

ABSTRACT
Disruption of cell cycle checkpoints has been well established as a hallmark of cancer. In particular, the G1-S transition mediated by the cyclin D-cyclin-dependent kinase 4/6 (CDK4/6) pathway is dysregulated in more than 90% of melanoma cases. Therefore, tumor cells mainly rely on the G2-M checkpoint to halt the cell cycle in order to repair DNA damage. Here, we review the promising method of cell cycle-mediated synthetic lethality for melanoma treatment, which entails exploiting somatically acquired mutations in the G1-S transition with inhibitors of the G2-M transition in order to specifically kill melanoma cells. The idea stems from the theory that melanoma cells lacking G1-S checkpoints are particularly vulnerable to mitotic catastrophe when presented with G2-M checkpoint inhibition in addition to DNA damage, whereas normal cells with intact G1-S checkpoints should theoretically be spared. This review explores the link between cell cycle dysregulation and synthetic lethality in melanoma cells and discusses potential future applications for this treatment.
KEYWORDS: G2-M transition, melanoma, synthetic lethality, G1-S transition, cell cycle checkpoint, CHK1, WEE1
Melanoma incidence is rising rapidly throughout the world and the five-year survival rate for newly diagnosed metastatic melanoma is under 25% [1]. Melanoma is usually a result of exposure to UV radiation, which causes a high mutational burden in melanocytes in the skin resulting in tumorigenesis. B-rapidly accelerated fibrosarcoma (BRAF) is a serine/threonine kinase that is mutated in more than 60% of malignant melanomas and causes over-proliferation of melanoma cells [2]. For this reason, BRAF inhibitors, along with surgery, radiation, and immunotherapy, are first-line treatments for metastatic melanoma [3]. However, despite remarkable initial response rates to BRAF inhibitors, acquired resistance is almost inevitable by way of mutation/upregulation of the mutant protein, reactivation of downstream factors in the pathway, or hyperactivation of alternative pathways [4]. Because of the increase in melanoma incidence, poor prognosis, and acquired resistance, there is a need for the development of novel treatments that suppress melanoma growth and counter BRAF inhibitor acquired resistance.
One approach to novel treatments is to establish synthetic lethality. The concept of synthetic lethality entails deficiencies in two or more genes leading to targeted cell death, whereas loss of one gene is innocuous [5]. These deficiencies can be caused by mutations, epigenetic alterations, or inhibitions (both natural and synthetic). In order for the cell to remain viable, deficiencies in one gene/pathway must be compensated for by another gene/pathway, leading to increased reliance on this other gene/pathway. The theory of synthetic lethality predicts that induced inhibition of the protein that these cells depend upon will preferentially kill them while sparing normal cells that have one or both genes unimpaired [6]. Synthetic lethality, therefore, is an attractive methodology for killing cancer cells that typically have a high mutational load, while non-mutated cells typically survive.
Synthetic lethality was first shown to be effective in treating breast cancer type 1 and 2 susceptibility protein (BRCA1/2)-deficient breast cancer. In individuals with this condition, base excision repair (BER) compensates for the loss of BRCA1/2-mediated homologous recombination repair (HRR) through poly (ADP-ribose) polymerase (PARP). Inhibiting BER using PARP inhibitors results in selective cytotoxicity of the BRCA1/2-deficient cancer cells, while BRCA1/2-proficient cells are protected due to the intact BRCA1/2-mediated HRR [7]. This synthetic lethality approach has also been approved for BRCA1/2-mutant ovarian cancers and breast cancers [8]. Although first described in breast and ovarian cancer, synthetic lethality may have its most profound impacts in melanoma, due to the excessive mutational burden in melanomas as a result of UV radiation [9]. In fact, PARP inhibitors have already been shown to be an effective strategy for achieving synthetic lethality in LIG4-deficient melanomas, which are deficient in non-homologous end joining (NHEJ) [10].
In addition to targeting NHEJ deficiencies in melanoma, another pathway to target in melanoma via synthetic lethality is cell cycle checkpoints. The G1-S transition mediated by the cyclin D-CDK4 pathway is dysregulated in more than 90% of melanoma cases [11]. The loss of cell cycle regulation leads to increased proliferation rates for the cancer cell, but also predisposes the cell to increased deleterious mutations [12]. These mutations also lead to increased dependence on the G2-M checkpoint to induce cell cycle arrest when exposed to DNA damage that requires repair [13]. This presents a unique opportunity to target and inhibit proteins responsible for maintaining the G2-M checkpoint in order to impede the ability of tumor cells to assess and repair DNA damage. Bypassing the G2-M checkpoint leads to aberrant mitosis and mitotic catastrophe due to the accumulation of DNA damage, ending in apoptosis [14,15]. Therefore, a simple approach presents itself: maximize DNA damage that is repaired by non-cancerous cells, prevent cell cycle arrest and DNA damage repair in melanoma cells by bypassing the G2-M checkpoint, and ensure the damage is taken through into mitosis where the effects, mainly apoptosis, will manifest (Figure 1).
Figure 1.
