

Abstract
The conversion of adenosine to inosine (A to I) by RNA editing represents a common posttranscriptional mechanism for diversification of both the transcriptome and proteome, and is a part of the cellular response for innate immune tolerance. Due to its preferential base-pairing with cytosine (C), inosine (I) is recognized as guanosine (G) by reverse transcriptase, as well as the cellular splicing and translation machinery. A-to-I editing events appear as A-G discrepancies between genomic DNA and cDNA sequences. Molecular analyses of RNA editing have leveraged these nucleoside differences to quantify RNA editing in ensemble populations of RNA transcripts and within individual cDNAs using high-throughput sequencing or Sanger sequencing-derived analysis of electropherogram peak heights. Here, we briefly review and compare these methods of RNA editing quantification, as well as provide experimental protocols by which such analyses may be achieved.
Keywords: Sanger sequencing, High-throughput sequencing, RNA processing, Posttranscriptional modifications, Illumina, Electropherogram
1. Introduction
The conversion of adenosine to inosine (A to I) by RNA editing is a posttranscriptional mechanism for increasing the diversity of the proteome and for modulating innate immune tolerance for host double-stranded RNAs formed by endogenous sequences [1, 2]. Common methods for the detection and quantification of A-to-I modifications have exploited the preferential base pairing of inosine with cytosine during reverse transcription (RT) to generate cDNA libraries or polymerase chain reaction (PCR) amplicons containing guanosine at the edited positions. These apparent guanosine (G) substitutions can be detected by various DNA sequencing methodologies. Pioneering studies on the RNA editing of transcripts encoding the GluA2 subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) subtype of ionotropic glutamate receptor and the 2C subtype of serotonin receptor (5HT2C) determined editing levels using molecular cloning and Sanger sequencing strategies [3, 4]. RT-PCR amplicons were subcloned into plasmid or phage vectors, transformed or transduced into microbial hosts, and then purified from single clonal colonies or plaques before being subjected to Sanger sequencing. Although such clone-based sequencing provided information about the relative proportion of distinct RNA isoforms, the accuracy and precision of this approach was limited by the number of clones sequenced.
Next-generation sequencing (NGS) revolutionized almost all aspects of modern molecular biology and genetics, permitting more efficient, sensitive, and accurate identification and quantification of transcriptome-wide RNA editing events. Pyrosequencing emerged as the first major NGS technology, but was later supplanted by Illumina NGS platforms. Illumina platforms rely on sequencing by synthesis chemistry that effectively eliminates the erroneous base calls in homopolymeric sequences that were a common pitfall of pyrosequencing [5]. A comparison of murine 5HT2C RNA editing profiles generated by each of these NGS technologies revealed decreased accuracy and greater experimental variability for pyrosequencing than Illumina sequencing [6]. As the most widely adopted NGS technology, Illumina platforms have been used in whole-transcriptome analyses as well as targeted RNA-sequencing studies. Although transcriptome profiling has been useful for identifying novel RNA editing sites, more targeted approaches increase the sequencing depth of transcripts expressed at low levels and therefore are better suited to accurately quantify editing of specific sites [7, 8]. High-throughput strategies for targeted RNA sequencing leverage single- or dual-indexed library preparations to simultaneously quantify the editing of many editing targets across multiple samples [7, 9].
Today, direct sequencing of RT-PCR products, by either Sanger sequencing or NGS, is the most common method of assessing RNA editing profiles [10]. Quantification of editing by NGS data analysis requires a bioinformatic pipeline involving a comparison of target sequences to edited and non-edited reference sequences [11, 12]. RNA editing can also be determined from Sanger sequencing-derived electropherograms where edited positions appear as mixed A/G peaks, and the relative height of each peak is proportional to the number of transcripts containing A or G at that position within the complex RNA population of a given sample [13, 14]. As it is not possible to use Sanger sequencing to differentiate between editing permutations when a transcript contains more than a single editing site, it is often used as an orthogonal approach to validate NGS analysis [8]. Here we describe and compare these methods for the quantification of RNA editing.
2. Materials
All reagents should be of molecular biology grade and prepared using nuclease-free water (seeNote 1). To minimize RNA degradation and PCR contamination, thoroughly and regularly clean laboratory benchtops and equipment with RNaseZap™ and DNA AWAY (seeNote 2), autoclave glassware, use nuclease-free and sterile filter tips on pipettes, and wear gloves while handling reagents. Place RNA samples on ice during reaction setup and at −80 °C for long-term storage. Thaw RNA samples on ice. All biohazardous materials should be disposed properly.
Although the methods described below are broadly applicable to quantify the editing of many sites that have been computationally or experimentally identified across different transcripts, cell types, and species, this example focuses on five editing events within serotonin 2C receptor transcripts isolated from the hypothalami of C57Bl/6J mice.
2.1. RNA Isolation
Invitrogen™ TRIzol™ Reagent.
Ultrasonic homogenizer.
Chloroform.
Isopropanol.
75% Ethanol.
Invitrogen™ TURBO™ DNase kit.
Thermo Scientific™ Nanodrop™.
