

Abstract
The Drug Design Data Resource (D3R) Grand Challenges present an opportunity to assess, in the context of a blind predictive challenge, the accuracy and the limits of tools and methodologies designed to help guide pharmaceutical drug discovery projects. Here, we report the results of our participation in the D3R Grand Challenge 4, which focused on predicting the binding poses and affinity ranking for compounds targeting the β-amyloid precursor protein (BACE-1). Our ligand similarity-based protocol using HYBRID (OpenEye Scientific Software) successfully identified poses close to the native binding mode for most of the ligands with less than 2 Å RMSD accuracy. Furthermore, we compared the performance of our HYBRID-based approach to that of AutoDock Vina and Dock 6 and found that using a reference ligand to guide the docking process is a better strategy for pose prediction and helped HYBRID to perform better here. We also conducted end-point free energy estimates on protein-ligand complexes on molecules dynamics based ensembles using molecular mechanics combined with generalized Born surface area method (MM-GBSA). We found that the binding affinity ranking based on MM-GBSA scores have poor correlation with the experimental values. Finally, the main lessons from our participation in D3R Grand Challenge 4 suggest that: i) the generation of the macrocycles conformers is a key step for successful pose prediction, ii) the protonation states of the BACE-1 binding site should be treated carefully, iii) the MM-GBSA method could not discriminate well between different predicted binding poses, and iv) the MM-GBSA method does not perform well at predicting protein-ligand binding affinities here.
Keywords: pose prediction, docking, binding affinity, ligand similarity, MM-GBSA
Introduction
One of the key challenges in the small-molecule drug discovery field is to find therapeutic compounds that bind to targeted biomacromolecules with high affinity. Typical questions that need to be answered during drug discovery projects include the following: How does a drug molecule interact with the residues in the binding pocket of the protein? What is the binding affinity of a molecule? How does the molecule compare to other prospective drug candidates in terms of strength of binding energy? Knowing the answers when conceptualizing a compound can help medicinal chemists make a more informed decision about the experiments that need to be done or the compounds that need to be synthesized, thus saving a lot of time, effort and money.
To address these questions, a huge amount of research has been focused into developing a broad spectrum of computational tools, ranging from fast and less accurate ones like docking [1] and scoring methods [2] to more computationally expensive, but ideally more accurate methods like relative/absolute binding free energy calculations [3].
Computational methods should be regularly tested on blind data sets to prevent researchers from exaggerating the performance of their methods by optimizing or over-fitting their algorithms to reproduce known data sets and to ensure we understand the relative performance of new versus established methods in a predictive context. To this purpose, the Grand Challenge organized by the Drug Design Data Resource (D3R) community was conceived to provide a platform to evaluate methods in a blind way and also to compare methods from different research groups.
One of the two biomolecular targets presented in the 2018 D3R Grand Challenge 4 (GC4) was BACE-1 (β-amyloid precursor protein cleaving enzyme 1). It is a transmembrane aspartyl protease and generates β-amyloid peptide by cleaving the amyloid precursor protein. β-amyloid peptides are involved in Alzheimer’s Disease (AD) [4], and BACE-1 has been seen as a possible target for the treatment and prevention of AD. Fig. 1 shows the binding site of BACE-1 characterized by the aspartyl dyad formed by the residues Asp32 and Asp228, which acts respectively as the acid and base during the proteolysis reaction [5].
Fig. 1.

Binding site of the BACE-1 protein containing the aspartyl dyad - Asp32 and Asp228. The PDB used in this figure is BA20 corresponding to the ligand BACE_20 released by the GC4 organizers. Other important residues in the active site that interact with the ligand are Tyr71 and Thr231.
For this challenge, the unpublished BACE-1 dataset was contributed by Novartis and consists of co-crystal structures along with experimental affinities. Unlike previous D3R challenges, this challenge is characterized by a high number of macrocyclic compounds and only a handful of peptide-like inhibitors (Fig. S1).
Our motivation behind taking part in the D3R Grand Challenge was to test a new workflow for binding mode and affinity predictions and to assess whether we could easily identify any way to improve the speed and accuracy of binding mode predictions. We report here the results of our participation, where we performed ligand similarity-based pose prediction and affinity predictions using MM-GBSA calculations for inhibitors of the target BACE-1.
For the pose prediction challenge, we took advantage of the available BACE-1 receptor structures co-crystallized with compounds of the same chemical families as the D3R query ligands and generated docked poses using a ligand-based design method called HYBRID [6]. However, instead of using the built-in scoring function in HYBRID, we selected the final pose based on inputs from structure visualization, stability in the binding site and binding scores generated using molecular mechanics and generalized Born surface area calculations (MM-GBSA) [7].
Overall, we found that our pose prediction protocol was able to select poses close to the native binding pose with acceptable accuracy. Moreover, we compared the performance of our HYBRID-based docking approach to two standard docking software, namely AutoDock Vina [8] and DOCK 6 [9] and found that HYBRID was more accurate and reliable for RMSD prediction. We also ranked binding affinities using MM-GBSA scores, but the results showed poor correlation with the experimental values.
Theory and Methods
D3R Grand Challenge 4 stages
The first stage of the challenge (Stage 1a) consisted of 20 ligands for pose prediction (Fig. S1) and 154 compounds for affinity ranking of which a subset of 34 ligands was included in the binding free energy calculations. For this stage, the organizers only provided a FASTA sequence for the protein BACE-1 and SMILES strings for the different compounds. Participants were free to use any of the available crystal structures. After this stage, 20 holo crystal structures of BACE-1 from the pose prediction dataset (without the ligands) were released for Stage 1b. Participants had the option of taking advantage of the released BACE-1 receptor structures and resubmitting their binding pose prediction for the same 20 ligands. In Stage 2, complete co-crystal structures of the 20 ligands were made available to test whether this additional knowledge about protein-ligand interactions could improve the affinity rankings and the binding free energies predictions.
Ligand similarity is widely used to predict binding modes
Ligand or molecular similarity is a commonly used concept for predicting different drug properties [10]. For binding mode prediction, molecular similarity translates to the idea that similar molecules are more likely to bind to a protein in a similar fashion.
There exists numerous commercial and academic software packages which have used ligand similarity to predict ligand binding modes. Fukunishi et al. ranked docked poses based on the degree of overlap with reference ligand(s) [11]. HYBRID and POSIT from OpenEye Scientific Software [6, 12] utilize a ligand-based scoring function which favorably scores similar shape and 3D alignment between query ligands and reference crystallographic ligands. Similar to HYBRID and POSIT, SDOCKER [13] has a modified CHARMM-based scoring function with an added similarity penalty. Alternatively, instead of modifying the scoring function in the above methods, PoPSS-Lite, developed by Kumar et al. [14] places query ligands in the binding pocket in the closest possible conformation and orientation as the reference ligand and performs grid-based energy minimization to remove bad contacts.
Interestingly, ligand similarity-based docking approaches were amongst the best-performing methods in the 2017 D3R Grand Challenge 3 [15]. For this work, we used HYBRID to perform the docking calculations.
We used available co- crystal structures of similar ligands to guide our docking protocol
We created a database of 320 BACE-1 co-crystal structures which we found in the RCSB Protein Data Bank (rcsb.org) [16]. We relied on visual inspection to select 9 protein crystal structures with co-crystallized ligands similar to the 20 target ligands - 2P83 [17], 2VIE [18], 3DV1 [19], 3DV5[19], 3K5C [20], 4DPF [21], 4DPI [21], 4GMI and 2QZL [22]). More details about the chosen receptor structures can be found in Table 1 and Fig. S2.
Table 1.
