

Abstract
Background:
Porokeratosis (PK) is a rare, heterogeneous group of keratinization disorders with an autosomal dominant inheritance pattern and is characterized by the presence of cornoid lamella. Disseminated superficial actinic PK is the most encountered subtype and typically manifests as multiple, small annular plaques with atrophic centers and slightly raised hyperkeratotic edges. Seven associated mutations (SSH1, SART3, MVKP, MVK, MVD, FDPS, and SLC17A9) have been reported in disseminated superficial actinic PK patients.
Aim:
We searched a Chinese disseminated superficial porokeratosis (DSAP) family to detect the causative genes. In the meantime, we reviewed the articles reported about DSAP in Chinese population, summarizing their clinical manifestations and discussing the incidence of DSAP in Chinese population.
Materials and Methods:
Sanger sequencing on the MVD and MVK genes was performed to identify the pathogenic mutation in a Chinese family with DSAP. Literature for DSAP cases reported in Chinese populations was searched by Sinomed and PubMed.
Results:
We identified the c. 875A > G (p. Asn292Ser) mutation in the MVD gene in the family.
Conclusions:
That mutation was a hotspot mutation. Literature review showed that the age of onset in DSAP family was earlier than that in sporadic patients; the lesion is common in the face in Chinese population which is distinct from studies in Caucasians; ultraviolet exposure is the main aggravating factor.
Key Words: Disseminated superficial actinic porokeratosis, MVD gene, mutation analysis, phenotypes
Introduction
Porokeratosis (PK) is a rare, heterogeneous group of keratinization disorders with an autosomal dominant inheritance pattern and is characterized by the presence of cornoid lamella.[1,2,3] Nearly 20 subtypes of PK defined by the size, location, and number of lesions have been reported in the literature,[4] such as PK of Mibelli, disseminated superficial PK, and disseminated superficial actinic porokeratosis (DSAP). DSAP is the most common subtype and is characterized by multiple small, annular plaques with atrophic centers and hyperkeratotic edges. The lesions occur mostly on sun-exposed body areas and can be exacerbated by ultraviolet exposure, infections, and immunosuppression.[5]
Although DSAP was first reported in 1967, the molecular mechanisms are not fully understood. DSAP has been recognized as a genetic disease that involves the abnormal differentiation of keratinocytes.[5] To date, five chromosomal loci (1p31.3–p31.1, 12q23.2–24.1, 12q24.1–q24.2, 15q25.1–26.1, and 16q24.1–24.3) have been associated with DSAP.[6] Mutations in MVK, MVD, PMVK, FDPS, and SLC17A9 are reported to be involved in DSAP,[5] and 47 mutations in the MVK gene have been identified in PK patients, especially in DSAP patients [Table 1]. Zhang et al. reported that approximately half of the patients carried MVD mutations,[2] although in the DSAP patients, there were fever mutations in MVD gene than in MVK. In this study, we performed Sanger sequencing and mutational analyses on the MVD and MVK genes to identify the pathogenic mutation in a Chinese family with DSAP.
Table 1.