2.2. RT-PCR
Applied Biosystems™ High-Capacity cDNA Reverse Transcription kit: 10× RT buffer, 100 mM dNTP mix, 10× RT random primers, MultiScribe™ Reverse Transcriptase, RNase inhibitor.
2× Phusion® High-Fidelity Mastermix with HF Buffer.
Sense and antisense target-specific primers: 10 μM Stocks of each diluted in water.
Sense and antisense universal primers: 10 μM Stocks of each diluted in water.
Exonuclease I (20,000 units/mL).
20× Sodium borate (SB) buffer: Measure 32 g NaOH and 220 g boric acid, and add water to a volume of 4 L.
Agarose gel: 1% Agarose diluted in 1× SB buffer, with 0.2–0.5 μg/mL ethidium bromide.
6× DNA gel loading dye (containing bromophenol blue and xylene cyanol FF).
Promega Wizard® SV Gel and PCR Clean-Up System.
2.3. Quantification of RNA editing
2.3.1. Analysis of Peak Heights from Sanger Sequencing Chromatograms
QSVanalyzer is a software application that provides relative peak height information for sequence variants from SCF, ABI, and Megabace trace files [15]. The application runs on Microsoft Windows using the Microsoft.NET Framework version 2.. Minimal hard disk space is required as the size of the application is 248 kb, and the total size of the output files generated in this example is about 300 kb. QSVanalyzer can be obtained from the Leeds Institute of Molecular Medicine website (http://dna-leeds.co.uk/qsv/download.php).
2.3.2. High-Throughput Sequencing Analysis
We developed a workflow for processing high-throughput sequencing data using commodity hardware and a Linux/UNIX operating system, including CentOS7 and Ubuntu 18.4. The workflow is written in Python 2.7 and leverages SeqKit [16] and FqTools [17]. To facilitate software environment management, we recommend using Anaconda2 to manage the required packages, including the aforementioned tools that are available via the Bioconda channel. To create the anaconda environment, first install Anaconda2 via https://www.anaconda.com/.
Once installed, open a terminal and execute the following commands to build the environment (note that the %> indicates the command-line prompt and # indicates the start of a comment):
%> conda create --name rna_editing python=2.7 # Create the conda environment
%> conda activate rna_editing # Activate the environment
%> conda install -c bioconda seqkit # Install SeqKit
%> conda install -c bioconda fqtools # Install FqTools
%> conda install -c bioconda fastx_toolkit # Install fastx_toolkit
%> pip install pandas matplotlib jupyter # Install packages using PIP
Alternatively, an Anaconda2 environment file is available at https://gitlab.com/jpcartailler/analysis-of-rna-editing-hts-data/blob/master/rna_editing.yaml which can be used via the following command to reproduce the environment:
%> conda env create -f rna_editing.yaml
3. Methods
3.1. RNA Isolation
Collect tissue sample in a 1.5 mL polypropylene tube and add 1 mL of Trizol reagent per 50–100 mg of tissue (seeNote 3).
Homogenize the sample in TRIzol reagent by sonication at ~140 W for 2–10 s (seeNote 4).
Add 200 μL of chloroform (per 1 mL of TRIzol reagent used in step 1) to the homogenized sample and shake the tube vigorously for 15 s; incubate for 2–3 min.
Centrifuge the sample at 12,000 × g for 15 min at 4 °C. The sample will separate into three distinct phases upon centrifugation.
Carefully remove the sample from the centrifuge without disturbing phases. Using a P200 micropipette, aspirate only the aqueous (top) layer and transfer the liquid to a new 1.5 mL polypropylene tube (seeNote 5). Discard the organic (pink) and interphase (white) layers.
To the isolated aqueous layer, add 0.5 mL isopropanol (per 1 mL of TRIzol reagent used in step 1). Mix thoroughly by inverting the tube 3–5 times. Incubate at room temperature for 10 min.
Centrifuge the sample at 12,000 × g for 10 min at 4 °C to precipitate the RNA.
Carefully decant or aspirate the isopropanol away from the RNA pellet. Resuspend the pellet with 1 mL 75% ethanol (per 1 mL of TRIzol reagent used in step 1), and vortex briefly for 1–2 s.
Centrifuge the sample at 12,000 × g for 5 min at 4 °C.
Carefully aspirate the ethanol away from the RNA pellet.
Allow the RNA pellet to dry for 5 min, and then use a P10 micropipette to carefully aspirate any residual ethanol, if necessary.
Resuspend the RNA in 20–50 μL of nuclease-free water.
Using a Nanodrop™, measure the concentration of the RNA sample.
Dilute an aliquot of the sample to 200 ng/μL (seeNote 6).
Add one-tenth sample volume of TURBO DNase™ buffer. Add 1 μL of TURBO DNase™ per 50 μL reaction and disperse by gently flicking the side of the tube. Incubate the reaction at 37 °C for 30 min.
Add one-fifth volume of DNase inactivation reagent, and disperse by flicking the tube once per minute for 5 min.
Centrifuge at 10,000 × g for 2 min.
Using a P10 micropipette, carefully aspirate the RNA sample without collecting any of the pelleted inactivation reagent and transfer into a new 1.5 mL polypropylene tube.
3.2. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
3.2.1. Reverse Transcription
In a 0.2 mL PCR strip tube, prepare a 20 μL reverse transcription reaction according to Table 1 (seeNote 7).
