxine (T4) (1.19 ng/dL) levels were normal (0.4–4.0 mU/L and 0.65–2.3 ng/dL, respectively). An underlying endocrinopathy was deemed unlikely, and the proband was referred to Medical Genetics.
At 3 yr and 5 mo, his height was 105 cm (90–95th centile), weight was 17.24 kg (75–90th centile), and head circumference was 55.7 cm (>97th centile). His facies were dysmorphic (Fig. 1). He was alert and responsive, with appropriate social interaction but became easily frustrated when trying to communicate with people, primarily using sign language. Hypotonia was evident. He wore ankle braces for tibial torsion and pronation and could walk on his own, but with an unsteady gait. His physical exam showed that in addition to macrocephaly he had a prominent forehead greater above the right than the left eye and a palpable coronal suture with the height of his skull higher anterior to the coronal suture. He also had very mild exophthalmos with esotropia, low-set ears of normal size, relatively large thumbs, and big toes. Rereview of a previous CT scan showed that imaging was not adequate to review the sutures, and review of systems was otherwise unremarkable. Altogether, his cranial shape and dysmorphic features were suggestive of craniosynostosis.
Proband 2 (King Edward Memorial Hospital; KEMH)
The proband was referred to the transitional Undiagnosed Disease Program (tUDP) of Western Australia for further investigation of apparent intellectual disability, macrocephaly, and dysmorphic features (Table 1; Fig. 1). She was the first child of nonconsanguineous European parents. Both parents were in good health, and there was no family history of development delay, psychiatric disorders, or birth defects. During the pregnancy, concerns were raised about lack of fetal movements; however, all scans were unremarkable. She was born full term at 41 wk with no concerns after delivery. Neonatal examination was normal. Birth parameters were length 51 cm (50–75th centile), weight 3.66 kg (50th centile), and head circumference 36 cm (85th centile). Initial development was on track: she smiled at 6 wk, sat at 9 mo, crawled at 9 mo, and walked independently at 16 mo. At 18 mo, she developed seizures, which included generalized tonic–clonic and absence seizures that were resistant to treatment. By 20 mo, her development had regressed significantly, and she was diagnosed with global developmental delay. She was diagnosed with alternating esotropia at 3 mo of age, patching was attempted, and at age 2 yr surgical correction of the right eye was performed. At 14 yr, she was diagnosed with significant intellectual disability. She also was noted to have an abnormal gait with femoral torsion requiring an osteotomy at age 13 yr. At 17 yr, she was noted to be of tall stature (75th centile), have hypermobile joints and dysmorphic features including relatively long face, everted lower lip, and a narrow jaw; there was no obvious asymmetry. Her head circumference was 57 cm (>97th centile) at age 17 yr (Fig. 1), and magnetic resonance imaging (MRI) showed a structurally normal brain.
Proband 3 (Rigshospitalet)
The proband was a 15-yr-old boy, who was followed for developmental delays, behavioral problems, and dysmorphic features. He was the only common child of a nonconsanguineous couple (Table 1). Both parents had mild learning difficulties and they managed to live independently, each with a new partner. The father had two healthy children with another partner, whereas the mother had a healthy daughter. The mother had three brothers and one sister who all attended special schools. The proband was born after an uncomplicated pregnancy at 39 gestational weeks by Cesarean delivery because of prolonged labor, and was admitted to the neonatal unit for respiratory support the first 2 d of life. According to the parents, it was because of a unilateral pneumothorax. Subsequently, the neonatal period was uneventful. Birth length was 52 cm (75–90th centile) and weight was 3642 g (50–75th centile).
From early on, there was a suspicion of delayed development. He walked independently after 18 mo of age and said his first word at 24 mo of age. His language development was markedly delayed. At the age of 3 yr, the proband was referred to a pediatric department because of global developmental delay. At the age of 5 yr, he could only say one-syllable words and sentences with two to three words. At his latest examination at 15 yr old, he had near-normal speech. He was seen by an otolaryngologist several times to evaluate his hearing, which was found to be normal. He had secretory otitis and had grommets placed in both ears twice but without any effect on language development. He had an adenoidectomy because of recurrent upper airway infections associated with sleep disturbances.