All exon mutations identified in mevalonate kinase, mevalonate decarboxylase, phosphomevalonate kinase, farnesyl diphosphate synthase, solute carrier family 17 member 9 for porokeratosis to date
| Gene | Exon | Mutation type | Nucleotide mutation | Protein alteration | Diease | Reference | |
|---|---|---|---|---|---|---|---|
| 1 | MVK | 2 | Start codon | c. 3G>C | p.Met1? | DSAP | [1] |
| 2 | MVK | 2 | Missense | c. 31C>T | p.Pro11Ser | DSAP | [2] |
| 3 | MVK | 2 | Missense | c. 34G>C | p.Gly12Arg | DSAP | [1] |
| 4 | MVK | 2 | Missense | c. 74G>T | p.Gly25Val | PK | [3] |
| 5 | MVK | 3 | Missense | c. 122T>C | p.Leu41Pro | DSAP | [1] |
| 6 | MVK | 3 | Missense | c. 122T>G | p.Leu41Arg | PK | [3] |
| 7 | MVK | 3 | Nonsense | c. 186G>A | p.Trp62* | DSAP | [4] |
| 8 | MVK | 3 | Missense | c. 205T>A | p.Ser69Thr | DSAP | [5] |
| 9 | MVK | 4 | Splice site | IVS4 + 1G>A | Facial PK | [6] | |
| 10 | MVK | 4 | Splice site | IVS4 + 2T>A | PP | [6] | |
| 11 | MVK | 4 | Missense | c. 235G>A | p.Asp79Asn | PK | [3] |
| 12 | MVK | 4 | Missense | c. 235G>T | p.Asp79Tyr | PK | [3] |
| 13 | MVK | 4 | Nonsense | c. 254C>G | p.Ser85* | PK | [3] |
| 14 | MVK | 4 | Splice site | c. 371 + 2T>A | p.Glu76Glyfs*9 | PK | [3] |
| 15 | MVK | 1-5 | Deletion | c.-1880_527 + 533del | p.? | PK | [3] |
| 16 | MVK | 5 | Deletion | c. 395del T | p.Val132Glufs*27 | DSAP/PK | [3,7] |
| 17 | MVK | 5 | Insertion | c. 417dup | p.Gly140Argfs*47 | DSAP/PK | [1,5,8,9] |
| 18 | MVK | 5 | Deletion | c. 437G>A | p.Ser146Asn | PK | [3] |
| 19 | MVK | 5 | Deletion | c. 451G>A | p.Val151Met | PK | [3] |
| 20 | MVK | 5 | Deletion | c. 481_482delTG | p.Cys161Argfs*25 | DSAP/PK | [1,3] |
| 21 | MVK | 5 | Deletion | c. 480_527del48 | p.Cys161_Arg176del | DSAP | [10] |
| 22 | MVK | 6 | Missense | c. 604G>A | p.Gly202Arg | DSAP/PK | [1,3,8] |
| 23 | MVK | 6 | Missense | c. 605G>A | p.Gly202Glu | PK | [3] |
| 24 | MVK | 7 | Deletion | c. 635_637delGAG | p.Gly212del | DSAP | [1] |
| 25 | MVK | 7 | Missense | c. 643C>G | p.Arg215Gly | DSAP | [9] |
| 26 | MVK | 7 | Missense | c. 650A>C | p.His217Pro | PK | [3] |
| 27 | MVK | 7 | Deletion | c. 671del | p.Leu224* | PK | [3] |
| 28 | MVK | 8 | Missense | c. 710C>A | p.Thr237Asn | PK | [3] |
| 29 | MVK | 8 | Missense | c. 764T>C | p.Leu255Pro | DSAP | [1] |
| 30 | MVK | 9 | Missense | c. 836T>C | p.Leu279Pro | DSAP | [1] |
| 31 | MVK | 9 | Deletion | c. 852dup | p.Ala285Serfs*15 | DSAP | [7] |
| 32 | MVK | 9 | Missense | c. 871T>G | p.Tyr291Asp | DSAP | [1] |
| 33 | MVK | 10 | Missense | c. 926G>T | p.Gly309Val | DSAP/PK | [3,11] |
| 34 | MVK | 10 | Deletion | c. 902del | p.Asn301Thrfs*5 | PK | [3] |
| 35 | MVK | 10 | Nonsense | c. 904C>T | p.Gln302* | PK | [3] |
| 36 | MVK | 10 | Missense | c. 935A>G | p.His312Arg | DSAP/PK | [1,3] |
| 37 | MVK | 10 | Missense | c. 965C>A | p.Thr322Asn | PK | [3] |
| 38 | MVK | 10 | Missense | c. 1004G>A | p.Gly335Asp | DSAP | [2] |
| 39 | MVK | 10 | Missense | c. 1012G>A | p.Gly338Ser | PK | [3] |
| 40 | MVK | 10 | Missense | c. 1024A>G | p.Thr342Ala | PK | [3] |
