

Abstract
Cefotaxime (CTX) is a third-generation cephalosporin (3GC) commonly used to treat infections caused by Escherichia coli . Two genetic mechanisms have been associated with 3GC resistance in E. coli . The first is the conjugative transfer of a plasmid harbouring antibiotic-resistance genes. The second is the introduction of mutations in the promoter region of the ampC β-lactamase gene that cause chromosome-encoded β-lactamase hyperproduction. A wide variety of promoter mutations related to AmpC hyperproduction have been described. However, their link to CTX resistance has not been reported. We recultured 172 cefoxitin-resistant E. coli isolates with known CTX minimum inhibitory concentrations and performed genome-wide analysis of homoplastic mutations associated with CTX resistance by comparing Illumina whole-genome sequencing data of all isolates to a PacBio sequenced reference chromosome. We mapped the mutations on the reference chromosome and determined their occurrence in the phylogeny, revealing extreme homoplasy at the −42 position of the ampC promoter. The 24 occurrences of a T at the −42 position rather than the wild-type C, resulted from 18 independent C>T mutations in five phylogroups. The −42 C>T mutation was only observed in E. coli lacking a plasmid-encoded ampC gene. The association of the −42 C>T mutation with CTX resistance was confirmed to be significant (false discovery rate <0.05). To conclude, genome-wide analysis of homoplasy in combination with CTX resistance identifies the −42 C>T mutation of the ampC promotor as significantly associated with CTX resistance and underlines the role of recurrent mutations in the spread of antibiotic resistance.
Keywords: ampC, bioinformatics, Escherichia coli, genomics, whole-genome sequencing
Data Summary
All data is available from the National Center for Biotechnology Information (NCBI) under BioProject number PRJNA592140. Raw Illumina sequencing data and metadata for all 171 Escherichia coli isolates used in this study is available from the NCBI Sequence Read Archive database under accession numbers SAMN15052485 to SAMN15052655. The full reference chromosome of ampC_0069 is available via GenBank accession number CP046396.1 and NCBI Reference Sequence NZ_CP046396.1. The scripts used to calculate homoplasy-based association analysis are available from GitHub (https://github.com/JordyCoolen/hombaampC) under MIT license.
Impact Statement.
In the past decades, the worldwide spread of extended-spectrum β-lactamases (ESBLs) has led to a substantial increase in the prevalence of resistant common pathogens, thereby restricting available treatment options. Although acquired resistance genes, e.g. ESBLs, get most attention, chromosome-encoded resistance mechanisms may play an important role as well. In Escherichia coli , chromosome-encoded β-lactam resistance can be caused by alterations in the promoter region of the ampC gene. To improve our understanding of how frequently these alterations occur, a comprehensive interpretation of the evolution of these mutations is essential. In the current study, we apply genome-wide homoplasy analysis to better perceive adaptation of the E. coli genome to antibiotics. Thereby, this study grants insights into how chromosome-encoded antibiotic resistance evolves and, by combining genome-wide association studies with homoplasy analyses, provides potential strategies for future association studies into the causes of antibiotics resistance.
Introduction
Escherichia coli is an important pathogen in both community and healthcare-associated infections [1, 2]. In the past decades, a substantial increase in resistance to third-generation cephalosporin (3GC) antibiotics in E. coli has been observed worldwide, mainly caused by the production of extended-spectrum β-lactamases (ESBLs) and AmpC β-lactamases, restricting available treatment options for common infections [3]. AmpC β-lactamases differ from ESBL as they hydrolyse not only broad-spectrum penicillins and cephalosporins, but also cephamycins. Moreover, AmpC β-lactamases are not inhibited by ESBL-inhibitors like clavulanic acid [3], limiting antibiotic treatment options even further. A widely used screening method for AmpC production is the use of susceptibility to cefoxitin (FOX), a member of the cephamycins [4].
Although ampC β-lactamase genes can be plasmid-encoded (plasmid-mediated ampC, pampC), they are also encoded on the chromosomes of numerous Enterobacterales . E. coli naturally carries a chromosome-mediated ampC (campC) gene but, unlike most other Enterobacterales , this gene is non-inducible due to the absence of the ampR regulator gene [3]. Chromosomal AmpC production in E. coli is exclusively regulated by promoter and attenuator mechanisms. This results in constitutive low-level campC expression that still allows the use of 3GC antibiotics, such as cefotaxime (CTX), to treat E. coli infections [3]. However, various mutations in the promoter/attenuator region of E. coli may cause constitutive hyperexpression of campC [5, 6], thereby increasing the minimum inhibitory concentrations (MICs) for broad-spectrum penicillins and cephalosporins and limiting appropriate treatment options.