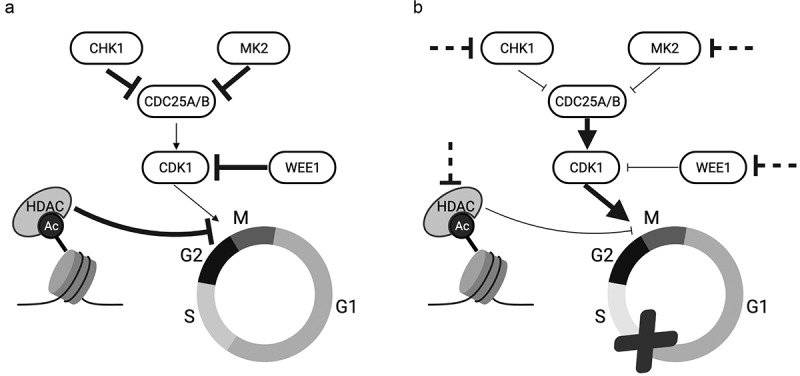
Achieving synthetic lethality by abrogating the G2-M checkpoint in G1-S checkpoint-deficient melanoma cells. (A) G2-M cell cycle transition is mediated by CHK1, MK2, WEE1, and HDACs in a normal cell. (B) G1-S checkpoint-defective melanoma cells can be targeted by several potential avenues for therapeutic inhibition (dashed inhibition lines). These cells progress uninhibited through the cell cycle and experience mitotic catastrophe. Thickness of arrows and inhibition lines represents relative pathway activity
CHK1 inhibition
Checkpoint kinase 1 (CHK1) is a serine/threonine-specific protein kinase that coordinates the DNA damage response (DDR) and cell cycle checkpoint response at several cellular stages. In the presence of DNA damage, CHK1 accumulates at the centromere and phosphorylates M-phase inducer phosphatase 1 (CDC25A) and M-phase inducer phosphatase 2 (CDC25B), indirectly leading to their degradation by cellular proteasomes [16]. In normal cells, CDC25A and CDC25B are responsible for the activation of cyclin-dependent kinase 1 (CDK1), a necessary enzyme for the cell to enter mitosis. Therefore, their indirect destruction via CHK1 causes cell cycle arrest at the G2-M transition [17].
Interestingly, high levels of CHK1 are correlated with worse prognosis of melanoma [18]. This may be advantageous for tumors as they are able to tolerate a higher level of DNA damage and possibly increase resistance to chemotherapy. Additionally, most melanoma cells are deficient in the G1-S checkpoint, thus relying on CHK1 to inhibit G2-M cell cycle progression to repair DNA damage [19]. For these reasons, CHK1 inhibitors are an intriguing option for achieving synthetic lethality in melanoma.
CHK1 inhibition has long been a field of interest for cancer therapy, particularly in combination with chemotherapeutics. In 1996, 7-hydroxy-staurosporine (UCN-01; Figure 2) was shown to abrogate the G2-M checkpoint and enhance cisplatin-induced cytotoxicity by 60-fold in Chinese hamster ovary cell lines [20]. That same year, UCN-01 was shown to enhance susceptibility of p53-deficient lymphoma and colon carcinoma cells to radiation [21]. It was not until 2000 that UCN-01 was shown to exert its cytotoxic effects by inhibiting CHK1 [22]. From then on, CHK1 inhibition was the primary method for achieving synthetic lethality by abrogating the G2-M checkpoint.
Figure 2.
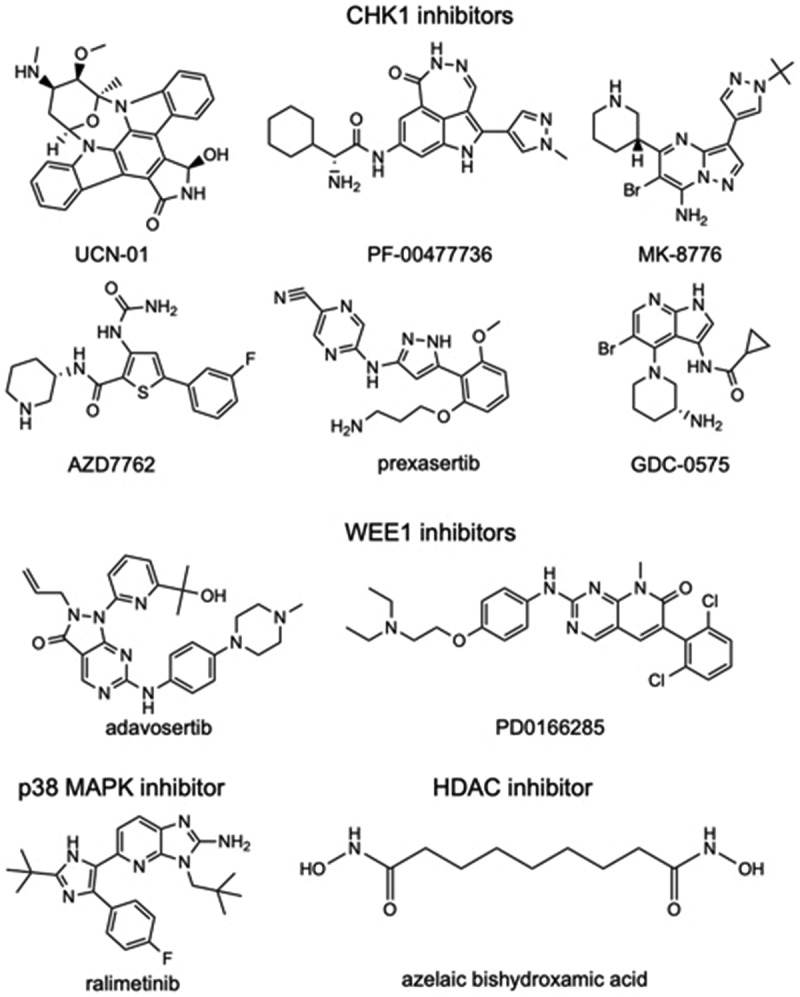
Chemical structures of G2-M checkpoint inhibitors. 2D structures available from PubChem (https://pubchem.ncbi.nlm.nih.gov) are provided for CHK1 inhibitors, UCN-01 (CID 72271), PF-00477736 (CID 135565545), AZD7762 (CID 11152667), MK-8776 (CID 16224745), GDC-0575 (CID 46917793), prexasertib (CID 46700756); WEE1 inhibitors, PD0166285 (CID 5311382) and adavosertib (CID 24856436); p38 MAPK inhibitor ralimetinib (CID 11539025); and HDAC inhibitor azelaic bishydroxamic acid (CID 65268). All CHK1, WEE1, and p38 MAPK inhibitors function as ATP competitive small-molecule inhibitors