Table 1.
Reverse transcription reaction setup
| Reagent | Volume (μL) | |
|---|---|---|
| + Reverse transcriptase (RT+) | − Reverse transcriptase (RT−) | |
| 10× RT buffer | 2 | 2 |
| 100 mM dNTP mix | 0.8 | 0.8 |
| 10× RT random primers | 2 | 2 |
| MultiScribe™ Reverse Transcriptase | 1 | 0 |
| RNase inhibitor | 1 | 1 |
| RNA (200–1000 ng) | Variable | Variable |
| Water | to 20 μL | to 20 μL |
Table 2.
Reverse transcription thermocycling protocol
| Step # | Temperature (°C) | Time |
|---|---|---|
| 1 | 25 | 10 min |
| 2 | 37 | 60 min |
| 3 | 85 | 5 min |
| 4 | 4 | ∞ |
3.2.2. Polymerase Chain Reaction
If possible, design target-specific sense and antisense primers flanking an edit site in separate exons to avoid amplification of genomic DNA and to generate amplicons between 75 and 300 base pairs in length (seeNote 9). For multiplex analyses of multiple RNA samples, add unique sequences to the 5′-ends of the target-specific sense and antisense primers during primer design. This approach allows amplification of each transcript with only two target-specific primers, followed by amplification with a set of “universal” primers allowing incorporation of index sequences and adapter sequences required for the Illumina NGS platform. In this example, we add T3 (ATTAACCCTCACTAAAGGGA) and T7 (TAATACGACTCACTATAGGG) RNA polymerase promoter sequences to the 5′-ends of the sense and antisense primers, respectively.
Design universal primers that include one of the two Illumina adapter sequences at the 5′-end, followed by a unique 6-nucleotide barcode, and sequences complementary to either T3 or T7 RNA polymerase promoter at the 3′-end (Fig. 1).
In a PCR strip tube, prepare a PCR reaction according to Table 3.
Run thermocycling program according to Table 4.
Remove unincorporated primers by adding 1 μL exonuclease I, and incubate at 37 °C for 20 min.
Incubate the reaction at 80 °C for 20 min to inactivate exonuclease I.
Add 1 μL each of 10 μM sense and antisense universal primers.
Run thermocycling program according to Table 5 (seeNote 10).
Fig. 1.
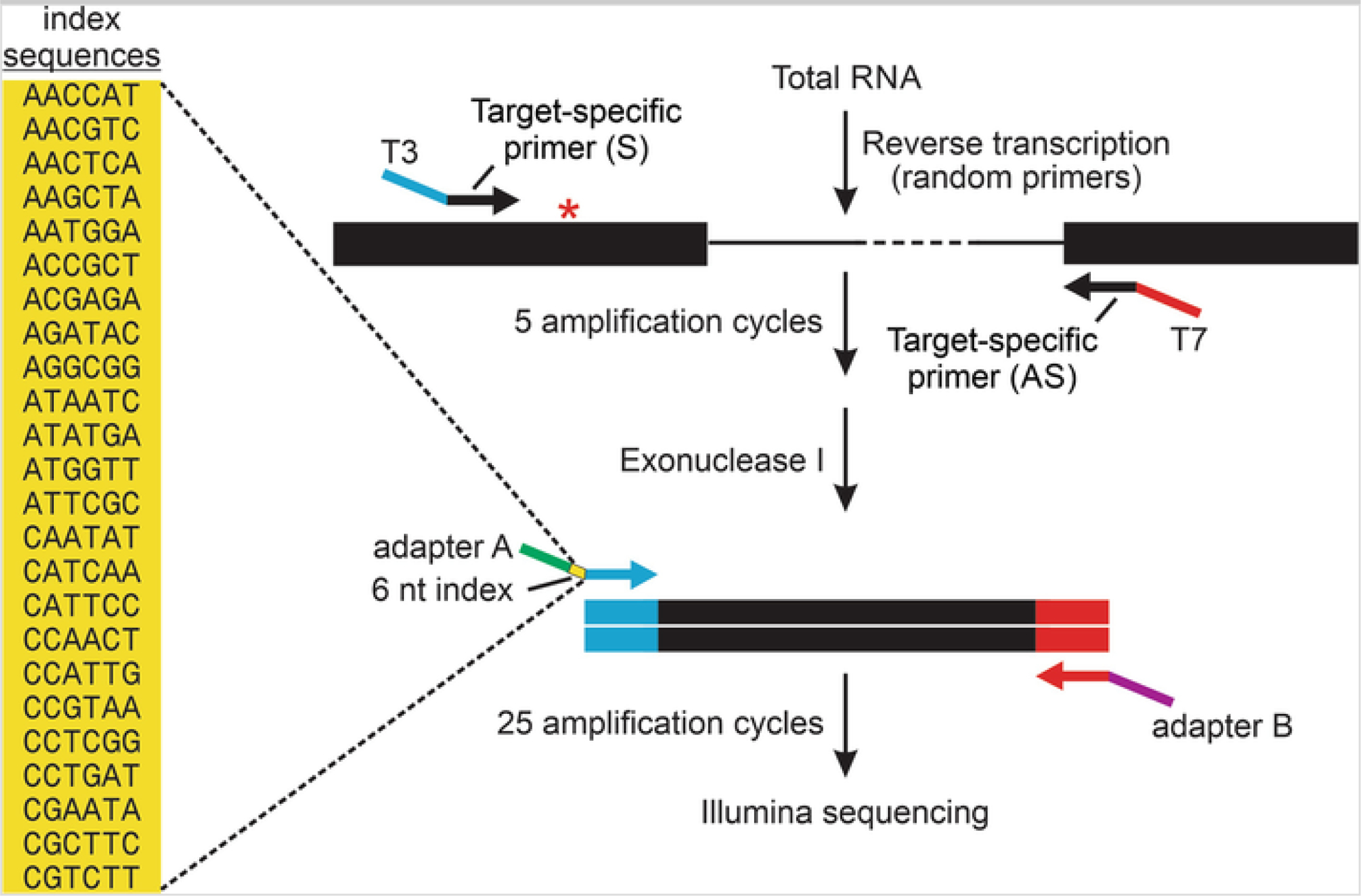
Next-generation sequencing strategy for multiplex quantification of RNA editing profiles. A schematic diagram is presented for RT-PCR amplification of a region of an mRNA target flanking an A-to-I editing site (asterisk). In general, target-specific primers are designed in adjacent exons containing either T3 (blue) or T7 (red) RNA polymerase promoter sequence extensions for the sense and antisense primers, respectively. These primers are used for PCR amplification (5 cycles) before digestion of the remaining single-stranded primers using exonuclease I. A second round of amplification (25 cycles) is performed with universal primers in which the oligonucleotide contains sequences matching the T3 promoter, one of 24 unique 6-nt index sequences (yellow) for sample identification, as well as an adapter sequence (Adapter A; green) or sequences matching the T7 and an adapter sequence (Adapter B; purple) for library preparation and high-throughput single-end sequencing on the Illumina platform
Table 3.
PCR setup for amplification using target-specific primers
| Reagent | Volume (μL) |
|---|---|
| Target-specific sense primer (10 μM) | 1 |
| Target-specific antisense primer (10 μM) | 1 |
| 2× Phusion® High-Fidelity Mastermix with HF Buffer | 25 |