Psychiatric evaluation was carried out because of behavioral anomalies. He was evaluated for attention deficit disorder but did not fulfill the diagnostic criteria. He was diagnosed with mild intellectual disability and behavioral problems. He was described as a sweet and sociable boy, but with mood changes and sudden outbursts, eventually leading to episodes of aggressive and violent behavior. As he grew older the behavioral problems worsened. He also had mild asthma. He was seen by an ophthalmologist a few times because it was suspected he had amblyopia, but vision was found to be normal. In childhood, he had obstipation/retention and incontinence problems both for urine and stools that lasted for many years. At 15 yr, he still had urine incontinence at night but rarely during daytime. No sleep problems were noted.
On physical examination, he had short stature, relative weight gain, and relative macrocephaly. Measurements were taken at the ages of 3 yr, 10 mo (length: 95 cm, 1st centile; weight: 18.3 kg, 75–90th centile; head circumference: 54 cm, >95th centile), 8 yr, 9 mo (length: 125 cm, <5th centile; weight: 32 kg, 50–75th centile), and 13 yr, 7 mo (length: 143 cm, <1st centile; weight: 46 kg, 25th centile). He presented with a large brachycephalic head, tall forehead, and slightly dysmorphic features such as full lower lip and absent Cupid's bow. The neck seemed short. He had no facial asymmetry or other signs of craniosynostosis. No brain CT/MRI scans had been performed.
Genomic Analyses
Proband 1 (ISMMS)
Clinical FMR1 trinucleotide repeat expansion testing for fragile X was normal (approximate 30 repeats), and a clinical craniosynostosis Sanger sequencing panel that included FGFR1, FGFR2, FGFR3, RAB23, EFNB1, MSX2, TWIST1, and POR did not detect any pathogenic variants. In addition, peripheral blood high-resolution chromosome analysis showed a normal 46, XY karyotype; however, chromosomal microarray (CMA) testing using the ISCA CGH + SNP 4 × 180K array (Agilent Technologies) identified a heterozygous 314.8 kb deletion on Chromosome 19q13.2 (Chr 19:42632535_42947378 [hg19]) (maximum size: 420.5 kb, Chr 19:42580845_43001360) (Table 1; Fig. 2). The identified deletion included 17 genes and transcripts (exon 1 of POU2F2, LOC100505622, MIR4323, DEDD2, ZNF526, GSK3A, ERF, CIC, PAFAH1B3, PRR19, TMEM145, MEGF8, CNFN, LOC101930071, LIPE, CXCL17, and exon 1 of LIPE-AS1), four of which are currently known Mendelian disease genes (ERF, CIC, MEGF8, and LIPE). Importantly, CMA testing was performed in 2015, but the CIC gene was not associated with dominantly inherited Mendelian disease by OMIM until 2017 (OMIM# 617600) (Lu et al. 2017). The heterozygous Chromosome 19q13.2 deletion was confirmed by fluorescence in situ hybridization (FISH), and familial CMA and FISH testing determined that neither parent was a carrier of the aberration, indicating that the deletion was apparently de novo in the proband (Fig. 2).
Figure 2.

(A) Illustration of the local genomic architecture of the Chromosome 19q13.2 region. The illustration is derived from the UCSC Genome Browser (https://genome.ucsc.edu/), which details location of known human genes from OMIM (dark green) and NCBI RefSeq (blue); identified deletion intervals for the three probands (red box plot, maximum size indicated by thin line); Proband 1 CMA probes (Agilent), Affymetrix CMA results (black: two copies; red: one copy), and fluorescence in situ hybridization (FISH) probe location (light green); Clinical Genome Reference (ClinGen) haploinsufficiency (HI) and triplosensitivity tracks (red for haploinsufficiency score 3, blue for triplosensitivity score 3, and gray for other evidence scores or not yet evaluated); ClinGen Pathogenic and Benign variants (red: deletion; blue: duplication); Database of Genomic Variants (DGV) Gold Standard variant and unmerged DGV structural variant tracks (red: deletion; blue: duplication); and copy-number variants (CNVs) cataloged in the DECIPHER database (red: deletion; blue: duplication). Note the overlap of deletions reported in the DGV and the identified proband deletions, suggesting incomplete penetrance and/or variable expressivity at this locus. (B) Confirmatory interphase FISH results of the ISMMS proband (Proband 1) and parental samples.
