

1. Genomic and Genetic Variation, Molecular Diagnosis, and Disease Traits
Clinical genomics may perhaps best be defined, from a retrospective historical and operational standpoint [1,2], as utilizing the variation inherent to the personal genome of an individual patient to formulate a molecular diagnosis that may be potentially clinically impactful. A molecular diagnosis is not a clinical diagnosis, but rather variation of a gene or genome that may potentially have contributory consequences for the patient’s disease process, either at present, or in the future. The predictive advantage of molecular diagnoses is particularly poignant in cases for which there is no family history of the clinical diagnosis or clinically observed phenotype to otherwise impart clinical suspicion. Like other clinical laboratory testing, the derived molecular diagnosis needs to be contextualized with the clinical observations. When the molecular and the clinical diagnoses are consistent with the patient’s assessment, i.e. clinical history and physical examination, and the emerging clinical picture for a given gene or variant allele matches the clinical synopsis of an online Mendelian Inheritance in Man (OMIM: https://www.omim.org/) defined phenotype, it may be clinically informative. Molecular diagnosis can sometimes help resolve clinical diagnostic ambiguities and be used to explore a differential diagnosis for a known rare disease [3], but should we begin to utilize it to assist in the formulation of a differential diagnosis? Moreover, how can we further elevate the individual clinical case ‘solved rate’ for molecular diagnoses achieved through genomics?
2. Copy-number Variation
2.1. Locus copy-number variation and disease, inherited variation and new mutation
The practice of clinical genetics has been utilizing measurement of some types of human genome variation, such as chromosome studies, as a clinical tool for decades. Down syndrome, caused by increased gene dosage (3 copies of all genes mapping to chromosome 21), is one illustrative example. The molecular mechanism leading to an extra copy of chromosome 21 and potential gene dosage effects – whether a nondisjunction trisomy 21 or an unbalanced Robertsonian translocation involving the acrocentric chromosomes [e.g. 46,XX,der(14;21)(q10;q10),+21 or 46,XX,der(21;21)(q10;q10),+21), or isochromosome 21 (e.g. 46,XY,i(21)(q10))] –– causes phenotypic expression of Down syndrome in the patient. The precise molecular mechanisms for the derivation of the three copies of chromosome 21 genomic sequence and whether the third copy is part of a derivative chromosome or resultant from a nondisjunction event, however, each have different clinically impactful meanings for the patient and family. Parental carrier status for balanced translocations further influences recurrence risk for families: a key component of the clinical evaluation which should not be overlooked for this condition that is most often conceptualized as ‘sporadic’ and due to new mutation nondisjunction trisomy 21. In this poignant clinical example associating genomic variation with a clinical phenotype of Down syndrome, the dosage of the chromosome 21 locus is equivalent whether occurring as new mutation nondisjunction trisomy 21 or potentially inherited Robertsonian t(14;21) and t(21;21) translocations or isochromosome i(21) from carrier unaffected parents. The number of resultant chromosomes in the inherited versus sporadic situation is different; 47 in nondisjunction with a de novo mutation event resulting in free trisomy 21, and 46 in the setting of an inherited balanced Robertsonian chromosome 21 translocation or isochromosome 21.
It is interesting to speculate about specific chromosome 21 gene dosage effects and clinical findings and phenotypes that can be observed in patients with Down syndrome: APP duplication [4,5] and early onset dementia, and an oncogene mapping to chromosome 21 and increased risk of leukemia. Or perhaps leukemia observed in a child with tetrasomy 21 [6,7]. It is also interesting to consider whether other well-known clinical observations for Down syndrome, implicate a potential dosage sensitive gene(s) or locus mapping to chromosome 21.
2.2. Copy-number as a biomarker for disease
Copy-number variation can be benign or cause disease. The concept of gene/locus dosage as a surrogate biomarker for the disease trait peripheral neuropathy, was perhaps one of the first inventions (United States Patent and Trade Office, USPTO#s: 5,306,616; 5,780,223) to argue this concept. The locus variation resulting in gene dosage abnormalities, which can be due to an autosomal dominant (AD) trait segregating with the Charcot-Marie-Tooth disease type 1A locus duplication [8], could be a biomarker used to elucidate a molecular diagnosis responsible for neuropathy – even before the actual ‘causative gene’, PMP22, was established as the likely dosage sensitive driver gene [9]. Of note, 20 years of clinical genetic testing, in >10,000 patients with clinical findings of peripheral neuropathy, has shown that ~80% of molecular diagnostic alleles are copy-number variants (CNV) at this PMP22 locus despite the observation that pathogenic variant alleles for dozens of genes, for this genetically heterogeneous disorder, can result in the CMT peripheral neuropathy trait [10].
2.3. Gene dosage and expression
Alterations in gene dosage can result from alterations in gene copy-number or gene expression levels or both. Moreover, optimal gene dosage to achieve biological homeostasis may depend on stage of organismal development or timing in lifecycle. Not all genes are ‘dosage sensitive’, i.e. haploinsufficient or triplosensitive, having one copy or three can be adequate to maintain biological balance and homeostasis of the diploid organism. Variant effects for dosage sensitive genes mapping to the X chromosome can be challenging to interpret in different sexes because contextualization can sometimes be confounded by X inactivation.
3. Rare Variation and Disease
3.1. Rare variation at a locus and perturbations of biological homeostasis
In general, four disease categories for pathology contributing genomic variation can be conceptualized. These group into four clinically observed types of genetic conditions that are associated with specific types of chromosome and/or genomic DNA variation [11,12]. These include: i) chromosomal disorders, ii) genomic disorders [1,13], iii) Mendelian disease traits, and iv) complex traits. Many common diseases are complex traits with genetic susceptibility and environmental contributory components.