| cDNA | 5 |
| Water | 18 |
Table 4.
Thermocycling protocol for PCR using target-specific primers
| Step # | Temperature (°C) | Time |
|---|---|---|
| 1 | 98 | 30 s |
| 2 | 98 | 10 s |
| 3 | 55 | 30 s |
| 4 | 72 | 45 s |
| 5 | Repeat steps 2–4 | 4× |
| 6 | 4 | ∞ |
Table 5.
Thermocycling protocol for PCR using universal primers
| Step # | Temperature (°C) | Time |
|---|---|---|
| 1 | 98 | 15 s |
| 2 | 56 | 30 s |
| 3 | 72 | 60 s |
| 4 | Repeat steps 1–3 | 24× |
| 5 | 4 | ∞ |
3.2.3. Gel Electrophoresis and Clean-Up
Add DNA gel loading dye to the PCR reaction and mix well by pipetting up and down several times.
Load the entire reaction onto a 1% agarose gel and resolve samples by electrophoresis in 1× SB buffer at 300 V until bottom dye band (bromophenol blue) migrates off the bottom of the gel.
Wearing amber-filtered goggles, image the gel using a blue-light DNA transilluminator. Check that the amplicon is of the correct size and that no bands are present in the RT− samples (indicative of PCR contamination), and then carefully use a single-edge razor blade to cut out the agarose containing the desired amplicon(s).
Purify the amplicons using Promega Wizard® SV Gel and PCR Clean-Up System according to the manufacturer’s instructions.
3.3. Sequencing
Sanger sequence RT-PCR amplicon with reverse primer (seeNote 11). Output should be collected in the “.ab1” chromatographic file format.
Subject “library-ready” RT-PCR amplicons to Illuminahigh-throughput sequencing.
3.4. Quantification of RNA Editing
3.4.1. Analysis of Peak Heights from Sanger Sequencing Chromatograms
Create a folder containing all the .ab1 traces to be analyzed (seeNote 12).
Open a single .ab1 file from the folder created in the previous step by going to File > Open in QSVanalyzer.
Once the trace is opened, go to QSV analysis > Select nucleotide.
Click on the position of the editing site within the chromatogram trace. Edited positions will appear as T/C mixed peaks in the sequence of the antisense strand. The “QSV information” window will automatically appear. One of the peaks will be designated as “Gene A” and the other will be designated as “Gene B” (seeNote 13). Verify that the 5′- and 3′-flanking sequences are correct.
Select all six reference bases (seeNote 14). T/C peak heights at the edited position will be adjusted relative to the heights of the reference peaks.
If only a single editing site is to be analyzed, select Create web pages and images, click Add, and then press Run batch. If more than one position is to be analyzed, select Add, and then select Find. A “QSV image” window will automatically open showing an abbreviated trace with the 5′- and 3′-flanking sequences on either side of the editing site bisected by a vertical red line. Close the “QSV image” window, then close the “QSV information” window, and repeat steps 4–6 until all sites for analysis within the chromatogram have been added (seeNote 15). When all sites have been selected, select Create web pages and images and then press run batch in the “QSV information” window (seeNote 16). In the “Browse for folder” window that automatically opens, select the folder created in Step 1 and click OK. A “QSV_data” subfolder will be created. This subfolder contains a web page with peak height information for each editing site within the trace file folder (seeNote 17).