Co-crystallized PDB structures used as references to guide our similarity-based docking protocol. Structures of the reference ligands are shown in Fig. S2.
| Reference structure | Target ligand |
|---|---|
| 2P83 | BACE_20 |
| 2QZL | BACE_20 |
| 2VIE | BACE_20 |
| 3DV1 | BACE_19 |
| 3DV5 | BACE_2, BACE_3, BACE_4, BACE_5, BACE_11 |
| 3K5C | BACE_12, BACE_13, BACE_14, BACE_15, BACE_16, BACE_17 |
| 4DPF | BACE_6, BACE_8, BACE_9, BACE_10 |
| 4DPI | BACE_1, BACE_7, BACE_9, BACE_10, BACE_18 |
| 4GMI | BACE 1, BACE 9, BACE 10, BACE 18 |
We prepared the chosen receptor PDB structures using the following steps. First, pdbfixer 1.4 was used to remove the ligand and the water molecules and to add the missing heavy atoms to the receptor structure. Next, we protonated the residues of the receptors at a pH of 4.5 and renamed the residue/atom names according to the AMBER naming scheme using PDB2PQR web server [23]. The resulting pqr files were converted to PDB files using Parmed 2.7.4.
Binding poses were generated using OMEGA and HYBRID
The ligand protonation states for the query ligands were calculated at pH 4.5 using pKa Plugin from ChemAxon and the Hydrogen atoms were added using Chimera 1.12 [24]. Out of the 20 ligands, BACE_1, BACE_17, BACE_18, BACE_19, and BACE_20 were modeled as neutral. The remaining 15 ligands had a charge of +1. We generated 1000 conformations for each ligand using OMEGA 3.0.8 [25] (Open-Eye Scientific Software). We next docked the target ligand into its corresponding receptor structure(s) using HYBRID 3.2.0.2 [6] with a docking resolution of 1.0 A. The receptor was treated as a rigid structure during docking.
Because docking typically does well at identifying reasonable potential binding modes, but not at correctly recognizing the single most likely binding mode [26], we chose to manually inspect well scoring poses and select those which seemed most reasonable. Specifically, the top 50 docked poses were visually inspected and poses similar to the reference PDB structures (Table 1 and Fig. S2) were selected for the following steps. This step seemed warranted in part because the majority of the top-performing submissions in past D3R grand challenges used visual inspection [15].
We selected binding poses based on stability in the binding pocket and MM-GBSA scores
The ligand binding site of BACE-1 is exposed to water, but, as is common in docking, we retained no ordered water molecules while docking. However, the active site of BACE-1 can either be wet or dry depending on the nature of the ligand [27]. In order to capture any structural changes in the protein-ligand complex, including changes in the water structure as different ligands bind, we simulated the selected binding poses from HYBRID in explicit solvent using molecular dynamics (MD) to have better conformational sampling. Ideally these simulations would also allow water to solvate the binding site and different ligands as is most suitable in each case.
The MD simulations were carried out using the
pmemd.cuda
module of the Amber18 simulation package [28]. We used Antechamber (from Amber 16 package [29]) to add partial charges to the ligand atoms using the AM1-BCC model [30]. Tleap, an auxiliary program of Amber16 was used to set up the simulation box and parameters. The protein and the ligands were modeled using the Amberff99sb [31] and GAFF version 1.8 [32] force fields respectively. The protein-ligand complex was then solvated in TIP3P water [33] in a cubic box with 10 Å padding. Na+ and Cl− counter ions were added to achieve a salt concentration of 0.1M with additional Cl− ions to ensure that the system was neutral. SHAKE was used to constrain the protein heavy atom-hydrogen bonds with a time step of 2 fs. Long-range electrostatic interactions were calculated using the particle mesh Ewald method, with a cutoff of 9.0 Å for the real space electrostatics and Lennard-Jones forces.
The ligand, water, and the ions were first minimized for 1000 steps keeping the protein fixed using 25 kcal/mol-Å2 positional restraints and then for another 1000 minimization steps with 10 kcal/mol-Å2 restraints. The system was next heated from 10 K to 300 K in NVT ensemble for 140 ps while restraining the protein-ligand complex using positional restraints of 10 kcal/mol-Å2. Next, the restraints were gradually released using 5 and 2 kcal/mol-Å2 restraints on the protein-ligand complex for 20 ps each, and then 2 kcal/mol-Å2 restraint only on the ligand for 20 ps. The temperature was regulated in the NVT simulations through Langevin dynamics with a collision frequency of 2 ps−1. The production run was performed in NPT ensemble for 14 ns with the first 4 ns being discarded as equilibration. Constant pressure was maintained in the simulation using isotropic pressure scaling with a relaxation time of 2 ps.
After generating trajectories using MD simulations, we used the MMPBSA.py program [34] in Amber16 to calculate the protein-ligand binding scores using the GBneck2 model [35] with a salt concentration of 0.1 nM. Coordinates were saved every 100 ps during MD, resulting in 100 frames which were analyzed for the MM-GBSA calculations for each ligand.
We would like to note here that even though MM-GBSA is often called an end-point binding free energy protocol, in our opinion, MM-GBSA scores are better regarded as scores rather than true free energies. The calculations are done on end-point trajectories and use a quasi-harmonic approximation to account for the entropy of the protein and the ligand. Also, the waters in the binding pocket are treated as a continuum solvent and hydrogen-bonds formed by water molecules in the binding pocket are neglected in the calculations.
In the last step of our pose prediction protocol, we picked the final poses based on both the stability of the ligand in the binding pocket and MM-GBSA scores. We assessed the stability of the poses in the binding site by calculating the RMSD of the docked poses at the start and the end of the MD simulations. Both these methods have been successfully used to discriminate between binding poses in literature. Liu et.al. [36] showed that stability of poses in MD simulations is a better metric compared to docking scores for discriminating between correct and decoy poses, while Kaus et al. [37] demonstrated better discriminating power with MM-GBSA scores against docking scores.
We first visualized the selected poses after MD to evaluate whether they drifted away from the initial docked poses. For the simulation initiated at each docked pose, we computed the RMSD at the end of the MD simulation with respect to the initial pose. After doing this for simulations begun from all poses, we selected the poses with the lowest final RMSD values. After this step, we compared the MM-GBSA scores of the selected poses and picked the pose with the most favorable MM-GBSA score as the top pose. Also, we submitted multiple binding pose predictions for the cases where the metrics (RMSD or MM-GBSA scores) were within 1 Å or 10 kcal/mol of each other for the docked poses. Even though the poses were selected after MD, the final submitted poses were the initial docked poses from HYBRID and were not minimized prior to submission.
We made predictions with AutoDock Vina and DOCK to compare against our ligand similarity-based protocol in Stage 1b
We did a reference test to check whether we could improve our docking accuracy if we swap HYBRID with other standard docking programs - AutoDock Vina 1.1.2 [8] and DOCK 6.8 [9]. Thus, we submitted two separate blind pose predictions with AutoDock Vina and DOCK 6 in Stage 1b. We used the same binding pocket described in Fig. 1. In addition, the final binding pose(s) was visually selected to be consistent with our protocol containing HYBRID. We didn’t do any additional MM-GBSA calculation to score the poses and relied on the in-built scoring functions.
AutoDock Vina requires the receptor and the ligand files to be provided in pdbqt format. We started with the same PDB files of the receptors and the mol2 files of the ligands which were used for HYBRID, and converted the structures to pdbqt format using scripts available in AutoDock Tools [38] —
prepare_receptor4.py
(v 1.13) and
prepare_ligand4.py
(v 1.10). The box size used for docking was 22 Å × 17 Å × 20 Å. Exhaustiveness and
num_modes
were both set to 50 and an
energy_range
value of 5 was used to ignore poorly docked poses.
For DOCK 6, both the ligand and the receptor files were prepared in Tripos mol2 format using Chimera. Partial charges were added to the atoms using AM1-BCC method [30] within Chimera. Next, we generated a molecular surface for the receptors without the hydrogen atoms using the dms functionality in Chimera, followed by the creation of the docking spheres using the sphgen program [39] within DOCK 6. A subset of the spheres which were within 7 Å of the binding site (Fig. 1) were selected using the accessory
program sphere_selector
. Then, we created a box around the spheres with a margin of 5 Å using the program showbox in the DOCK suite. Next, the energy interactions were evaluated between a dummy atom and all protein atoms within 0.3 Å resolution using the DOCK accessory program GRID [40]. Van der Waals interactions were modeled using 6-12 Lennard-Jones potential and electrostatic interactions using Coulomb potential with a dielectric coefficient of ε=4r, where r is the distance. The resulting grid was used to score the binding poses using grid-score [40]. For the docking algorithm, we used standard parameters mentioned by Allen et al. [9].