Proband 2 (KEMH)
Genetic testing prior to enrollment in the tUDP included fragile X testing, CMA using an Illumina HumanCytoSNP-12 BeadChip, and gene panel testing for disorders that overlap with Angelman syndrome and conditions associated with absent speech (gene list available on request) using massively parallel sequencing (MPS) and a primary Illumina TruSight One Expanded Sequencing Panel, which did not identify any variants of clinical significance.
After enrollment in the tUDP, we organized for multiple additional gene panels (genes associated with epilepsy, X-linked intellectual disability, and cohesinopathies; lists available on request) using reanalyses of the MPS data and a higher-resolution CMA (Illumina CytoSNP-850K BeadChip v1.2), which both showed a likely pathogenic heterozygous 583.2-kb deletion of Chromosome 19q13.2 (Chr 19:42433934_43017156; [hg19]) (maximum size: 592.7 kb, Chr 19:42430503_43023219) (Table 1; Fig. 2). The deletion contained five OMIM disease–associated genes including ATP1A3, ERF, CIC, MEGF8, and LIPE and at least 19 additional RefSeq genes (LOC100505585, RABAC1, GRIK5, ZNF574, POU2F2, LOC100505622, MIR4323, DEDD2, ZNF526, GSK3A, PAFAH1B3, PRR19, TMEM145, MIR8077, CNFN, LOC101930071, CXCL17, exon 1 of LIPE-AS1, and exons 6–9 of CEACAM1). The identified deletion was not detected by the previous standard-resolution CMA because of a limited number of markers in the deleted region, resulting in reduced sensitivity. Parental CMA testing using the higher-resolution microarray did not show the 19q13.2 deletion in either parent, and it was concluded to be apparently de novo.
Proband 3 (Rigshospitalet)
The proband was tested for fragile X syndrome, which identified a normal repeat length for FMR1 trinucleotide repeat expansion testing. However, CMA testing identified a heterozygous 63.7 kb deletion on Chromosome 19q13.2 (Chr 19:42696659_42760365 [hg19], maximum size: 75.5 kb, Chr 19: 42690571_42766082) (Table 1; Fig. 2). The identified deletion included four genes (DEDD2, ZNF526, GSK3A, and ERF), and of these, ERF is currently the only known gene associated with Mendelian disease. The deletion was confirmed by quantitative polymerase chain reaction (qPCR), and testing of the parents showed that the deletion was maternally inherited. Although the proband's mother was not available for a complete medical history and clinical evaluation, she did report being otherwise healthy but with a history of learning disabilities that required special education. The proband's maternal half-sister did not have the deletion, and no other information regarding further genetic testing in the mother's family was available.
DISCUSSION
We report three unrelated probands with macrocephaly, craniofacial dysmorphology, dysmorphic facies, and global developmental delays, with apparently de novo deletions of Chromosome 19q13.2 that include ERF and CIC in two of the probands and an inherited deletion of Chromosome 19q13.2 that includes only ERF in one proband (Table 1). This case series adds to the previously reported patients with heterozygous ERF and/or CIC sequence variants (Supplemental Material) and directly supports haploinsufficiency of these genes as a cause of a syndromic cranial phenotype with autosomal dominant inheritance, variable expressivity, and incomplete penetrance.
A direct role for ERF in craniosynostosis has previously been established as mice with reduced functional ERF exhibited multiple-suture synostosis, and fibroblasts or lymphoblastoid cells derived from affected individuals with loss-of-function ERF variants showed lower expression of full-length ERF (Twigg et al. 2013). Of note, no structural variants were identified among more than 270 variant-negative individuals in this study. Moreover, variable expressivity and incomplete penetrance have also been reported among families with ERF-related craniosynostosis syndrome (Glass et al. 2019). Other variable features associated with ERF include widely spaced eyes, shortening and/or vertical displacement of the nose, prominent orbits and forehead, and behavioral and learning problems (e.g., concentration and language acquisition), which is consistent with the speech delays observed in our probands.