| 41 | MVK | 10 | Missense | c. 1028T>C | p.Leu343Pro | DSAP | [4] |
| 42 | MVK | 10 | Splice site | c. 1039 + 2T>C | p.Leu348Ilefs*17 | DSAP | [1,2] |
| 43 | MVK | 11 | Deletion | c. 1058_1071del | p.Val353Alafs*9 | DSAP | [7] |
| 44 | MVK | 11 | Missense | c. 1067C>G | p.Thr356Arg | PK | [3] |
| 45 | MVK | 11 | Missense | c. 1093T>A | p.Phe365Ile | PK | [3] |
| 46 | MVK | 11 | Missense | c. 1094T>C | p.Phe365Ser | DSAP/PK | [1,3] |
| 47 | MVK | 11 | Missense | c. 1126G>A | p.Gly376Ser | DSAP/PK | [1,2,3] |
| 48 | MVD | 1 | Start codon | c. 1A>G | p.Met1? | PK | [3] |
| 49 | MVD | 1 | Missense | c. 44C>G | p.Pro15Arg | DSAP/DSP | [8] |
| 50 | MVD | 1 | Splice site | c. 70 + 2T>G | p.? | PK | [3] |
| 51 | MVD | 4 | Missense | c. 302C>G | p.Pro101Arg | PK | [3] |
| 52 | MVD | 4 | Missense | c. 383C>T | p.Ala128Val | PK | [3] |
| 53 | MVD | 5 | Missense | c. 482G>T | p.Arg161Leu | PK | [3] |
| 54 | MVD | 5 | Missense | c. 482G>A | p.Arg161Gln | PK | [3] |
| 55 | MVD | 6 | Splice site | c. 678 + 1G>T | p.? | PK | [3] |
| 56 | MVD | 7 | Missense | c. 682C>T | p.Arg228Trp | PK | [3] |
| 57 | MVD | 7 | Missense | c. 683G>A | p.Arg228Gln | PK/DSAP | [3,8] |
| 58 | MVD | 7 | Missense | c. 746T>C | p.Phe249Ser | PK/DSAP/DSP | [3,8] |
| 59 | MVD | 7 | Missense | c. 875A>G | p.Asn292Ser | PK/DSAP/DSP | [3,8] this study |
| 60 | MVD | 9 | Deletion | c. 1111_1113del | p.Ile371del | PK | [3] |
| 61 | MVD | 10 | Missense | c. 1126G>A | p.Gly376Arg | PK/DSAP/DSP | [3,8] |
| 62 | PMVK | 1 | Start codon | c. 1A>G | p.Met1? | PK | [3] |
| 63 | PMVK | 1 | Missense | c. 94A>T | p.Arg32* | PK | [3] |
| 64 | PMVK | Missense | c. 143G>A | p.Lys48Arg | PK | [8] | |
| 65 | PMVK | 3 | Missense | c. 205A>G | p.Lys69Glu | PK | [3] |
| 66 | PMVK | 3 | Nonsense | c. 312G>A | p.Trp104* | PK | [3] |
| 67 | PMVK | 4 | missense | c. 314T>C | p. Leu105Pro | PM | [12] |
| 68 | PMVK | 4 | Nonsense | c. 412C>T | p.Arg138* | PK/DSP | [3,13] |
| 69 | PMVK | 5 | Deletion | c. 550del | p.Leu184* | PK | [3] |
| 70 | FDPS | 4 | Missense | c. 338G>A | p.Arg113Gln | PK | [3] |
| 71 | FDPS | 1-2 | Deletion | c.-1129_141 + 994del | p.? | PK | [3] |
| 72 | FDPS | 4-7 | Deletion | c. 283-1776_649-143del | p.? | PK | [3] |
| 73 | FDPS | 5 | Splice site | c. 486 + 1G>A | p.Ser163_Lys353delins13 | PK | [3] |
| 74 | FDPS | Splice site | c. 773 + 1 G>A | DSAP/DSP | [8] | ||
| 75 | SLC17A9 | 2 | Missense | c. 25C>T | p.Arg9Cys | DSAP | [14] |
| 76 | SLC17A9 | 10 | Missense | c. 932G>A | p.Arg311Gln | DSAP | [14] |
PK: Porokeratosis, DSAP: Disseminated superficial actinic porokeratosis, DSP: Disseminated superficial porokeratosis, PM: Porokeratosis of Mibelli, MVK: Mevalonate kinase, MVD: Mevalonate decarboxylase, PMVK: Phosphomevalonate kinase, FDPS: Farnesyl diphosphate synthase, SLC17A9: Solute carrier family 17 member 9. Reference:
Zhang SQ, Jiang T, Li M, Zhang X, Ren YQ, Wei SC, et al. Exome sequencing identifies MVK mutations in disseminated superficial actinic porokeratosis. Nat Genet 2012;44:1156-60.