In 2006, researchers found that normally poor inducers of apoptosis such as excess thymidine, hydroxyurea, and camptothecin significantly increased lethality of a colon cancer cell line with concurrent treatment of siRNA-mediated depletion of CHK1 [23]. In 2008, two CHK1 inhibitors (PF-00477736 and AZD7762; Figure 2) were shown to abrogate cell cycle arrest and induce apoptosis in HT-29 colorectal adenocarcinoma cells when combined with the cytotoxic agent gemcitabine, selectively sparing healthy cells [24,25]. AZD7762 was also shown to impede Rad51 foci formation and subsequently HRR repair when combined with gemcitabine and IR in pancreatic cancer cell lines [26]. Specifically, the inability to stop at the G2-M checkpoint in the presence of induced DNA damage led to aberrant mitosis and apoptosis. These researchers concluded that CHK1 inhibitors require an exogenous source of DNA damage, and radiation treatment sensitized the cells to CHK1 inhibition [26]. Indeed, the CHK1 inhibitor MK-8776 has been shown to radiosensitize triple-negative breast cancer by enhancing DNA damage and inhibiting autophagy, suggesting an adjunctive role for CHK1 inhibition [27].
CHK1 inhibitors have also been extensively researched in combination with several different pharmaceutical therapies in order to maximize its efficacy against melanoma. For example, a study showed that treatment with either CHK1 inhibitor GDC-0575 or GNE-323 (Figure 2) in combination with gemcitabine drastically reduced proliferation of healthy cells as well as melanoma cells, whereas combination with subclinical hydroxyurea preferentially killed melanoma cells while retaining the proliferative capacity of healthy cells [28]. In addition, CHK1 inhibitors have been tested as an alternate therapy for BRAF inhibitor-resistant melanomas. Interestingly, PF-00477736 has been shown to re-sensitize melanomas to BRAF inhibitors and act synergistically in tumor suppression [18]. The authors propose that melanoma cells use a feedback loop between BRAF and CHK1 to manage replication stress, minimize DNA damage, and sustain cell proliferation. Inhibition of both of these proteins leads to impeded cell cycle arrest, aberrant mitosis, and apoptosis [18]. This study suggests the possibility of combination treatment of CHK1 and BRAF inhibitors in melanoma. Although it is evident that optimization of adjunctive treatments is not yet known, these studies suggest the intriguing potential for CHK1 inhibitors to act synergistically or additively with other small-molecule inhibitors and/or chemotherapy in melanoma patients.
However, some researchers have suggested that CHK1 inhibitors AR323 and AR678 can function, although with less potency, as single-agent therapies specifically targeting melanomas with high levels of endogenous DNA damage [29]. These researchers theorized that melanomas with extreme replicative stress are more dependent on the G2-M transition to repair DNA damage, and thus are more susceptible to CHK1 inhibition [29]. In addition, it has been shown that CHK1 inhibition requires hypoxic conditions to suppress melanoma tumor growth, which may explain why initial research indicated that CHK1 inhibition alone was not a sufficient therapy in vitro, but in vivo results supported CHK1 inhibition as a viable stand-alone treatment option [30]. The requirement for hypoxic conditions may enable CHK1 inhibitors to be even more selective in their targeting of melanoma cells, as oxygen deprivation is common in tumor environments. Recent evidence supports this theory, as AZD7762 paired with the anti-angiogenic drug bevacizumab yielded synergistic repression of melanoma growth [30].