Two of the identified deletions also included the MEGF8 and LIPE genes, which have been associated with Mendelian disease. However, the phenotypic presentations of the probands were not consistent with Carpenter syndrome (e.g., acrocephalopolysyndactyly) or familial partial lipodystrophy, which are caused by biallelic pathogenic variants in MEGF8 and LIPE, respectively (Twigg et al. 2013; Farhan et al. 2014). Furthermore, the two probands did not present with any characteristic features of Chitayat syndrome (e.g., hyperphalangism, respiratory involvement), which is associated with a specific heterozygous missense variant in ERF c.266A > G (p.Tyr89Cys) (Balasubramanian et al. 2017). In addition, the heterozygous deletion identified in Proband 2 also included ATP1A3, which has been associated with Mendelian disease. Although heterozygous missense and small in-frame deletion/insertion ATP1A3 variants have been associated with various ATP1A3-related neurologic disorders inherited in an autosomal dominant pattern, including dystonia and CAPOS syndrome (cerebellar ataxia, areflexia, pes cavus, optic atrophy, sensorineural hearing loss), haploinsufficiency has not been proposed as a pathogenic mechanism of disease (Brashear et al. 1993; Miyatake et al. 2021). However, a contiguous gene deletion effect of the heterozygous 583.2-kb deletion in Proband 2 cannot be ruled out.
Importantly, the CIC gene has recently been classified by OMIM as associated with Mendelian disease inherited in an autosomal dominant pattern (OMIM# 617600). This was driven by the 2017 exome sequencing discovery of pathogenic CIC variants in patients with neurobehavioral phenotypes, including intellectual disability, developmental delay, seizures, attention deficit/hyperactivity disorder, and autism spectrum disorder (Lu et al. 2017). The reported pathogenic CIC sequence variants included missense, nonsense, frameshift, and splice-site alterations, together suggesting loss of function as a primary disease mechanism. Similar to the mutational spectrum of pathogenic ERF variants, structural variants that affect CIC have yet to be reported. However, given that ERF and CIC are <15 kb apart from each other on Chromosome 19q13.2, structural alleles that disrupt both genes would conceivably result in a more complex syndromic phenotype that includes craniosynostosis and neurobehavioral traits.
It is also notable that the identified Chromosome 19q13.2 deletions have low-frequency overlapping copy-number variants (CNVs) reported among healthy individuals in the Database of Genomic Variants (DGV) (Fig. 2; MacDonald et al. 2014). Although this may be antithetical to a classification of likely pathogenic for these aberrations, it further supports that heterozygous loss of ERF and/or CIC has variable expressivity and/or incomplete penetrance, which is consistent with previous reports on ERF and/or CIC sequence variants (Glass et al. 2019). Despite the observation of deletion alleles in the DGV, their pathogenicity is supported by the ERF and CIC constraint scores (observed/expected) in the gnomAD database for loss-of-function variants (0.06; 90%CI: 0.02–0.26 and 0.08; 90%CI: 0.04–0.18, respectively). Both genes also currently have a ClinGen haploinsufficiency score of 2, which is defined as “some evidence for dosage pathogenicity.” In addition, a much larger deletion of the region has been reported in a patient with Diamond–Blackfan anemia, who also had craniosynostosis, delayed speech, and cranial deformities, among other clinical features (Yuan et al. 2016).
In conclusion, these informative individuals add to those previously reported with heterozygous ERF sequence variants and craniosynostosis (Shen et al. 2010; Topf et al. 2014; Lu et al. 2016; Sun et al. 2016; Liu et al. 2019) and those with heterozygous CIC variants and neurobehavioral disorders (Vissers et al. 2010; Athanasakis et al. 2014; Iossifov et al. 2014; C Yuen et al. 2017; Lu et al. 2017; Kim et al. 2019). The identified heterozygous deletions are smaller than any previously reported structural aberration at this locus, which also overlap low-frequency deletion CNVs in the DGV. Taken together, these individuals and their heterozygous deletions strongly support haploinsufficiency of ERF (with or without CIC) as a cause of a syndromic cranial phenotype with autosomal dominant inheritance, variable expressivity, and incomplete penetrance that can present with macrocephaly, abnormal skull morphology or craniosynostosis, and developmental delays.