We used MM-GBSA scores to rank ligands and to predict relative binding free energies
We used the same MM-GBSA protocol described above to rank the 154 ligands provided for the affinity prediction challenge. The initial binding pose was manually created in Chimera based on a reference structure. The reference was chosen either from the available co-crystal structures or from binding poses we predicted using HYBRID for the 20 ligands in the pose prediction challenge depending on which reference ligand had the highest molecular similarity. More details about the selected reference structures can be found in Table S1 and S2.
For the relative binding free energy prediction portion of the challenge, BACE_10 was specified as the reference ligand by GC4 organizers. We subtracted the MM-GBSA score of BACE_10 from our MM-GBSA scores and submitted the results as relative binding free energy predictions in Stage 1a and Stage 2. The predictions aren’t necessarily relative binding free energies, but we wanted to see whether there might be any correlation with relative binding free energies with this approximation. Although we had initially planned to do alchemical free energy calculations for the binding free energy predictions, the short time frame of this challenge and the complexity of handling macrocycles in alchemical calculations [41] made us favor MM-GBSA calculations.
Results and Discussion
We utilized our approach for ligand similarity-based placement and re-scoring with MM-GBSA to participate in all the three stages for the biomolecular target BACE-1 in D3R GC4. As detailed in the methods, Stage 1a of the challenge involved cross-docking prediction for 20 ligands, affinity ranking for 154 ligands and binding affinity prediction for a subset of 34 ligands. Stage 1b included self-docking challenge for the same 20 ligands as in Stage 1a. For Stage 2, the co-crystal structures corresponding to the 20 BACE-1-ligand complexes from the pose prediction challenge were released and we were asked to recompute the affinity ranking as well as the free energies.
For the cross-docking challenge in Stage 1a, we identified co-crystal structures in the RCSB database containing similar ligands to the 20 target ligands provided for the blind pose prediction challenge and used these structures for docking using HYBRID. The top pose(s) was determined using a combination of visual inspection, end-point free energy estimates (MM-GBSA scores) and stability of the ligands in the binding pocket. For the self-docking challenge in Stage 1b, we directly used the receptor structures provided to perform docking and kept the rest of the protocol the same as in stage 1a. We also used MM-GBSA scores for ligand affinity ranking and relative binding free energy predictions in both Stage 1a and Stage 2.
We achieved <2 Å for both mean and median RMSD with our HYBRID-based protocol
The performance of our ligand similarity-based docking protocol was reasonably good in both the cross-docking and self-docking challenges. Fig. 3, 4, S3 and Table S3 summarize the performance of our docking predictions.
Fig. 3.
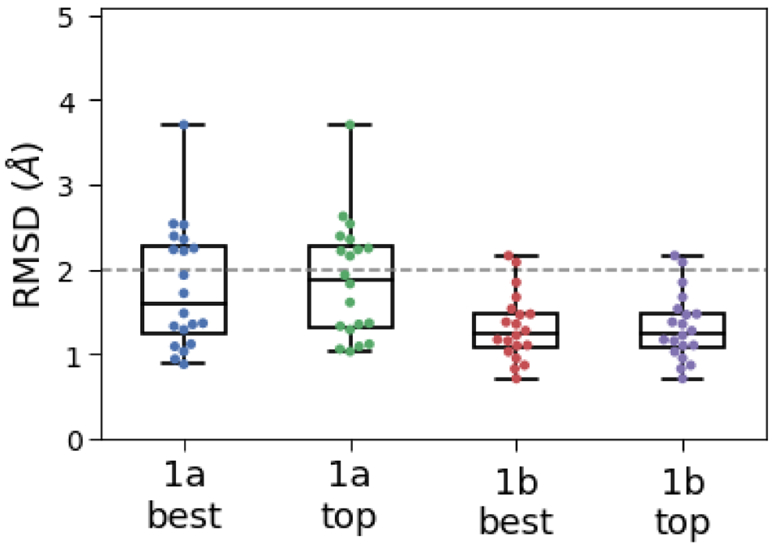
Boxplot comparing RMSD values of the ‘best’ and the ‘top’ pose based on our pose prediction protocol. 1a and 1b refers to the two pose prediction stages in GC4. Each dot represents the RMSD for a different ligand. The boxplot shows the location of the median RMSD values, and the 75th and 25th percentile values. The variations in the x values do not have any significance.
Fig. 4.

Comparison of our Stage 1a ‘best’ ranking poses (in blue) with the corresponding crystal structure (in pink). RMSD values are reported for each pose. Compounds underscored by a red line have RMSD values less than 1 Å, those underscored by a light blue line have RMSD values less than 2 Å and those underscored by a navy blue line have RMSD values higher than 2 Å.
For some of the BACE-1 inhibitors, we had submitted multiple pose predictions. We will be referring to the pose that we ranked first in our submission as the ‘top’ pose and the pose with the lowest RMSD as the ‘best’ pose. In Stage 1a, our ‘best’ pose predictions achieved a mean and median RMSD of 1.60 Å and 1.79 Å, whereas our ‘top’ pose predictions did not do that much worse, with a mean and median RMSD of 1.87 Å and 1.88 Å . 12 out of 20 structures had an RMSD of < 2 Å , which is generally considered as a successful docking prediction [26,42].
For a majority of the ligands, we were able to distinguish between good and bad docking poses based on their stability in the binding pocket during the MD simulations and had submitted multiple poses for only 8 ligands in Stage 1a. The ‘top’ pose and the ‘best’ pose were close to each other as seen from the mean and median RMSD (Table S3), usually within 0.5 Å RMSD, demonstrating that our protocol was able to separate the good from the bad poses in most cases. The superposition of the ‘best’ pose for each ligand in Stage 1a on to the respective crystal structure provided by the GC4 organizers is reported in Fig. 4.
For the self-docking in Stage 1b, the presence of correct binding site conformation in the receptor helped to improve our overall docking predictions. For example, we used the PDB structures 3DV5 and BA04 for cross-docking in Stage 1a and self-docking in Stage 1b respectively for the ligand BACE_4. There were subtle differences in the side-chain orientations of the residues Gln12, Leu30, Gln73, Asn233 and Arg235 in the two structures (Fig. S4). Having the correct side-chain packing in the self-docking challenge improved the RMSD from 2.23 Å in Stage 1a to 0.87Å in Stage 1b.
In Stage 1b, we were able to narrow down to only a single binding mode for all but one ligand. Our ‘top’ and ‘best’ predictions in this stage consisted of the same twenty structures with identical RMSD distributions (Fig. 3). We achieved a RMSD accuracy of <2 Å for 18 out of 20 structures for the ‘best’ pose (also the ‘top’ pose here) with the remaining two being very close to 2 Å as seen in Fig. 3 and Fig. S3. For the ‘best’ pose and the ‘top’ pose, we got a mean RMSD of 1.32 Å and a median RMSD of 1.25 Å.
Relative to other participants, we ranked in the middle in both Stage 1a and Stage 1b pose prediction challenges (Fig. S5 and S6), with the top groups having about 0.5 Å median RMSD, which is about 1 Å better than us. Thus, we have a lot of room to improve our protocol.
While doing post-analysis of our results after the experimental data was released, we noticed a few places where we could have done a better job in implementing our pose prediction protocol and in improving its accuracy.
First, unlike some other standard docking software, HYBRID doesn’t minimize the docked poses after placing them in the binding pocket. We had directly submitted the outputs from HYBRID in the pose prediction challenge. Minimizing the poses before submission would have helped relieve any bad contacts or torsional strains in the macrocycles.
Second, we used an older version of OMEGA to generate conformers for docking, which, even though was able to handle macrocycles, was not optimized to handle macrocycles well. As a result, for some of the ligands (Table S4) we did not sample conformations close to the native co-crystal pose, which in turn decreased our pose prediction accuracy. Recently a new distance geometry-based sampling algorithm was introduced in (released in Oct 2018 after D3R GC4), which is tailored towards sampling macrocycle conformations [43] and will possibly help sampling the ligand conformational space in a more robust manner.
