

Abstract
Many recent disease outbreaks in humans had a zoonotic virus etiology. Bats in particular have been recognized as reservoirs to a large variety of viruses with the potential to cross-species transmission. In order to assess the risk of bats in Switzerland for such transmissions, we determined the virome of tissue and fecal samples of 14 native and 4 migrating bat species. In total, sequences belonging to 39 different virus families, 16 of which are known to infect vertebrates, were detected. Contigs of coronaviruses, adenoviruses, hepeviruses, rotaviruses A and H, and parvoviruses with potential zoonotic risk were characterized in more detail. Most interestingly, in a ground stool sample of a Vespertilio murinus colony an almost complete genome of a Middle East respiratory syndrome-related coronavirus (MERS-CoV) was detected by Next generation sequencing and confirmed by PCR. In conclusion, bats in Switzerland naturally harbour many different viruses. Metagenomic analyses of non-invasive samples like ground stool may support effective surveillance and early detection of viral zoonoses.
Introduction
Bats belong to the order Chiroptera, a group of mammals with 21 families and more than 1’300 species of which approximately 1’236 are classified by the International Union for Conservation of Nature (IUCN) [1–4]. Bats are one of the most diverse and abundant groups of animals, living all over the world except the Arctic and Antarctic [5, 6]. Nevertheless, nearly a third of all bat species are endangered [1, 7–9]. Bat species have adapted to various food sources such as arthropods, fruit, nectar, pollen, small mammals, fish, frogs, and blood, and they found a niche to navigate and hunt in darkness with the ability of echolocation and flight [10]. Bats play a key role in the ecosystem by pollinating flowers and dispersing seeds [10–12]. Five different families of insectivorous microbats are endemic in Europe, including Vespertilionidae, Molossidae, Rhinolophidae, Pteropodidae and Emballonuridae. Of the approx. 53 bat species identified in Europe, 30 are also endemic in Switzerland [5–7, 13, 14].
Bats have unique biological, physiological, and immunological characteristics that seem to make them ideal candidates to host and spread viruses. First of all, they are the only mammals capable of powered flight; some species migrate several hundred miles for winter hibernation enabling viral spread over long distances [15–17]. Also some species native to Switzerland, including Nyctalus noctula, N. leisleri and N. lasiopterus, Vespertilio murinus and Pipistrellus nathusii, are known to fly long distances [7]. Second, the longevity, i.e. up to 41 years (Myotis brandtii), facilitates long virus persistence [18–21]. Third, bats roost in colonies with a size of up to a million animals per colony which supports virus transmission between individuals [16]. Moreover, as bats are phylogenetically the oldest mammals on earth, their immune system was able to adapt and cope with viral infections over a long time of co-evolution. Bats seem to control viral replication more effectively than other mammals [22, 23]. It has been shown that due to the development of flight, the immune system adapted to the high metabolic rates i.e., increased metabolism and high body temperature during flight, through genetic changes. Flight would therefore lead to a daily activation of the immune system and could explain why bats can host viruses with no signs of illness [16, 22, 24, 25]. The large number of different bat species may also contribute at least in part to the large number of different bat-associated zoonotic viruses [26, 27].
Bats are not only ideal hosts and reservoirs for many different viruses, but in addition several factors also support the efficient transmission of viruses. These include intrinsic factors such as body condition, sex, or social and reproductive status as well as extrinsic factors influencing the habitat of the bats such as drought, cave destruction, and bush fires [16, 18, 28–30]. Stress (e.g., starving, mating, breeding, fighting, high population density) amplifies virus transmission from bat to bat even further as it downregulates the immune system and makes the animals more susceptible to viral infections, which has a direct impact on the epidemiology of viral diseases [16, 18, 28–30]. Habitat loss and lack of food often forces bats to move closer to domestic areas, thereby increasing the chance of virus transmission to other species including humans and farm animals [18].
Many different viruses have been detected in bats using a variety of assays, including serological tests, polymerase chain reaction (PCR) and virus isolation [16, 31–41]. Among those, countless viruses from insects, plants, fungi, and bacteria mainly reflect the dietary habits of the bats [42–44]. But bats are also the natural hosts for many different vertebrate viruses including emerging zoonotic viruses such as Ebola virus, Marburg hemorrhagic fever filoviruses, rabies virus, severe acute respiratory syndrome coronavirus (SARS-CoV), Middle-East respiratory syndrome coronavirus (MERS-CoV), Nipah (NiV) and Hendra virus (HeV) [14, 18, 24, 32, 38, 45–48]. Up to date, 20 out of 29 virus families associated with bats were detected in Europe, and the different viruses were compiled in the database DBatVir [49, 50]. Viruses from at least 12 families found in Europe have zoonotic potential, but direct bat-to-human transmission has been reported only for lyssaviruses and so far with a low prevalence [14, 32]. For transmission of bat borne viruses to humans often an intermediate host is needed [47, 51, 52]. For example in the coronavirus outbreaks SARS-CoV-1 and MERS-CoV, the viruses originated from bats but were transmitted to humans through an intermediate host i.e. civets and dromedary, respectively [46, 48, 53, 54]. The pandemic caused by SARS-CoV-2 presumably originated from a bat coronavirus with a 96% nucleotide similarity to the SARS-CoV-2 detected in a bat from the province Yunann (SARSr-CoV; RaTG13) [55, 56]. Coronaviruses circulate widely in wildlife and the bat-to-human transmission and the role of the intermediate host in transmission is still unclear [57]. However, some reports suggest that the Malayan pangolin (Manis javanica) may serve as an intermediate host of SARS-CoV-2 [55, 56, 58, 59].
While the virus diversity of bats has been determined in several countries including China, Japan, Myanmar, the USA and France [5, 43, 60–63], no such data is available from Switzerland. Therefore, in a collaboration with the foundation for bat conservation Switzerland and local care takers of bat colonies, more than 7’000 bats were sampled for the metagenomic study presented here.
Material and methods
Animals and sample collection
Samples were collected between 2015 and 2020 from bats presented at care centers in the cantons Zurich, Basel, Berne, Grisons, Jura, Neuchatel, Lucerne, Schaffhausen, and St. Gallen and from selected bat colonies in the cantons Aargau, Jura, Lucerne, Obwalden, Schaffhausen, Schwyz, Solothurn, St. Gallen, and Zurich (S1 Fig). The geographic coordinates of bat colonies can be provided by the Swiss Bat Conservation Foundation and bat care centers upon formal request. On living bats, only non-invasive sampling procedures (feces collection from the floor) were performed. Fecal samples of individual bats were collected at the bat care center located at the Zoo Zurich, where injured animals or animals that were disturbed during hibernation are housed in boxes. During routine cleaning of the boxes, fresh fecal samples were collected and stored in 2 ml tubes. Fecal samples from bat colonies were collected from the floor and stored in 50 ml tubes during routine inspection by authorized persons when the animals were present. Organs were collected from bats which were found dead or brought to the bat care centers and due to injuries had to be euthanized by veterinarians. For this, the animals were anesthetized with 20 mg/kg Alfaxalone (Alfaxan) s.c. in the neck area and, after 10min, euthanized with Pentobarbital (Esconarkon; 0.1–0.2 ml / animal) i.p. From necropsy, brain, heart, lung, intestine, and spleen combined with liver were collected and stored separately in 2-ml tubes. The samples of individual animals were pooled if they matched concerning three specific criteria: (i) the species, (ii) the sample type and (iii) the location (canton). All other samples from individual animals were prepared separately. Ground stool samples of colonies were not pooled. All samples were stored at -20°C. Table 1 shows an overview of the samples analyzed in this study; for more detail see S1–S3 Tables. Ethical approval was not required because only non-invasive sampling procedures on living bat were performed, and corpses were from natural deaths or from bats that had to be euthanized due to injuries. The euthanasia, thus, was not part of this study.
Table 1. Place of collection, bat species, numbers of bats, and samples collected in this study.
| Numbers of animals | |||
|---|---|---|---|
| Canton | Bat species | Feces | Organs |
| Aargau | Myotis daubentonii | 200a | |
| Myotis myotis | 2’454a | 11 | |
| Myotis mystacinus | 50a | 2 | |
| Myotis nattereri | 50a | ||
| Nyctalus noctula | 200a | ||
| Pipistrellus nathusii | 4 | ||
| Pipistrellus kuhlii | 1 | ||
| Pipistrellus pipistrellus | 1 | ||
| Pipistrellus sp. | 1 | ||
| Rhinolophus ferrumequinum | 5a | ||
| Vespertilio murinus | 200a | ||
| Basel | Pipistrellus kuhlii | 7 | |
| Pipistrellus nathusii | 10 | ||
| Pipistrellus pipistrellus | 1 | 16 | |
| Berne | Plecotus auritus | 1 | |
| Grisons | Eptesicus nilssonii | 3 | |
| Myotis nattereri | 1 | ||
| Nyctalus leisleri | 2 | ||
| Pipistrellus nathusii | 1 | ||
| Pipistrellus pipistrellus | 2 | ||
| Plecotus auritus | 2 | ||
| Plectous macrobullaris | 1 | ||
| Rhinolophus ferrumequinum | 186a | ||
| Rhinolophus hipposideros | 12a | ||
| Jura | Myotis myotis | 600a | |
| Pipistrellus pipistrellus | 1 | ||
| Lucerne | Myotis daubentonii | 1 | |
| Myotis myotis | 778a | 1 | |
| 2 | |||
| Pipistrellus nathusii | 1 | ||
| Pipistrellus pygmaeus | 1 | 1 | |
| Neuchâtel | Myotis myotis | 2 | |
| Pipistrellus nathusii | 2 | ||
| Pipistrellus pipistrellus | 2 | 1 | |
| Obwalden | Myotis myotis | 250a | |
| Schaffhausen | Plecotus austriacus | 1 | |
| Myotis myotis | 370a | ||
| Schwyz | Myotis myotis | 212a | |
| Solothurn | Myotis myotis | 208a | |
| St. Gallen | Myotis myotis | 310a | |
| Plecotus auritus | 1 | ||
| Pipistrellus kuhlii | 1 | ||
| Uri | Myotis myotis | 300a | |
| Zurich | Myotis daubentonii | 4 | 3 |
| Myotis myotis | 568a | ||
| 1 | |||
| Myotis mystacinus | 1 | 1 | |
| Nyctalus noctula | 72 | ||
| Pipistrellus kuhlii | 8 | 11 | |
| Pipistrellus nathusii | 7 | 5 | |
| Pipistrellus pipistrellus | 13 | 14 | |
| Pipistrellus sp | 7 | 4 | |
| Plecotus auritus | 4 | 2 | |
| Vespertilio murinus | 1 | 2 | |
| Liechtenstein | Myotis myotis | 93a | |
| Total | 7’183 | 108 | |
a ground stool sample of colony.