Go to the QSV_data subfolder and open the web page for each editing site (seeNote 18). For each trace, record the relative peak height data for “Variant A” and “Variant B” under the “Trace images and peak heights” section of the web page.
- Calculate the percentage of transcripts containing an edited nucleotide by using the peak heights obtained from step 7 as shown (seeNote 19):
3.4.2. High-Throughput Sequencing Analysis
Clone or download the software repository from https://gitlab.com/jpcartailler/analysis-of-rna-editing-hts-data. Enter this working directory (if set up, activate the anaconda environment).
Place your FastQ file in the data/directory and ensure that it is in GZIP format.
-
Enter the src/ directory and create a file called config.yaml, which contains all the configuration parameters expected from the workflow script. The “targets” represent specific genes targeted in the analysis, since more than one may have been sequenced and need to be demultiplexed. In this example, there is only one target:
paths:
data: ../path/to/sample.fastq.gz
results: ../results
targets:
5HT2C:
name: Mus musculus 5-hydroxytryptamine (serotonin) receptor 2C (mRNA)
analysis_criteria:
seqkit_degenerate: no # allow degenerates
seqkit_max_mismatch: 0 # no mismatch
sequences:
barcodes:
sample_1: AACGTC
sample_2: ATATGA
sample_3: CGTCTT
promoter: GCTGGACCGGTATGTAGCA
# reference -- sequence(s) of variants to search for within reads
reference:
# seq_help: editing site (A/G) indicated with |
# seq_help: |-|---||-----------------------------------------------------------
sequence_1: ATACGTAATCCTATTGAGCATAGCCGGTTCAATTCGCGGACTAAGGCCATCATGAA
sequence_2: GTACGTAATCCTATTGAGCATAGCCGGTTCAATTCGCGGACTAAGGCCATCATGAA
sequence_3: ATGCGTAATCCTATTGAGCATAGCCGGTTCAATTCGCGGACTAAGGCCATCATGAA (etc…)
sequence_32:GTGCGTGGTCCTGTTGAGCATAGCCGGTTCAATTCGCGGACTAAGGCCATCATGAA
# variant_position -- nucleotide position where RNA editing var…
variant_position: 13
# variant_extended_by_nt -- for exact matching, number of nucle…
variant_extended_by_nt: 20
-
Execute the workflow from the src/ directory:
%> python workflow.py
-
Then, execute the bash scripts generated from the workflow.py output:
%> bash workflow_step1.bash
%> bash workflow_step2.bash
-
The results/ directory will have files in .seq format, for each barcode and sequence target (in this case, 3 barcodes and 32 sequences per barcode):
results/job-01.5HT2C.AACGTC.sequence_1.seq
results/job-01.5HT2C.AACGTC.sequence_2.seq
results/job-01.5HT2C.AACGTC.sequence_3.seq
…
results/job-01.5HT2C.AACGTC.sequence_32.seq
-
Within the results/ directory, run the following command to quantify the number of reads per barcode-sequence combination:
%> wc -l results/statistics_barcode_search_exact/*.seq
These results can then be tabulated manually into a worksheet.
3.4.3. Comparison of Sanger Sequencing and NGS Strategies
In agreement with previous reports [18], the levels of RNA editing measured by peak height analysis and high-throughput sequencing do not differ significantly (Fig. 2). While Sanger sequencing-derived methods of RNA editing quantification can be used to confirm the extent of editing determined by high-throughput sequencing, the accuracy of peak height analysis may be compromised when sequencing chromatograms are noisy or when levels of editing are very low (<5%). High-throughput sequencing analysis offers an additional benefit over peak height analysis in that it can differentiate between editing permutations of multiple sites within a single RNA transcript (Fig. 3).
Fig. 2.