Clinical application of examining and interpreting variation of the entire human genome has been used extensively for patient genome characterization using classical chromosome analyses provided by clinical cytogenetic studies, in essence single cell genomics. Whereas it is an incredibly important tool for the identification of disease category number (i), i.e. chromosomal disorders, clinical cytogenetic studies do not allow visualization of submicroscopic DNA rearrangements (e.g. deletion and duplication CNV) or DNA sequence variation (i.e. SNV) involving gene(s). However, the tremendous clinical utility of the haploid human genome reference sequence is as an adjuvant clinical genomics tool to assess DNA variation genome-wide.
Clinically impactful variant types can involve chromosomal changes, DNA structural variants (SV) including CNV and copy-number neutral SV such as inversions, insertion/deletions or indels of < 50 bp variant alleles, and SNV or Watson-Crick (W-C) base pair (bp) changes that may introduce protein structural changes. In general, clinical genome-wide arrays identify submicroscopic pathogenic CNV and chromosome abnormalities while genomic DNA sequencing (cES and WGS) can identify pathogenic SNV and indels. cES has limited ability to detect pathogenic CNV given variation of read depth introduced by exome capture; WGS is better than cES for CNV, particularly those of smaller size and that map outside coding regions, but clinical experience is thus far limited due to availability and cost.
For SNV, the potential predicted protein structure or functional change introduced by variation of a gene may be interpreted by ‘conceptual translation’ via the genetic code. Standards and guidelines are available for variant interpretation to facilitate molecular diagnosis [14]. However, these apply only to known Mendelian disease genes. A pilot study of ~75 unsolved clinical exomes (cES) demonstrated more than half of the unsolved could be solved, i.e. a plausible molecular diagnosis established, by a further research analyses and elucidating novel disease genes by identifying multiple cases with a similar clinical phenotype and having the same gene harboring a damaging variant allele(s) [15].
Examining the patient’s personal genome in a family trio (proband + parents) exome sequencing (ES) manner, with computational filtering of rare variants inherited from one parent or the other, can enable the identification of the new mutation(s), and potential pathological variation contributing to the sporadic disease process as compared with inherited variant alleles [16,17]. The disease associated new mutation can be: i) an entire chromosome as in nondisjunction trisomy 21 causing Down syndrome, ii) SV/CNV perturbing a gene’s dosage or expression or structure of a gene, perhaps APP gene dosage associated with early onset dementia and an oncogene causing leukemia both as endophenotypes of Down syndrome, or iii) a W-C bp change, SNV, resulting in a pathogenic rare variant allele such as for example a DVL1 or DVL3 gene −1 frameshift allele causing autosomal dominant Robinow syndrome [18].
Variation of the haploid reference human genome, which is 3×109 base pairs of DNA (A, C, G, or T), can consist of changes of a single nucleotide in a uniquely defined coding or non-coding nucleotide sequence that can vary into any one of the other three bases at that particular W-C base pair position. Variation of nucleotides mapping within repetitive sequences (Alu, LINE) and low-copy repeats (LCRs) or segmental duplications provides a particular challenge to detecting individual locus variation - the ‘non-unique’ map position of the DNA sequence that is varying within the repeat. Each personal genome has an abundance of rare variation, but through systematic computational filtering one can narrow the potential disease contributing clinically impactful variation to a small number of rare variants that can potentially constitute medically actionable variation and thus, formulate a molecular diagnosis in a locus or gene specific manner. Some types of rare variation, e.g. copy-number neutral inversions, balanced translocations, and repeat expansions are not detectable by current clinical genomics assays.
3.2. Clinical exomes
Initial clinical exome sequencing (cES) studies on significant numbers of consecutive patients/cases, N=814 [16] and N=3,386 [17], undergoing genome-wide analyses by cES in a clinical diagnostic laboratory, revealed a molecular diagnosis explaining part/all of the observed clinical disease phenotype in about 25–30% of patients. Also of interest was the identification by cES of multi-locus pathogenic variation, i.e. two or more molecular diagnoses, often resulting in a blended phenotype [19]. In all, about 1 in 20 clinically affected individuals that have a molecular diagnosis concluded by cES can have multi-locus pathogenic variation. A blended phenotype can result from two or more molecular diagnoses that can have either overlapping or distinct clinical phenotypic features that have been associated with each of the individual genes/loci [20]. The resultant ‘blended phenotypes’ for multilocus pathogenic variation can be particularly challenging to clinical diagnosis for the physician observing the patient because the patient’s phenotype may not be perceived as a mixture of disease traits and thus sometimes including both clinical diagnoses, or parts of the phenotypic trait features, within the formulated differential diagnoses.
Systematic reanalysis of cES cases in which no molecular diagnosis was concluded showed that over time the case/cohort ‘molecular diagnosis(es) solved rate’ can substantially increase [21]. Currently, much of this observed increase in ‘case molecular diagnoses solved rate’ reflects the new ‘disease gene’ discoveries that transpired during the time interval between initial cES and re-analyses of extant data; variant associated disease gene discovery is occurring at a remarkable pace [21]!