Enrichment of viral particles and nucleic acid isolation
Homogenization and filtration
To homogenize fecal samples from individual animals or small groups, 360 μl of phosphate buffered saline (PBS) was added to 30 mg of feces. To homogenize organs, 300–500 μl of PBS was added to 30 mg of tissue from up to 10 individual animals grouped according to species, geographical location, and organ type. Complete homogenization of tissue samples was achieved by adding a stainless-steel bead of 5 mm diameter (Qiagen, Hombrechtikon, Switzerland) into each tube. Then, samples were homogenized in the TissueLyser II (Qiagen, Hombrechtikon, Switzerland) at 20 Hz for 2 min, followed by 5 min of centrifugation at 16’060 x g (Biofuge Fresco, Heraeus Instruments, Hanau, Germany). The supernatants were collected with a 5-ml syringe (Injekt F, B. Braun, Sempach, Switzerland) and a 22 G needle (0.7 x 32 mm, AGANI NEEDLE, Terumo, Eschborn, Germany) and filtered through a 0.45 μm syringe filter (Puradisc 13 mm, Whatman GE Healthcare, Chicago, Illinois, USA) to remove bacterial and host cells. If clogging occurred, filtration was repeated. After the filtration step, pools of tissues from animals of the same species and geographical location were combined in 1.5-ml Eppendorf tubes. Ground stool samples from colonies were placed in a petri dish and cut into pieces with a scalpel blade (carbon steel sterile surgical blade#18, B. Braun, Sempach, Switzerland), and the sample material was divided evenly into two new 15-ml tubes. One aliquot was stored at -20°C as backup and the other used for homogenization. For this, 12 ml of PBS was added, the sample mixed with a vortex, and then centrifuged for 10 min at 21’890 x g (Hereus Multifuge 3 S-R, Thermo Fisher, Waltham, Massachusetts, USA). The supernatant was transferred into a 2-ml tube and centrifuged a second time for 8 min at 16’060 x g. Then, the supernatant was taken out with a 5-ml syringe (Injekt F, B. Braun, Sempach, Switzerland) and 22 G needle (0.7 x 32 mm, AGANI NEEDLE, Terumo, Eschborn, Germany) and filtered first through a 0.8 μm syringe filter (13 mm Supormembrane, Pall Corporation, New York, USA) and then through a 0.45 μm syringe filter (Puradisc 13 mm, Whatman, GE Healthcare, Chicago, Illinois, USA) into 1.5-ml Eppendorf tubes [64].
Nuclease treatment
To remove nucleic acids not protected by a virus coat, 134 μl of each filtered homogenate from the step above was treated with 1 μl of micrococcal nuclease (2 x 106 gel U/ml; New England Biolabs, Ipswich, Massachusetts, USA), 14 μl of 10 X micrococcal nuclease buffer and 1 μl of Ribonuclease A (30mg/ml; Sigma-Aldrich, St. Louis, Missouri, USA). Then, the samples were incubated for 15 min at 45°C and for 1 h at 37°C [64].
Nucleic acid isolation
Viral RNA and DNA was prepared by using the QIAmp Viral RNA Mini kit (Qiagen, Hombrechtikon, Switzerland) as described by the manufacturer with modifications: the RNA carrier was omitted and, as a first step, 594 μl of AVL buffer was mixed with 6 μl of 1% β-mercaptoethanol (Bio-rad, Hercules, California, USA). The nucleic acid was eluted with 20 μl of RNase free water and 20 μl of Tris-EDTA buffer (TE) [64, 65].
Reverse transcription and second strand synthesis
For cDNA synthesis, 2.5 μM of a random primer with a known 20-nt tag sequence (SISPA-N, 5’-GCT GGA GCT CTG CAG TCA TCN NNN NN-3’) was added to the nucleic acid samples prepared in the step above, incubated at 97°C for 3 min, and cooled on ice. Then 20 μl of cDNA-mix (RevertAid First Strand H minus cDNA Synthesis kit; Thermo Fisher, Waltham, Massachusetts, USA), consisting of 10 μl of 5 X reaction buffer, 5 μl of 10 mM dNTP mix, 2.5 μl of 20U/ μl Ribolock RNase inhibitor, and 2.5 μl of 200U/ μl RevertAid H minus RT, was added to each sample, and the suspension was incubated for 10 min at 25°C, 90 min at 45°C, and 5 min at 70°C. Finally, 1 μl of RNase H (New England Biolabs, Ipswich, Massachusetts, USA) was added to degrade residual RNA, and the sample was incubated for 20 min at 37°C. For second strand synthesis, 45.5 μl of cDNA, 0.6 μl of 10 X Klenow buffer (Thermo Fisher, Waltham, Massachusetts, USA), 0.4 μl of 100 μM SISPA-N primer, and 1 μl of 10 mM dNTP were mixed, denatured for 1 min at 95°C and cooled on ice. Then, 2.5 μl of Klenow Fragment 3’→5’ exo- (Thermo Fisher, Waltham, Massachusetts, USA) was added, and the mixture was incubated for 15 min at 25°C and for 1 h at 37°C and subsequently denatured for 1 min at 95°C. Following cooling on ice, another 1.25 μl of Klenow Fragment 3’→5’ exo- was added and the reaction was allowed to proceed for 15 min at 25°C and 1 h at 37°C. Samples were purified using the PureLink® PCR Micro kit (Invitrogen, Thermo Fisher, Waltham, Massachusetts, USA) according to the manufacturer’s protocol. Finally, dsDNA was eluted with 12 μl of buffer E1 (Invitrogen, Thermo Fisher, Waltham, Massachusetts, USA) [64].
Random amplification
Before library preparation, dsDNA was amplified using a sequence-independent single primer (SISPA primer, 5’-GTT GGA GCT CTG CAG TCA TC-3’) and HotStarTaq DNA polymerase (5U/ μl; Qiagen, Hombrechtikon, Switzerland) as described previously [66]. Then, the samples were purified using the QIAquick PCR Purification kit (Qiagen, Hombrechtikon, Switzerland) according to manufacturer’s protocol and eluted with 30 μl of elution buffer (EB). The end concentrations of dsDNA in the samples were measured using a Qubit™ 2.0 Fluorometer (Invitrogen, Carlsbad, California, USA) [64].
Library preparation
For fragmentation of the dsDNA and ligation of specific adaptors to the DNA fragments, the samples were diluted to a final concentration of 3 ng of DNA in 50 μl of EB buffer (or 1 ng per 50 μl for samples with low initial concentrations). The diluted samples were transferred into microTUBES (Covaris, Massachusetts, USA) and sonicated using an S220 Ultrasonicator System (Covaris, Massachusetts, USA) to obtain 500 bp fragments for paired end NGS runs, or 300 bp fragments for single end NGS runs, according to the manufacturer’s manual. For library preparation, the NEBNext Ultra II DNA Library Prep kit for Illumina (New England Biolabs, Ipswich, Massachusetts, USA) was employed according to the manufacturer’s manual. AMPure XP Beads (Beckman Coulter, Brea, California, USA) were used to clean up the adaptor-ligated DNA, and the NEBNext Multiplex Oligos (96 Unique Dual Index Primer Pairs; New England Biolabs, Ipswich, Massachusetts, USA) were used for the barcoding of the samples.
Quality control and sequencing
The Agilent 2200 TapeStation (Agilent Technologies, Santa Clara, California, USA) was used with a D1000 HS ScreenTape (Agilent Technologies, Santa Clara, California, USA) to measure the size distribution and molarity of each sample. Sample denaturation, dilution, and sequencing were performed at the Functional Genomics Center Zurich (FGCZ) on either the NextSeq 500 system using the high-output flow cell with a paired-end NGS run and 2 x 150 nucleotide read length or the Illumina NovaSeq 6000 system using a single end NGS run with 100 nucleotide read length according to the protocols provided by the manufacturer (Illumina, San Diego, California, USA). The PhiX Control v3 Library as run quality control according to the manual (Illumina, San Diego, California, USA).
Electron microscopy
For electron microscopy, selected fecal and tissue samples were subtilized in 15-ml tubes with 5 to 6 ml of PBS. Then, the same volume of 1-butanol was added prior to vortexing for 3 min and centrifugation for 30 min at 500–700 x g (Heraeus™ Multifuge™ X3, Thermo Fisher, Waltham, Massachusetts, USA). After incubation at 4°C for 5 h, the aqueous phase was transferred into a new 15-ml tube and topped up to 5 ml with PBS if the volume was smaller. Then, 5 ml (minimal ratio 1:1) of 1-butanol was added, and the mixture was vortexed for 3 min, centrifugated for 30 min at 500–700 x g (Heraeus™ Multifuge™ X3, Thermo Fisher, Waltham, Massachusetts, USA), and incubated for 5 h at 4°C. Again, the aqueous phase was transferred into a new 15-ml tube for mid-speed centrifugation, topped up with PBS to a final volume of 10 ml, and centrifugated for 20 min at 10’000 x g (Sorvall RC 5C PLUS using the HB-4 Rotor, Marshall Scientific, Hampton, New York, USA). The supernatant was transferred into an ultra-clear tube (14x95 mm, Beckmann Coulter, Brea, California, USA) and centrifugated for 2 h at 20’000 rpm and 4°C (60’000 x g Sorvall WX 100 ultra-series centrifuge using the SW40 Rotor, Thermo Fisher, Waltham, Massachusetts, USA). Then, the supernatant was discarded, and the pellet containing the viral particles was resuspended in 20 μl of PBS. For negative staining, a parafilm strip (Bemis Company, Inc., Neenah, Wisconsin, USA) was placed onto a smooth surface. Then, 10 μl of the resuspended virus particles, a drop of double distilled water (ddH2O filtered with 0.22 μm pore size), and a drop of 2% phosphotungstic acid (PTA; H3(P(W3O10)4)xH2O), pH 7.0 were pipetted side by side onto the parafilm. A grid (carbon coated parlodion film mounted on a 300 mesh/inch copper grid), which was glow discharged to make it hydrophilic, was placed with the carbon coated side onto the top of the sample drop. After 10 min, the grid was placed onto the top of the ddH2O drop for a few seconds, and finally, onto the PTA for 1 min. Then, PTA was gently removed using a filter paper, and the grid was placed into a transmission electron microscope (CM12, Philips, Eindhoven, Netherlands) equipped with a CCD camera (Ultrascan 1000, Gatan, Pleasanton, California, USA) for analysis at 100 kV.
PCR
Selected viral contigs detected by NGS were confirmed as follows: RNA was prepared by adding 270 μl of PBS to 30 mg of fecal or tissue sample, followed by vortexing and centrifugation for 3 min at 16’060 x g (Biofuge Fresco, Heraeus Instruments, Hanau, Germany). Then, 150 μl of the supernatant was used for RNA extraction using the Qiagen RNA Mini kit (Qiagen, Hombrechtikon, Switzerland) according to the manufacturer’s manual. DNA from tissue and fecal samples was prepared using the Qiagen DNA Mini kit or the Qiagen DNA Stool kit, respectively, according to the manufacturer’s manual (Qiagen, Hombrechtikon, Switzerland).
Pan-corona OneStep RT-PCR
The Pan-corona OneStep RT-PCR was performed as described by Vijgen et al. [67]. Additionally, two set of primers covering different region of polymerase gene were designed based on obtained sequence. The following primers were used: set one: forward 5’- GTTGATGGCGTGCCATTTGT-3’, reverse 5’- ATTGAGGCAACCACCGTCAT-3’ and set two: forward 5’- GGTGATGCCACTACCGCATA-3’, reverse 5’- TGAGCAGAACTCGTGTGGAC-3’. As a positive control the porcine epidemic diarrhea virus isolate CV777 was used and as a negative control nuclease free water. The PCR product was analyzed by agarose gel electrophoresis. The expected band of approx. 251, 465 and 396 base pairs (bp), for pancorona, set 1 and set 2, respectively, were cut out with a scalpel blade, and the DNA was extracted using the Gel extraction kit (Qiagen, Hombrechtikon, Switzerland) according to the manufacturer’s manual and sequenced at Microsynth (Balgach, Switzerland).