Liu Y, Wang J, Qin Y, Huang C, Archacki S, Ma J, et al. Identification of three mutations in the MVK gene in six patients associated with disseminated superficial actinic porokeratosis. Clin Chim Acta 2016;454:124-9.
Zhang Z, Li C, Wu F, Ma R, Luan J, Yang F, et al. Genomic variations of the mevalonate pathway in porokeratosis. Elife 2015;4:e06322.
Dai J, Chen M, Fu X, Yu Y, Shi Z, Yu C, et al. Mutation analysis of the MVK gene in Chinese patients with disseminated superficial actinic porokeratosis. J Dermatol Sci 2013;72:320-2.
Lu WS, Zheng XD, Yao XH, Zhang LF, Wang MQ, Jiang FX, et al. A novel MVK missense mutation in one Chinese family with disseminated superficial actinic porokeratosis. Mol Biol Rep 2014;41:7229-33.
Sun RF, Chen H, Zhu W, Lian S. Dermoscopic features and gene mutation in the mevalonate pathway of five sporadic patients with porokeratosis. Chin Med J (Engl) 2017;130:1747-8.
Zhou Y, Liu J, Fu X, Yu Y, Shi B, Yu G, et al. Identification of three novel frameshift mutations of the MVK gene in four Chinese families with disseminated superficial actinic porokeratosis. Br J Dermatol 2013;169:193-5.
Li M, Li Z, Wang J, Ni C, Sun Z, Wilson NJ, et al. Mutations in the mevalonate pathway genes in Chinese patients with porokeratosis. J Eur Acad Dermatol Venereol 2016;30:1512-7.
Lu WS, Zheng XD, Yao XH, Zhang LF, Hu B, Lu YJ, et al. Detection of a novel missense mutation in the mevalonate kinase gene in one Chinese family with DSAP. Int J Clin Exp Pathol 2014;7:728-32.
Li CX, Sun SL, Liang JY, Yuan YQ, Zhang SQ, Chen PJ, et al. A novel non-frameshift deletion in MVK gene responsible for disseminated superficial actinic porokeratosis in one Chinese family. J Eur Acad Dermatol Venereol 2017;31:e510-2.
Zhou MS, Xie HF, Chen ML, Jian D, Liu FF, Chen X, et al. A novel mutation for disseminated superficial actinic porokeratosis in the MVK gene. Br J Dermatol 2014;171:427-9.
Song NJ, Luan J, Zhang ZH. Updating and identifying a novel mutation in the PMVK gene in classic porokeratosis of mibelli. Clin Exp Dermatol 2017;42:910-1.
Wang J, Liu Y, Liu F, Huang C, Han S, Lv Y, et al. Loss-of-function mutation in PMVK causes autosomal dominant disseminated superficial porokeratosis. Sci Rep 2016;6:24226.
Cui H, Li L, Wang W, Shen J, Yue Z, Zheng X, et al. Exome sequencing identifies SLC17A9 pathogenic gene in two Chinese pedigrees with disseminated superficial actinic porokeratosis. J Med Genet 2014;51:699-704.
Materials and Methods
Sample
Nineteen individuals (8 males and 11 females) in five generations of a Chinese family with DSAP from Anhui Province of China were recruited [Figure 1a]. The proband (II 4), a 79 years old male presented diffuse annular plaque with trophic centers and brownish hyperkeratotic edges that were partially fused on the face, trunk and extremities over 10 years [Figure 1b]. The mother of the proband (I 1, died) was described as an affected individual. The daughters (III3, III5, III7 and III9), a grandson (IV5), a granddaughter (IV6) and a nephew (III2) were also affected at various ages of onset with lesions [Table 2, Figure 1c-d]. The diagnosis was performed by two experienced dermatologists. Histopathology from the proband showed a cornoid lamellae presenting as columns of PK [Figure 1e], which confirmed the diagnosis. No medical history was documented for the individuals. Written informed consent was obtained from the volunteers, followed by 17 venous blood samples obtained from the family. The study was approved by the Ethics Committee of Anhui Medical University and managed in accordance with the Declaration of Helsinki principles.