4. Functional Annotation of Human Genes
The current build of the haploid reference human genome contains ~20,000 predicted protein-coding genes (e.g. exons and introns, etc.), that can be annotated computationally given their features or gene structure characteristics. However, for at least some 75%, or > 15,000 or more computationally annotated genes, it remains to be determined whether variation in that gene results in expression of a phenotype in the human organism; over 18,000 human genes remain to be clinically curated at https://clinicalgenome.org/. While disease gene discovery is proceeding at a remarkable pace, it still requires a tremendous research and clinical effort to ‘functionally annotate’ what may clinically be observed with variation in these computationally predicted genes. This clinical contextualization is important to subsequent variant interpretation. Some of that functional annotation of gene variation will likely take place through model organism research studies, and here the mouse, fruitfly, and zebrafish have provided tremendous biological insights into clinically observed phenotypic features such as microcephaly [22–25]. Nonetheless, a lot of human disease traits, particularly neurobehavioral and neurocognitive traits [26,27] not readily assayed in model organisms, manifest extensive genetic heterogeneity and may require genome-wide assays, such as ES, of clinical populations rather than studies on ‘disease focused’ research and large populations of patients with one disease category or ‘clinical phenotypic diagnosis’. Both disease gene discovery research, and implementation of ES and Whole Genome Sequence (WGS) based molecular diagnosis in clinical situations, are also informed by construction of an allelic series for a given gene or locus [28–31]; this too will require better integration of research genomics and cES data. Data sharing of such gene locus variant information and clinical phenotypic information will likely benefit all stakeholders, including patients, families, extended family members and even populations, around the globe, as well as physicians caring for their patients and families.
5. Multi-locus Pathogenic Variation or Dual Molecular Diagnoses and Disease
Initial hints that dual molecular diagnoses, resulting in blended phenotypes with distinct or overlapping phenotypic features, might occur more frequently than perhaps anticipated by geneticists has derived from clinical implementation of molecular karyotyping [32], multi-gene panel testing [33], and pilot clinical ES [19] studies. A case study revealed a de novo mutation causing a genomic disorder (PTLS; MIM: 610883) in an individual with an inherited CNV allele causing an autosomal dominant (AD) peripheral neuropathy (HNPP; MIM: 162500) in the family [32]. Pilot studies of a small case series (N=250) revealed two or more molecular diagnoses in about 5–6% of cases for which a molecular diagnosis (25% of consecutive cases) was concluded [19]. A similar observation, of about 1 in 20 (~ 5%) of the 25% solved clinical cases, with two or more molecular diagnoses [20] was found in a larger study (N=7,374 consecutive cases). For this latter study, all combinations of Mendelian patterns (autosomal dominant, AD; autosomal recessive, AR; X-linked, XL) were observed for the individual single gene disease traits, (traits found at https://www.omim.org): AD + AD, AD + AR, AD + XL, AR + AR, AR + XL, XL + XL, with a small minority of AR + AR disease trait combinations (10.9%). Moreover, for essentially all dual molecular diagnosis with multi-locus pathogenic variation, with the exception of AR + AR traits, combinations of inherited and de novo mutation alleles were observed.