Pan-adeno PCR
The Pan-adeno nested PCR was performed as described by Wellehan et al. [68] using 5 μl DNA, 0.4 μl of HotStarTaq Polymerase (5U/ μl, Qiagen, Hombrechtikon, Switzerland), 0.5 μl dNTP mix (10 mM), 2.5 μl of PCR-Buffer (10X), 0.25 μl each of Panadeno outer forward and reverse primer (100 μM) and 16.1 μl nuclease free water. For the second amplification, 2 μl of the reaction mixture from the first amplification were mixed with 0.4 μl of HotStarTaq Polymerase (5U/ μl, Qiagen, Hombrechtikon, Switzerland), 0.5 μl dNTP mix (10 mM), 2.5 μl of PCR-Buffer (10X), 0.25 μl each of Panadeno inner forward and reverse primer (100 μM) and 19.1 μl nuclease free water. As a positive control the canine adenovirus 1 was used and as a negative control nuclease free water. The PCR product was then analyzed by agarose gel electrophoresis. The expected band of approx. 318–324 bp was cut out with a scalpel blade, and the DNA was extracted and sequenced as described above.
Rotavirus H NSP5 RT-PCR
To amplify the NSP5 segment of Rotavirus H (RVH), specific primers were designed using Primer3Plus [69] with manual inspection in Clonemanager 9 (Sci-Ed Software, Cary, North Carolina, USA). As a negative control nuclease free water was used. The following primers were used: forward 5’-GGAACTAAAAACTTCAATCGTTGCTG-3’ and reverse 5’-GTTTTTATTGATGACCTCAGGGGC-3’. Before amplification, an initial denaturation step with 10 μl of extracted RNA for 5 min at 97°C was performed. To the denatured RNA 40 μl of the PCR mix containing 10 μl of One Step RT-PCR Buffer (5X; Qiagen, Hombrechtikon Switzerland), 1.6 μl forward Primer (100 μM) and 1.6 μl reverse Primer (100 μM), 2 μl dNTP Mix (10 mM each), 2.0 μl One Step Enzyme Mix (Qiagen Hombrechtikon, Switzerland), 22.6 μl nuclease free water, and 0.2 μl RNasin (Promega, Madison, Wisconsin, USA) was added. The PCR conditions were as follows: 30 min at 50°C, 15 min at 95°C, 40 cycles of 50 sec at 94°C, 50 sec at 55°C and 60 sec at 72°C, a final extension step for 10 min at 72°C. The PCR product was analyzed by agarose gel electrophoresis. The expected band of approx. 666 bp was cut out with a scalpel blade, and the DNA was extracted and sequenced as described above.
Data analysis
Sequencing data was used in reference guided analysis and de novo assembly pipelines as described previously [65]. The PCR primers, sequencing adaptors, and low-quality ends in raw reads were trimmed using Trimmomatic (version 0.36) and cutadapt (version 2.9). Quality controlled reads were aligned using Bowtie2 (version 2.4.1) to the human genome to remove contamination introduced during sample preparation. This was followed by the assembly of un-mapped reads in a reference guided analysis to detect even viruses with low numbers of reads. Reads were first aligned to an inhouse database containing 37’400 full length viral genomes downloaded from the NCBI database and bat-associated viruses from DBatVir database [49] using Bowtie2 (parameters: -a—very-sensitive—no mixed—no discordant -X 1000), and mapped reads and mapped bases per viral genome were calculated using bedtools (version 2.29.2).The same inhouse database was used subsequently to align reads in the metagenomic pipeline of the SeqMan NGen v17 (DNAStar, Lasergene, Madison, Wisconsin, USA) in order to visualize and confirm assembled reads. Finally, to build up contigs in the de-novo analysis, quality-controlled raw reads were assembled using megahit (version 1.1.3) with multiple k-mers [70]. To annotate contigs taxonomically, assembled contigs were compared against the NCBI none-redundant nucleotide nt database [71] using default parameter settings of BLASTN (version 2.6.0+) [72] and taxonomically annotated. Hits were sorted by bit scores, and hits with e value ≤ 1 × 10−5 and bit score ≥ 100 were used for contig taxonomy annotation. The naïve best-hit-method was then used to obtain the specific taxa assignment per contig after manual inspection of the alignments. The resulting viral contigs were further investigated and aligned using MEGA X and Clone manager ver. 9 (Sci-Ed). Phylogenetic trees were constructed in Mega X using the Maximum Likelihood method with 1’000 bootstrap value and a cut-off of 70% [73].
Accession numbers
The nucleotide sequences of selected viruses identified in this study have been registered at the GenBank under the following accession numbers: MT815927-MT815982 and MT818221. All raw sequencing data generated during this study were uploaded to the Sequence Read Archive (SRA) under accession number PRJNA693645.
Results
Fresh fecal samples of bats living in Switzerland (individual animals, colonies) were collected between 2018 and 2020, and tissue samples were taken from dead or diseased bats which had to be euthanized between 2015 and 2020 (Table 1 and S1 Fig). The samples were grouped into 174 sample pools (69 feces and 105 organ pools) according to sample type, bat species and collection location and then sequenced by NGS (Table 1, S1–S3 Tables). The data will be presented in the following order (i) a general overview of the virome of Swiss bats, (ii) the virome composition of different bat species, (iii) differences in the virome composition according to the sample types, (iv) an overview of viral genome abundance, and (v) a focus on selected virus families of vertebrates including Coronaviridae, Adenoviridae, Reoviridae, Parvoviridae, Circoviridae, and Hepeviridae.
Overview of the virome of Swiss bats
The raw sequence data consisting of 1.28 x 109 reads (0.5 x 106 to 3.7 x 107 from each pool) was analyzed using two different pipelines: (i) de novo assembly and (ii) reference-based assembly. The de novo assembly generated a total of 7’477 contigs matching to viral genomes. Using an inhouse data base, 1.68 x 107 of the 1.28 x 109 sequenced reads were assembled to virus genomes. Of these, 1.69 x 106 (10%) reads matched to vertebrate viruses and 1.52 x 107 (90%) to non-vertebrate viruses (Fig 1A and 1B). The generated sequencing reads were assembled to 39 virus families, i.e., 16 (41%) families of viruses from vertebrates and 23 (59%) families of viruses from non-vertebrates which include 11 (28.2%) families of insect-, 11 (28.2%) families of plant/fungal- and 1 (2.6%) family of environmental viruses.
Fig 1. Overview of the virome of Swiss bats.

A. Heatmap of viral reads from 16 virus families of vertebrates and 23 virus families of non-vertebrates and their distribution among all 18 bat species investigated shown as percentage of the total number of reads generated in each bat species. Number in parentheses indicate the numbers of different bat species in which sequences of a virus family have been detected. B. Read abundance of the different virus families.
Vertebrate viruses
Reads assembled to the genomes of viruses of vertebrates included mainly the families of the Circoviridae (84.2%), Genomoviridae (3.6%), Coronaviridae (3.1%) and Adenoviridae (2.6%) (Fig 1A and 1B). Electron microscopy of a ground stool sample of a Myotis myotis colony with 1.3 x 106 reads assembled to a circovirus sp. indeed revealed particles with a size (approx. 20nm) and shape (icosahedral/round) resembling that of circoviruses (Fig 2A) and an estimated particle concentration of > 1012 particles per ml (Fig 2A).
Fig 2. Electron microscopy of selected samples.

A. Negative stain electron micrograph of a ground stool pool homogenate from a Myotis myotis colony. B. Negative stain electron micrograph of a pooled liver and spleen homogenate from Pipistrellus nathusii.
Insect viruses
Reads assembled to virus families infecting insects included mainly Parvoviridae (54.3%), Iflaviridae (37%) and Polycipiviridae (6.6%) (Fig 1A and 1B). More than 6 x 106 reads were assembled to the subfamily Densovirinae, i.e., parus major densovirus, which was detected in 141 samples. A full-length genome of 5’164 nucleotides (nt) assembled from 6 x 105 sequencing reads of an intestine pool of Pipistrellus sp. had a 97% nt similarity to a previously described parus major densovirus (GenBank acc. NC_031450). Interestingly, reads assembled to parus major densovirus have been detected also in several tissue pools including lung, liver/spleen, and intestine. Electron microscopy of a pooled liver/spleen homogenate from Pipistrellus nathusii tissue revealed particles with a size (approx. 22nm) and shape (icosahedral/round) resembling that of densovirus particles (Fig 2B). Additionally, several bee viruses were detected in ground stool samples of Myotis myotis colonies.
Plant/Fungal viruses
Reads assembled to genomes of viruses infecting plants or fungi included mainly the families of the Solemoviridae (88.1%), Tymoviridae (9%) and Alphaflexiviridae (1.8%) (Fig 1A and 1B).
The virome composition of the different bat species
The sequences of two different virus families were detected in all 18 bat species, the Circoviridae and Densovirinae. The sequences of Iflaviridae were identified in 17, of Genomoviridae and vertebrate associated Parvoviridae in 12, and of Reoviridae in 10 bat species. Pipistrellus pipistrellus was the bat species with the largest variety of different virus families detected (11 virus families of vertebrates/11 virus families of non-vertebrates; 11/11), followed by Myotis myotis (8/10), Pipistrellus nathusii (9/7), Myotis daubentonii (8/4) and Plecotus auritus (7/5) (Fig 1A). Among the 18 species of bats, Myotis myotis represented the largest sample group with 87.4% of all sampled animals and was the species with the highest number of reads assembled to vertebrate viruses (0.5% of total generated sequencing reads), as well as the species with the largest number, 33, of different viruses detected (Fig 1A and S2 Fig). The second largest number of different viruses, 24, was detected in Pipistrellus pipistrellus (of which 12 were viruses of vertebrates and 12 of non-vertebrates), followed by Pipistrellus nathusii and Myotis mystacinus with 17 different viruses each. The lowest number of vertebrate viral reads were detected in Plecotus austriacus and of non-vertebrate viral reads in Rhinolophus ferrumequinum i.e., 1.45 x10-5 and 6.8 x 10−3 of total generated reads, respectively.
Five bat species in Switzerland are known to migrate i.e., Nyctalus leisleri, Nyctalus lasiopteros, Nyctalus noctula, Pipistrellus nathusii and Vespertilio murinus. Samples from all migrating species except Nyctalus lasiopteros were collected. Between migrating and non-migrating bats only minor differences in the virus genome diversity and the numbers of reads were observed (S2 Fig). The average numbers of different viruses detected in migrating bats was lower than in non-migrating bats. On the other hand, the average number of different viruses of vertebrates was higher in migrating than in non-migrating bats (Fig 1A and 1B, and S2 Fig). Nyctalus noctula was the species with highest percentage of non-vertebrate viral reads detected (7.27% of the total generated reads) (S2 Fig).
Differences in the virome composition according to the sample types
In general, more viral reads were detected in fecal and intestine samples of individual animals than in tissue samples (brain, lung, and combined liver/spleen). The largest number of viral reads assembled to vertebrate virus families were detected in ground stool samples from colonies, whereas reads assembled to non-vertebrate viruses were most abundant in fecal samples of individual animals. Specifically, Retroviridae and Polyomaviridae sequences were detected mainly in tissue samples while Nairoviridae sequences were only detected in tissue pools and Astroviridae, Caliciviridae and Coronaviridae sequences were not detected in tissue samples. In the intestine pool and tissue pool, families of vertebrate viruses were the most abundant, 13 and 11, respectively. In the ground stool samples, families of non-vertebrate viruses were the most abundant, 14. All samples had in common that more reads were assembled to non-vertebrate than to vertebrate viruses (Table 2).