METHODS
Clinical Evaluation
Clinical evaluations were performed at the Genetics Clinic at the Icahn School of Medicine at Mount Sinai (ISMMS), New York (M.A. and E.W.J.); the transitional Undiagnosed Disease Program WA, Genetic Services of Western Australia (C.P., G.B.); and the Copenhagen University Hospital, Rigshospitalet (T.D.H.).
Clinical Molecular and Cytogenomic Testing
Proband 1 (ISMMS)
Clinical molecular genomic analyses included FMR1 trinucleotide repeat expansion testing for fragile X using triplet repeat primed PCR (AmplideX® PCR/CE FMR1 Reagents; Asuragen) and Southern blot. In addition, testing included a clinical craniosynostosis Sanger sequencing panel that interrogated FGFR1, FGFR2, FGFR3, RAB23, EFNB1, MSX2, TWIST1, and POR. All molecular testing was performed at Mount Sinai Genomics Inc. (DBA Sema4; previously known as the Mount Sinai Genetic Testing Laboratory), according to standard operating procedures.
Clinical cytogenomic testing included peripheral blood Giemsa-banded chromosome and CMA analyses. High-resolution karyotyping (550-band-level) was performed on peripheral blood using standard laboratory protocols. DNA from the proband was tested on the SurePrint G3 ISCA CGH + SNP 4x180K array (Agilent Technologies), as per manufacturer instructions and as described (Botton et al. 2019; Cohen et al. 2020). Higher-resolution CMA testing of the proband on the CytoScan HD platform (Affymetrix) was performed per manufacturer instructions as reported (Reiner et al. 2017, 2018; Khan et al. 2019). Interphase FISH confirmation of the identified deletion was performed using standard laboratory protocols and a BAC probe that hybridized to the 19q13.2 region (Empire Genomics RP11-317E13) and a control probe specific to the subtelomeric region of Chromosome 19q (TelVysion 19q, Abbott Molecular D19S238E). All cytogenomic testing was performed at Mount Sinai Genomics Inc. (DBA Sema4; previously known as the Mount Sinai Genetic Testing Laboratory), according to standard operating procedures.
Proband 2 (KEMH)
For the KEMH proband, all cytogenomic testing was performed at the Department of Diagnostic Genomics, PathWest, Western Australia. Single-nucleotide polymorphism (SNP) microarray was performed using both an Infinium HumanCytoSNP-12 v2.1 BeadChip assay and an Infinium Human CytoSNP-850K v1.2 BeadChip assay according to manufacturer's instructions (Illumina). Analysis was carried out with KaryoStudio (v4.1), and only CNVs containing RefSeq genes were interrogated. MPS was performed using the Illumina TruSight One Expanded Sequencing panel (FC-141-2007) and Illumina NextSeq 550 instrument with High Output kit (FC-404-2004) according to manufacturer's instructions (Illumina). Secondary analyses were performed in Illumina BaseSpace using BWA enrichment pipeline (v2.1.1). Tertiary analyses and small sequence variant filtering were performed in Alissa Interpret (v5.1; Agilent Technologies). CNV analysis of MPS data was performed using an in-house pipeline.
Proband 3 (Rigshospitalet)
CMA analysis of a peripheral blood sample of the patient was performed using the Agilent SurePrint G3 Human CGH Microarray kit 2x400K (Agilent Technologies) as described (Schejbel et al. 2011). qPCR was used for confirmation of the identified deletion in the proband and parental samples as described (Roos et al. 2009).
ADDITIONAL INFORMATION
Data Deposition and Access
The data sets generated and/or analyzed during the current study are not publicly available because these data were derived from clinical genetic testing. De-identified genomic information for Proband 1 can be found in LOVD (https://databases.lovd.nl/shared/individuals/00373335) under individual ID 00373335. De-identified genomic information for Probands 2 and 3 can be found in DECIPHER (https://www.deciphergenomics.org/) under Patient IDs 421530 and 251589.
Ethics Statement
This study was approved by the Institutional Review Board (IRB) of the Icahn School of Medicine at Mount Sinai (ISMMS) and the families provided written informed consent for research participation and publication.
Acknowledgments
The authors thank the families for supporting publication of this series. This study was supported in part by Mount Sinai Genomics, Inc. (DBA Sema4) and the Icahn School of Medicine at Mount Sinai. This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from contact@deciphergenomics.org. Funding for the DECIPHER project was provided by Wellcome.