Remarkably, with monoallelic variants at two loci; i.e. AD + AD, AD + XL or XL + XL disease traits, the pathogenic variants were identified as de novo mutational events in 44.7% of patients when both parents were available for testing (Figure 1) [20]. These dual molecular diagnosis pathogenic alleles could be different variant types, such as SNV or CNV, and AR traits could result from a biallelic combination of an SNV and a CNV allele at a single locus. These observations support the clinical utility of a combined SNV + CNV detection approach using cES and CMA [34,35].
Figure 1.
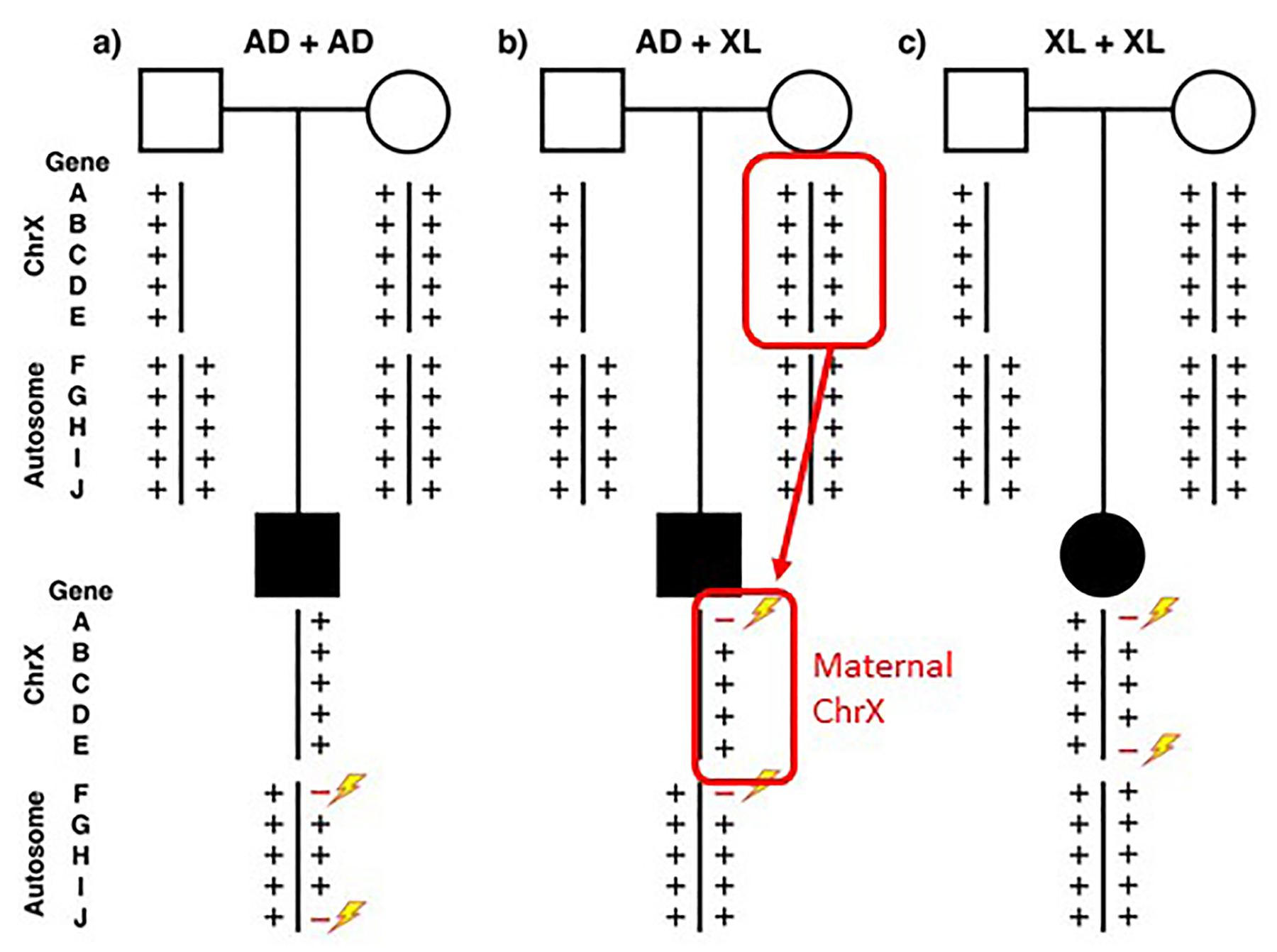
Multilocus pathogenic variation and de novo mutation Standard pedigree symbols are used with squares representing males and circles females; filled squares and circles depict an individual with a clinical disease phenotype. Below these pedigree symbols are shown representative X chromosome and 1 representative of the 22 autosomes each with 5 different gene loci A through J. The plus (+) symbol refers to wild type and variant alleles at the gene locus. Rare variant pathogenic alleles are represented as red minus signs and new mutation depicted by a lightning bolt. AD, autosomal dominant; XL, X-linked.
For AR disease traits with biallelic variation segregating from carrier parents, 52.8% had homozygous alleles. For AR + AR combinations of dual molecular diagnosis, it usually occurs by transmission from carrier parents (Figure 2a). However, with uniparental isodisomy (UPD), the two chromosomes can result in homozygosity for all recessive carrier state loci on that chromosome (Figure 2b), or a large deletion CNV could potentially unmask a recessive carrier state for genes at linked loci (Figure 2c). It is interesting to note that ES studies from an arthrogryposis clinical diagnosis cohort from Turkey, primarily from a population with consanguinity and admixture, had a shift in the ratio of presumptive dual diagnoses for AR + AR disease traits [36] as both loci with homozygous alleles showing a higher rate for such AR + AR (88.9% both homozygous) trait combinations for dual molecular diagnosis – an observation that could perhaps be somewhat related to the population substructure. With consanguinity, large haplotype blocks of the genome may become autozygous due to identity-by-descent (IBD) (Figure 2d).
Figure 2.
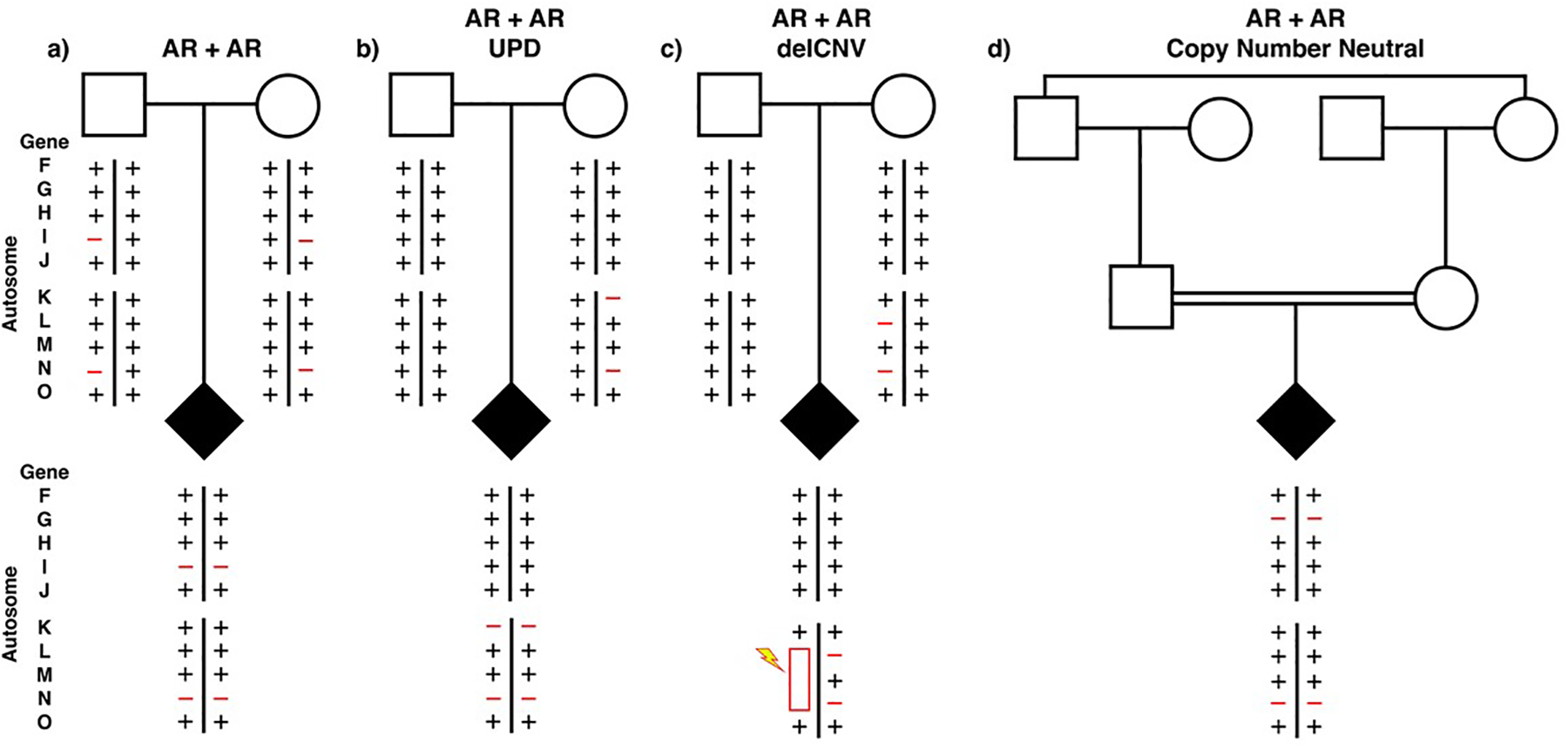
Multilocus pathogenic variation and biallelic AR disease traits Filled diamond indicates affected individual of either sex. Two representative autosomes from the 22 are shown. Four different ways to obtain a homozygous disease gene locus are shown: (a) Mendelian segregation from carrier unrelated parents, (b) uniparental isodisomy (UPD), (c) deletion CNV, and d) identity-by-descent from consanguineous parentage. Rare variant pathogenic alleles are represented as red minus signs and new mutation depicted by a lightning bolt.
Regarding future clinical genomics studies and implementation work it will potentially be informative to explore whether WGS provides further molecular diagnostic variants; SV/CNV and repeat expansion pathogenic alleles are of particular note here. WGS may better detect heterozygous smaller sized CNV (<50 kb) and noncoding variation affecting gene function. WGS could potentially yield a higher rate of dual molecular diagnoses cases than cES because it can better detect smaller sized and noncoding pathogenic CNV; particularly, for dual molecular diagnoses constituted by de novo mutation at a locus. Studies from comparative molecular genome-wide approaches with cES (N = 11,000 consecutive ES studies) suggest that some 30% of clinically significant CNV detected with a combination of SNV + CNV technologies (cES + CMA) may evade detection by current ES alone [34]. Moreover, the finding of homozygous SV haplotypes in cES, mainly exon deletion CNV embedded in a homozygous haplotype, suggest such rare variant alleles may represent de novo mutation, on a recently evolved haplotype in a family or clan [34].