Table 2. Overview of reads assembled to different virus families categorized by sample type.
| Sample type | ||||||
|---|---|---|---|---|---|---|
| Virus family | Feces (individual animals) | Ground stool (colonies) | Intestine pool | Tissue pool | ||
| Lung pool | Liver/Spleen pool | Brain pool | ||||
| Vertebrate | ||||||
| Adenoviridae | 1.4 x 104 | 0 | 2.5 x 104 | 0 | 3.7 x 103 | 0 |
| Astroviridae | 1.3 x 104 | 0 | 3 | 0 | 0 | 0 |
| Caliciviridae | 2 x 103 | 0 | 1.9 x 103 | 0 | 0 | 0 |
| Circoviridae | 970 | 1.4 x 106 | 1.1 x 103 | 80 | 1.4 x 103 | 351 |
| Coronaviridae | 3.6 x 104 | 1.7 x 104 | 377 | 0 | 0 | 0 |
| Genomoviridae | 207 | 6 x 104 | 69 | 14 | 3 | 0 |
| Hepeviridae | 1.3 x 104 | 0 | ||||
| Nairoviridae | 0 | 0 | 0 | 0 | 1 x 104 | 0 |
| Parvoviridae | 2.2 x 103 | 2.6 x 104 | 4.1 x 103 | 109 | 2.9 x 103 | 0 |
| Peribunyaviridae | 0 | 353 | 0 | 0 | 0 | 0 |
| Picobirnaviridae | 0 | 0 | 99 | 0 | 0 | 0 |
| Picornaviridae | 0 | 0 | 2 | 0 | 0 | 0 |
| Polyomaviridae | 0 | 386 | 17 | 0 | 1 x 103 | 0 |
| Reoviridae | 1.8 x 104 | 2.5 x 103 | 8.7 x 103 | 2 | 271 | 0 |
| Retroviridae | 6 | 0 | 87 | 1.4 x 103 | 487 | 233 |
| Unclassified | 66 | 620 | 47 | 0 | 0 | 3 |
| Total | 1 x 105 | 1.5 x 106 | 4.1 x 104 | 1.6 x 103 | 2 x 104 | 587 |
| Non-vertebrate | ||||||
| Alphaflexiviridae | 0 | 1.5 x 103 | 0 | 0 | 0 | 0 |
| Betaflexviridae | 0 | 51 | 0 | 0 | 0 | 0 |
| Birnaviridae | 0 | 799 | 0 | 0 | 0 | 0 |
| Circovirdiae | 593 | 123 | 5 | 0 | 0 | 0 |
| Dicistroviridae | 0 | 4.4 x 104 | 1.4 x 105 | 81 | 299 | 0 |
| Endornaviridae | 0 | 0 | 23 | 0 | 0 | 0 |
| Genomoviridae | 21 | 1.6 x 104 | 0 | 0 | 0 | 0 |
| Iflaviviridae | 4.8 x 106 | 8.7 x 104 | 6.4 x 105 | 1.9 x 104 | 2 x 104 | 17 |
| Iridoviridae | 0 | 1.1 x 105 | 0 | 0 | 0 | 0 |
| Nodaviridae | 0 | 330 | 0 | 0 | 0 | 0 |
| Partitiviridae | 1 | 0 | 0 | 0 | 0 | 0 |
| Parvoviridae | 5 x 106 | 5.8 x 105 | 2.4 x 106 | 4 x 104 | 1.3 x 105 | 165 |
| Polycipiviridae | 83 | 2.6 x 104 | 9.6 x 105 | 31 | 1.5 x 103 | 8 |
| Rhabdoviridae | 0 | 0 | 0 | 0 | 100 | 0 |
| Secoviridae | 0 | 0 | 0 | 13 | 0 | 0 |
| Solemoviridae | 0 | 7.6 x 104 | 7 | 0 | 0 | 0 |
| Tombusviridae | 33 | 0 | 0 | 0 | 0 | 0 |
| Tymoviridae | 0 | 7.8 x 103 | 0 | 0 | 0 | 0 |
| unclassified | 25 | 1.9 x 103 | 6 x 103 | 0 | 0 | 6 |
| Virgaviridae | 653 | 0 | 114 | 0 | 48 | 0 |
| Total | 9.8 x 106 | 9.6 x 105 | 4.2 x 106 | 6 x 104 | 1.5 x 105 | 196 |
| Total vertebrate/non-vertebrate | 9.9 x 106 | 2.5 x 106 | 4.2 x 106 | 6.1 x 104 | 1.7 x 105 | 783 |
The total numbers of reads assembled to each virus family was categorized according to the different sample types including fecal samples of individual animals or pools of fecal samples from individual animals, ground stool samples of colonies, intestine pools, and tissue pools.
Abundance of the different genome classes
Virus families from all genome classes were detected. Most virus families, 19, belong to the ssRNA viruses (19), followed by ssDNA viruses (8), and dsRNA viruses (5). Of the 19 families of ssRNA viruses, 7 infect vertebrates, 5 insects and 7 plants. Concerning the number of assembled reads the ssDNA viruses were the most abundant with 9.7 x 106 assembled reads (57.7% of all assembled reads), followed by ssRNA viruses with 6.9 x 106 assembled reads (41.2% of all assembled reads), and dsDNA viruses with 1.5 x 105 assembled reads (0.92% of all assembled reads) (Fig 3). Sequences of ssDNA viruses were detected in all 18 bat species investigated, those of ssRNA viruses in 17, and those of dsRNA viruses in 11.
Fig 3. Distribution of viral reads according to genome classes.

Because of the large differences in read numbers, the y-axis was divided into three parts to facilitate visualization of all virus genome classes in a single graph: part one from 0 to 1x 104 reads, part two from 1x 104 to 1.5x 106 reads, and part three from 2x 106 to >4.x 106 reads.
Selected viruses of vertebrates
Viruses from 6 different virus families were studied in more detail i.e., coronaviruses (CoV), adenoviruses, rotaviruses A and H (RVA and RVH), parvoviruses (PV) and hepeviruses due to their potential zoonotic impact, and circoviruses because they were the most abundant among the viruses of vertebrates detected in the bat samples. Viruses mentioned above were detected in the single-end run on the Illumina NovaSeq 6000 system using a single end NGS run with 100 nucleotide read length.
Coronaviridae
Coronaviridae are enveloped viruses with a virion size of 120–160 nm for spherical morphology and of 75–90 nm x 170–200 nm for bacilliform morphology. The coronavirus genome consists of a single, linear segment of 26 to 32 kb of +RNA [74]. In our study reads assembled to Coronaviridae were identified in 17/174 (9.8%) sample pools. In ground stool pools of 13 Myotis myotis colonies and one Vespertilio murinus colony, in two fecal pools of Nyctalus noctula, and in one intestine pool of Pipistrellus pipistrellus, contigs of 614 to 20’189 nt in length were assembled that showed a high similarity (>87%) to different bat coronavirus genomes, mainly of the genus alphacoronavirus.
Additionally, in a ground stool sample of one Vespertilio murinus colony a contig of a bat coronavirus (GenBank acc. number MT818221) with a length of 20’189 nt was assembled that showed 86% nt similarity to a Middle East respiratory syndrome-related coronavirus (MERS-CoV) genome from China (GenBank acc. number MG021451) (Fig 4), a member of the genus betacoronavirus, subgenus Merbecovirus. The contig covered 3 ORF’s, including ORF1ab, ORF1a, and the ORF coding for the spike protein (S4 Table). Sanger sequencing of products obtained from pan-corona RT-PCR [67] and RT-PCR with primer sets 1 and 2 from the polymerase gene generated sequences of 158 nt, 443 nt and 392 nt, respectively, with 100% similarity to the contig generated by de novo assembly (GenBank acc. number MT818221).
Fig 4. Phylogenetic analysis of the RNA dependent RNA polymerase gene of the bat betacoronaviruses.
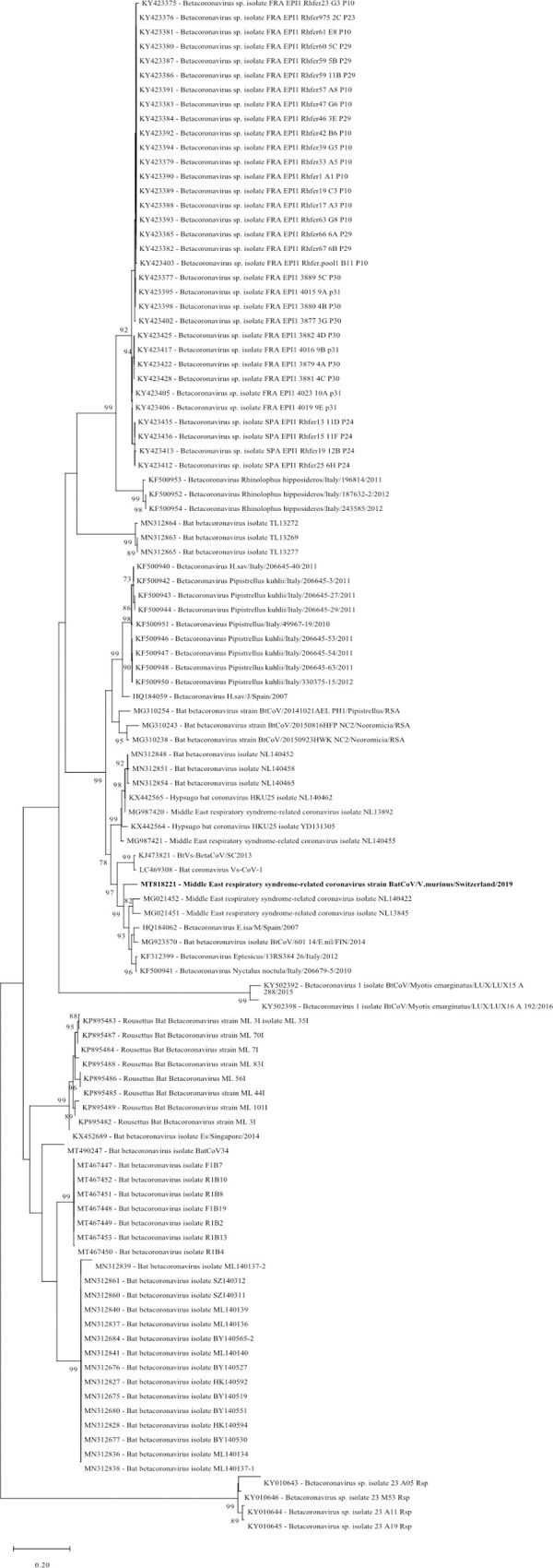
The sequence obtained in this study (GenBank acc. number MT818221) is shown in bold. Sequences from alphacoronaviruses are marked with a purple background and those of betacoronaviruses with a blue background. Sequences were aligned using Muscle. For phylogenetic analysis, the Maximum likelihood tree with 1’000 bootstraps was used. Only values ≥ 70% are displayed.
Adenoviridae
Adenoviridae are non-enveloped viruses with an icosahedral capsid morphology and a virion size of 70–90 nm. The genome consists of a single segment of a linear ds-DNA of 26 to 48 kbp length [74]. Of the five genera of Adenoviridae only the genus mastadenovirus was detected in a fecal pool of Pipistrellus nathusii, a fecal pool of Nyctalus noctula, an intestine pool of Pipistrellus pipistrellus, an intestine pool of Pipistrellus nathusii, and a combined liver/spleen pool of Pipistrellus pipistrellus. Six assembled contigs (lengths between 2’184 and 14’493 nt) had 83 to 99% nt identity to the genome of a bat adenovirus 2 (GenBank acc. number JN252129) (S4 Table, Fig 5).
Fig 5. Phylogenetic analysis of the adenovirus DNA polymerase gene.

The sequences obtained in this study (GenBank acc. numbers MT8159528-29, MT815933-35) are shown in bold. Sequences of non-bat associated viruses are marked with a red background and those of bat associated viruses with a blue background. The pictograms on the right side represent the species in which the virus was detected. Sequences were aligned using Muscle. For phylogenetic analysis, the Maximum likelihood tree with 1’000 bootstraps was used. Only values ≥ 70% are displayed.