A wide variety of promoter and attenuator mutations have been related to AmpC hyperproduction [6]. AmpC hyperproduction is primarily caused by alterations of the ampC promoter region, leading to a promoter sequence that more closely resembles the E. coli consensus σ70 promoter with a TTGACA −35 box separated by 17 bp from a TATAAT −10 box. These alterations can be divided into different variants associated with, for example, an alternate displaced promoter box, a promoter box mutation or an alternate spacer length due to insertions [6]. Furthermore, mutations of the attenuator sequence can lead to changes in the hairpin structure that strengthen the effect of promoter alterations on AmpC hyperproduction. In the study by Tracz et al. on FOX-resistant E. coli isolated from Canadian hospitals, 52 variants of the promoter and attenuator region were described [6]. Tracz et al. used a two-step quantitative reverse-transcriptase PCR (qRT-PCR) to determine the effect of promoter/attenuator variants on ampC expression. Various mutations were related to different delta–delta cycle threshold values in the qRT-PCR and corresponding variations in FOX resistance. An interesting observation that emerged from this study was that the −32T>A and the −42C>T mutation were the major alterations that strengthened the ampC promoter. Both result in a consensus −35 box. Although it is known that AmpC hyperproduction leads to FOX resistance, as studied by Tracz et al., the effects of various mutations on resistance to a 3GC antibiotic such as CTX have not been explored. This is relevant because CTX is commonly used in the treatment of patients with severe E. coli infections, often in combination with selective digestive tract decontamination in intensive care units [7, 8].
While previous research mainly focused on the chromosomal AmpC resistance mechanism and the impact of AmpC hyperproduction, there is a lack in knowledge and understanding of the evolutionary origin of these promoter/attenuator variants. Notably, it is unexplored how the two most prominent promoter mutations, −32T>A and −42C>T, are distributed over the E. coli phylogeny and therewith how often they occur. More precisely, literature shows selective pressure can lead to convergent evolution that results in the reoccurrence of a mutation in multiple isolates independently and in separate lineages [9]. This phenomenon is named homoplasy [10]. A consistency index can be calculated to quantify homoplasy by dividing the minimum number of changes on the phylogeny by the number of different nucleotides observed at that site minus one [11], effectively quantifying how often the same mutation occurred in a phylogenetic tree. One can use the consistency index to recognize genomic locations subjected to homoplasy, and relate the SNP positions that are inconsistent with the phylogeny to antibiotic resistance, as has been done, for example, in multiple studies on Mycobacteria spp. [12–15].
In the present study, we hypothesize that some of the mutations in the ampC promoter/attenuator region are homoplastic and are associated with CTX resistance. To test our hypothesis, we performed genome-wide homoplasy analysis and combined it with a genome-wide analysis of polymorphisms associated with CTX resistance by constructing an E. coli reference chromosome and combining it with whole-genome sequencing (WGS) data of 172 both FOX resistant and ESBL-negative E. coli isolates from human and animal origin previously collected by our research group [16].
Methods
Isolate selection, DNA isolation, library preparation and DNA sequencing
One hundred and seventy-two both FOX resistant (MIC >8 mg l−1) and ESBL-negative E. coli isolates previously used by our study group [16] were selected in the present study (Table S1, available with the online version of this article). To summarize the methods, DNA isolation was performed as previously described [16], library preparations were performed using an Illumina Nextera XT library preparation kit, and DNA sequencing was performed using an Illumina NextSeq 500 to generate 2×150 bp paired-end reads or 2×300 bp reads on an Illumina MiSeq. De novo assembly was also performed identically to the method as described previously [16] using SPAdes version 3.11.1 [17].
Phylogroup and multilocus sequence typing (MLST)
Phylogroup stratification was performed using ClermonTyping version 1.4.0 [18]. MLST sequence types (STs) were derived from the contigs using Mlst version 2.5 PubMLST (31 October 2017) [19, 20].