Figure 1.
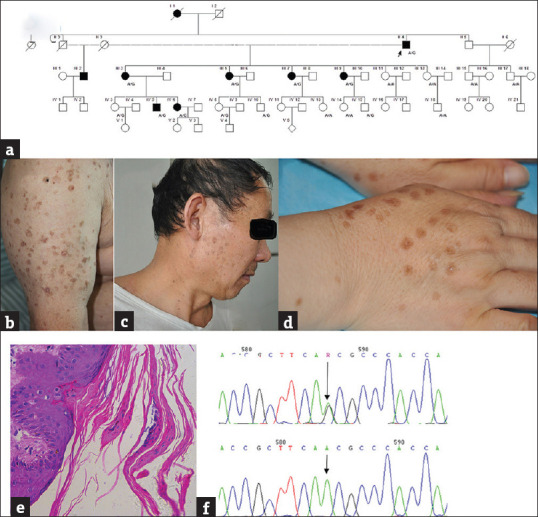
Clinical features of the family with disseminated superficial porokeratosis and DNA sequencing of MVD gene sequence. (a) Pedigree of the family. Arrow, the proband of the family. A/G and A/A indicate individuals genotype. (b) Diffuse small, annular, atrophic, keratotic lesions on the proband's (II 4) upper limb. (c) The lesion presented on the face of III 2. (d) The lesion located on opisthenar of III 9. (e) Histological examination of the proband (×40). (f) Sanger sequencing chromatograms of affected and unaffected at the c. 875A > G mutation site indicated by arrow
Table 2.
Summary of the clinical features and mutation in the family
| Subjects | Relation | Sex | Age | Onset age | Affected Regions | Exacerbation | Number of lesions | Genotype |
|---|---|---|---|---|---|---|---|---|
| II4 | Proband | Male | 79 | 60 | Face, neck, trunk, limbs | Sun-exposed | >1000 | A/G |
| III1 | Nephew | Male | 60 | 45 | Face, upper limb, neck | 50-80 | ||
| III3 | Daughter | Female | 55 | 44 | Face, neck, upperlimb, crus | Sun-exposed | >100 | A/G |
| III5 | Daughter | Female | 52 | 42 | Face, neck, opisthenar, trunk, right crus | 30-50 | A/G | |
| III7 | Daughter | Female | 49 | 40 | Opisthenar | 10-30 | A/G | |
| III9 | Daughter | Female | 45 | 25 | Face, neck, Opisthenar, trunk, limbs | Sun-exposed | 50-80 | A/G |
| III11 | Daughter | Female | 43 | A/A | ||||
| III13 | Daughter | Female | 40 | A/A | ||||
| III15 | Nephew | Male | 49 | A/A | ||||
| IV17 | Nephew | Male | 47 | A/A | ||||
| IV3 | Granddaughter | Female | 34 | A/G | ||||
| IV5 | Grandson | male | 32 | 25 | Forehead, neck, trunk, upper limbs | Sun-exposed, tired | 10-30 | A/G |
| IV6 | Granddaughter | female | 30 | 18 | Face, neck, right opisthenar | Sun-exposed, post partum | 10-30 | A/G |
| IV8 | Granddaughter | female | 26 | A/G | ||||
| IV10 | Grandson | male | 23 | A/G | ||||
| IV13 | Granddaughter | female | 17 | A/A | ||||
| IV14 | Granddaughter | female | 21 | A/A | ||||
| IV-15 | Grandson | male | 14 | A/G | ||||
| IV-18 | Grandson | male | 17 | A/A |
Sanger sequencing and mutation analysis
DNA was extracted from 2 mL venous blood according to the QIAamp DNA Blood Mini Kit (Qiagen, Germany). DNA samples were amplified using a polymerase chain reaction with specially designed primer pairs covering the all exon and the exon–intron boundary regions of MVK and MVD. Primers were designed using primer3. The sequencing products were analyzed by an ABI3730XL DNA Analyzer. The results were analyzed by the PolyPhred software and revised manually.