One contig with a length of 1’673 nt had 97% nt similarity to the DNA polymerase gene of a bat adenovirus (GenBank acc. number JX065126). Three assembled contigs had 71 to 83% nt similarity to the genomes of two bat mastadenoviruses (GenBank acc. number KX961096; MK625182) (S4 Table, Fig 5). Sanger sequencing of a pan-adeno PCR [68] product of the DNA polymerase gene generated sequences between 247–252 nt with a similarity between 79 and 100% to the contigs generated by de novo assembly of the sequencing reads.
Reoviridae
Reoviridae are non-enveloped viruses with an icosahedral capsid morphology and a virion size of 60–85 nm. The genome consists of 9 to 12 segments of dsRNA with a total genome size of 19 to 32 kbp. The Reoviridae includes viruses that infect vertebrates as well as viruses that infect non-vertebrate hosts [74]. In the bat samples RVA and RVH, which belong to the genus rotavirus, have been detected [75–77].
In three samples (two fecal pools, one intestine pool) of Myotis daubentonii 22 contigs were assembled to RVH each covering one to two segments, i.e. NS1 to NS5 and VP1 to VP4 and VP6 (S4 Table, Figs 6 and 7). Three contigs had a nt similarity between 92 and 93% to the NSP5 segment of a porcine RVH (GenBank acc. number KT962037) from South Africa (Fig 7). Sanger sequencing of a 666 bp RT-PCR product from the NSP5 segment generated sequences of 530–543 nt with 99 to 100% similarity to the contigs generated by de novo assembly of the sequencing reads.
Fig 6. Phylogenetic analysis of specific regions of the rotavirus H genome.

The sequences obtained in this study (GenBank acc. numbers MT815961-69) are shown in bold. Sequences of non-bat associated viruses are marked with a red background and those of bat associated viruses with a blue background. The pictograms on the right side represent the species in which the virus was detected. Sequences were aligned using Muscle. For phylogenetic analysis, the Maximum likelihood tree with 1’000 bootstraps was used. Only values ≥ 70% are displayed.
Fig 7. Phylogenetic analysis of specific regions of the rotavirus H genome.

The sequences obtained in this study (GenBank acc. numbers MT815948-60) are shown in bold. Sequences of non-bat associated viruses are marked with a red background and those of bat associated viruses with a blue background. The pictograms on the right side represent the species in which the virus was detected. Sequences were aligned using Muscle. For phylogenetic analysis, the Maximum likelihood tree with 1’000 bootstraps was used. Only values ≥ 70% are displayed.
Furthermore, in five samples 11 contigs were assemble to RVA, each covering one to two segments of VP1 to VP4 (S4 Table). The samples included a ground stool sample of a Rhinolophus hipposiderus colony and a Rhinolophus ferrumequinum colony, two intestine pools of Pipistrellus pipistrellus and one combined liver/spleen pool of Pipistrellus kuhlii. Three contigs (2’8328 bp, 1’617bp, 2’687 bp) were 96% or 73% similar to a bat RVA VP2 segment (GenBank acc. number KJ020892) or a human RVA VP2 segment (GenBank acc. number KF835897), respectively. Four contigs had 74 to 95% nt similarity to different RVA VP1 segments from bats, humans, and pigs (GenBank acc. number MN433617; MH238214; KF690125; EF583033) (Fig 8).
Fig 8. Phylogenetic analysis of specific regions of the rotavirus A genome.

The sequences obtained in this study (GenBank acc. numbers MT815937-47) are shown in bold. Sequences of non-bat associated viruses are marked with a red background and those of bat associated viruses with a blue background. The pictograms on the right side represent the species in which the virus was detected. Sequences were aligned using Muscle. For phylogenetic analysis, the Maximum likelihood tree with 1’000 bootstraps was used. Only values ≥ 70% are displayed.
Parvoviridae
Parvoviridae are non-enveloped viruses with a ssDNA genome of either positive or negative polarity. The virion has an icosahedral morphology and a size of 21–26 nm. The genome consists of one linear DNA segment of 4 to 6.3 kb [74]. In total, nine contigs with lengths between 746 and 2’908 nt from different samples and bat species were assembled to one bat PV genome (GenBank acc. number KJ641683) with nt similarity between 74 and 85% (Fig 9) belonging to the subfamily Parvovirinae and are unclassified Parvovirinae. The contigs covered the ORFs coding for the nonstructural protein 1 (NS1) and viral protein 1 (VP1) (S4 Table). The nine contigs originated from an intestine pool of Pipistrellus pipistrellus, an intestine pool of Plecotus auritus, a fecal pool of Nyctalus noctula, two fecal pools of Pipistrellus nathusii, a combined liver/spleen pool of Pipistrellus kuhlii, and a combined liver/spleen pool of Plecotus auritus.
Fig 9. Phylogenetic analysis of the NS1 region of parvoviruses.

The sequences obtained in this study (GenBank acc. numbers MT815971-73, MT815975, 77, 78) are shown in bold. Sequences of non-bat associated viruses are marked with a red background and those of bat associated viruses with a blue background. The pictograms on the right side represent the species in which the virus was detected. Sequences were aligned using Muscle. For phylogenetic analysis, the Maximum likelihood tree with 1’000 bootstraps was used. Only values ≥ 70% are displayed.
Circoviridae
Circoviridae was the most abundant vertebrate virus family in this study (84% of all reads assembled to vertebrate viruses). Circoviridae are small non-enveloped viruses with a ssDNA genome of either positive or negative polarity. The virion has an icosahedral morphology and a size of 12–27 nm. The genome is a circular DNA with a length of 1.7–2.3 kb. These viruses infect mainly vertebrate hosts [74]. Three samples revealed full genomes, including the two ORFs coding for the replicase and the capsid protein (S4 Table) of bat-associated circoviruses belonging to the family Circoviridae and unclassified Circoviruses. In one fecal sample of Vespertilio murinus a contig with a length of 2’134 nt and 90% nt similarity to a bat circovirus (GenBank acc. number KX756991) from China was assembled (Fig 10). Two contigs with lengths of 1’641 bp and 1’668 nt, originating from a brain sample of a Myotis myotis bat, a ground stool sample of a Myotis myotis colony and a ground stool sample of a Rhinolophus hipposideros colony, with 99% nt similarity to two different circovirus sp. (GenBank acc. number KY302869; KY302864) from Hungary were assembled (Fig 10).
Fig 10. Phylogenetic analysis of the full genome of circoviruses.

The sequences obtained in this study (GenBank acc. numbers MT815980-82) are shown in bold. Sequences of non-bat associated viruses are marked with a red background and those of bat associated viruses with a blue background. The pictograms on the right side represent the species in which the virus was detected. Sequences were aligned using Muscle. For phylogenetic analysis, the Maximum likelihood tree with 1’000 bootstraps was used. Only values ≥ 70% are displayed.
Hepeviridae
Hepeviridae are small non-enveloped viruses with an icosahedral capsid of 27–34 nm. The genome consists of a linear, positive sensed ssRNA of 6.6 to 7.2 kb. Hepeviridae have so far been detected only in vertebrate hosts [74].
In a fecal pool of Pipistrellus nathusii, an orthohepevirus D contig with a length of 6’647 nt was assembled and showed 75% nt similarity to a bat hepevirus genome from China (GenBank acc. number KX513953) belonging to the genus orthohepevirus. The contig covered two ORF’s coding for the nonstructural polyprotein and the capsid protein (S4 Table). In the phylogenetic analysis of Fig 11, the separate clade of bat hepevirus is shown.
Fig 11. Phylogenetic analysis of full and partial genome of hepeviruses.

The sequence obtained in this study (GenBank acc. numbers MT815970) is shown in bold. Sequences of non-bat associated viruses are marked with a red background and those of bat associated viruses with a blue background. The pictograms on the right side represent the species in which the virus was detected. Sequences were aligned using Muscle. For phylogenetic analysis, the Maximum likelihood tree with 1’000 bootstraps was used. Only values ≥ 70% are displayed.
Discussion
This study included samples from 7’291 bats of 18 different species. Specifically, organ or fecal samples from 245 individual animals and ground stool samples from 36 different bat colonies (8 different species) with approximately 7’046 individual animals were analyzed by NGS. This metagenomic analysis revealed sequences of a total of 39 different virus families including 16 virus families of vertebrates (Fig 1A and 1B). Similar viral metagenomic studies of bats from Singapore, China, and Myanmar have yielded comparable virus diversities [6, 60, 78, 79]. However, direct comparison of the virus genome diversity of bats in Switzerland with those from other countries is difficult because of different sample types, sample sizes, species, pooling strategies, and health conditions of the animals. Furthermore, due to different sample preparation methods as well as pipelines used for analysis the output may introduce bias to the final results. Since this is the first pilot study on the virome of bats from Switzerland, a large number of different bat species and samples were included in order to obtain a first overview. In Croatia, the virome of 455 bats from seven bat species (Myotis myotis, Miniopterus schreibersii, Rhinolophus ferrumequinum, Eptesicus serotinus, Myotis blythii, Myotis nattereri and Myotis emarginatus) was determined from saliva, fecal and guano samples, and showed a dominance of viruses from invertebrates [80], similar to our study. Moreover, as reported previously the potential zoonotic viruses in European bats include viruses from the families Adenoviridae, Astroviridae, Coronaviridae, Hepeviridae, Reoviridae and Retroviridae [5, 32, 81–97], which have all been detected in our study as well. Interestingly, the data obtained in a study conducted in France is strikingly different. While the number of different virus families of vertebrates detected was much smaller (8), it included many virus families that were not detected in our study, such as Bunyaviridae, Flaviviridae, Herpesviridae, and Orthomyxoviridae [5]. These differences may be due to the different populations sampled in that study (bats with behavioral changes and close human contact) compared to our study (mainly healthy bats) as well as the larger numbers of different bat species (18) and sample types (fecal samples, liver, spleen, lung, brain and intestine) investigated in our study.
The question whether native bat species harbor CoVs was of special interest in the present investigation. Such virus sequences were indeed detected and belonged to either the alpha- or betacoronaviruses while no gamma- or deltacoronaviruses were detected in the samples. This finding was consistent with the results of previous studies in other geographical locations [32, 40, 48, 54, 89, 98, 99] and the common knowledge about host preferences of the different coronaviruses; alpha- and betacoronaviruses mainly infect mammals including humans while the gamma- and deltacoronaviruses infect birds [6, 100]. However, so far none of the coronaviruses detected in European bats were 100% identical to human pathogenic viruses such as SARS-CoV-1, MERS-CoV and SARS-CoV-2 [32]. Nevertheless, closely related CoVs have been detected i.e., in Italy, where the genomes of two viruses closely related to MERS-CoV (80% nt similarity [89]) have been detected in Pipistrellus pipistrellus and Hypsugo savii. Similarly, in a ground stool sample of a Vespertilio murinus colony sampled in our study, a contig of 20 kb with a nt similarity of 86% to a MERS-like CoV genome from China was assembled [101].
Rhabdovirus sequences were not identified in our study, although rabies lyssaviruses have previously been detected in Myotis daubentonii bats in Switzerland, albeit at low prevalence (0.36%) [38]. Rhabdoviruses, especially viruses from the genus Lyssavirus, are zoonoses. In Europe, several lyssaviruses were detected in bats including the European bat lyssavirus 1 and 2 (EBLV-1/2). It is known that bats can transmit rabies/lyssaviruses to humans by biting and scratching [32]. In the present study neither EBLV-1 nor EBLV-2 genomes were detected. Eptesicus serotinus and Myotis daubentonii, both endemic in Switzerland, are known to be the main hosts of EBLV-1 or 2, respectively [7, 32]. However, in our study only samples of Myotis daubentonii were collected.