Author Contributions
R.S., S.A.S., and E.W.J. conceived the case series, reviewed the literature, analyzed and interpreted all data, and drafted and/or revised the manuscript; A.S.A.C. reviewed the literature, analyzed and interpreted all data, and drafted and revised the manuscript; M.A. and E.W.J. oversaw the clinical workup of Proband 1; G.M., W.A.K., L.S., and L.E. contributed data, analyzed and interpreted all data, and revised the manuscript; C.P., D.N.A., K.J.W., and G.B. oversaw the clinical workup of Proband 2, contributed data, analyzed and interpreted all data, and revised the manuscript; T.D.H., and M.K. oversaw the clinical workup of Proband 3, contributed data, analyzed and interpreted all data, and revised the manuscript.
Funding
This was funded in part by the National Institutes of Health/National Institute of Child Health and Human Development (NIH/NICHD) P01 HD078233 (E.W.J.).
Competing Interest Statement
R.S., G.M., L.S., L.E., and S.A.S. are or were paid employees of Sema4, Stamford, CT.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Athanasakis E, Licastro D, Faletra F, Fabretto A, Dipresa S, Vozzi D, Morgan A, d'Adamo AP, Pecile V, Biarnes X, et al. 2014. Next generation sequencing in nonsyndromic intellectual disability: from a negative molecular karyotype to a possible causative mutation detection. Am J Med Genet A 164A: 170–176. 10.1002/ajmg.a.36274 [DOI] [PubMed] [Google Scholar]
- Balasubramanian M, Lord H, Levesque S, Guturu H, Thuriot F, Sillon G, Wenger AM, Sureka DL, Lester T, Johnson DS, et al. 2017. Chitayat syndrome: hyperphalangism, characteristic facies, hallux valgus and bronchomalacia results from a recurrent c.266A>G p.(Tyr89Cys) variant in the ERF gene. J Med Genet 54: 157–165. 10.1136/jmedgenet-2016-104143 [DOI] [PubMed] [Google Scholar]
- Botton MR, Lu X, Zhao G, Repnikova E, Seki Y, Gaedigk A, Schadt EE, Edelmann L, Scott SA. 2019. Structural variation at the CYP2C locus: characterization of deletion and duplication alleles. Hum Mutat 40: e37–e51. 10.1002/humu.23855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brashear A, Sweadner KJ, Cook JF, Swoboda KJ, Ozelius L. 1993. ATP1A3-related neurologic disorders. In GeneReviews® (ed. Adam MP, Ardinger HH, Pagon RA, et al. ). University of Washington, Seattle, WA: [updated 2018 Feb. 22.] [PubMed] [Google Scholar]
- Chaudhry A, Sabatini P, Han L, Ray PN, Forrest C, Bowdin S. 2015. Heterozygous mutations in ERF cause syndromic craniosynostosis with multiple suture involvement. Am J Med Genet A 167A: 2544–2547. 10.1002/ajmg.a.37218 [DOI] [PubMed] [Google Scholar]
- Clarke CM, Fok VT, Gustafson JA, Smyth MD, Timms AE, Frazar CD, Smith JD, Birgfeld CB, Lee A, Ellenbogen RG, et al. 2018. Single suture craniosynostosis: identification of rare variants in genes associated with syndromic forms. Am J Med Genet A 176: 2522. 10.1002/ajmg.a.38846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ASA, Simotas C, Webb BD, Shi H, Khan WA, Edelmann L, Scott SA, Singh R. 2020. Haploinsufficiency of the basic helix-loop-helix transcription factor HAND2 causes congenital heart defects. Am J Med Genet A 182: 1263–1267. 10.1002/ajmg.a.61537 [DOI] [PubMed] [Google Scholar]