Results
Mutation detected
Four patients (II 4, III 3, III 9, and IV 5) and three controls (III 11, III 13, and III 15) were sequenced for MVK gene using Sanger sequencing. However, no associated mutations were identified. Then, we sequenced the MVD gene and identified a c. 875A > G (p. Asn292Ser) mutation in exon 7 in the four patients; no changes were observed in the three unaffected individuals [Figure 1f]. The study was repeated in other family members of the family, and the results indicated that other three affected individuals (III 5, III 7, and IV 6) and four predicted patients (IV 3, IV 8, IV10, and IV 15) carried this mutation. We did not find the mutations in other individuals in the family [Table 2]. Pathogenicity analysis of the mutation indicated that the SIFT Score was 0 and the PolyPhen2 score was 0.996, which implied that this mutation might be harmful.
Literature review
We searched the literature for DSAP cases reported in Chinese populations from January 2000 to October 2017 using Sinomed and PubMed. Fifty-two articles describing > 100 families and over 100 sporadic DSAP cases, were selected. The earliest age of onset ranged from 0 to 76 years old, of which the earliest age at onset was 0–52 years in the families and 15–76 years in sporadic patients. DSAP lesions presented on the face were reported in 37 studies, accounting for 71.1% of the total literature. The lesions were aggravated by sun exposure in 18 studies (34.6%) mostly in summer and reduced in winter.
Discussion
Several DSAP family reports have been previously published, suggesting that genetic factors are involved in its pathogenesis. According to Table 1, the number of mutations associated with PK in MVK, MVD, PMVK, FDPS, and SLC17A9 was 47, 14, 8, 5, and 2, respectively. This includes 44 missense (57.8%), 3 start codon (3.9%), 5 nonsense (6.6%), 15 deletion (19.7%), 1 insertion (1.3%), and 8 splicing defects (10.5%). The MVK and MVD genes were two mutational hotspots. Two mutations (c. 746T > C and c. 875A > G) in the MVD gene were identified in 50 unrelated patients and accounted for 81% of all patients with MVD mutations.[2] Li et al. reported that 13.6% of all patients carried the mutation (c. 875A > G) in the study.[6] In our study, we confirmed the mutation c. 875A > G (p. Asn292Ser) in the MVD gene, which is a mutational hotspots.
The MVD gene in the 16q24.1–24.3 region encodes mevalonate 5-diphosphate decarboxylase, an important enzyme in the mevalonate pathway that catalyzes the ATP-dependent decarboxylation of mevalonate 5-diphosphate to isopentenyl diphosphate, which is a ubiquitous precursor for cholesterol, heme A, ubiquinones, dolichol, and isoprenylated proteins.[7] Isoprenylated proteins regulate cell growth, division, and differentiation, and they are probably related to retained nuclei in the stratum corneum. The abnormal generation of isoprenoids might predispose patients to idiopathic inflammation of the skin and result in the development of PK.[2]
Patients in this family exhibited the widest range of phenotypes in terms of both lesion number and distributions. The age of onset spanned from 18 to 60 years, and the diameter of the lesions was generally 0.5–1 cm, which is consistent with an earlier study.[2] In the second, third, and fourth generation, the mean age of onset was 60, 39, and 21 years old, respectively, suggesting that the age of onset was becoming increasingly early. However, it is also possible that the younger individuals in the family have asked for medical assistance earlier in life, which needs further confirmation. Seven patients, except III 7, exhibited face lesions including the eyes and the surrounding skin, supporting the conclusion that MVD mutations are linked to PK around the eyes.[2] Patients of II 4, III 3, III 9, IV 5, and IV 6 reported exacerbation after sun exposure. Patient IV 6 experienced the exacerbation of DSAP during pregnancy. We identified that IV 3, IV 8, IV 10, and IV 15, who carried the mutation, had no lesions to date. Their ages ranged from 14 to 34 years old. Other members of the family showed DSAP-related clinical manifestations at this age, and although they are currently still asymptomatic, we cannot deny that they might develop DSAP in the future. The potential explanation for various clinical manifestations is a combination of genetic factors and environmental exposure.