In Switzerland, five bat species, Nyctalus leisleri, N. noctula, N. lasiopterus, Pipistrellus nathusii and Vespertilio murinus, migrate several hundred kilometers between summer and winter quarter [7]. Migration habits may lead to harboring more viruses due to broader contact with various surroundings, e.g. other bat populations, other living areas, and insects. However, our data reveal that only in two migrating species i.e., Pipistrellus nathusii and Vespertilio murinus, the number of different virus families detected, 16 and 10, respectively, was higher than the average 9.6 virus families per bat species. The observation that the virome of migrating bats is not more diverse than that of non-migrating bats has been made in previous studies as well [16, 17, 102, 103].
In the present study, 90% of all assembled reads belonged to virus families of non-vertebrates. This observation is not consistent with other reports where vertebrate viruses clearly dominated the virome [5, 6, 42, 43, 60, 61, 104]. This difference may be explained by the different sample material used, only feces or only tissue samples or both as in our study [5, 6, 42, 43, 60, 61, 104]. The viromes of bat guano from Texas, California, and Singapore consisted of mainly non-vertebrate viruses, particularly insect viruses, thereby supporting the hypothesis that dietary habits are a likely explanation for the large numbers of insect viruses detected in fecal samples [42–44, 60, 104]. Although most of our samples were pooled organs, more reads were assembled to non-vertebrate than vertebrate viruses, since all sampled bat species were insectivorous, and because a higher number of reads was obtained from fecal pools. Another interesting observation was the high number of reads assembled to honeybee viruses in fecal pools of Myotis myotis colonies. Bee-associated viruses were found also in different bat species from other countries [42–44, 104]. The detected sequences were mostly related to the deformed wing virus (Varrora destructor virus) which causes either asymptomatic infection in western honeybees (Apis mellifera) or can lead to wing deformity, behavioral changes, and early mortality in other bee species [42, 105].
Adenoviruses have been detected in bats from several countries including Italy, Germany, China, and Hungary, however, the number of different adenoviruses detected in bats is small [36, 40, 81, 82, 106, 107]. The genus Mastadenovirus has been thought to infect only mammals, including humans [36, 108, 109]. However, cross-species transmission has nevertheless been observed with adenoviruses, and circulation between different animal species and transmission from animals to humans is possible [36, 110, 111].
Most metagenomic studies of bats use feces as sample material. As Parvoviridae can be detected in high concentrations in human blood samples [112], Canuti et al. sampled EDTA-blood of two bat species and showed a relatively low prevalence of PVs in bats from West Africa and Central America [39]. In our study nearly a quarter of the samples had reads assembled to Parvovirinae, mainly bat PVs. For the emergence of new viruses, their evolutionary potential and mutation rate is of particular importance. RNA viruses are known to mutate much more frequently than DNA viruses, however, among the DNA viruses the PVs have a relatively high mutation rate, and host switching has been observed [113, 114]. On the other hand, PVs are known to be coevolving with their host [115].
Circoviridae was the most abundant vertebrate virus family in this study (84%) while in other studies it was the Parvoviridae [6]. In addition, the prevalence of circovirus genomes in this study was higher than that revealed in previous reports [40, 44, 116, 117].
RVA and RVH have been detected in metagenomic analyses of bat samples from different geographical locations [75–77]. However, it is not known whether RV infections in bats lead to disease [76, 118]. RVs have been detected also in several other wild animals [119].
Bats are known hosts for different hepeviruses of the genus Orthohepevirus [96, 120, 121]. Bat hepeviruses, including the contig revealed in this study, seem to cluster with one another and generate a separate clade withing the Orthohepeviruses, the Orthohepevirus D [96, 120]. This indicates that bats may not serve as reservoirs for hepeviruses infecting other mammals including humans [96].
The most interesting finding in this study was an almost complete genome of a MERS-like CoV detected in a ground stool sample of a Vespertilio murinus colony. It will be interesting to study the zoonotic potential of this bat betacoronavirus and to monitor the colony in which it was detected over time as this would allow to assess the accumulation of mutations in the CoV genome in a natural reservoir host.
Human interaction with wildlife is one of the major contributors to zoonotic spillover and this impact should be reassessed [57]. Metagenomic analysis of ground stool samples of bat colonies represents an ideal non-invasive high throughput method to monitor the complexity of the viral genome diversity. It allows also to detect viruses with zoonotic potential and to assess the potential risk for their transmission to other species including humans. This first assessment of the virome of Swiss bats forms a platform for future in-depth studies to investigate changes in virus prevalence, virus biology, virus-host interaction, and virus emergence.
Supporting information
(TIF)
Numbers in parentheses represent the numbers of virus families from vertebrates/non-vertebrates; *migrating bat species.
(TIF)
7’046 animals of 8 different bat species and from 36 different bat colonies were sampled.
(DOCX)
137 individual animals from 16 bat species were sampled.
(DOCX)
108 individual animals were dissected and the lung, liver combined with spleen, intestine and brain collected.
(DOCX)
(DOCX)
Acknowledgments
We want to thank the Bat Foundation Switzerland, the cantonal commissioners for bat protection, the care takers of bat colonies for their help with the sample collection, and Elisabeth Schraner for assistance with electron microscopy.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
C.F. was supported by the Foundation for Research in Science and the Humanities at the University of Zurich (Grant number STWF-19-013)(https://www.research.uzh.ch/en/funding/researchers/stwf.html). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Frick WF, Kingston T, Flanders J. A review of the major threats and challenges to global bat conservation. Annal of the New York Academy of Sciences. 2019. Epub 2019/04/02. doi: 10.1111/nyas.14045 [DOI] [PubMed] [Google Scholar]
- 2.Simmons NBaALC. Bat Species of the World: A taxonomic and geographic database. c2020 [cited 2020 Apr 28]. Available from: https://batnames.org/home.html.
- 3.Amador LI, Arévalo RLM, Almeida FC, Catalano SA, Giannini NP. Bat systematics in the light of unconstrained analyses of a comprehensive molecular supermatrix. J Mamm Evol. 2018;25(1):37–70. doi: 10.1007/s10914-016-9363-8 [DOI] [Google Scholar]
- 4.Burgin CJ, Colella JP, Kahn PL, Upham NS. How many species of mammals are there? J Mammal 2018;99(1):1–14. doi: 10.1093/jmammal/gyx147 [DOI] [Google Scholar]
- 5.Dacheux L, Cervantes-Gonzalez M, Guigon G, Thiberge JM, Vandenbogaert M, Maufrais C, et al. A preliminary study of viral metagenomics of French bat species in contact with humans: identification of new mammalian viruses. PLoS One. 2014;9(1):e87194. Epub 2014/01/29. doi: 10.1371/journal.pone.0087194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu D, Zhu C, Wang Y, Ai L, Yang L, Ye F, et al. Virome analysis for identification of novel mammalian viruses in bats from Southeast China. Sci Rep. 2017;7(1):10917. Epub 2017/09/07. doi: 10.1038/s41598-017-11384-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.fledermausschutz.ch [Internet]. Switzerland: Bat conservation; c2020 [cited 2020 Apr 16]. Available from: https://fledermausschutz.ch/.
- 8.iucn.org [Internet]. Switzerland: International Union for Conservation of Nature; c2020 [cited 2020 Apr 16]. Available from: https://www.iucn.org/content/microchiropteran-bats-global-status-survey-and-conservation-action-plan.
- 9.batlife-europe.info [Internet]. England: BatLife Europe: Bat Conservation; c2020 [cited 2020 Apr 30]. Available from: https://www.batlife-europe.info/.
- 10.Teeling EC, Vernes SC, Dávalos LM, Ray DA, Gilbert MTP, Myers E, et al. Bat Biology, Genomes, and the Bat1K Project: To Generate Chromosome-Level Genomes for All Living Bat Species. Annu Rev Anim Biosci. 2018;6:23–46. Epub 2017/11/20. doi: 10.1146/annurev-animal-022516-022811 [DOI] [PubMed] [Google Scholar]
- 11.Riccucci M, Lanza B. Bats and insect pest control: a review. Vespertilio. 2014;17:161–9. [Google Scholar]
- 12.Bumrungsri S, Lang D, Harrower C, Sripaoraya E, Kitpipit K, Racey PA. The dawn bat, Eonycteris spelaea Dobson (Chiroptera: Pteropodidae) feeds mainly on pollen of economically important food plants in Thailand. Acta Chiropterologica. 2013;15(1):95–104. doi: 10.3161/150811013x667894 [DOI] [Google Scholar]
- 13.Lina PH. Common names of European bats: UNEP/EUROBATS Secretariat; 2016.