6. Improving Solved Rates for Molecular Diagnosis
Recent work shows that re-analysis of extant cES data can increase molecular diagnostic rates of a given clinical cohort over time [21]. Systematic reanalysis of these extant data suggest that the increased molecular diagnostic ‘case solved rate’ is majorly due to the definition of novel molecularly diagnosable disease genes and by new disease gene discovery and clinical annotation at https://clinicalgenome.org/ of the human haploid genome reference build – the latter yet another testimonial to this haploid human reference genome as a robust and rich resource. Nevertheless, some fraction of these molecular diagnoses could relate to the detection of structural variant (SV/CNV) alleles; about 5 – 10% of the incremental increased molecular diagnostic solved rate in one study [34]. Structural variant alleles of small sizes (< 50 Kb) will be more likely to be detected by WGS versus ES. Moreover, it is not clear whether WGS for clinical molecular diagnosis, and the detection of SV/CNV molecular diagnostic alleles, should be better performed with short read DNA sequencing technologies (more accurate when coverage at the single W-C bp level > 20x) or long read DNA sequencing technologies. The long read technologies may help further resolve novel breakpoint junctions or ‘join points’ that may have been formed during structural variant mutagenesis at least in one study [37]. Remarkably, a hybrid mixture’ of short read + long read provides additional breakpoint/join point information of rearranged genomic DNA and information regarding de novo SNV generation due to hypermutation near SV breakpoint junctions. Those experimental observations enable potential insights into SV mechanism(s), such as microhomology-mediated break-induced replication (MMBIR), as well as SNV formation [37]. The finding of breakpoint and/or join-point microhomology, or alternatively microhomeology, is particularly relevant to resolving complex genomic rearrangements events [38] which can result from iterative template switches during replicative repair [39–41].
Widespread clinical implementation has not been yet adopted for any form of WGS in a clinical diagnostic laboratory. The absence of the ability to perform WGS as a clinically available high throughput test may potentially be a missed opportunity for both cancer molecular diagnoses, and possibly precision medicine guided treatments, as well as in rare Mendelian disease traits, genomic disorders, and chromosomal syndromes. Emerging knowledge suggests that chromothripsis and chromoanasynthesis [42–44], ways in which the human genome is shuffled into a massively changed order and arrangement of base pairs of DNA sequences that may map to a singular chromosome seem to have important clinical ramifications. The potential clinical utility of the chromothripsis/chromoanasynthesis phenomenon in cancer molecular diagnosis [45], guiding chemotherapy dosing and duration of treatment, and as a prognostic indicator regarding potential cancer recurrence risk, are all currently unknown and all remain to be studied in many different cancer types and in many patients.
Moreover, the ability to detect copy-number, chromosome number, CNV at a specific locus, of a diploid genome in gene coding or noncoding regions as provided by some forms of WGS, can be important in clinical applications of genomics on a single cell level and perhaps for some forms of noninvasive prenatal testing (NIPT) – the latter when performed to noninvasively detect from cell free DNA (cfDNA) specific aneuploidies in the developing fetus [46].