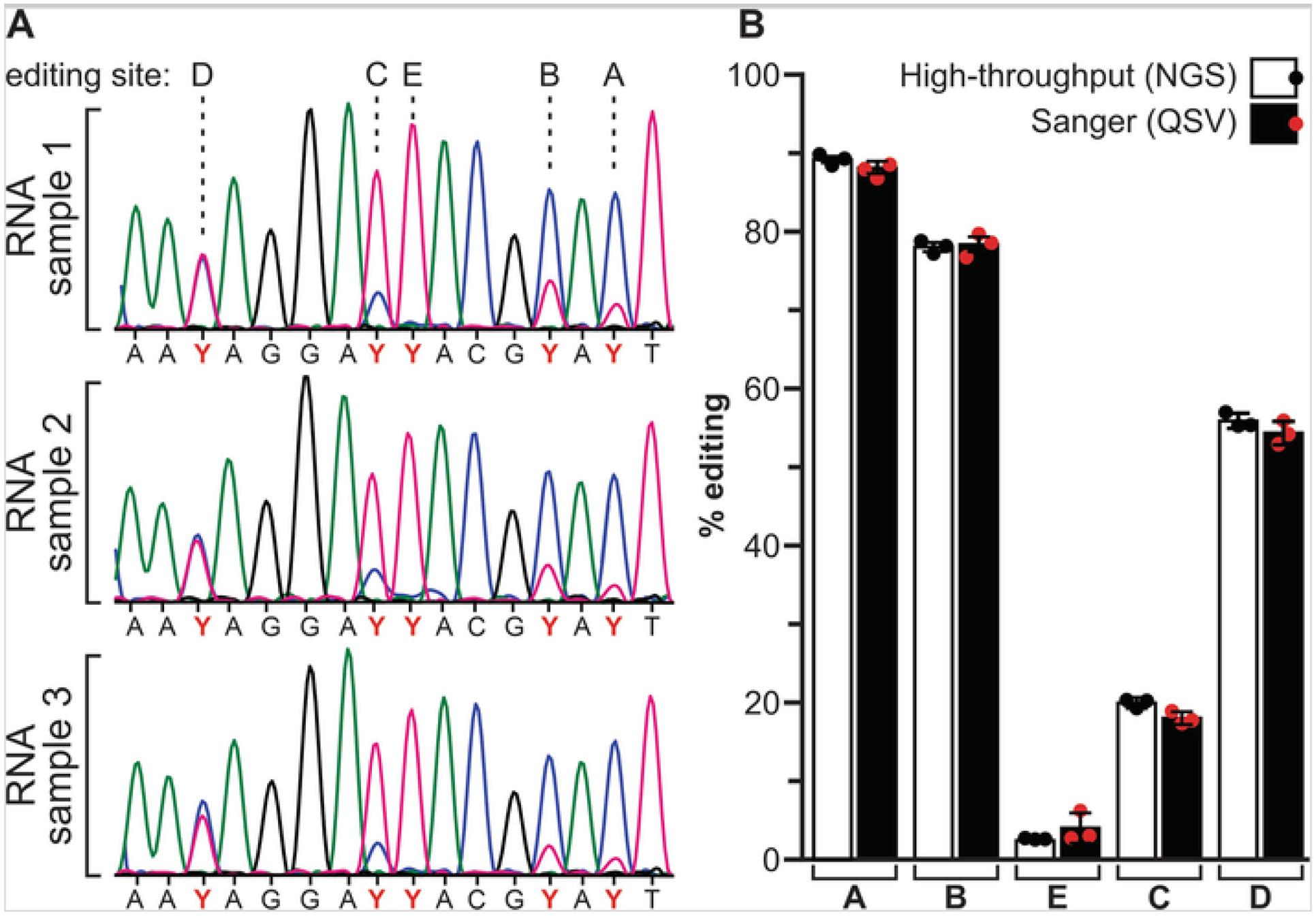
Analysis of hypothalamic 5HT2C RNA editing profiles in C57Bl/6J mice. (a) Representative electropherogram traces from Sanger sequencing of cDNAs generated from hypothalamic RNA (n = 3) are presented. The positions of the editing sites (A–E) are shown and the mixed T/C peaks from the antisense strand are designated as pyrimidine (Y). (b) Comparisons of RNA editing quantification using high-throughput (NGS) and Sanger (QSV) sequencing revealed no significant differences; mean ± SEM, n = 3
Fig. 3.
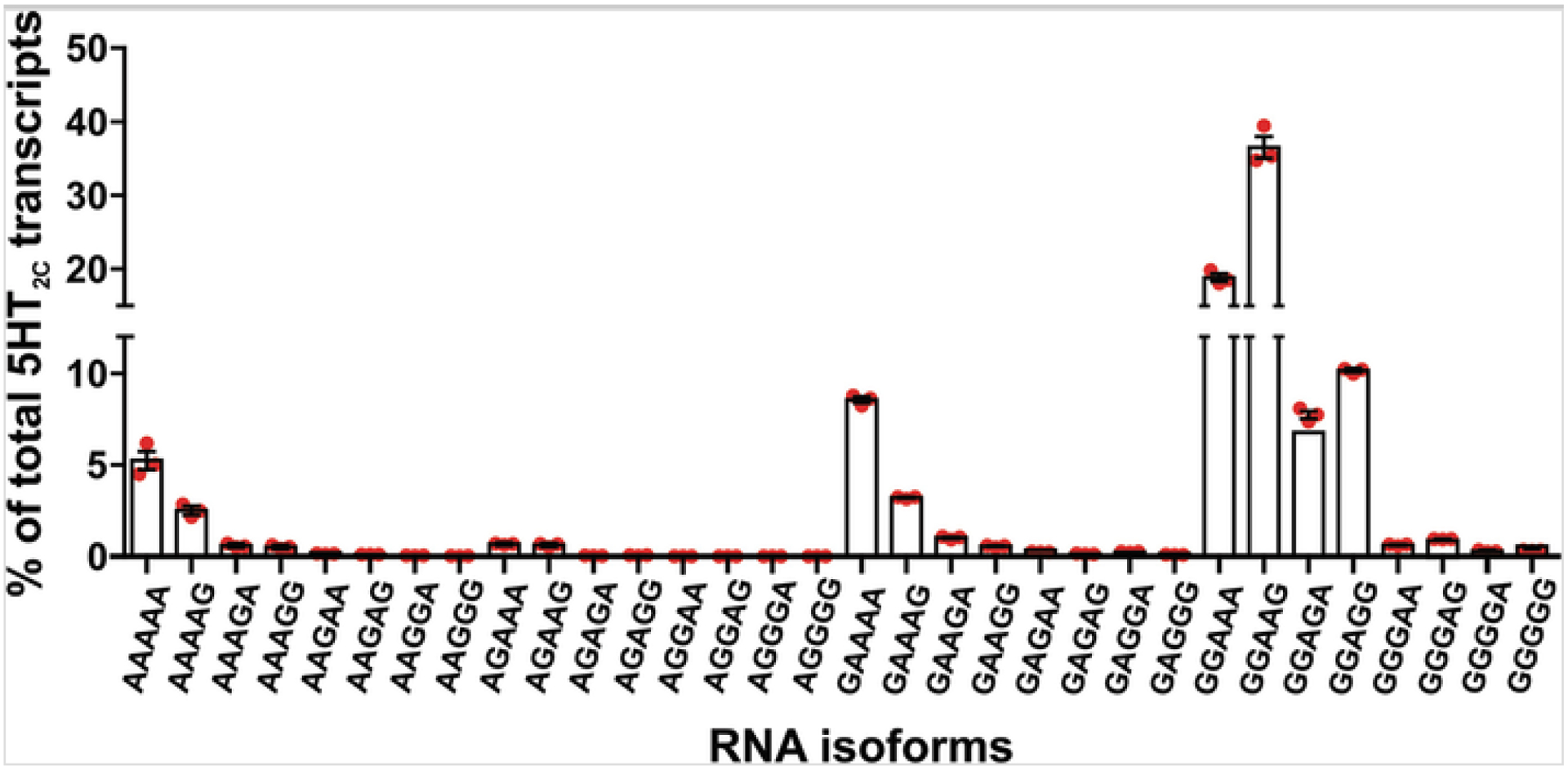
Quantification of 5HT2C RNA editing profiles in mouse hypothalamus using high-throughput sequencing. The relative expression for all possible 5HT2C isoforms generated by RNA editing is represented as the percentage of total 5HT2C sequence reads; mean ± SEM, n = 3. The 32 possible permutations of A-to-I editing are designated according to the presence of either an adenosine (A) or a guanosine (G) at each of the five distinct editing sites within the analyzed cDNA library (e.g., AAAAA = non-edited, GGGGG = fully edited)
4. Notes
To prepare all reagents, stocks, and reactions directly contacting nucleic acids (RNA samples, primer stocks, PCR reactions, etc.), we recommend using commercially available autoclaved, 0.22 μM sterile filtered, nuclease- and protease-free water. All other materials (SB buffer, agarose gels, etc.) can be prepared using high-quality laboratory water (e.g., Milli-Q® water).
To prevent RNA degradation and PCR contamination, we recommend working with RNA and DNA samples in a dedicated PCR hood equipped with a UV sterilizer. Do not work in the hood while the UV light is on.
For very small samples such as tissue punches, homogenize the sample in 300 μL of Trizol, and then perform a scaled-down RNA extraction procedure through step 5 of Subheading 3.1. Follow the directions in the RNeasy® Micro Kit handbook beginning at step 4 of the handbook protocol for purifying total RNA from animal and human tissues.