- 14.Shipley R, Wright E, Selden D, Wu G, Aegerter J, Fooks AR, et al. Bats and Viruses: Emergence of Novel Lyssaviruses and Association of Bats with Viral Zoonoses in the EU. Trop Med Infect Dis. 2019;4(1). Epub 2019/02/07. doi: 10.3390/tropicalmed4010031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffin DR. 1970. Migrations and homing in bats, p. 233–264. In Wimsatt W. A.(ed.), Biology of bats. Academic Press, New York, N.Y. 1970:233–64. doi: 10.1016/B978-0-12-758001-2 [DOI] [Google Scholar]
- 16.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19(3):531–45. doi: 10.1128/CMR.00017-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brosset A. The migrations of Pipistrellus nathusii, in France. Possible implication on the spreading of rabies. Mammalia. 1990. doi: 10.1515/mamm-1990-0205 [DOI] [Google Scholar]
- 18.Smith I, Wang LF. Bats and their virome: an important source of emerging viruses capable of infecting humans. Curr Opin Virol. 2013;3(1):84–91. Epub 2012/12/21. doi: 10.1016/j.coviro.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkinson GS, South JM. Life history, ecology and longevity in bats. Aging cell. 2002;1(2):124–31. doi: 10.1046/j.1474-9728.2002.00020.x [DOI] [PubMed] [Google Scholar]
- 20.Wilkinson GS, Adams DM. Recurrent evolution of extreme longevity in bats. Biol Lett. 2019;15(4):20180860. doi: 10.1098/rsbl.2018.0860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Podlutsky AJ, Khritankov AM, Ovodov ND, Austad SN. A new field record for bat longevity. J Gerontol A Biol Sci Med Sci. 2005;60(11):1366–8. doi: 10.1093/gerona/60.11.1366 [DOI] [PubMed] [Google Scholar]
- 22.Zhang G, Cowled C, Shi Z, Huang Z, Bishop-Lilly KA, Fang X, et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 2013;339(6118):456–60. Epub 2012/12/20. doi: 10.1126/science.1230835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker ML, Schountz T, Wang LF. Antiviral immune responses of bats: a review. Zoonoses Public Health. 2013;60(1):104–16. Epub 2012/08/01. doi: 10.1111/j.1863-2378.2012.01528.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Shea TJ, Cryan PM, Cunningham AA, Fooks AR, Hayman DT, Luis AD, et al. Bat flight and zoonotic viruses. Emerg Infect Dis. 2014;20(5):741–5. doi: 10.3201/eid2005.130539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sulkin SE, Allen R. Virus infections in bats. Monogr Virol. 1974;8(0):1–103. doi: 10.1159/000395514 [DOI] [PubMed] [Google Scholar]
- 26.Mollentze N, Streicker DG. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proceedings of the National Academy of Sciences. 2020;117(17):9423–30. doi: 10.1073/pnas.1919176117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olival KJ, Epstein JH, Wang L-F, Field HE, Daszak P. Are bats exceptional viral reservoirs. New directions in conservation medicine: Applied cases of ecological health. 2012:195–212. doi: 10.1002/9781118818824.ch11 [DOI] [Google Scholar]
- 28.Plowright RK, Field HE, Smith C, Divljan A, Palmer C, Tabor G, et al. Reproduction and nutritional stress are risk factors for Hendra virus infection in little red flying foxes (Pteropus scapulatus). Proc. Royal Soc. B. 2008;275(1636):861–9. doi: 10.1098/rspb.2007.1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leroy E, Gonzalez J, Pourrut X. Ebolavirus and other filoviruses. Wildlife and Emerging Zoonotic Diseases: The biology, circumstances and consequences of cross-species transmission: Springer; 2007. p. 363–87. doi: 10.1007/978-3-540-70962-6_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wimsatt WA. Biology of bats Chapter 1—Bat Origins and Evolution. New York, N.Y.: Elsevier Inc. All; 1970. 1–64 p. doi: 10.1002/aja.1001290107 [DOI] [Google Scholar]
- 31.Kuno G. Persistence of arboviruses and antiviral antibodies in vertebrate hosts: its occurrence and impacts. Rev Med Virol. 2001;11(3):165–90. doi: 10.1002/rmv.314 [DOI] [PubMed] [Google Scholar]
- 32.Kohl C, Kurth A. European bats as carriers of viruses with zoonotic potential. Viruses. 2014;6(8):3110–28. Epub 2014/08/13. doi: 10.3390/v6083110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sendow I, Field HE, Curran J. Henipavirus in Pteropus vampyrus bats, Indonesia. Emerg Infect Dis. 2006;12(4):711. doi: 10.3201/eid1204.051181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reynes J-M, Counor D, Ong S, Faure C, Seng V, Molia S, et al. Nipah virus in Lyle’s flying foxes, Cambodia. Emerg Infect Dis. 2005;11(7):1042. doi: 10.3201/eid1107.041350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philbey AW, Kirkland PD, Ross AD, Davis RJ, Gleeson AB, Love RJ, et al. An apparently new virus (family Paramyxoviridae) infectious for pigs, humans, and fruit bats. Emerg Infect Dis. 1998;4(2):269. doi: 10.3201/eid0402.980214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diakoudi G, Lanave G, Moreno A, Chiapponi C, Sozzi E, Prosperi A, et al. Surveillance for Adenoviruses in Bats in Italy. Viruses. 2019;11(6). Epub 2019/06/06. doi: 10.3390/v11060523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monath TP. Ecology of Marburg and Ebola viruses: speculations and directions for future research. The Journal of infectious diseases. 1999;179(Supplement_1):S127–S38. doi: 10.1086/514281 [DOI] [PubMed] [Google Scholar]
- 38.Megali A, Yannic G, Zahno ML, Brügger D, Bertoni G, Christe P, et al. Surveillance for European bat lyssavirus in Swiss bats. Arch Virol. 2010;155(10):1655–62. Epub 2010/08/28. doi: 10.1007/s00705-010-0750-9 [DOI] [PubMed] [Google Scholar]
- 39.Canuti M, Eis-Huebinger AM, Deijs M, de Vries M, Drexler JF, Oppong SK, et al. Two novel parvoviruses in frugivorous New and Old World bats. PLoS One. 2011;6(12). doi: 10.1371/journal.pone.0029140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han HJ, Wen HL, Zhao L, Liu JW, Luo LM, Zhou CM, et al. Novel coronaviruses, astroviruses, adenoviruses and circoviruses in insectivorous bats from northern China. Zoonoses and public health. 2017;64(8):636–46. doi: 10.1111/zph.12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Male MF, Kraberger S, Stainton D, Kami V, Varsani A. Cycloviruses, gemycircularviruses and other novel replication-associated protein encoding circular viruses in Pacific flying fox (Pteropus tonganus) faeces. Infect Genet Evol. 2016;39:279–92. doi: 10.1016/j.meegid.2016.02.009 [DOI] [PubMed] [Google Scholar]
- 42.Ge X, Li Y, Yang X, Zhang H, Zhou P, Zhang Y, et al. Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J Virol. 2012;86(8):4620–30. Epub 2012/02/15. doi: 10.1128/JVI.06671-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donaldson EF, Haskew AN, Gates JE, Huynh J, Moore CJ, Frieman MB. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat. J Virol. 2010;84(24):13004–18. Epub 2010/10/06. doi: 10.1128/JVI.01255-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Victoria JG, Wang C, Jones M, Fellers GM, Kunz TH, et al. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol. 2010;84(14):6955–65. Epub 2010/05/12. doi: 10.1128/JVI.00501-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hayman DT. Bats as Viral Reservoirs. Annu Rev Virol. 2016;3(1):77–99. Epub 2016/08/22. doi: 10.1146/annurev-virology-110615-042203 [DOI] [PubMed] [Google Scholar]
- 46.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310(5748):676–9. Epub 2005/09/29. doi: 10.1126/science.1118391 [DOI] [PubMed] [Google Scholar]
- 47.Wang LF, Anderson DE. Viruses in bats and potential spillover to animals and humans. Curr Opin Virol. 2019;34:79–89. Epub 2019/01/18. doi: 10.1016/j.coviro.2018.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan Y, Zhao K, Shi ZL, Zhou P. Bat Coronaviruses in China. Viruses. 2019;11(3). Epub 2019/03/02. doi: 10.3390/v11030210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L, Liu B, Yang J, Jin Q. DBatVir: the database of bat-associated viruses. Database (Oxford). 2014;2014:bau021. Epub 2014/03/18. doi: 10.1093/database/bau021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.mgc.ac.cn/DBatVir/ [Internet]. China: DBatVir Database of bat-associated viruses.; c2020 [cited 2020 Apr 20]. Available from: http://www.mgc.ac.cn/DBatVir/.
- 51.Halpin K, Hyatt AD, Fogarty R, Middleton D, Bingham J, Epstein JH, et al. Pteropid bats are confirmed as the reservoir hosts of henipaviruses: a comprehensive experimental study of virus transmission. Am J Trop Med Hyg. 2011;85(5):946–51. doi: 10.4269/ajtmh.2011.10-0567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Field H, Young P, Yob JM, Mills J, Hall L, Mackenzie J. The natural history of Hendra and Nipah viruses. Microbes Infect. 2001;3(4):307–14. doi: 10.1016/s1286-4579(01)01384-3 [DOI] [PubMed] [Google Scholar]
- 53.Zaki AM, Van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–20. doi: 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- 54.Hu B, Ge X, Wang L-F, Shi Z. Bat origin of human coronaviruses. Virol J. 2015;12(1):221. doi: 10.1186/s12985-015-0422-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3. Epub 2020/02/03. doi: 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paraskevis D, Kostaki EG, Magiorkinis G, Panayiotakopoulos G, Sourvinos G, Tsiodras S. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect Genet Evol. 2020;79:104212. doi: 10.1016/j.meegid.2020.104212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frutos R, Serra-Cobo J, Chen T, Devaux CA. COVID-19: Time to exonerate the pangolin from the transmission of SARS-CoV-2 to humans. Infect Genet Evol. 2020;84:104493. doi: 10.1016/j.meegid.2020.104493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. 2020;26(4):450–2. doi: 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the origin and continuing evolution of SARS-CoV-2. National Science Review. 2020. Natl Sci Rev. 2020; 7(6):1012–23. doi: 10.1093/nsr/nwaa036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He B, Li Z, Yang F, Zheng J, Feng Y, Guo H, et al. Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses. PLoS One. 2013;8(4):e61950. Epub 2013/04/22. doi: 10.1371/journal.pone.0061950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu Z, Ren X, Yang L, Hu Y, Yang J, He G, et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J Virol. 2012;86(20):10999–1012. Epub 2012/08/01. doi: 10.1128/JVI.01394-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang F, Wang Y, Zheng W, He B, Jiang T, Li Y, et al. Metagenomic analysis of bat virome in several Chinese regions. Sheng Wu Gong Cheng Xue Bao. 2013;29(5):586–600. doi: 10.1128/msphere.00587-20 [DOI] [PubMed] [Google Scholar]
- 63.Zheng XY, Qiu M, Guan WJ, Li JM, Chen SW, Cheng MJ, et al. Viral metagenomics of six bat species in close contact with humans in southern China. Arch Virol. 2018;163(1):73–88. Epub 2017/10/05. doi: 10.1007/s00705-017-3570-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kubacki J, Fraefel C, Bachofen C. Implementation of next-generation sequencing for virus identification in veterinary diagnostic laboratories. J Vet Diagn Invest. 2020; 1040638720982630. doi: 10.1177/1040638720982630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kubacki J, Flacio E, Qi W, Guidi V, Tonolla M, Fraefel C. Viral Metagenomic Analysis of Aedes albopictus Mosquitos from Southern Switzerland. Viruses. 2020;12(9):929. doi: 10.3390/v12090929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bachofen C, Kubacki J, Jermini M, Giannini P, Martinetti G, Ripellino P, et al. New subcluster of HEV genotype 3 strains linked to the first confirmed Swiss case of foodborne hepatitis E infection. Case Rep. J. 2017;1(1):003. doi: 10.5167/uzh-148669 [DOI] [Google Scholar]
- 67.Vijgen L, Moës E, Keyaerts E, Li S, Van Ranst M. A pancoronavirus RT-PCR assay for detection of all known coronaviruses. Methods Mol Biol. 2008;454:3–12. doi: 10.1007/978-1-59745-181-9_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wellehan JF, Johnson AJ, Harrach B, Benkö M, Pessier AP, Johnson CM, et al. Detection and analysis of six lizard adenoviruses by consensus primer PCR provides further evidence of a reptilian origin for the atadenoviruses. J Virol. 2004;78(23):13366–9. doi: 10.1128/JVI.78.23.13366-13369.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM. Primer3Plus, an enhanced web interface to Primer3. Nucleic acids research. 2007;35(Web Server issue):W71–W4. Epub 2007/05/07. doi: 10.1093/nar/gkm306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674–6. doi: 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- 71.NCBI, database n [Internet]. United States of America: National Center for Biotechnology Information database n; c2020 [cited 2020 Nov 01]. Available from: ftp://ftp.ncbi.nlm.nih.gov/blast/db/