- C Yuen RK, Merico D, Bookman M, Howe J L, Thiruvahindrapuram B, Patel RV, Whitney J, Deflaux N, Bingham J, Wang Z, et al. 2017. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 20: 602–611. 10.1038/nn.4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan SM, Robinson JF, McIntyre AD, Marrosu MG, Ticca AF, Loddo S, Carboni N, Brancati F, Hegele RA. 2014. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can J Cardiol 30: 1649–1654. 10.1016/j.cjca.2014.09.007 [DOI] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. 2009. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet 84: 524–533. 10.1016/j.ajhg.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass GE, O'Hara J, Canham N, Cilliers D, Dunaway D, Fenwick AL, Jeelani NO, Johnson D, Lester T, Lord H, et al. 2019. ERF-related craniosynostosis: the phenotypic and developmental profile of a new craniosynostosis syndrome. Am J Med Genet A 179: 615–627. 10.1002/ajmg.a.61073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuze Y, Holmes G, Peter I, Richtsmeier JT, Jabs EW. 2014. Closing the gap: genetic and genomic continuum from syndromic to nonsyndromic craniosynostoses. Curr Genet Med Rep 2: 135–145. 10.1007/s40142-014-0042-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. 2014. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515: 216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan WA, Cohen N, Scott SA, Pereira EM. 2019. Familial inheritance of the 3q29 microdeletion syndrome: case report and review. BMC Med Genomics 12: 51. 10.1186/s12920-019-0497-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Lee MH, Park HS, Park MH, Lee SW, Kim SY, Choi JY, Shin HI, Kim HJ, Ryoo HM. 2003. Erk pathway and activator protein 1 play crucial roles in FGF2-stimulated premature cranial suture closure. Dev Dyn 227: 335–346. 10.1002/dvdy.10319 [DOI] [PubMed] [Google Scholar]
- Kim SH, Kim B, Lee JS, Kim HD, Choi JR, Lee ST, Kang HC. 2019. Proband-only clinical exome sequencing for neurodevelopmental disabilities. Pediatr Neurol 99: 47–54. 10.1016/j.pediatrneurol.2019.02.017 [DOI] [PubMed] [Google Scholar]
- Korberg I, Nowinski D, Bondeson ML, Melin M, Kolby L, Stattin EL. 2020. A progressive and complex clinical course in two family members with ERF-related craniosynostosis: a case report. BMC Med Genet 21: 90. 10.1186/s12881-020-01015-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Le T, Zhu Y, Elakis G, Turner A, Lo W, Venselaar H, Verrenkamp CA, Snow N, Mowat D, et al. 2018. A craniosynostosis massively parallel sequencing panel study in 309 Australian and New Zealand patients: findings and recommendations. Genet Med 20: 1061–1068. 10.1038/gim.2017.214 [DOI] [PubMed] [Google Scholar]
- le Gallic L, Sgouras D, Beal G Jr, Mavrothalassitis G. 1999. Transcriptional repressor ERF is a Ras/mitogen-activated protein kinase target that regulates cellular proliferation. Mol Cell Biol 19: 4121–4133. 10.1128/mcb.19.6.4121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Xu YJ, Li RG, Wang ZS, Zhang M, Qu XK, Qiao Q, Li XM, Di RM, Qiu XB, et al. 2019. HAND2 loss-of-function mutation causes familial dilated cardiomyopathy. Eur J Med Genet 62: 103540. 10.1016/j.ejmg.2018.09.007 [DOI] [PubMed] [Google Scholar]
- Lu CX, Gong HR, Liu XY, Wang J, Zhao CM, Huang RT, Xue S, Yang YQ. 2016. A novel HAND2 loss-of-function mutation responsible for tetralogy of Fallot. Int J Mol Med 37: 445–451. 10.3892/ijmm.2015.2436 [DOI] [PubMed] [Google Scholar]
- Lu HC, Tan Q, Rousseaux MW, Wang W, Kim JY, Richman R, Wan YW, Yeh SY, Patel JM, Liu X, et al. 2017. Disruption of the ATXN1-CIC complex causes a spectrum of neurobehavioral phenotypes in mice and humans. Nat Genet 49: 527–536. 10.1038/ng.3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. 2014. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 42: D986–D992. 10.1093/nar/gkt958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake S, Kato M, Kumamoto T, Hirose T, Koshimizu E, Matsui T, Takeuchi H, Doi H, Hamada K, Nakashima M, et al. 2021. De novo ATP1A3 variants cause polymicrogyria. Sci Adv 7: eabd2368. 10.1126/sciadv.abd2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner J, Karger L, Cohen N, Mehta L, Edelmann L, Scott SA. 2017. Chromosomal microarray detection of constitutional copy number variation using saliva DNA. J Mol Diagn 19: 397–403. 