One form of long read WGS may also have important clinical utility for measuring gene dosage/expression or copy-number changes (SV/CNV) at a specific locus or chromosome, and potentially for enabling genome-wide assessment of abnormal genomic imprinting and assaying for disease phenotypes resulting from perturbations of imprinted loci [47]. The ability to determine methylated W-C bp by Nanopore long range sequencing may eventually be helpful in detecting clinically relevant epigenetic changes and in integrating haplotypes and dosage information as judged by either epigenetic silencing or expression quantitative trait loci (eQTL) detected from genome-wide association studies (GWAS). The convergence of rare variant analyses from ES with variant information from common variant studies and other genome-wide studies is potentially possible with the noncoding variant information provided by WGS and may be clinically useful for precision medicine – as perhaps illustrated through the conceptual idea of the compound inheritance and gene dosage model and the study of birth defects due to perturbations of developmental processes such as can occur in congenital scoliosis, congenital anomalies of the kidney and urinary tract (CAKUT), and lungs [48–51].
7. Conclusions and Hypothesis Testing
These research observations, and many other clinical examples too numerous to list, suggest that a multitude of potential utilities for genomics and molecular diagnosis in clinical practice are emerging. These include contemplating the many clinical applications of detection of mosaicism [52–54], uniparental disomy as a mediator of AR traits [55], chromosome analyses in somatic tissue mutagenesis, e.g. cancer genome pathology studies of genomic DNA from biopsied material, and gene/genome integrity in SV mutagenesis [56]. Moreover, genomics and molecular diagnosis can potentially provide further testable hypotheses to explore genetic models for disease, including Mendelian disease traits, genomic disorders, and even somatic genomic and chromosomal changes. Nevertheless, current clinical genomics assays each have their own limitations and experimental challenges stimulating exploration of hybrid approaches and additional technologies for assaying clinically relevant variation. The computational and analytical challenges of genomics BigData also require the development of artificial intelligence (AI) approaches to genome analyses. Much work remains and it is likely that as scientists and physicians there will be many lessons to learn with each clinical implementation. Genomics and molecular diagnosis will continue to build a logic of disease simultaneously providing insights into human biology and refining medical practice [57].
8. Expert Opinion
The partnership among clinical geneticists, molecular geneticists, bioinformaticians, and human disease scientists will remain critical as the field of clinical genomics and molecular diagnostics continues to evolve. Despite the increasing use of unbiased genetic and genomic testing (karyotyping, CMA, cES, WGS) to survey genome-wide for molecular aberrations, clinical acumen paired with an understanding of the limitations of each testing type will be needed to achieve molecular diagnoses. At the same time, novel disease gene and variant discoveries driven by research will inform technology and clinical testing and analysis development (e.g. inclusion of newly established disease genes on a clinical array; development of cES capture designs to incorporate the most current human genome haploid reference sequence assembly; application of UPD and AOH analyses to cES and WGS).
In a patient suspected of having a genetic condition, assaying for pathogenic variation in his/her genome using clinical genomics molecular diagnostic test can help clarify a clinical diagnosis. Chromosomal syndromes were defined by changes in chromosome number and or structure. Clinically, chromosome studies by G-banded karyotypes can be very important for medical management and as illustrated for Down syndrome, a frequently observed genetic event occurring in ~1/700 live births, confirm the clinical diagnosis and guide recurrence risk estimates for family counseling. CMA defined genomic disorders by delineating submicroscopic genomic rearrangements and the associated clinical phenotypes. Exon focused microarray designs further refined the resolution and extent of human genome structural variation, mapped and delineated dosage sensitive genes contributing to clinically relevant phenotypes, determined ‘driver genes’ for trait manifestation, and further revealed complex genomic rearrangements and multigenic mechanisms for disease. ES has helped further determine contributions of inherited versus de novo SNV alleles to disease. Combining cES and exon focused CMA data on clinical cases can detect biallelic exonic deletion CNV and CNV + SNV compound heterozygous states for AR disease trait loci [34,35,58,59]. Calculating B-allele frequency from ES data to delineate genomic intervals of absence of heterozygosity (AOH) can reveal autozygous regions implicating UPD, founder alleles, and evidence for consanguinity and IBD of a new pathogenic variant arising in a previous generation within a clan [60].
Thus far, the data suggest a clear path forward for molecular diagnosis and clinical genomics; i.e. a ‘gene-centric’ focus to understand human biology and disease by ES and family based genomics - and to enable variant interpretation and contextualization to the clinical question. The more genes in the human genome for which we understand their function, the more disease biology we will learn and the more readily one can interpret variation and predict medically actionable potentially pathogenic variation enabling one to render a molecular diagnosis. Currently > 90% of the ~20,000 computationally annotated genes in the human genome remain to be ‘clinically annotated’ at https://clinicalgenome.org/.
From the perspective of disease gene discovery, since we seek to understand variation underlying disease traits, a study design and data analysis approach for 100,000 genomes that investigates 1000 different disease phenotypes in 100 unrelated individuals for each phenotype may identify more ‘disease genes’ than an approach that studies 10,000 genomes in individuals with 10 distinct clinical diagnoses. As the application of cES continues to scale up combining genome data from research on rare disease families with different disease traits from around the world with the data generated from cES, facilitated by gene matching of worldwide researchers and clinicians through the MatchMaker Exchange [61], we will continue to functionally annotate the genes in the human genome providing many insights into the biology of disease.