Third, in this work, the query ligands in the pose prediction challenge had very similar reference ligands already deposited in the RCSB PDB database with co-crystal structures. POSIT [12] might have been a better docking tool than HYBRID for the given dataset. For cross-docking studies with TanimotoCombo ligand similarity greater than 0.9, POSIT has been shown to have significantly better accuracy than other docking tools. It employs a shape-matching algorithm to overlay the query ligand on a reference ligand and optimizes the alignment of the query ligand and the induced strain.
Fourth, initially we tried to identify similar ligands in our PDB database using shape and color similarity search. We searched the database using OMEGA and OEShape (Open-Eye Toolkits version 3.0.8, OpenEye Scientific Software) to identify ligands that are similar to the 20 ligands. However, we were unsuccessful in identifying close ligand structures with this approach, probably because the version of OMEGA we were using was not particularly good for macrocycles, resulting in low similarity scores even for macrocycles which in fact were quite similar. As a result, we had to identify similar ligands manually. Additionally other similarity tools like TanimotoCombo similarity or Tversky coefficient, which has been successfully used by Kumar et al. [14] in earlier Grand Challenges could have done a better job at selecting the reference ligands and in turn, automating the process.
Fifth, instead of relying on the final snapshot at the end of MD simulations to select the predicted binding pose, we could also select the cluster heads of the MD trajectories. The final snapshot might not necessarily be the representative conformation for that simulation.
Lastly, another manual part of our protocol was visually selecting the poses after HYBRID docking. As mentioned before, HYBRID does not minimize or cluster the docked poses in the binding pocket. Having an additional step where we minimized and clustered the poses by a RMSD filter could have reduced the final number of docked poses, and thus could have saved us time.
Then, MD and MM-GBSA calculations could be performed on all the selected poses to pick the top ones based on RMSD and MM-GBSA scores. We could also select the cluster heads we obtain during MD simulations instead of relying on the final snapshot at the end of MD simulations to select the predicted binding pose.
We expect that incorporating the above changes will improve the pose prediction accuracy of our protocol and make it less human intensive and easier to scale-up for a large number of ligands at the same time .
Equilibrated poses from MD simulations have higher RMSD in general
As our predictions, we submitted the outputs from HYBRID as our predicted poses during the challenge even though we used MD simulations to assess the stability of these poses. The poses were not minimized or equilibrated to relax any steric or Coulombic clashes prior to submission. An alternate approach would have been to submit poses relaxed by MD. Hence, in Stage 1b, we decided to have two separate submissions: i) The initial binding poses before MD corresponding to the outputs generated by HYBRID. Similar to Stage 1a, these poses were not minimized or equilibrated to relax any steric or Coulombic clashes prior to submission. and ii) the final binding poses at the end of our 14 ns long MD trajectories. Our objective was to see how the relaxed binding poses from the MD simulations fared compared to the initial starting conformations.
Overall the quality of the pose prediction degraded mildly when we considered the binding poses after MD. The mean and the median increased from 1.32 and 1.25 Å before MD to 1.75 and 1.70 Å , respectively after MD. Although the RMSD of 9 out of the 20 poses either improved or didn’t change significantly after MD, the rest of them differed by at least 0.5 Å or more.
This finding is not surprising since if the starting pose in MD is not stable, it is highly likely that the ligand will try to explore around the binding pocket during the simulation to find favorable and/or metastable binding modes. Also, the proteins are crystallized at very low temperatures, whereas we use room temperature in our MD simulations. This is also a possible contributor to the higher RMSD values achieved after MD for some of the ligands – thermal motion ought to result in some change in binding mode, but additionally, the low temperature binding mode might be different (perhaps only subtly) from that at room temperature.
The worst RMSD after MD was reported for the BACE_2 ligand. Based on the MD trajectory, the BACE_2 ligand drifted away from the binding site after 14 ns of MD. Therefore, we decided to perform MD simulations on the released BACE_2 crystal structure (Fig. 6 and Fig. S7.A) to find the cause of the drifting of the predicted pose - whether it was because of poor pose prediction or because of any force field errors. Our results show that starting with the crystal structure, the ligand stays in the binding site after 14 ns of MD (Fig. S7.B). However, the non-macrocyclic part of the ligand moved from its initial position. Furthermore, if we compare the BACE_2 crystal structure to our ‘best’ pose before MD, we notice that these are two different BACE_2 conformers with many structural differences (Fig. S7.C, circled regions). This finding means that our BACE_2 docked ligand left the binding site during MD simulations, likely because of its poor starting conformation. This highlights the importance of correct conformer generation for accurate pose prediction.
Fig. 6.

Comparison of the crystal structures of BACE_2, BACE_3, BACE_18 and BACE_20 (in pink) with our Stage 1b ‘best’ ranking poses before MD simulations (in blue) and our Stage 1b ‘best’ ranking poses after MD simulations (in blue), respectively. MD affected our performance in different ways for different ligands. For example, the worst RMSD value was reported for the BACE_2 ligand, the RMSD value corresponding to the BACE_18 ligand increased after MD, and the RMSD values corresponding to the BACE_3 and the BACE_20 ligands decreased after MD. Compounds underscored by a red line have RMSD values less than 1 Å, those underscored by a light blue line have RMSD values less than 2 Å and those underscored by a navy blue line have RMSD values higher than 2 Å.
Moreover, the RMSD value of the BACE_18 ligand was 2.08 Å and increased to 2.34 Å after MD. The analysis of the corresponding MD trajectory shows that the established interactions between the ligand and the aspartyl dyad are different from those before MD (Fig. S8). This may be due to a wrong protonation of the aspartyl dyad residues at pH 4.5. The activity of BACE-1 is known to be pH-dependent, with a reported activity peak between pH 3.5 and 5.5 [44, 45], which is why we protonated our proteins as appropriate for pH 4.5.
In order to investigate whether we correctly protonated the aspartyl dyad or not, we changed the protonation states of Asp32 and Asp228 for the BACE_18 protein structure and performed separate MD simulations on the modified protein-ligand complexes. Thus, in addition to the BACE_18 complex bearing a protonated Asp32 and a deprotonated Asp228 (32p, 228d), we designed four BACE_18 complexes (Fig. 7) with different orientations of the ligand’s hydroxyl groups that interact with the aspartyl dyad for complex - A) protonated Asp32 and deprotonated Asp228 (32p1, 228d1) and tried all the reasonable protonation states of the aspartyl dyad at pH 4.5 on complexes: B) deprotonated Asp32 and protonated Asp228 (32d, 228p), C) protonated Asp32 and protonated Asp228 (32p1, 228p1), and D) protonated Asp32 and protonated Asp228 (32p2, 228p2). C and D are different by the orientation of the hydrogens and the oxygen (OD2) of the carboxyl group on their Asp32 and Asp228.
Fig. 7.

Interactions occurring in the BACE_18 active site between the ligand and the aspartyl dyad Asp32/Asp228 at the end of 14 ns of MD simulations. The protonation states of the four complexes were held fixed during MD. A) BACE_18 with protonated Asp32 and deprotonated Asp228 (32p1, 228 d1), B) BACE_18 with deprotonated Asp32 and deprotonated Asp228 (32d, 228d), C) BACE_18 with protonated Asp32 and protonated Asp228 (32p1, 228p1), and D) BACE_18 with protonated Asp32 and protonated Asp228 (32p2, 228p2). C and D are different by the orientation of the hydrogens and the oxygen (OD2) of the carboxyl group on their Asp32 and Asp228. The calculated distances are in Å. More details about the specific atoms involved in the labeled hydrogen bond interactions are reported in Table S6.