Only sonicate enough so that the sample is homogenous and no particulates are visible. Additional sonication may result in RNA degradation.
Be careful not to transfer any of the interphase layer when aspirating the aqueous layer. Collection of the interphase into the RNA sample increases the risk of genomic DNA contamination during subsequent RT-PCR amplification.
If it is not possible to dilute the sample to 200 ng/μL, add 2–3 μL DNase directly to the sample.
When synthesizing cDNA, it is important to include a negative control that lacks reverse transcriptase (RT−) to assess PCR contamination from genomic DNA and cDNA products within the laboratory environment.
Reverse transcription at 37 °C can be carried out for 2 h to increase the cDNA yield of low-abundance transcripts.
Amplicon size and relative positions of multiple editing site(s) within a single transcript will determine whether single-read or paired-end reads are required during Illumina sequencing.
PCR cycle number may be increased to amplify low-abundance transcripts; however, increasing cycle number may increase potential errors introduced by PCR.
The antisense primer used for Sanger sequencing should anneal at least 50 base pairs downstream of the editing site(s) because the first ~30 base pairs of sequence are generally of poor quality. It is important to generate antisense Sanger sequencing reads because subsequent QSV analysis depends on the relative peak heights, and T/C peak heights are more consistent than A/G peak heights [13].
Include only high-quality Sanger sequencing data in the QSV analysis. Ensure that all electropherograms to be analyzed have high signal-to-noise ratios (i.e., electropherograms should be virtually free of “background” peaks; double peaks should only appear at the edited positions). Include all replicates for a single transcript in the same folder.
Usually the variant with the larger peak height will be designated as “Gene A,” and the minor variant will be designated as “Gene B.” Occasionally, QSVanalyzer will miscall the minor variant. If this happens, manually enter the correct nucleotide into the “Gene B” box.