10.1016/j.jmoldx.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner J, Pisani L, Qiao W, Singh R, Yang Y, Shi L, Khan WA, Sebra R, Cohen N, Babu A, et al. 2018. Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of a Bardet–Biedl syndrome 9 (BBS9) deletion. NPJ Genom Med 3: 3. 10.1038/s41525-017-0042-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos L, Jonch AE, Kjaergaard S, Taudorf K, Simonsen H, Hamborg-Petersen B, Brøndum-Nielsen K, Kirchhoff M. 2009. A new microduplication syndrome encompassing the region of the Miller–Dieker (17p13 deletion) syndrome. J Med Genet 46: 703–710. 10.1136/jmg.2008.065094 [DOI] [PubMed] [Google Scholar]
- Schejbel L, Schmidt IM, Kirchhoff M, Andersen CB, Marquart HV, Zipfel P, Garred P. 2011. Complement factor H deficiency and endocapillary glomerulonephritis due to paternal isodisomy and a novel factor H mutation. Genes Immun 12: 90–99. 10.1038/gene.2010.63 [DOI] [PubMed] [Google Scholar]
- Sharma VP, Fenwick AL, Brockop MS, McGowan SJ, Goos JA, Hoogeboom AJ, Brady AF, Jeelani NO, Lynch SA, Mulliken JB, et al. 2013. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet 45: 304–307. 10.1038/ng.2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Li XF, Shen AD, Wang Q, Liu CX, Guo YJ, Song ZJ, Li ZZ. 2010. Transcription factor HAND2 mutations in sporadic Chinese patients with congenital heart disease. Chin Med J (Engl) 123: 1623–1627. [PubMed] [Google Scholar]
- Sun YM, Wang J, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Shi HY, Hou XM, Huang RT, et al. 2016. A HAND2 loss-of-function mutation causes familial ventricular septal defect and pulmonary stenosis. G3 (Bethesda) 6: 987–992. 10.1534/g3.115.026518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timberlake AT, Furey CG, Choi J, Nelson-Williams C; Yale Center for Genome Analysis, Loring E, Galm A, Kahle KT, Steinbacher DM, Larysz D, et al. 2017. De novo mutations in inhibitors of Wnt, BMP, and Ras/ERK signaling pathways in non-syndromic midline craniosynostosis. Proc Natl Acad Sci 114: E7341–E7347. 10.1073/pnas.1709255114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topf A, Griffin HR, Glen E, Soemedi R, Brown DL, Hall D, Rahman TJ, Eloranta JJ, Jungst C, Stuart AG, et al. 2014. Functionally significant, rare transcription factor variants in tetralogy of Fallot. PLoS One 9: e95453. 10.1371/journal.pone.0095453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Vorgia E, McGowan SJ, Peraki I, Fenwick AL, Sharma VP, Allegra M, Zaragkoulias A, Sadighi Akha E, Knight SJ, et al. 2013. Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat Genet 45: 308–313. 10.1038/ng.2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, et al. 2010. A de novo paradigm for mental retardation. Nat Genet 42: 1109–1112. 10.1038/ng.712 [DOI] [PubMed] [Google Scholar]
- Wilkie AOM, Johnson D, Wall SA. 2017. Clinical genetics of craniosynostosis. Curr Opin Pediatr 29: 622–628. 10.1097/MOP.0000000000000542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Meng Z, Liu L, Deng X, Hu X, Liang L. 2016. A de novo 1.6 Mb microdeletion at 19q13.2 in a boy with Diamond–Blackfan anemia, global developmental delay and multiple congenital anomalies. Mol Cytogenet 9: 58. 10.1186/s13039-016-0268-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated and/or analyzed during the current study are not publicly available because these data were derived from clinical genetic testing. De-identified genomic information for Proband 1 can be found in LOVD (https://databases.lovd.nl/shared/individuals/00373335) under individual ID 00373335. De-identified genomic information for Probands 2 and 3 can be found in DECIPHER (https://www.deciphergenomics.org/) under Patient IDs 421530 and 251589.