The literature review presented additional information about the DSAP phenotype. The earliest age of onset in familial patients was 0–52 years and was 15–76 years in sporadic patients, supporting an earlier study that these nonfamilial cases belonged to a subtype of DSAP with an unusually late onset.[8] DSAP lesions usually have been exposed to sunlight but are rarely on the face. However, Gu et al. identified that 82.4% of cases of DSAP with lesions presented on the face,[9] and 71.1% of the studies described the lesion involving the face. This suggests that lesions presenting on the face are common in Chinese populations, which is distinct from studies in Caucasians. Sun exposure, radioactivity, viral infection, renal transplantation, and bone marrow transplantation have been reportedly involved in DSAP development.[10] Excluding sun exposure, few of the above risk factors were reported in Chinese populations, which is likely due to ethnic and geographical differences in DSAP.
Conclusions
We confirmed a c. 875A > G mutation of MVD gene in a Chinese family with DSAP, which was a hotspot mutation. Literature review showed that the age of onset in DSAP family was earlier than that in sporadic patients; the lesion is common in the face in Chinese population which is distinct from studies in Caucasians; ultraviolet exposure is the main aggravating factor. These observations enriched our understanding of DSAP in China. Further functional studies of the candidate protein are needed to define a diagnostic strategy for DSAP mutation screening.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Li CX, Sun SL, Liang JY, Yuan YQ, Zhang SQ, Chen PJ, et al. A novel non-frameshift deletion in MVK gene responsible for disseminated superficial actinic porokeratosis in one chinese family. J Eur Acad Dermatol Venereol. 2017;31:e510–2. doi: 10.1111/jdv.14360. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Z, Li C, Wu F, Ma R, Luan J, Yang F, et al. Genomic variations of the mevalonate pathway in porokeratosis. Elife. 2015;4:e06322. doi: 10.7554/eLife.06322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cui H, Li L, Wang W, Shen J, Yue Z, Zheng X, et al. Exome sequencing identifies SLC17A9 pathogenic gene in two chinese pedigrees with disseminated superficial actinic porokeratosis. J Med Genet. 2014;51:699–704. doi: 10.1136/jmedgenet-2014-102486. [DOI] [PubMed] [Google Scholar]
- 4.Song NJ, Luan J, Zhang ZH. Updating and identifying a novel mutation in the PMVK gene in classic porokeratosis of mibelli. Clin Exp Dermatol. 2017;42:910–1. doi: 10.1111/ced.13197. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Wang J, Qin Y, Huang C, Archacki S, Ma J, et al. Identification of three mutations in the MVK gene in six patients associated with disseminated superficial actinic porokeratosis. Clin Chim Acta. 2016;454:124–9. doi: 10.1016/j.cca.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Li M, Li Z, Wang J, Ni C, Sun Z, Wilson NJ, et al. Mutations in the mevalonate pathway genes in chinese patients with porokeratosis. J Eur Acad Dermatol Venereol. 2016;30:1512–7. doi: 10.1111/jdv.13653. [DOI] [PubMed] [Google Scholar]
- 7.Rossoni L, Hall SJ, Eastham G, Licence P, Stephens G. The putative mevalonate diphosphate decarboxylase from picrophilus torridus is in reality a mevalonate-3-kinase with high potential for bioproduction of isobutene. Appl Environ Microbiol. 2015;81:2625–34. doi: 10.1128/AEM.04033-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang ZH, Huang W, Niu ZM, Liu WD, Xiang LH, Yuan WT, et al. Two closely linked variations in actin cytoskeleton pathway in a chinese pedigree with disseminated superficial actinic porokeratosis. J Am Acad Dermatol. 2005;52:972–6. doi: 10.1016/j.jaad.2005.01.099. [DOI] [PubMed] [Google Scholar]
- 9.Gu CY, Zhang CF, Chen LJ, Xiang LH, Zheng ZZ. Clinical analysis and etiology of porokeratosis. Exp Ther Med. 2014;8:737–41. doi: 10.3892/etm.2014.1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu P, Zhang S, Yao Q, Liu X, Wang X, Huang C, et al. Identification of a genetic locus for autosomal dominant disseminated superficial actinic porokeratosis on chromosome 1p31.3-p31.1. Hum Genet. 2008;123:507–13. doi: 10.1007/s00439-008-0504-x. [DOI] [PubMed] [Google Scholar]