The observed hydrogen bond interactions between the ligand and the aspartyl dyad residues in these four different cases after MD simulations are reported in Fig. 7. More details about the specific atoms involved in these hydrogen bond interactions are reported in the caption of Table S6. Among the four cases, only the double protonated aspartyl dyad of BACE_18 do not interact with the ligand (Fig. 7 C and D). To verify whether the interactions shown in Fig. 7 are preserved during the MD simulations, we measured two different average distances; one between Asp32 and the ligand BACE_18, and another between Asp228 and the ligand BACE_18. We did this for each of the four complexes A, B, C, and D (Table S6). The calculated average distances confirm that Asp32 and Asp228 of complexes A and B establish more stable interactions with the BACE_18 ligands than the aspartyl dyad residues of complexes D and C. The superim-position of the BACE_18 ligand pose extracted from each of the modified complexes after MD with the released BACE_- 18 crystal structure (Fig. S9) shows that all the four ligand poses are different. This analysis suggests that the binding mode of BACE_18 depends on the protonation states of the aspartyl dyad in BACE-1 and is consistent with other studies that evaluate the pH-dependent activity of BACE-1 [46,27, 47].
At pH 4.5, the aspartyl dyad of apo state of BACE-1 is most likely to be mono-protonated based on the pKa values of the aspartyl dyad (protonated Asp32, deprotonated Asp228). In this work, we protonated the aspartyl dyad of the protein-ligand complex at the same state as the apo state, which might not be a good approach. Several computational studies have shown that the pKa of the aspartyl dyad changes when inhibitors are present in the binding pocket [27,48]. Constant pH molecular dynamics might be able to capture the effect of ligand binding on the protonation states of the receptors, but is computationally expensive.
The PDB2PQR server we used for protonating the proteins utilizes PROPKA 3.1 [49] as a back end. While analyzing our results, we found that the server-selected protonation state was not the same across different receptor structures with a few of them having both Asp32 and Asp228 in the protonated state. Treating the protonation states more carefully by adding a step to check whether the protein was protonated as desired could have improved our predictions. Also, the existing pKa tools needs to be improved for more accurate prediction.
MM-GBSA scores are not efficient in discriminating between crystal pose and poorly predicted poses
During our post-analysis after the release of the experimental data, we decided to test the power of MM-GBSA in discriminating between potential binding poses for the same ligand. We evaluated the MM-GBSA scores of the released crystal pose and compared them against the score for the ‘best’ pose predicted by us in Stage 1b. Table 2 shows the calculated MM-GBSA scores for the two poses and also the RMSD of our predicted pose with the crystal pose as a reference.
Table 2. Table showing MM-GBSA scores for the crystal structures released by the GC4 oragnizers and the binding poses from our Stage 1b ‘best’ prediction.
The reported RMSD values are with respected to the crystal pose. All RMSD values are reported in Å. In an ideal scenario, MM-GBSA score for the crystal pose should be lower than that of Stage 1b ‘best’ pose, especially for the ligands with high RMSD values. Rows highlighted with red are for the ligands where MM-GBSA score predicted the crystal pose to be poorer and those highlighted with gray are for cases where MM-GBSA score for the crystal pose and our predicted pose are almost the same.
| Crystal Pose | Stage 1b ‘Best Pose’ | |||
|---|---|---|---|---|
| Ligand ID | MM-GBSA score | MM-GBSA score | RMSD before MD | RMSD after MD |
| BACE_1 | −68.8±0.5 | −67.3±0.4 | 2.16 | 1.45 |
| BACE_2 | −56.1±0.5 | −24.6±0.4 | 1.38 | 3.85 |
| BACE_3 | −69.4±0.9 | −68.9±0.7 | 0.95 | 0.68 |
| BACE_4 | −62.9±0.4 | −60.9±0.8 | 0.87 | 1.81 |
| BACE_5 | −57.6±0.6 | −48.6±0.3 | 1.17 | 1.68 |
| BACE_6 | −54.0±0.5 | −46.2±0.5 | 0.71 | 1.58 |
| BACE_7 | −51.3±0.5 | −45.0±0.5 | 1.10 | 1.16 |
| BACE_8 | −38.4±0.4 | −44.1 ±0.4 | 1.03 | 1.19 |
| BACE_9 | −52.7±0.5 | −44.8±0.4 | 1.22 | 2.06 |
| BACE_10 | −47.5±0.4 | −44.5±0.6 | 0.83 | 1.56 |
| BACE_11 | −45.5±0.4 | −26.1±0.3 | 1.28 | 2.84 |
| BACE_12 | −56.4±0.5 | −35.4±0.4 | 1.35 | 1.25 |
| BACE_13 | −29.8±0.5 | −29.0±0.9 | 1.10 | 1.97 |
| BACE_14 | −50.4±0.6 | −40.6±0.5 | 1.47 | 2.47 |
| BACE_15 | −51.4±0.4 | −37.8±0.5 | 1.46 | 1.74 |
| BACE_16 | −46.3±0.8 | −37.6±0.5 | 1.85 | 2.15 |
| BACE_17 | −46.2±0.4 | −51.7±0.5 | 1.53 | 1.73 |
| BACE_18 | −46.6±0.6 | −49.1±0.5 | 2.08 | 2.34 |
| BACE_19 | −58.2±0.4 | −48.9±0.4 | 1.16 | 1.02 |
| BACE_20 | −66.8±0.5 | −66.1±0.5 | 1.67 | 0.56 |
Lower MM-GBSA scores for the crystal pose indicate that MM-GBSA was successful in discriminating between the poses, particularly for the cases with RMSD close to or higher than 2 Å for our predicted pose. For six out of twenty ligands, MM-GBSA scores ranked our predicted pose better or almost similar to the crystal pose even when the RMSD value of our predicted pose is high or increases after MD or both indicating that the pose is unstable. Thus, MM-GBSA scores alone might not be sufficient in discriminating between different binding poses.
Our protocol worked better than standard AutoDock Vina and DOCK 6 docking software
Each docking software is associated with a unique docking algorithm and scoring function. Since we are visually selecting the final pose, the scoring function is not that important in our protocol. However, the search algorithm is of crucial importance. If the docking algorithm is not able to generate poses close to the correct binding pose (and score these well enough that they are retained by the algorithm), then there is no good pose to choose from and, the selected final pose(s) will have high RMSD.
To test whether we can generate better poses, we decided to make predictions using AutoDock Vina and DOCK 6 along with HYBRID in Stage 1b. HYBRID uses pre-genera conformations with Omega and places them in the binding pocket in different positions and orientations to have similar 3D conformation as the reference crystallographic ligand. On the other hand, AutoDock Vina uses a stochastic global search algorithm [8], while DOCK 6 uses a more systematic search using the ‘anchor-and-grow’ algorithm [9]. Both of them do not need any reference binding pose to perform the search.
Fig. 8 compares the RMSD distribution and the median values which we obtained from the predictions made with the three pieces of docking software. The RMSD values corresponding to the HYBRID-based protocol were much more compact and consistent compared to AutoDock Vina and DOCK 6.
Fig. 8.

Boxplot comparing RMSD values of the ‘best’ poses obtained using different docking software in Stage 1b of the blind pose prediction challenge. HYBRID performed better than AutoDock Vina and DOCK 6 as a docking software in our protocol.
Overall, AutoDock Vina has good RMSD accuracy with 18 poses having ≤ 2 Å RMSD. AutoDock Vina automatically clusters the docked poses, unlike HYBRID. As a result, the poses were far apart in RMSD, and we were able to narrow down on a single pose during pose selection for most of the ligands. Thus, we submitted multiple pose predictions for only four ligands in the AutoDock Vina submission, and for all of them, the ‘top’ pose and the ‘best’ pose were the same. Also, the median RMSD of 1.27 Å for AutoDock Vina was almost the same as HYBRID (1.28 Å). The docking algorithm failed for the two ligands BACE_13 and BACE_18 and generated poses were far from the expected. This resulted in a high mean RMSD of 1.55 Å.
For DOCK 6, there were seven ligands where the search algorithm completely fell through, resulting in poses either out of the binding site or improperly oriented. Surprisingly DOCK 6 had the best median RMSD value at 1.12 Å for the ‘best’ pose among the three programs. We tried tuning different parameters to make the search algorithm more robust but were not able to generate good docked poses.