While CHK1 inhibitors may be a potential therapy in the future of melanoma treatment, CHK1 inhibition does not come without its drawbacks. One study reported that the status of p53 does not predict the efficacy of CHK1 inhibitors in combination with chemotherapy in inducing aberrant mitosis and apoptosis [31]. This would suggest that CHK1 inhibitors do not, in fact, preferentially target tumor cells that are deficient in p53. However, most other studies have reported CHK1 inhibitors sparing healthy cells in vivo. Dietlein et al. also proposed the potential for acquired resistance to CHK1 inhibition through overactivation of MK2 [32]. If this avenue is easily achieved by melanoma cells, CHK1 inhibitors may not be an adequate long-term treatment for melanoma.
Clinical trials combining CHK1 inhibition with other forms of chemotherapy in other cancers have yielded mixed results. After positive results in a phase I clinical trial, MK-8776 in combination with cytosine arabinoside offered no significant benefit in the treatment of acute myelogenous leukemia in a randomized phase II clinical trial [33] (Figure 2). Another selective CHK1 inhibitor known as prexasertib (LY2606368; Figure 2) has the unique capability of not only inhibiting CHK1, but also inducing double-stranded DNA breaks, leading to rapid mitotic catastrophe and xenografted lung adenocarcinoma cell death [34]. Prexasertib has also exhibited synergism with cisplatin and talazoparib, a PARP inhibitor, in reducing viability of pediatric osteosarcoma cell lines [35]. It has since displayed modest clinical activity and tolerability in increasing progression-free survival in both high-grade serous ovarian carcinoma and advanced triple-negative breast cancer [36,37]. However, a phase II clinical trial involving prexasertib in combination with cisplatin and pemetrexed in the treatment of non-small cell lung cancer was halted prematurely due to an increased incidence of thromboembolic events in the experimental group [38]. Although there are several ongoing clinical trials involving CHK1 inhibition with positive preliminary results [39], there are currently none looking into treatment for melanoma.
WEE1 inhibition
WEE1 is a nuclear serine/threonine protein kinase that directly phosphorylates tyrosine residues on CDK1, thereby inactivating it and effectively inducing cell cycle arrest at the G2-M transition [40]. Similar to CHK1, this protein is essential for tumor cells to induce cell cycle arrest when presented with DNA damage. In addition, high WEE1 expression increases with melanoma tumor progression and is associated with a shorter relapse-free period [41]. WEE1 expression is also linked to a positive correlation with thicker primary tumors, ulceration, and poor disease-free survival in patients with melanoma [41]. For these reasons, WEE1 inhibitors are actively being researched as synthetic lethal drugs in melanoma therapy.
Inhibition of WEE1 has been shown to reduce melanoma cell viability by promoting DNA damage and apoptosis [41]. This is likely due to the accumulation of endogenous DNA damage manifesting in aberrant mitosis, as WEE1 inhibition prevents cell cycle arrest and DNA damage repair. Indeed, administration of WEE1-targeted miRNA (miR-195) in combination with the chemotherapy doxorubicin significantly reduced stress-induced G2-M cell cycle arrest, which was restored by stable overexpression of WEE1 [42]. In addition, a selective WEE1 inhibitor pyridopyrimidine molecule known as PD0166285 (Figure 2) was shown to abrogate the G2-M checkpoint, leading to p53-dependent radiosensitization in ovarian and colon cancer cell lines, as well as melanoma cell death in a murine model [43,44].
Another small molecular inhibitor of WEE1 known as adavosertib (MK-1775, AZD-1775; Figure 2) has been shown to selectively sensitize p53-deficient lung, breast, and prostate cancer cell lines to several DNA-damaging agents and radiation therapy and work synergistically with these agents in suppressing tumor growth in vivo while sparing p53-proficient cells [45,46]. Accordingly, a phase I clinical trial reported the safety and tolerability of adavosertib in monotherapy and as an adjunct to chemotherapy in patients with refractory solid tumors, including melanomas [47]. A recent clinical trial has also shown the increased efficacy of adavosertib in combination with gemcitabine and radiation in newly diagnosed, advanced pancreatic cancer when compared to gemcitabine and radiation alone [48]. In addition, several clinical trials are currently recruiting patients for combination treatment of a WEE1 inhibitor with different chemotherapies in various cancers, but it has not yet been tested in the treatment of melanoma.
Despite the potential strengths of this form of therapy, attempts to achieve synthetic lethality through WEE1 inhibition have yielded controversial results. It has also been reported that miR-195 administration increased melanoma proliferation [42]. In addition, preclinical data have suggested that WEE1 is downregulated in melanoma metastases as compared with primary melanoma [19]. Correspondingly, miR-195 overexpression also enhanced migration and invasiveness of melanoma cells [42]. This may indicate that WEE1 may have secondary effects outside of impeding the G2-M transition, including inhibiting propagative and/or migratory signaling. These studies could also be the result of inadequate administration of exogenous DNA damage, which would lead to unrestricted proliferation without a need to repair DNA damage and induce cell cycle arrest. Regardless, more research is needed to delineate the positive and negative effects of WEE1 inhibition alone and in combination with other cytotoxic agents in vivo.