The application of genomic approaches in human genetic research and the clinic has also documented a prominent role for new mutation in disease, and is beginning to evolve testable models for multilocus pathogenic variation in disease. Data from clinical genomics studies is growing at a logarithmic rate. This is an immensely exciting time for clinical genomics and the international spirit of collaboration and cooperation that emboldened the human genome project, but much work remains [62].
Highlights:
Explores the role of clinical genomics and molecular diagnosis in clinical practice and disease research
Delineates types of genetic and genomic variation and assays for detection
Emphasizes rare variation and new mutation in disease
Describes ways to improve case solved rates for molecular diagnosis
Advocates use of clinical data for disease research
References
[* = of importance ** = of considerable importance]
- 1.Lupski JR. Genomic disorders ten years on. Genome Med. 2009. April 24;1(4):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lupski JR. Clinical genomics: from a truly personal genome viewpoint. Hum Genet. 2016. June;135(6):591–601. [DOI] [PubMed] [Google Scholar]
- 3.Posey JE. Genome sequencing and implications for rare disorders. Orphanet J Rare Dis. 2019. June 24;14(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delabar JM, Goldgaber D, Lamour Y, et al. Beta amyloid gene duplication in Alzheimer’s disease and karyotypically normal Down syndrome. Science. 1987. March 13;235(4794):1390–2. [DOI] [PubMed] [Google Scholar]
- 5.Rovelet-Lecrux A, Hannequin D, Raux G, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006. January;38(1):24–6. [DOI] [PubMed] [Google Scholar]
- 6.Shin MG, Choi HW, Kim HR, et al. Tetrasomy 21 as a sole acquired abnormality without GATA1 gene mutation in pediatric acute megakaryoblastic leukemia: a case report and review of the literature. Leuk Res. 2008. October;32(10):1615–9. [DOI] [PubMed] [Google Scholar]
- 7.Jabs EW, Stamberg J, Leonard CO. Tetrasomy 21 in an infant with Down syndrome and congenital leukemia. Am J Med Genet. 1982. May;12(1):91–5. [DOI] [PubMed] [Google Scholar]
- 8.Lupski JR, de Oca-Luna RM, Slaugenhaupt S, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991. July 26;66(2):219–32. [DOI] [PubMed] [Google Scholar]
- 9.Roa BB, Garcia CA, Suter U, et al. Charcot-Marie-Tooth disease type 1A. Association with a spontaneous point mutation in the PMP22 gene. N Engl J Med. 1993. July 8;329(2):96–101. [DOI] [PubMed] [Google Scholar]
- 10.DiVincenzo C, Elzinga CD, Medeiros AC, et al. The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med. 2014. November;2(6):522–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stankiewicz P, Lupski JR. The genomic basis of medicine. In: Firth J, Conlon C, Cox T, editors. The Oxford Text Book of Medicine. 6th ed: Oxford University Press; 2020. pp. 1–49, doi: 1093/med/9780198746690.001.0001. [Google Scholar]
- 12.Lupski JR. 2018 Victor A. McKusick Leadership Award: Molecular Mechanisms for Genomic and Chromosomal Rearrangements. Am J Hum Genet. 2019. March 7;104(3):391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harel T, Lupski JR. Genomic disorders 20 years on-mechanisms for clinical manifestations. Clin Genet. 2018. March;93(3):439–449. [DOI] [PubMed] [Google Scholar]
- 14.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015. May;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eldomery MK, Coban-Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017. March 21;9(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014. November 12;312(18):1880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *17.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014. November 12;312(18):1870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes clinical exome and molecular diagnoses on initial >2000 consecutive cases.
- 18.White JJ, Mazzeu JF, Coban-Akdemir Z, et al. WNT Signaling Perturbations Underlie the Genetic Heterogeneity of Robinow Syndrome. Am J Hum Genet. 2018. January 4;102(1):27–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013. October 17;369(16):1502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **20.Posey JE, Harel T, Liu P, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N Engl J Med. 2017. January 5;376(1):21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]; Delineates the contribution of multi-locus pathogenic variation and dual molecular diagnosis to disease phenotypes.
- **21.Liu P, Meng L, Normand EA, et al. Reanalysis of Clinical Exome Sequencing Data. N Engl J Med. 2019. June 20;380(25):2478–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrates increased molecular diagnosis solved rate by reanalysis of extant data showing new disease gene discovery is a major contributor to new molecular diagnoses.