As noted, DOCK 6 performed worst, but it seems possible this could be due to specific issues with its search methodology as it relates to this challenge. Particularly, the ‘anchor- and-grow’ search algorithm used for conformer generation may not be well suited for macrocycles. More work is needed to be done to investigate whether DOCK 6 is efficient for macrocycle docking or not.
Hand-engineered poses performed better compared to those from pose prediction protocol for ligands with high similarity
Some of the reference structures that we used in our docking protocol were very similar to the target ligands. For example, BACE_3 and the ligand co-crystallized with BACE-1 in the PDB structure 3DV5 differ only by a single hydroxyl group. For such simple cases, we directly made the structural changes using Chimera (listed in Table S5) and submitted the binding pose to evaluate how well docking search algorithms perform compared to educated human guesses. However, human guesses were not always possible, especially when the changes involved adding or modifying nonaromatic rings (BACE_2 and BACE_7) or macrocycles with different ring sizes (BACE_19 and 3DV1).
We had submitted six hand-engineered predictions in Stage 1a and for all of them we obtained equal or better RMSD compared to our HYBRID-based protocol. In particular, for the ligand BACE_5 and BACE_16, the hand-engineered poses have an RMSD < 2 Å, while the respective RMSD of HYBRID-based poses are > 2 Å (Fig. 9). This highlights that a lot of work is still needed in order to improve search algorithms to achieve better accuracy in binding mode predictions.
Fig. 9.

Plot comparing RMSD of predicted poses using ligand similarity-based protocol to RMSD of the respective hand-engineered poses in Stage 1a. We had submitted multiple predictions for the BACE_6 and BACE_20 ligands and hence, have multiple dots showing the RMSD of our different predictions. Overall, the hand-engineered predictions were better than the ligand similarity-based protocol.
MM-GBSA scores have poor correlation with experimental affinity ranking
Affinity ranking.
Stage 1a and Stage 2 focused on affinity ranking of a set of 154 ligands. As detailed in the methods, we used a structure-based protocol for the affinity ranking. We docked the ligands into the proteins and performed short 14 ns explicit solvent MD simulations. Finally, we scored the ligands using MM-GBSA calculations. At the end of the challenge, the organizers reported quality metrics (the Kendall’s τ and the Spearman’s ρ) to assess the correlation between the experimental and predicted affinity rankings for the 154 compounds.
The overall correlation between our predicted binding affinity ranking and the experimental affinity ranking is rather poor for both the Kendall’s τ and the Spearman’s ρ. We have obtained a Kendall’s τ of 0.17 and a respective Spearman ρ of 0.26 for our Stage 1a MM-GBSA based submission. Out of the 26 structure-based affinity ranking submissions in Stage 1a, our MM-GBSA based submission was classified as the 7th (Fig. S11), with the best submission in this category having a Kendall’s coefficient and a Spearman’s ρ of 0.38 and 0.54, respectively. There was also no improvement for the structure-based ranking between Stage 1a and Stage 2 indicating that our MM-GBSA protocol is not performing well in ranking the ligands from this affinity ranking set. For Stage 2, our MM-GBSA based submission was ranked 19th among 48 submissions using the Kendall’s τ correlation coefficients (Fig. S12) and 18th using the Spearman’s ρ. The top submission scored a Kendall’s τ of 0.39 and a Spearman’s ρ of 0.54.
The poor performance of MM-GBSA is consistent with our findings in the Stage 2 ligand affinity ranking challenge in GC4 where we predicted the same affinity ranking for a different set of binding poses generated using AutoDock-GPU and obtained comparably poor performance between MM-GBSA scores and AutoDock-GPU docking scores based on the Kendall’s τ and Spearman’s ρ metrics [50].
Relative affinity prediction.
In Stage 1a and Stage 2, we performed MD simulations on the 34 ligands of the free energy set, then computed end-point free energy approximates with MM-GBSA method, and submitted the results as relative binding free energy predictions. Knowing that MM-GBSA is not a rigorous method for reliable relative free energy calculations, we wanted to explore the predictive power of this approximation method.
For free energy predictions, the correlation metrics (Kendal τ, Spearman’s ρ and Pearson’s r) disclosed by the GC4 organizers show that our submitted ranking does not reflect the experimental ranking of binding affinities satisfactorily.
Comparing to the experimental binding free energies, the MM-GBSA results led to correlation coefficients near 0 in Stage 1a (a Kendall’s τ of 0.17, a Spearman’s ρ of 0.22, and a Pearson’s r of 0.2), meaning a disagreement between our predicted values and the experimental binding data. Similarly in Stage 2, the evaluation of the results shows that the correlation coefficients values are still low (a Kendall’s τ of 0.01, a Spearman’s ρ of 0.02, and a Pearson’s r of 0.05) for this stage. In Stage 1a, our submission based on MM-GBSA method was ranked 8th among 22 submissions (Fig. S13). Regarding Stage 2, our submission was ranked 21st among 31 submitted predictions (Fig. S14). Also, the reported RMSE values are high for both Stage 1a (8.17 kcal/mol) and Stage 2 (8.14 kcal/mol). Consequently, the employed MM-GBSA scoring method is not correlated to the experimental relative binding affinity of the ligands.
While we find MM-GBSA performs poorly here, a variety of previous literature results have reported reasonable performance and positive correlations between calculated binding scores and experimental affinities [51,52]. The GB-Neck2 model used in this work is a relatively new GBSA model and has not been widely tested for protein-ligand binding affinity rankings. However, Su et al. used it successfully in their QM-MM/GBSA work on benzimidazole inhibitors [53]. It is also possible that successes of MM-GBSA may be published more often than failures so our sampling of the literature could be subject to publication bias. Moreover, participants in previous D3R challenges found a poor correlation between MM-GBSA free energy estimates and experimental binding affinities [54,55,56]. In the best case scenario, MM-GBSA is expected to be consistent with the results generated using the MM-PBSA method (molecular mechanics - Poisson-Boltzmann surface area), which itself has been shown to be an unreliable method [57,58,59].
The failure of MM-GBSA in this work can be attributed to many factors. MM-GBSA might not be suited for macrocycles or the receptor BACE-1, or fundamental limitations of the method and the approximations it makes might be a problem here. Additionally, the ligand dataset had both positive and neutral molecules and MM-GBSA has been shown to have poor correlation for ligands with different net charges [51].
In our calculations, we used a ‘single trajectory protocol’, where the unbound ligand and receptor structures are extracted from the same protein-ligand complex trajectory. Even though this is a computationally efficient way of performing the calculations, the simulations did not sample the complete unbound conformational space explored by the receptor and the ligand separately. This might be especially ‘important for macrocycles, which have lots of flexible bonds and have a vast conformational space. A ‘multiple trajectory protocol’ where all the three end-states are simulated separately might have been a better approach.
Additionally, we ran a 14 ns long MD for generating structures for MM-GBSA calculations, which might not have been necessary. Running MM-GBSA on single minimized structures or very short MD trajectories ( < 1 ns long) have been shown to be equally efficient as long MD trajectories [51,53], saving both time and cost in our workflow.
Conclusions
The 2018 Grand Challenge was a great learning opportunity, especially since this was the first challenge in which the majority of the dataset consisted of macrocycles of different sizes and charges. Macrocycles are difficult to model because of the presence of a large number of flexible bonds and conformational restraints. The free energy prediction component was particularly challenging because the dataset involved both macrocycles of different sizes and also a few linear ligands. Breaking macrocycles requires special approximations to get good convergences in free energy calculations [41]. In addition to this, the active site of the BACE-1 target is also challenging in nature because of the presence of the aspartyl dyad.
The challenge helped us to identify many of the short-comings in our pose prediction protocol, as well as several ways in which we can improve it. We could have potentially made our protocol less tedious by minimizing the docked poses from HYBRID and using better similarity metrics for identifying reference ligands from available co-crystal PDB structures. Using better macrocylic conformer sampling and protein protonation tools can potentially improve the pose prediction accuracy.
A strong point for our protocol was using ligand similarity for pose prediction. At the same time, it is also a limitation. Since BACE-1 inhibitors have been widely studied and there exists a substantial amount of deposited co-crystal structures of them, we were able to find reference ligands with very high molecular similarity to our query ligands in this work. Researchers might not have this edge for newly discovered protein targets or ligands with unique scaffolds. Methods like FRED [6], AutoDock Vina and Dock6 which do not require any reference ligand might be more well-suited for those cases.