Combination of CHK1 and WEE1 inhibition
Recently, WEE1 inhibitors have been utilized in combination with CHK1 inhibitors to combat tumor growth. AR458323, a CHK1 inhibitor (Figure 2), was utilized in a combination siRNA screen of 195 different genes involved in the cell cycle and DNA repair to determine any synergistic anti-cancer effects in prostate and lung cancer lines. The most prominent synergistic effect was the combination of the CHK1 inhibitor and a WEE1 siRNA [49]. Combined treatment of CHK1 and WEE1 inhibitors have since yielded synergistic antitumor effects in head and neck cancer, ovarian cancer, mantle cell lymphoma, and neuroblastoma [50–53]. Low dose CHK1 inhibition and gemcitabine combined with hhigh-dose WEE1 inhibition have also been shown to synergistically suppress pancreatic adenocarcinoma growth while maintaining tolerability by limiting toxic adverse effects [54].
The synergistic effect of combined CHK1 and WEE1 inhibition was also seen in vitro in malignant melanoma cells with the small-molecule inhibitors AZD7762 and adavosertib. Concurrent treatment decreased cellular viability and increased apoptosis as a result of the accumulation of DNA damage and premature mitosis of S-phase cells [55]. In addition, combined treatment reduced spheroid growth and led to greater tumor growth inhibition in melanoma xenografts compared to either inhibitor used as single agents [55]. Importantly, combination treatment of these two inhibitors was found to be equally effective in BRAF-inhibitor sensitive, naturally BRAF-inhibitor resistant, and treatment-induced BRAF-inhibitor resistant melanoma cell lines [56]. This combined treatment did not require an external source of DNA damage and very modestly affected normal fibroblast and melanocyte cell lines, and therefore suggests a low risk of adverse effects in vivo. The absence of a requirement for adjunctive chemotherapy and restriction of off-target effects bodes well for the future development of this combination therapy in the treatment of malignant melanoma, for which there are very few effective treatments.
More research must be done to elucidate the synergistic effects and potential adverse events of combination CHK1 and WEE1 inhibition in melanoma therapy. For example, although many studies have shown that CHK1 and WEE1 inhibitors each induce selective p53-deficient cell death due to abrogation of the G2-M checkpoint, other studies have shown that combination therapy of dual CHK1 and WEE1 inhibition exerted their cytotoxic effects independently of p53 status [51,53]. This could mean that combined CHK1 and WEE1 inhibition could exert detrimental off-target effects in healthy cells. Future studies should look into the safety and tolerability of combined CHK1 and WEE1 inhibition, although no clinical trials are currently researching this promising dual therapy in any cancer type.
MK2 inhibition
MAP kinase-activated protein kinase 2 (MK2) is a serine/threonine kinase activated by p38 MAPK that is involved in several cellular processes including inflammatory response and cellular proliferation. It has also been shown that the G2-M transition is directly mediated by MK2 inhibition of CDC25B in the context of DNA damage or stress, similarly to CHK1 [57]. However, MK2 activation in response to low-dose chemotherapy was independent of CHK1 activation (and vice-versa), suggesting that targeting the p38 MAPK/MK2 pathway may add to the synergistic lethality of CHK1 inhibition [58]. Indeed, CHK1 inhibition has been shown to work synergistically with inhibition of MK2, as combination therapy in BRAF-mutant lung adenocarcinoma and KRAS-mutant pancreatic adenocarcinoma cell lines leads to mitotic catastrophe and massive apoptosis, whereas single-agent treatment did not result in any significant cytotoxicity [32]. MK2 inhibition has also been shown to selectively target p53-deficient mouse embryonic fibroblasts (MEFs) in combination with low-dose chemotherapy and spare p53-proficient cells, suggesting that this therapy may have very little adverse effects [58]. This study also showed that the known CHK1 inhibitor UCN-01 also potently inhibits MK2, suggesting that its efficacy is due to the dual inhibition of both pathways [58].
Clinical trials have focused largely on the inhibition of p38 MAPK in order to reduce downstream activation of MK2. A recent phase 1b/2 clinical trial of the p38 MAPK inhibitor ralimetinib (LY2228820; Figure 2) in combination with gemcitabine and carboplatin in patients with ovarian cancer showed a modest improvement in progression-free survival [59]. The CHK1 and MK2 dual inhibitor UCN-01, although well tolerated, did not show any clinical benefit in the treatment of metastatic melanoma in a phase II clinical trial [60]. More research will need to be done regarding MK2 inhibition with adjunctive therapies in order to determine if this therapy could be utilized to achieve synthetic lethality in melanoma.
HDAC inhibition
Histone deacetylases (HDACs) remove acetyl groups from lysine residues of exposed histone tails. This process alters chromatin structure, thereby changing the transcription levels of nearby genes and contributing to the downregulation of several cell cycle regulators and tumor suppressors. HDACs also reverse the deactivation of nonhistone substrates, such as heat shock protein 90 (HSP90) and nuclear factor kappa B (NF-κB), both of which have prominent tumor-promoting effects [61]. Correspondingly, HDACs are overexpressed in malignant melanomas and confer enhanced resistance to typical therapies [62]. Indeed, siRNA-mediated knockdown of HDAC1 led to cell cycle arrest in G1, mitotic catastrophe, and apoptosis in human osteosarcoma cell lines [63]. Therefore, research has recently focused on histone deacetylase inhibitors (HDACIs), such as valproic acid and trichostatin A, as potential anti-tumor therapies.
