

Sir,
Xeroderma pigmentosum (XP) is a group of rare inherited cancer-prone skin conditions characterized by sever sensitivity to the DNA damaging effects of ultraviolet (UV) radiations and results in hyperpigmentation, accelerated aging, ocular manifestation, cutaneous photosensitivity, and high risk of skin carcinoma.[1] There are eight different types of XP, consistent with eight different proteins, which if become mutant, can result in XP phenotypes. These proteins are involved in the repair of UV-induced DNA damage. Seven of these proteins (XPA, XPB, XPC, XPD, XPE, XPF, and XPG) are associated with the removal of UV-induced damage from the DNA. The eighth group (XPV) is associated with replication of DNA containing unrepaired damage.[2]
In this study, two Pakistani consanguineous kindreds, inheriting autosomal recessive XP [Figure 1a and b], were ascertained from Balochistan Province of Pakistan. The study was approved by BUITEMS Institutional Review Board. The clinical features of affected individuals in both the families were photosensitivity, photophobia, poikiloderma, xeroderma, hypo- and hyperpigmentation, and ocular manifestations (corneal ulceration and blurred vision) [Figure 1c-h], which were consistent with XP.[3,4] After informed written consent, blood samples for the current study were collected from nine individuals including four affected [Figure 1a and b] and subjected to genomic DNA extraction was carried out by standard phenol-chloroform method. Genomic DNA of the two patients; IV-1 from family A and V-2 from family B was subjected to whole exom sequencing (WES) following the previously described protocol by Ijaz et al., (2019).[5]
Figure 1.

Pedigrees and clinical presentation of the studied families segregating autosomal recessive xeroderma pigmentosum. (a) Pedigrees of family A and (b) family B. Members whose blood was available for the study are shown with *. (c and d) clinical pictures of affected individual IV-2 of family A; and (g and h) clinical pictures of affected individuals V-2 and V-3 of family B
WES generated 108,035 and 105,406 variants for patient IV-1 of family A and V-2 of family B, respectively. Filtering of the data revealed two homozygous frameshift variants c.1382_1383insG in exon 9 and c.1934delC in exon 10 of XPC gene in patients IV-1 of family A and V-2 of family B, respectively. Sanger sequencing confirmed the co-segregation of frameshift insertion mutation, c.1382_1383InsG; (p.P462Tfs*32), in exon 9 of XPC gene in family A and c.1934delC (p.P645Lfs*5) in exon 10 of XPC in family B [Figure 2]. None of these sequence variant (c.1382_1383InsG and c.1934delC) has previously been reported in public databases, such as Exome Aggregation Consortium (http://exac.broadinstitute.org/), genome aggregation database (gnomAD) (https://gnomad.broadinstitute.org/) and 1000 genomes. The sequence variants were also not found in our in-house exome data of additional 50 Pakistani unrelated individuals from the same ethnicity. These two novel mutations, c.1382_1383insG and c.1934delC, in XPC probably results in a frameshift and premature termination codon p.P462Tfs*32 and p.P645Lfs*5, respectively.
Figure 2.

Sequencing chromatograms of XPC gene showing co-segregation of the two frameshift variants in family A and B
Bioinformatic analysis of normal and mutant proteins was performed through GalaxyTBM [Figures 3 and 4]. In case of p.P462Tfs*32, the XPC protein will lose its normal interactions from 465 to 490, which are involved in proper folding of the protein through hydrogen bonding [Figure 3e and f], so, perturbing the protein structure. In addition, the mutant protein will also lose its RAD23B binding region (496–734), damage recognition region (607-766), DNA-binding region (607-831), ERCC2 and GTF2H1 interactions region (816-940) and CETN2 interaction residues (847-866). In case of p.P645Lfs*5 [Figure 4c], the proline-645 responsible for special backboned conformation is replaced by less rigid, bigger in size leucine which leads to bumps in the structure consequently altering the special conformation. The truncated protein (p.P645Lfs*5) lacking the residues required for damage recognition and DNA binding, the mutant protein will be unable to interact with ERCC2, GTF2H1, and CETN2 proteins, so losing majority of its functions.
Figure 3.

Surface view of the surrounding residues in normal (a and c), p.P462Tfs*32, (b) and p.P645Lfs*5 (d) XPC. (e) Hydrogen bonding among the residues (465 to 490) of normal and (f) mutant (p.P462Tfs*32) XPC
Figure 4.
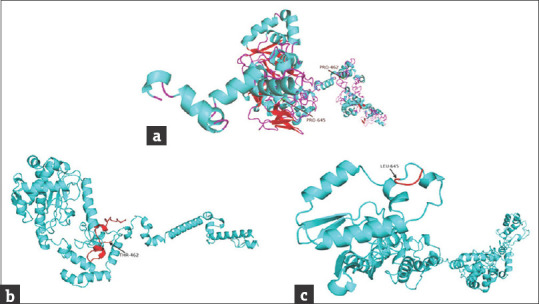
Modeled structures of normal and mutant XPC proteins. (a) Structure of wild type XPC protein, the arrows show the position of frameshift start in diseased individuals; (b) structure of p.P462Tfs*32 mutant protein, the miss-sequence residues (461-491) are shown in red color; (c) structure of p.P645Lfs*5 mutant protein, the miss-sequence residues (645-648) are shown in red
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
The authors would like to thank members of both families for their cooperation and participation in the study. The work presented here was funded by Higher Education Commission (HEC), Islamabad, Pakistan through a project (5265/NRPU/HEC/2016) to AW. This work was further supported by the Alexander-von-Humboldt Foundation, Germany through a return fellowship to AW.
References
- 1.Ide F, Oda H, Nakatsuru Y, Kusama , Sakashita H, Tanaka K, et al. Xeroderma pigmentosum group A gene action as a protection factor against 4-nitroquinoline 1-oxide-induced tongue carcinogenesis. Carcinogenesis. 2001;22:567–72. doi: 10.1093/carcin/22.4.567. [DOI] [PubMed] [Google Scholar]
- 2.Kakumu E, Nakanishi S, Shiratori HM, Kato A, Kobayashi W, Machida S, et al. Xeroderma pigmentosum group C protein interacts with histones: Regulation by acetylated states of histone H3. Genes Cells. 2017;22:310–27. doi: 10.1111/gtc.12479. [DOI] [PubMed] [Google Scholar]
- 3.Ijaz A, Basit S, Gul A, Batool L, Hussain A, Afzal S, et al. XPC gene mutations in families with xeroderma pigmentosum from Pakistan; Prevalent founder effect. Congenit Anom. 2018;59:18–21. doi: 10.1111/cga.12281. [DOI] [PubMed] [Google Scholar]
- 4.Shah K, Mehmood S, Jan A, Abbe I, Hussain Ali R, Khan A, et al. Sequence variants in nine different genes underlying rare skin disorders in 10 consanguineous families. Int J Dermatol. 2017;56:1406–13. doi: 10.1111/ijd.13778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ijaz A, Basit S, Gul A, Batool L, Hussain A, Afzal S, et al. UV-sensitive syndrome: Whole exome sequencing identified a nonsense mutation in the gene UVSSA in two consanguineous pedigrees from Pakistan. J Dermatol Sci. 2019;95:113–8. doi: 10.1016/j.jdermsci.2019.08.003. [DOI] [PubMed] [Google Scholar]