- 22.Link N, Chung H, Jolly A, et al. Mutations in ANKLE2, a ZIKA Virus Target, Disrupt an Asymmetric Cell Division Pathway in Drosophila Neuroblasts to Cause Microcephaly. Dev Cell. 2019. December 16;51(6):713–729 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lloyd KCK, Adams DJ, Baynam G, et al. The Deep Genome Project. Genome Biol. 2020. February 3;21(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karaca E, Weitzer S, Pehlivan D, et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell. 2014. April 24;157(3):636–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaffer AE, Eggens VR, Caglayan AO, et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell. 2014. April 24;157(3):651–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mannik K, Magi R, Mace A, et al. Copy number variations and cognitive phenotypes in unselected populations. JAMA. 2015. May 26;313(20):2044–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lupski JR. Cognitive phenotypes and genomic copy number variations. JAMA. 2015. May 26;313(20):2029–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Assia Batzir N, Kishor Bhagwat P, Larson A, et al. Recurrent arginine substitutions in the ACTG2 gene are the primary driver of disease burden and severity in visceral myopathy. Hum Mutat. 2020. March;41(3):641–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng H, Capponi S, Wakeling E, et al. Missense variants in TAF1 and developmental phenotypes: challenges of determining pathogenicity. Hum Mutat. 2019. October 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang Y, Wangler MF, McGuire AL, et al. The phenotypic spectrum of Xia-Gibbs syndrome. Am J Med Genet A. 2018. June;176(6):1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan B, Neira J, Pehlivan D, et al. Clinical exome sequencing reveals locus heterogeneity and phenotypic variability of cohesinopathies. Genet Med. 2019. March;21(3):663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potocki L, Chen KS, Koeuth T, et al. DNA rearrangements on both homologues of chromosome 17 in a mildly delayed individual with a family history of autosomal dominant carpal tunnel syndrome. Am J Hum Genet. 1999. February;64(2):471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saporta AS, Sottile SL, Miller LJ, et al. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol. 2011. January;69(1):22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dharmadhikari AV, Ghosh R, Yuan B, et al. Copy number variant and runs of homozygosity detection by microarrays enabled more precise molecular diagnoses in 11,020 clinical exome cases. Genome Med. 2019. May 17;11(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan B, Wang L, Liu P, et al. CNVs cause autosomal recessive genetic diseases with or without involvement of SNV/indels. Genet Med. 2020. June 24: 10.1038/s41436-020-0864-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pehlivan D, Bayram Y, Gunes N, et al. The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance. Am J Hum Genet. 2019. July 3;105(1):132–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beck CR, Carvalho CMB, Akdemir ZC, et al. Megabase Length Hypermutation Accompanies Human Structural Variation at 17p11.2. Cell. 2019. March 7;176(6):1310–1324 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carvalho CM, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016. April;17(4):224–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carvalho CM, Ramocki MB, Pehlivan D, et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet. 2011. October 2;43(11):1074–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu P, Yuan B, Carvalho CMB, et al. An Organismal CNV Mutator Phenotype Restricted to Early Human Development. Cell. 2017. February 23;168(5):830–842 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bahrambeigi V, Song X, Sperle K, et al. Distinct patterns of complex rearrangements and a mutational signature of microhomeology are frequently observed in PLP1 copy number gain structural variants. Genome Med. 2019. December 9;11(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011. January 7;144(1):27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu P, Erez A, Nagamani SC, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011. September 16;146(6):889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maher CA, Wilson RK. Chromothripsis and human disease: piecing together the shattering process. Cell. 2012. January 20;148(1–2):29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cortes-Ciriano I, Lee JJ, Xi R, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet. 2020. March;52(3):331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vossaert L, Wang Q, Salman R, et al. Validation Studies for Single Circulating Trophoblast Genetic Testing as a Form of Noninvasive Prenatal Diagnosis. Am J Hum Genet. 2019. December 5;105(6):1262–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carvalho CMB, Coban-Akdemir Z, Hijazi H, et al. Interchromosomal template-switching as a novel molecular mechanism for imprinting perturbations associated with Temple syndrome. Genome Med. 2019. April 23;11(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu N, Ming X, Xiao J, et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. 2015. January 22;372(4):341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verbitsky M, Westland R, Perez A, et al. The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat Genet. 2019. January;51(1):117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren X, Yang N, Wu N, et al. Increased TBX6 gene dosages induce congenital cervical vertebral malformations in humans and mice. J Med Genet. 2019. December 30;57:371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karolak JA, Vincent M, Deutsch G, et al. Complex Compound Inheritance of Lethal Lung Developmental Disorders Due to Disruption of the TBX-FGF Pathway. Am J Hum Genet. 2019. February 7;104(2):213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lupski JR. Genetics. Genome mosaicism--one human, multiple genomes. Science. 2013. July 26;341(6144):358–9. [DOI] [PubMed] [Google Scholar]
- 53.Liu Q, Karolak JA, Grochowski CM, et al. Parental somatic mosaicism for CNV deletions - A need for more sensitive and precise detection methods in clinical diagnostics settings. Genomics. 2020;112(5):2937–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gambin T, Liu Q, Karolak JA, et al. Low-level parental somatic mosaic SNVs in exomes from a large cohort of trios with diverse suspected Mendelian conditions. Genetics in Medicine. 2020;doi: 10.1038/s41436-020-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakka P, Pattillo Smith S, O’Donnell-Luria AH, et al. Characterization of Prevalence and Health Consequences of Uniparental Disomy in Four Million Individuals from the General Population. Am J Hum Genet. 2019. November 7;105(5):921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song X, Beck CR, Du R, et al. Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res. 2018. August;28(8):1228–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Childs B Genetic Medicine: A Logic of Disease. Baltimore and London: The Johns Hopkins University Press; 1999. [Google Scholar]
- 58.Gambin T, Akdemir ZC, Yuan B, et al. Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res. 2017. February 28;45(4):1633–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gambin T, Yuan B, Bi W, et al. Identification of novel candidate disease genes from de novo exonic copy number variants. Genome Med. 2017. September 21;9(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzaga-Jauregui C, Yesil G, Nistala H, et al. Functional biology of the Steel syndrome founder allele and evidence for clan genomics derivation of COL27A1 pathogenic alleles worldwide. European Journal of Human Genetics. 2020;2:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Azzariti DR, Hamosh A. Genomic Data Sharing for Novel Mendelian Disease Gene Discovery: The Matchmaker Exchange. Annu Rev Genomics Hum Genet. 2020. April 27. [DOI] [PubMed] [Google Scholar]
- 62.Bamshad MJ, Nickerson DA, Chong JX. Mendelian Gene Discovery: Fast and Furious with No End in Sight. Am J Hum Genet. 2019. September 5;105(3):448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]