In addition to increasing the transcription of vital tumor suppressor genes such as p21 and p16, HDAC inhibition through treatment of azelaic bishydroxamic acid (ABHA; Figure 2) has been shown to induce the loss of the G2-M transition in tumor cells, whereas healthy cells exhibit cell cycle arrest [64]. Loss of this G2-M checkpoint results in aberrant mitosis, fractured multinuclei and micronuclei, and eventually cell death in melanoma cells, while sparing normal neonatal foreskin fibroblasts [64]. The researchers confirmed that the killing of the tumor cells was specifically due to HDAC inhibitor-induced uninterrupted progression through the G2-M checkpoint by pharmacologically inducing the G2-M checkpoint, thereby saving the tumor cells [64]. These results were also reproduced with several other HDAC inhibitors with similar results, as tumor cells experienced failure of chromosomal pairing before mitosis, leading to incorrect segregation of chromosomes and apoptosis [65]. One of the main benefits of HDAC inhibitor treatment is the obviation of an external stressor, such as chemotherapy or radiation, as cell death is caused by endogenous stresses [66].
HDAC inhibitors have been proposed as a novel anti-cancer drug, but there are drawbacks with the institution of this therapy in a clinical setting. HDACs have a wide range of downstream effectors, and therefore HDAC inhibition may have unintended consequences outside of tumor growth repression. Indeed, the most common HDAC inhibitors under preclinical and clinical evaluation are broad-spectrum and nonselective [61]. Normal cells also require a balance of histone acetylation and deacetylation for properly functioning cellular processes, and disturbances of this balance may be detrimental to the overall health of the patient. In addition, HDAC inhibitors have not yet been demonstrated to work in vivo against melanoma, which is notoriously resistant to most cancer therapies. Combination therapy is likely a future direction for research of HDAC inhibitors, but interactions with other ssmall-molecule inhibitors and cytotoxic agents have not yet been fully elucidated.
Conclusion and future directions
The G2-M transition is a promising target for synthetic lethality in melanoma (Figure 1). However, there are some drawbacks with this form of treatment. The requirement for p53-deficiency for lethality allows for selective targeting of tumor cells, but also offers no therapeutic benefits for melanomas that have not lost G1-S checkpoint functionality. This requires tumor genome sequencing and institutionalization of personalized medicine, which can be cost-ineffective. In addition, off-target effects and toxicity associated with combination therapies using cell cycle inhibitors must be more thoroughly researched during development of novel therapies in order to determine whether they can be used in a clinical setting. Furthermore, “modulator” drugs, such as CHK1 and WEE1 inhibitors, that only function by enhancing or synergizing with chemotherapy or other treatments are typically overlooked in drug development as funding is not sufficient [67].
Because of the G1-S checkpoint loss in many melanomas, achieving synthetic lethality in melanoma may be best accomplished by targeting the G2-M checkpoint. Importantly, this form of therapy provides selective targeting of p53-deficient cells, thus sparing healthy cells and leaving minimal detrimental side effects in patients. In addition, ssmall-molecule inhibitors of the G2-M checkpoint have been shown to function in coordination, and sometimes synergistically, with standard treatments of chemotherapy, radiation, and BRAF inhibitors. This treatment may also be a viable alternative to BRAF inhibition in melanoma after acquiring resistance to the drug, an important feature considering most melanomas eventually develop resistance at some point.
Still, there is still much research to be done regarding manipulation of the G2-M checkpoint to achieve synthetic lethality in melanoma. Inhibition of the G2-M checkpoint may be a sound strategy, but results may not be as promising in a clinical setting. For this reason, determining the most effective and lowest risk combination therapy of ssmall-molecule inhibitors and DNA-damaging agents may very well be the next step in this field. In addition, because the loss of the G1-S transition via deregulation of p53 is characteristic of most cancers, the strategies of synthetic lethality demonstrated in this review may also apply to other cancers. More research is required to fully understand the breadth of effectiveness of facilitating the G2-M transition in tumor cells. Synthetic lethality is a quickly growing field in cancer biology, and targeting the G2-M transition may be another tool in the arsenal of melanoma repression.
Acknowledgments
This work was supported by the Georgetown University Biology Department (N.B.). Figure development was facilitated by BioRender (https://BioRender.com). 2D chemical structures were derived with ChemDraw 20.0 (PerkinElmer Informatics).