We recommend including all six upstream bases as reference bases, even if one or more of them are edited nucleotides.
It is necessary to click Find before selecting additional nucleotides, but this step may be omitted after addition of the first nucleotide.
QSV information window may be accessed at any time by going to QSV analysis > QSV ratios.
Image (.png) and spreadsheet (.xls) files are also created for each analysis, but are not used in the analysis here.
Web pages for each edit site are automatically named in the following format: “QSV_data_5′ flanking sequence_Variant A_Variant B_3′ flanking sequence_−1-2-3-4-5-6_.htm,” where the 5′- and 3′-flanking sequences are the 10 nucleotides up- and downstream of the edit site, Variant A and Variant B are the nucleotide variants at the edit site, and the trailing numbers are reference bases. Web pages may be renamed if desired.
Although web page output files created by QSVanalyzer include relative adjusted peak heights and peak height ratios for each trace, we have found that calculating % editing using uncorrected peak heights provides a more reliable estimate of % editing as determined by high-throughput sequencing.
Acknowledgments
This work was supported by the Joel G. Hardman and Mary K. Parr Endowed Chair in Pharmacology, NIH/NIDDK (R01 DK119508), and Vanderbilt Technologies for Advanced Genomics [VANTAGE, Vanderbilt Ingram Cancer Center (P30 CA68485), the Vanderbilt Vision Center (P30 EY08126), and NIH/NCRR (G20 RR030956)].
References
- 1.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, Jantsch MF, Dorin J, Adams IR, Scadden AD, Ohman M, Keegan LP, O’Connell MA (2014) The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9(4):1482–1494. 10.1016/j.celrep.2014.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenthal JJ (2015) The emerging role of RNA editing in plasticity. J Exp Biol 218(Pt 12):1812–1821. 10.1242/jeb.119065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sommer B, Kohler M, Sprengel R, Seeburg PH (1991) RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67(1):11–19 [DOI] [PubMed] [Google Scholar]
- 4.Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB (1997) Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387(6630):303–308. 10.1038/387303a0 [DOI] [PubMed] [Google Scholar]
- 5.Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17(6):333–351. 10.1038/nrg.2016.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morabito MV, Ulbricht RJ, O’Neil RT, Airey DC, Lu P, Zhang B, Wang L, Emeson RB (2010) High-throughput multiplexed transcript analysis yields enhanced resolution of 5-hydroxytryptamine 2C receptor mRNA editing profiles. Mol Pharmacol 77(6):895–902. 10.1124/mol.109.061903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang R, Li X, Ramaswami G, Smith KS, Turecki G, Montgomery SB, Li JB (2014) Quantifying RNA allelic ratios by microfluidic multiplex PCR and sequencing. Nat Methods 11(1):51–54. 10.1038/nmeth.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM (2009) Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324(5931):1210–1213. 10.1126/science.1170995 [DOI] [PubMed] [Google Scholar]
- 9.Hood JL, Morabito MV, Martinez CR 3rd, Gilbert JA, Ferrick EA, Ayers GD, Chappell JD, Dermody TS, Emeson RB (2014) Reovirus-mediated induction of ADAR1 (p150) minimally alters RNA editing patterns in discrete brain regions. Mol Cell Neurosci 61:97–109. 10.1016/j.mcn.2014.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jinnah H, Ulbricht RJ (2019) Using mouse models to unlock the secrets of non-synonymous RNA editing. Methods 156:40–45. 10.1016/j.ymeth.2018.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porath HT, Carmi S, Levanon EY (2014) A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun 5:4726. 10.1038/ncomms5726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diroma MA, Ciaccia L, Pesole G, Picardi E (2019) Elucidating the editome: bioinformatics approaches for RNA editing detection. Brief Bioinform 20(2):436–447. 10.1093/bib/bbx129 [DOI] [PubMed] [Google Scholar]
- 13.Eggington JM, Greene T, Bass BL (2011) Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2:319. 10.1038/ncomms1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jepson JE, Reenan RA (2007) Genetic approaches to studying adenosine-to-inosine RNA editing. Methods Enzymol 424:265–287. 10.1016/S0076-6879(07)24012-1 [DOI] [PubMed] [Google Scholar]
- 15.Carr IM, Robinson JI, Dimitriou R, Markham AF, Morgan AW, Bonthron DT (2009) Inferring relative proportions of DNA variants from sequencing electropherograms. Bioinformatics 25(24):3244–3250. 10.1093/bioinformatics/btp583 [DOI] [PubMed] [Google Scholar]
- 16.Shen W, Le S, Li Y, Hu F (2016) SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One 11(10):e0163962. 10.1371/journal.pone.0163962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Droop AP (2016) fqtools: an efficient software suite for modern FASTQ file manipulation. Bioinformatics 32(12):1883–1884. 10.1093/bioinformatics/btw088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Song Y, Shi X, Liu J, Xiong S, Chen W, Fu Q, Huang Z, Gu N, Zhang R (2018) The landscape of miRNA editing in animals and its impact on miRNA biogenesis and targeting. Genome Res 28(1):132–143. 10.1101/gr.224386.117 [DOI] [PMC free article] [PubMed] [Google Scholar]