Our pose prediction protocol includes human expertise. Many past challenges (including D3R and SAMPL)[15] have had greater success with manual workflows than fully automated ones, and this has significant similarity to how modeling tools are still often employed in the pharmaceutical industry. Thus, one major challenge for the field is to progress to where fully automated workflows outperform manual ones.
In this work, MM-GBSA performed poorly for both discriminating between different binding poses and for affinity ranking. This may be due to the challenging BACE-1 active site or the difficulty of the ligand series, consisting of macrocycles with different net charges or both. One could also explore using only static minimized structures or ‘multiple trajectory protocol’ for improving MM-GBSA performance.
To sum up, we used ligand similarity to predict binding conformations for the cross-docking and self-docking challenge and MM-GBSA scores to rank ligand affinities. Our ligand similarity-based protocol was able to identify binding poses close to the native poses with an accuracy of 2 Å for most of the ligands. On the other hand, our predicted MM-GBSA scores had a weak correlation with the experimental affinities.
Supplementary Material
Fig. 2.

A schematic description of our ligand similarity-based protocol used for pose prediction in Stage 1a. For Stage 1b, we directly used the receptor structures provided by the organizers to dock the ligands using HYBRID. The down arrow represents the input files provided by the D3R GC4 organizers and the up arrow represents our pose prediction submissions.
Fig. 5.

Comparison of RMSD of ligand binding poses before and after MD in Stage 1b. Overall, the binding poses after MD have higher RMSD values than the poses selected before MD: the RMSD values of 9 ligands either improved or did not change significantly after MD and the RMSD values of the remaining 11 ligands changed significantly after MD.
Acknowledgements
We particularly appreciate Christopher I. Bayly (OpenEye Scientific Software) for his insight on BACE-1 protonation states and different modeling techniques. We would like to thank Open-Eye Scientific Software for providing us (via an academic license) with many of the pieces of software used in this work. Molecular graphics used in the paper were generated using Chimera (University of California, San Francisco) [24]. LEK and SS thanks Caitlin Bannan for helpful discussions on pKa of small molecules. SS also thanks Mark McGann (OpenEye Scientific Software) for discussing macrocycle conformer generation using OMEGA. We also appreciate financial support from the National Institutes of Health (1R01GM108889-01 and 1R01GM124270-01A1). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplementary Materials
The Supporting Information is available free of charge on https://github.com/MobleyLab/D3R_2018 and includes all the analysis scripts and input files used for this work. We have also provided Table 2, S1, S2 and our BACE-1 co-crystal PDB database in csv format in the github repository.
Contributor Information
Sukanya Sasmal, Department of Pharmaceutical Sciences, University of California, Irvine..
Léa El Khoury, Department of Pharmaceutical Sciences, University of California, Irvine..
David L. Mobley, Departments of Pharmaceutical Sciences and Chemistry, University of California, Irvine.
References
- 1.Sousa SF, Fernandes PA, Ramos MJ, Proteins Struct Funct Bioinf 65(1), 15 (2006). DOI 10.1002/prot.21082 [DOI] [PubMed] [Google Scholar]
- 2.Gilson MK, Zhou HX, Annu Rev Biophys Biomol Struct 36 (2007). DOI 10.1146/annurev.biophys.36.040306.132550 [DOI] [PubMed] [Google Scholar]
- 3.Chodera JD, Mobley DL, Shirts MR, Dixon RW, Branson K, Pande VS, Curr Opin Struct Biol 21(2), 150 (2011). DOI 10.1016/j.sbi.2011.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC, Nat Neurosci 4(3), 233 (2001). DOI 10.1038/85064 [DOI] [PubMed] [Google Scholar]
- 5.Toulokhonova L, Metzler WJ, Witmer MR, Copeland RA, Marcinkeviciene J, J Biol Chem 278(7), 4582 (2003). DOI 10.1074/jbc.M210471200 [DOI] [PubMed] [Google Scholar]
- 6.McGann M, J Comput Aided Mol Des 26(8), 897 (2012). DOI 10.1007/s10822-012-9584-8 [DOI] [PubMed] [Google Scholar]
- 7.Genheden S, Ryde U, Expert Opin Drug Discov 10(5), 449 (2015). DOI 10.1517/17460441.2015.1032936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trott O, Olson AJ, J Comp Chem 31(2), 455 (2010). DOI 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, Rizzo RC, J Comp Chem 36(15), 1132 (2015). DOI 10.1002/jcc.23905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maggiora G, Vogt M, Stumpfe D, Bajorath J, J Med Chem 57(8), 3186 (2013). DOI 10.1021/jm401411z [DOI] [PubMed] [Google Scholar]
- 11.Fukunishi Y, Nakamura H, J Mol Graph Model 26(6), 1030 (2008). DOI 10.1016/j.jmgm.2007.07.001 [DOI] [PubMed] [Google Scholar]
- 12.Kelley BP, Brown SP, Warren GL, Muchmore SW, J Chem Inf Model 55, 1771 (2015). DOI 10.1021/acs.jcim.5b00142 [DOI] [PubMed] [Google Scholar]
- 13.Wu G, Vieth M, J Med Chem 47(12), 3142 (2004). DOI 10.1021/jm040015y. PMID: 15163194 [DOI] [PubMed] [Google Scholar]
- 14.Kumar A, Zhang KY, J Comput Aided Mol Des 0(0), 0 (2018). DOI 10.1007/s10822-018-0142-x [DOI] [Google Scholar]
- 15.Gaieb Z, Parks CD, Chiu M, Yang H, Shao C, Walters WP, Lambert MH, Nevins N, Bembenek SD, Ameriks MK, Mirzadegan T, Burley SK, Amaro RE, Gilson MK, J Comput Aided Mol Des 0(0), 0 (2019). DOI 10.1007/s10822-018-0180-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilliland G, Berman HM, Weissig H, Shindyalov IN, Westbrook J, Bourne PE, Bhat TN, Feng Z, Nucleic Acids Res 28(1), 235 (2000). DOI 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kortum SW, Benson TE, Bienkowski MJ, Emmons TL, Prince DB, Paddock DJ, Tomasselli AG, Moon JB, LaBorde A, TenBrink RE, Bioorg Med Chem Lett 17(12), 3378 (2007). DOI 10.1016/j.bmcl.2007.03.096 [DOI] [PubMed] [Google Scholar]
- 18.Clarke B, Demont E, Dingwall C, Dunsdon R, Faller A, Hawkins J, Hussain I, MacPherson D, Maile G, Matico R, Milner P, Mosley J, Naylor A, OäĂŹBrien A, Redshaw S, Riddell D, Rowland P, Soleil V, Smith KJ, Stanway S, Stemp G, Sweitzer S, Theobald P, Vesey D, Walter DS, Ward J, Wayne G, Bioorg Med Chem Lett 18(3), 1017 (2008). DOI 10.1016/j.bmcl.2007.12.019 [DOI] [PubMed] [Google Scholar]
- 19.Machauer R, Laumen K, Veenstra S, Rondeau JM, Tintelnot-Blomley M, Betschart C, Jaton AL, Desrayaud S, Staufenbiel M, Rabe S, Paganetti P, Neumann U, Bioorg Med Chem Lett 19(5), 1366 (2009). DOI 10.1016/j.bmcl.2009.01.055 [DOI] [PubMed] [Google Scholar]
- 20.Lerchner A, Machauer R, Betschart C, Veenstra S, Rueeger H, McCarthy C, Tintelnot-Blomley M, Jaton AL, Rabe S, Desrayaud S, Enz A, Staufenbiel M, Paganetti P, Rondeau JM, Neumann U, Bioorg Med Chem Lett 20(2), 603 (2010). DOI 10.1016/j.bmcl.2009.11.092 [DOI] [PubMed] [Google Scholar]