Funding Statement
This work was supported by the National Institute of General Medical Sciences [1R15GM129628].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Howlader N, Noone A, Krapcho M, et al. (eds). SEER Cancer Statistics Review, 1975–2016. Bethesda, MD: National Cancer Institute. https://seer.cancer.gov/csr/1975_2016/. 2019. [Google Scholar]
- [2].Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. [DOI] [PubMed] [Google Scholar]
- [3].Gibney GT, Atkins MB.. Choice of first-line therapy in metastatic melanoma. Cancer. 2019;125:666–669. [DOI] [PubMed] [Google Scholar]
- [4].Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. [DOI] [PubMed] [Google Scholar]
- [6].Reinhardt HC, Jiang H, Hemann MT, et al. Exploiting synthetic lethal interactions for targeted cancer therapy. Cell Cycle. 2009;8:3112–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. [DOI] [PubMed] [Google Scholar]
- [8].Federal Drug Administration. FDA approves olaparib for germline BRCA-mutated metastatic breast cancer [press release] . Retrieved from. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-germline-brca-mutated-metastatic-breast-cancer. 2018. [Google Scholar]
- [9].Thompson N, Adams DJ, Ranzani M. Synthetic lethality: emerging targets and opportunities in melanoma. Pigment Cell Melanoma Res. 2017;30:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Czyz M, Toma M, Gajos-Michniewicz A, et al. PARP1 inhibitor olaparib (lynparza) exerts synthetic lethal effect against ligase 4-deficient melanomas. Oncotarget. 2016;7:75551–75560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Walker GJ, Flores JF, Glendening JM, et al. Virtually 100% of melanoma cell lines harbor alterations at the DNA level within CDKN2A, CDKN2B, or one of their downstream targets. Genes Chromosomes Cancer. 1998;22:157–163. [DOI] [PubMed] [Google Scholar]
- [12].Broustas CG, Lieberman HB. DNA damage response genes and the development of cancer metastasis. Radiat Res. 2014;181:111–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dasika GK, Lin SJ, Zhao S, et al. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene. 1999;18:7883–7899. [DOI] [PubMed] [Google Scholar]
- [14].Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. [DOI] [PubMed] [Google Scholar]
- [15].Erenpreisa J, Cragg MS. Mitotic death: a mechanism of survival? A review. Cancer Cell Int. 2001;1:1,2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Krämer A, Mailand N, Lukas C, et al. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol. 2004;6:884–891. [DOI] [PubMed] [Google Scholar]
- [17].Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci. 2013;70:4009–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hwang B, Adhikary G, Eckert RL, et al. Chk1 inhibition as a novel therapeutic strategy in melanoma. Oncotarget. 2018;9:30450–30464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee B, Sandhu S, McArthur G. Cell cycle control as a promising target in melanoma. Curr Opin Oncol. 2015;27:141. [DOI] [PubMed] [Google Scholar]
- [20].Bunch RT, Eastman A. Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin Cancer Res. 1996;2:791–797. [PubMed] [Google Scholar]
- [21].Wang Q, Fan S, Eastman A, et al. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst. 1996;88:956–965. [DOI] [PubMed] [Google Scholar]
- [22].Graves PR, Yu L, Schwarz JK, et al. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem. 2000;275:5600–5605. [DOI] [PubMed] [Google Scholar]
- [23].Rodriguez R, Meuth M. Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress. Mol Biol Cell. 2006;17:402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Blasina A, Hallin J, Chen E, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7:2394–2404. [DOI] [PubMed] [Google Scholar]
- [25].Zabludoff SD, Deng C, Grondine MR, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7:2955–2966. [DOI] [PubMed] [Google Scholar]
- [26].Morgan MA, Parsels LA, Zhao L, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70:4972–4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhou Z, Yang Z, Wang S, et al. The Chk1 inhibitor MK-8776 increases the radiosensitivity of human triple-negative breast cancer by inhibiting autophagy. Acta Pharmacol Sin. 2017;38:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Oo ZY, Proctor M, Stevenson AJ, et al. Combined use of subclinical hydroxyurea and CHK1 inhibitor effectively controls melanoma and lung cancer progression, with reduced normal tissue toxicity compared to gemcitabine. Mol Oncol. 2019;13:1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Brooks K, Oakes V, Edwards B, et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene. 2013;32:788–796. [DOI] [PubMed] [Google Scholar]
- [30].Possik P, Müller J, Gerlach C, et al. Parallel in vivo and in vitro melanoma RNAi dropout screens reveal synthetic lethality between hypoxia and DNA damage response inhibition. Cell Rep. 2014;9:1375–1386. [DOI] [PubMed] [Google Scholar]
- [31].Zenvirt S, Kravchenko-Balasha N, Levitzki A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene. 2010;29:6149–6159. [DOI] [PubMed] [Google Scholar]
- [32].Dietlein F, Kalb B, Jokic M, et al. A synergistic interaction between Chk1- and MK2 inhibitors in KRAS-mutant cancer. Cell. 2015;162:146–159. [DOI] [PubMed] [Google Scholar]
- [33].Webster JA, Tibes R, Morris L, et al. Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk Res. 2017;61:108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].King C, Diaz HB, McNeely S, et al. LY2606368 causes replication catastrophe and antitumor effects through CHK1-dependent mechanisms. Mol Cancer Ther. 2015;14:2004–2013. [DOI] [PubMed] [Google Scholar]
- [35].Heidler CL, Roth EK, Thiemann M, et al. Prexasertib (LY2606368) reduces clonogenic survival by inducing apoptosis in primary patient-derived osteosarcoma cells and synergizes with cisplatin and talazoparib. Int J Cancer. 2020;147:1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lee JM, Nair J, Zimmer A, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018;19:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gatti-Mays ME, Karzai FH, Soltani SN, et al. A phase II single arm pilot study of the CHK1 inhibitor prexasertib (LY2606368) in BRCA wild-type, advanced triple-negative breast cancer. Oncologist. 2020;25:1013–e1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wehler T, Thomas M, Schumann C, et al. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer. 2017;108:212–216. [DOI] [PubMed] [Google Scholar]
- [39].National Cancer Institute . Intervention dynamic trial listing page. Accessed 2021, April 1. Available from: https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/prexasertib.