- 72.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 73.Kumar S SG, Li M, Knyaz C, and Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Elsevier Mol Biol Evol. 2018;35:1547–9. doi: 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.King AM, Lefkowitz E, Adams MJ, Carstens EB. Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses: Elsevier; 2011. [Google Scholar]
- 75.Esona MD, Mijatovic-Rustempasic S, Conrardy C, Tong S, Kuzmin IV, Agwanda B, et al. Reassortant group A rotavirus from straw-colored fruit bat (Eidolon helvum). Emerg Infect Dis. 2010;16(12):1844. doi: 10.3201/eid1612.101089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yinda CK, Ghogomu SM, Conceição-Neto N, Beller L, Deboutte W, Vanhulle E, et al. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018;4(1):vey008. Epub 2018/03/30. doi: 10.1093/ve/vey008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim HK, Yoon SW, Kim DJ, Koo BS, Noh JY, Kim J, et al. Detection of Severe Acute Respiratory Syndrome‐Like, Middle East Respiratory Syndrome‐Like Bat Coronaviruses and Group H Rotavirus in Faeces of Korean Bats. Transbound Emerg Dis. 2016;63(4):365–72. doi: 10.1111/tbed.12515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu Z, Yang L, Ren X, He G, Zhang J, Yang J, et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016;10(3):609–20. Epub 2015/08/11. doi: 10.1038/ismej.2015.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mendenhall IH, Wen DLH, Jayakumar J, Gunalan V, Wang L, Mauer-Stroh S, et al. Diversity and Evolution of Viral Pathogen Community in Cave Nectar Bats. Viruses. 2019;11(3). Epub 2019/03/12. doi: 10.3390/v11030250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Šimić I, Zorec TM, Lojkić I, Krešić N, Poljak M, Cliquet F, et al. Viral Metagenomic Profiling of Croatian Bat Population Reveals Sample and Habitat Dependent Diversity. Viruses. 2020;12(8):891. doi: 10.3390/v12080891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sonntag M, Mühldorfer K, Speck S, Wibbelt G, Kurth A. New adenovirus in bats, Germany. Emerg Infect Dis. 2009;15(12):2052. doi: 10.3201/eid1512.090646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jánoska M, Vidovszky M, Molnár V, Liptovszky M, Harrach B, Benkő M. Novel adenoviruses and herpesviruses detected in bats. Vet J. 2011;189(1):118–21. doi: 10.1016/j.tvjl.2010.06.020 [DOI] [PubMed] [Google Scholar]
- 83.Kohl C, Vidovszky MZ, Mühldorfer K, Dabrowski PW, Radonić A, Nitsche A, et al. Genome analysis of bat adenovirus 2: indications of interspecies transmission. J Virol. 2012;86(3):1888–92. Epub 2011/12/02. doi: 10.1128/JVI.05974-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Drexler JF, Corman VM, Wegner T, Tateno AF, Zerbinati RM, Gloza-Rausch F, et al. Amplification of emerging viruses in a bat colony. Emerg Infect Dis. 2011;17(3):449–56. Epub 2011/03/12. doi: 10.3201/eid1703.100526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kemenesi G, Dallos B, Görföl T, Boldogh S, Estók P, Kurucz K, et al. Novel European lineages of bat astroviruses identified in Hungary. Acta Virol. 2014;58(1):95–8. Epub 2014/04/11. doi: 10.4149/av_2014_01_95 [DOI] [PubMed] [Google Scholar]
- 86.Gloza-Rausch F, Ipsen A, Seebens A, Göttsche M, Panning M, Drexler JF, et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg Infect Dis. 2008;14(4):626–31. Epub 2008/04/11. doi: 10.3201/eid1404.071439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Falcón A, Vázquez-Morón S, Casas I, Aznar C, Ruiz G, Pozo F, et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch Virol. 2011;156(10):1883–90. Epub 2011/07/19. doi: 10.1007/s00705-011-1057-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lelli D, Papetti A, Sabelli C, Rosti E, Moreno A, Boniotti MB. Detection of coronaviruses in bats of various species in Italy. Viruses. 2013;5(11):2679–89. Epub 2013/11/05. doi: 10.3390/v5112679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moreno A, Lelli D, De Sabato L, Zaccaria G, Boni A, Sozzi E, et al. Detection and full genome characterization of two beta CoV viruses related to Middle East respiratory syndrome from bats in Italy. Virol J. 2017;14(1):239. doi: 10.1186/s12985-017-0907-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.August TA, Mathews F, Nunn MA. Alphacoronavirus detected in bats in the United Kingdom. Vector Borne Zoonotic Dis. 2012;12(6):530–3. Epub 2012/01/27. doi: 10.1089/vbz.2011.0829 [DOI] [PubMed] [Google Scholar]
- 91.Drexler JF, Gloza-Rausch F, Glende J, Corman VM, Muth D, Goettsche M, et al. Genomic characterization of SARS-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J Virol. 2010; 84(21): 11336–49. doi: 10.1128/JVI.00650-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rihtaric D, Hostnik P, Steyer A, Grom J, Toplak I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch Virol. 2010;155(4):507–14. Epub 2010/03/11. doi: 10.1007/s00705-010-0612-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Annan A, Baldwin HJ, Corman VM, Klose SM, Owusu M, Nkrumah EE, et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg Infect Dis. 2013;19(3):456–9. Epub 2013/04/30. doi: 10.3201/eid1903.121503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reusken CB, Lina PH, Pielaat A, de Vries A, Dam-Deisz C, Adema J, et al. Circulation of group 2 coronaviruses in a bat species common to urban areas in Western Europe. Vector Borne Zoonotic Dis. 2010;10(8):785–91. Epub 2010/01/09. doi: 10.1089/vbz.2009.0173 [DOI] [PubMed] [Google Scholar]
- 95.De Benedictis P, Marciano S, Scaravelli D, Priori P, Zecchin B, Capua I, et al. Alpha and lineage C betaCoV infections in Italian bats. Virus Genes. 2014;48(2):366–71. Epub 2013/11/19. doi: 10.1007/s11262-013-1008-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Drexler JF, Seelen A, Corman VM, Tateno AF, Cottontail V, Zerbinati RM, et al. Bats worldwide carry hepatitis E virus-related viruses that form a putative novel genus within the family Hepeviridae. J Virol. 2012;86(17):9134–47. doi: 10.1128/JVI.00800-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kohl C, Lesnik R, Brinkmann A, Ebinger A, Radonić A, Nitsche A, et al. Isolation and characterization of three mammalian orthoreoviruses from European bats. PLoS One. 2012;7(8):e43106. Epub 2012/08/21. doi: 10.1371/journal.pone.0043106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kivistö I, Tidenberg E-M, Lilley T, Suominen K, Forbes KM, Vapalahti O, et al. First report of coronaviruses in Northern European bats. Vector Borne Zoonotic Dis. 2020;20(2):155–8. doi: 10.1089/vbz.2018.2367 [DOI] [PubMed] [Google Scholar]
- 99.Bonny TS, Driver JP, Paisie T, Salemi M, Morris JG, Shender LA, et al. Detection of Alphacoronavirus vRNA in the Feces of Brazilian Free-Tailed Bats (Tadarida brasiliensis) from a Colony in Florida, USA. Diseases. 2017;5(1):7. doi: 10.3390/diseases5010007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol. 2012;86(7):3995–4008. doi: 10.1128/JVI.06540-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Luo C-M, Wang N, Yang X-L, Liu H-Z, Zhang W, Li B, et al. Discovery of novel bat coronaviruses in South China that use the same receptor as Middle East respiratory syndrome coronavirus. J Virol. 2018;92(13). doi: 10.1128/JVI.00116-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barbour R, Davis W. Bats of America. Lexington. Kentucky University of Kentucky Press; p. 1969. [Google Scholar]
- 103.Shankar V, Orciari LA, Mattos CD, Kuzmin IV, Pape WJ, O’Shea TJ, et al. Genetic divergence of rabies viruses from bat species of Colorado, USA. Vector Borne Zoonotic Dis. 2005;5(4):330–41. doi: 10.1089/vbz.2005.5.330 [DOI] [PubMed] [Google Scholar]
- 104.Zana B, Kemenesi G, Urbán P, Földes F, Görföl T, Estók P, et al. Metagenomic analysis of bat guano samples revealed the presence of viruses potentially carried by insects, among others by Apis mellifera in Hungary. Acta Vet Hung. 2018;66(1):151–61. doi: 10.1556/004.2018.014 [DOI] [PubMed] [Google Scholar]
- 105.Grozinger CM, Flenniken ML. Bee viruses: Ecology, pathogenicity, and impacts. Annu. Rev. Entomol. 2019;64:205–226. doi: 10.1146/annurev-ento-011118-111942 [DOI] [PubMed] [Google Scholar]
- 106.Finoketti F, Dos Santos RN, Campos AAS, Zani ALDS, Barboza CM, Fernandes MES, et al. Detection of adenovirus, papillomavirus and parvovirus in Brazilian bats of the species Artibeus lituratus and Sturnira lilium. Arch Virol. 2019;164(4):1015–25. Epub 2019/02/10. doi: 10.1007/s00705-018-04129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vidovszky MZ, Kohl C, Boldogh S, Görföl T, Wibbelt G, Kurth A, et al. Random sampling of the Central European bat fauna reveals the existence of numerous hitherto unknown adenoviruses. Acta Vet Hung. 2015;63(4):508–25. doi: 10.1556/004.2015.047 [DOI] [PubMed] [Google Scholar]
- 108.Scott MK, Chommanard C, Lu X, Appelgate D, Grenz L, Schneider E, et al. Human adenovirus associated with severe respiratory infection, Oregon, USA, 2013–2014. Emerg Infect Dis. 2016;22(6):1044. doi: 10.3201/eid2206.151898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pauly M, Hoppe E, Mugisha L, Petrzelkova K, Akoua-Koffi C, Couacy-Hymann E, et al. High prevalence and diversity of species D adenoviruses (HAdV-D) in human populations of four Sub-Saharan countries. Virol J. 2014;11:25. Epub 2014/02/11. doi: 10.1186/1743-422X-11-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Benkö M, Harrach B. Molecular evolution of adenoviruses. Curr Top Microbiol Immunol. 2003;272:3–35. Epub 2003/05/16. doi: 10.1007/978-3-662-05597-7_1 [DOI] [PubMed] [Google Scholar]
- 111.Davison AJ, Benkő M, Harrach B. Genetic content and evolution of adenoviruses. J Gen Mol Virol. 2003;84(11):2895–908. doi: 10.1099/vir.0.19497-0 [DOI] [PubMed] [Google Scholar]
- 112.Anderson M, Higgins P, Davis L, Willman J, Jones S, Kidd I, et al. Experimental parvoviral infection in humans. J Infect Dis. 1985;152(2):257–65. doi: 10.1093/infdis/152.2.257 [DOI] [PubMed] [Google Scholar]
- 113.Shackelton LA, Parrish CR, Truyen U, Holmes EC. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc Natl Acad Sci. 2005;102(2):379–84. doi: 10.1073/pnas.0406765102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shackelton LA, Hoelzer K, Parrish CR, Holmes EC. Comparative analysis reveals frequent recombination in the parvoviruses. J Gen Virol. 2007;88(Pt 12):3294. doi: 10.1099/vir.0.83255-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lukashov VV, Goudsmit J. Evolutionary relationships among parvoviruses: virus-host coevolution among autonomous primate parvoviruses and links between adeno-associated and avian parvoviruses. J Virol. 2001;75(6):2729–40. doi: 10.1128/JVI.75.6.2729-2740.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ge X, Li J, Peng C, Wu L, Yang X, Wu Y, et al. Genetic diversity of novel circular ssDNA viruses in bats in China. J Gen Virol. 2011;92(11):2646–53. doi: 10.1099/vir.0.034108-0 [DOI] [PubMed] [Google Scholar]
- 117.de Sales Lima FE, Cibulski SP, dos Santos HF, Teixeira TF, Varela APM, Roehe PM, et al. Genomic characterization of novel circular ssDNA viruses from insectivorous bats in Southern Brazil. PLoS One. 2015;10(2):e0118070. doi: 10.1371/journal.pone.0118070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shi Z. Bat and virus. Protein & cell. 2010;1(2):109–14. doi: 10.1007/s13238-010-0029-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Matthijnssens J, Bilcke J, Ciarlet M, Martella V, Bányai K, Rahman M, et al. Rotavirus disease and vaccination: impact on genotype diversity. Future Microbiol. 2009;4(10):1303–16. doi: 10.2217/fmb.09.96 [DOI] [PubMed] [Google Scholar]
- 120.Wang B, Yang X-L, Li W, Zhu Y, Ge X-Y, Zhang L-B, et al. Detection and genome characterization of four novel bat hepadnaviruses and a hepevirus in China. Virol J. 2017;14(1):1–10. doi: 10.1186/s12985-016-0669-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kobayashi T, Murakami S, Yamamoto T, Mineshita K, Sakuyama M, Sasaki R, et al. Detection of bat hepatitis E virus RNA in microbats in Japan. Virus Genes. 2018;54(4):599–602. doi: 10.1007/s11262-018-1577-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
Numbers in parentheses represent the numbers of virus families from vertebrates/non-vertebrates; *migrating bat species.
(TIF)
7’046 animals of 8 different bat species and from 36 different bat colonies were sampled.
(DOCX)
137 individual animals from 16 bat species were sampled.
(DOCX)
108 individual animals were dissected and the lung, liver combined with spleen, intestine and brain collected.
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.