- 21.Sandgren V, Agback T, Johansson PO, Lindberg J, Kvarnström I, Samuelsson B, Belda O, Dahlgren A, Bioorg Med Chem 20(14), 4377 (2012). DOI 10.1016/j.bmc.2012.05.039 [DOI] [PubMed] [Google Scholar]
- 22.Coburn CA, Stachel SJ, Jones KG, Steele TG, Rush DM, DiMuzio J, Pietrak BL, Lai MT, Huang Q, Lineberger J, Jin L, Munshi S, Katharine Holloway M, Espeseth A, Simon A, Hazuda D, Graham SL, Vacca JP, Bioorg Med Chem Lett 16(14), 3635 (2006). DOI 10.1016/j.bmcl.2006.04.076 [DOI] [PubMed] [Google Scholar]
- 23.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA, Nucleic Acids Res 32(suppl_2), W665 (2004). DOI 10.1093/nar/gkh381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, J Comp Chem 25(13), 1605 (2004). DOI 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 25.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT, J Chem Inf Model 50(4), 572 (2010). DOI 10.1021/ci100031x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS, J Med Chem 49(20), 5912 (2006). DOI 10.1021/jm050362n [DOI] [PubMed] [Google Scholar]
- 27.Ellis CR, Tsai CC, Hou X, Shen J, J Phys Chem Lett 7(6), 944 (2016). DOI 10.1021/acs.jpclett.6b00137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Case D, Brozell S, Cerutti D, Cheatham TI, Cruzeiro V, Darden T, Duke R, Ghoreishi D, Gohlke H, Goetz A, Greene D, Harris R, Homeyer N, Izadi S, Kovalenko A, Lee T, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Mermelstein D, Merz K, Miao Y, Monard G, Nguyen H, Omelyan I, Onufriev A, Pan F, Qi R, Roe D, Roitberg A, Sagui C, Schott-Verdugo S, Shen J, Simmerling C, Smith J, Swails J, Walker R, Wang J, Wei H, Wolf R, Wu X, Xiao L, York D, Kollman P. Amber 2018, university of california, san francisco: (2018) [Google Scholar]
- 29.Case D, Cerutti D, Cheateham T, Darden T, Duke R, Giese T, Gohlke H, Goetz A, Greene D, Homeyer N, Simmerling C, Botello-Smith W, Swail J, Walker R, Wang J, Wolf R, Wu X, Xiao L, Kollman P. Amber 2016, university of california, san francisco: (2016) [Google Scholar]
- 30.Jakalian A, Jack DB, Bayly CI, J Comput Chem 23(16), 1623 (2002). DOI 10.1002/jcc.10128 [DOI] [PubMed] [Google Scholar]
- 31.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C, Proteins Struct Funct Bioinf 65, 712 (2006). DOI 10.1002/prot.21123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA, J Comp Chem 25(9), 1157 (2004). DOI 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, J Chem Phys 79(2), 926 (1983). DOI 10.1063/1.445869 [DOI] [Google Scholar]
- 34.Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE, J Chem Theory Comput 8(9), 3314 (2012). DOI 10.1021/ct300418h [DOI] [PubMed] [Google Scholar]
- 35.Nguyen H, Roe DR, Simmerling C, J Chem Theory Comput 9(4), 2020 (2013). DOI 10.1021/ct3010485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu K, Kokubo H, J Chem Inf Model 57(10), 2514 (2017). DOI 10.1021/acs.jcim.7b00412 [DOI] [PubMed] [Google Scholar]
- 37.Kaus JW, Harder E, Lin T, Abel R, McCammon JA, Wang L, J Chem Theory Comput 11(6), 2670 (2015). DOI 10.1021/acs.jctc.5b00214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ, J Comput Chem 30(16), 2785 (2009). DOI 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DesJarlais RL, Sheridan RP, Seibel GL, Dixon JS, Kuntz ID, Venkataraghavan R, J Med Chem 31(4), 722 (1988). DOI 10.1021/jm00399a006 [DOI] [PubMed] [Google Scholar]
- 40.Meng EC, Shoichet BK, Kuntz ID, J Comput Chem 13(4), 505 (1992). DOI 10.1002/jcc.540130412 [DOI] [Google Scholar]
- 41.Yu HS, Deng Y, Wu Y, Sindhikara D, Rask AR, Kimura T, Abel R, Wang L, J Chem Theory Comput 13(12), 6290 (2017). DOI 10.1021/acs.jctc.7b00885 [DOI] [PubMed] [Google Scholar]
- 42.Plewczynski D, Łaźniewski M, Augustyniak R, Ginalski K, J Comput Chem 32(4), 742 (2011). DOI 10.1002/jcc.21643 [DOI] [PubMed] [Google Scholar]
- 43.Poongavanam V, Danelius E, Peintner S, Alcaraz L, Caron G, Cummings MD, Wlodek S, Erdelyi M, Hawkins PCD, Ermondi G, Kihlberg J, ACS Omega 3(9), 11742 (2018). DOI 10.1021/acsomega.8b01379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V, Nature 402(6761), 537 (1999). DOI 10.1038/990114 [DOI] [PubMed] [Google Scholar]
- 45.Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, Nukina N, Mol Cell Biol 28(11), 3663 (2008). DOI 10.1128/MCB.02185-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mermelstein DJ, McCammon JA, Walker RC, J Mol Recognit 32(3), e2765 (2019). DOI 10.1002/jmr.2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ellis CR, Shen J, J Am Chem Soc 137(30), 9543 (2015). DOI 10.1021/jacs.5b05891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim MO, Blachly PG, McCammon JA, PLoS Comput Biol 11(10), 1 (2015). DOI 10.1371/journal.pcbi.1004341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH, J Chem Theory Comput 7(2), 525 (2011). DOI 10.1021/ct100578z [DOI] [PubMed] [Google Scholar]
- 50.El Khoury L, Santos-Martins D, Sasmal S, Eberhardt J, Bianco G, Ambrosio F, Solis-Vasquez L, Koch A, Mobley DL, Forli S, ChemRxiv (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rastelli G, Del Rio A, Degliesposti G, Sgobba M, J Comput Chem 31(4), 797 (2010). DOI 10.1002/jcc.21372 [DOI] [PubMed] [Google Scholar]
- 52.Lyne PD, Lamb ML, Saeh JC, J Med Chem 49(16), 4805 (2006). DOI 10.1021/jm060522a [DOI] [PubMed] [Google Scholar]
- 53.Su PC, Tsai CC, Mehboob S, Hevener KE, Johnson ME, J Comput Chem 36(25), 1859 (2015). DOI 10.1002/jcc.24011. URL https://onlinelibrary.wiley.com/doi/abs/10.1002/jcc.24011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Réau M, Langenfeld F, Zagury JF, Montes M, J Comput Aided Mol Des 32(1), 231 (2018). DOI 10.1007/s10822-017-0063-0. URL 10.1007/s10822-017-0063-0 [DOI] [PubMed] [Google Scholar]
- 55.Misini Ignjatović M, Caldararu O, Dong G, Muñoz-Gutierrez C, Adasme-Carreño F, Ryde U, J Comput Aided Mol Des 30(9), 707 (2016). DOI 10.1007/s10822-016-9942-z. URL 10.1007/s10822-016-9942-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salmaso V, Sturlese M, Cuzzolin A, Moro S, J Comput Aided Mol Des 32(1), 251 (2018). DOI 10.1007/s10822-017-0051-4. URL 10.1007/s10822-017-0051-4 [DOI] [PubMed] [Google Scholar]
- 57.Shirts MR, Mobley DL, Brown SP, Drug design: structure-and ligand-based approaches pp. 61–86 (2010) [Google Scholar]
- 58.Mikulskis P, Genheden S, Rydberg P, Sandberg L, Olsen L, Ryde U, J Comput Aided Mol Des 26(5), 527 (2012). DOI 10.1007/s10822-011-9524-z. URL 10.1007/s10822-011-9524-z [DOI] [PubMed] [Google Scholar]
- 59.Thompson DC, Humblet C, Joseph-McCarthy D, J Chem Inf Model 48(5), 1081 (2008). DOI 10.1021/ci700470c. URL 10.1021/ci700470c. PMID: 18465849 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.