- [40].McGowan CH, Russell P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. Embo J. 1993;12:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Magnussen GI, Holm R, Emilsen E, et al. High expression of Wee1 is associated with poor disease-free survival in malignant melanoma: potential for targeted therapy. PloS One. 2012;7:e38254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bhattacharya A, Schmitz U, Wolkenhauer O, et al. Regulation of cell cycle checkpoint kinase WEE1 by miR-195 in malignant melanoma. Oncogene. 2013;32:3175–3183. [DOI] [PubMed] [Google Scholar]
- [43].Wang Y, Li J, Booher RN, et al. Radiosensitization of p53 mutant cells by PD0166285, a novel G2 checkpoint abrogator. Cancer Res. 2001;61:8211–8217. [PubMed] [Google Scholar]
- [44].Hashimoto O, Shinkawa M, Torimura T, et al. Cell cycle regulation by the Wee1 inhibitor PD0166285, pyrido [2,3-d] pyimidine, in the B16 mouse melanoma cell line. BMC Cancer. 2006;6:292,2407-6-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bridges KA, Hirai H, Buser CA, et al. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;17:5638–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hirai H, Iwasawa Y, Okada M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. [DOI] [PubMed] [Google Scholar]
- [47].Leijen S, Van Geel RM, Pavlick AC, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016;34:4371–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cuneo KC, Morgan MA, Sahai V, et al. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol. 2019;37:2643–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Davies KD, Cable PL, Garrus JE, et al. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol Ther. 2011;12:788–796. [DOI] [PubMed] [Google Scholar]
- [50].Van Harten AM, Buijze M, Van Der Mast R, et al. Targeting the cell cycle in head and neck cancer by Chk1 inhibition: a novel concept of bimodal cell death. Oncogenesis. 2019;8:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Carrassa L, Chilã R, Lupi M, et al. Combined inhibition of Chk1 and Wee1: in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle. 2012;11:2507–2517. [DOI] [PubMed] [Google Scholar]
- [52].Russell MR, Levin K, Rader J, et al. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013;73:776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chilá R, Basana A, Lupi M, et al. Combined inhibition of Chk1 and Wee1 as a new therapeutic strategy for mantle cell lymphoma. Oncotarget. 2015;6:3394–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Koh S, Wallez Y, Dunlop CR, et al. Mechanistic distinctions between CHK1 and WEE1 inhibition guide the scheduling of triple therapy with gemcitabine. Cancer Res. 2018;78:3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Magnussen GI, Emilsen E, Giller Fleten K, et al. Combined inhibition of the cell cycle related proteins Wee1 and Chk1/2 induces synergistic anti-cancer effect in melanoma. BMC Cancer. 2015;15. DOI: 10.1186/s12885-015-1474-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Margue C, Philippidou D, Kozar I, et al. Kinase inhibitor library screening identifies synergistic drug combinations effective in sensitive and resistant melanoma cells. J Exp Clin Cancer Res. 2019;38. DOI: 10.1186/s13046-019-1038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Manke IA, Nguyen A, Lim D, et al. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell. 2005;17:37–48. [DOI] [PubMed] [Google Scholar]
- [58].Reinhardt HC, Aslanian AS, Lees JA, et al. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Vergote I, Heitz F, Buderath P, et al. A randomized, double-blind, placebo-controlled phase 1b/2 study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine and carboplatin versus gemcitabine and carboplatin for women with recurrent platinum-sensitive ovarian cancer. Gynecol Oncol. 2020;156:23–31. [DOI] [PubMed] [Google Scholar]
- [60].Li T, Christensen SD, Frankel PH, et al. A phase II study of cell cycle inhibitor UCN-01 in patients with metastatic melanoma: a California cancer consortium trial. Invest New Drugs. 2012;30:741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Garmpis N, Damaskos C, Garmpi A, et al. Targeting histone deacetylases in malignant melanoma: a future therapeutic agent or just great expectations? Anticancer Res. 2017;37:5355–5362. [DOI] [PubMed] [Google Scholar]
- [62].Krumm A, Barckhausen C, Kucuk P, et al. Enhanced histone deacetylase activity in malignant melanoma provokes RAD51 and FANCD2-triggered drug resistance. Cancer Res. 2016;76:3067–3077. [DOI] [PubMed] [Google Scholar]
- [63].Senese S, Zaragoza K, Minardi S, et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol. 2007;27:4784–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Qiu L, Burgess A, Fairlie DP, et al. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol Biol Cell. 2000;11:2069–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Warrener R, Beamish H, Burgess A, et al. Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints. Faseb J. 2003;17:1550–1552. [DOI] [PubMed] [Google Scholar]
- [66].Gabrielli B, Brooks K, Pavey S. Defective cell cycle checkpoints as targets for anti-cancer therapies. Front Pharmacol. 2012;3. DOI: 10.3389/fphar.2012.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Dent P, Tang Y, Yacoub A, et al. CHK1 inhibitors in combination chemotherapy. Mol Interv. 2011;11:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]