Obtaining the ampC promoter/attenuator region
To detect the promoter/attenuator region, a custom blast database [21] was created using the 271 bp fragment as described by Peter-Getzlaff et al. [22] using E. coli K-12 strain ER3413 (accession no. CP009789.1). ABRicate version 0.8.9 [23] was used to locate matching regions per sample and these were extracted and converted into multi-fasta format using a custom Python script. Strains were labelled AmpC putative hyperproducer when promoter mutations were found, as previously identified by Caroff et al. [24], Siu et al. [25] and Tracz et al. [6].
pampC detection
Detection of pampC genes was performed by using ABRicate version 0.5 [23] and ResFinder database (16/02/2018) as described by Coolen et al. [16].
PacBio single-molecule real-time (SMRT) sequencing of an E. coli isolate
To enable an accurate SNP analysis, a reference chromosome of an E. coli isolate from our collection (ampC_0069) was constructed using PacBio SMRT sequencing. For sequencing, genomic DNA (gDNA) was extracted using a bacterial gDNA isolation kit (Norgen Biotek). A single E. coli isolate was subjected to DNA shearing using Covaris g-TUBEs for 30 s at 11 000 r.p.m. Each DNA sample was separated into two aliquots. Size selection was performed using a 0.75 % agarose cassette and marker S1 on the BluePippin system (Sage Science) to obtain either 4–8 kb or 4–12 kb DNA fragments. This size selection was chosen to maintain all DNA fragments, including these originating from plasmids (data not used in this study). Library preparation was performed using the PacBio SMRTbell template prep kit 1.0 (Pacific Biosciences). For cost-effectiveness, samples were barcoded and pooled with other samples that are not relevant for this study. Sequencing was conducted using the PacBio Sequel I (Pacific Biosciences) on a Sequel SMRT Cell 1M v2 (Pacific Biosciences) with a movie time of 10 h (and 186 min pre-extension time). Subreads per sample were obtained by extracting the bam files using SMRT Link version 5.1.0.26412 (Pacific Biosciences).
Chromosomal reconstruction using de novo hybrid assembly
To obtain a full-length chromosome, Unicycler version 0.4.7 [26] (settings: --mode bold) was used, combining Illumina NextSeq 500 2×150 bp paired-end reads with PacBio Sequel SMRT subreads. Because Unicycler requires fasta reads as input, the subreads in bam format were converted to fasta by using bam2fasta version 1.1.1 from pbbioconda (https://github.com/PacificBiosciences/pbbioconda) prior to de novo hybrid assembly. The full circular chromosome was uploaded to the National Center for Biotechnology Information (NCBI) database and annotated using NCBI Prokaryotic Genome Annotation Pipeline (PGAP) version 4.10 [27, 28].
SNP analysis using E. coli reference ampC_0069
Alignment of Illumina reads and SNP calling was performed for all isolates to the reference chromosome of E. coli isolate ampC_0069 using Snippy version 4.3.6 (https://github.com/tseemann/snippy). A full-length alignment (fullSNP) and a coreSNP alignment containing SNP positions shared among all isolates were generated by using snippy-core version 4.3.6 (https://github.com/tseemann/snippy).
Inferring of phylogeny
A phylogenetic tree was inferred by using the coreSNP alignment as input for FastTree(MP) version 2.1.3 SSE3 (settings: -nt -gtr) [29].
Detection of homoplasy
The consistency index for all nucleotide positions on the chromosome was calculated using HomoplasyFinder version 0.0.0.9000 [10]. The coreSNP phylogeny was used as true phylogeny and the consistency index was calculated using the multiple sequence alignments fullSNP alignment as input.
Relating mutations to CTX resistance
To assess whether certain mutations were linked to CTX resistance, all non-plasmid-harbouring ampC E. coli isolates were used. CTX resistance was defined using European Committee on Antimicrobial Susceptibility Testing (EUCAST) guideline standards of CTX MIC>2 mg l−1 [30]. CTX MIC results were obtained from our previous study [16]. For each nucleotide position on the reference chromosome, the numbers of resistant and sensitive isolates were counted and tested for adenine versus all other nucleotides, thymine versus all other nucleotides, cytosine versus all other nucleotides, and guanine versus all other nucleotides, creating a contingency table and performing a Fisher's exact test in R 3.6.1 [31]. To correct for multiple testing, P values were adjusted using false discovery rate (FDR) [32].
Selection of genomic positions of interest
Genomic positions with potential roles in CTX resistance were identified based on FDR ≤0.05 for CTX and a consistency index of ≤0.05882353. Annotation of mutation positions was obtained by using the genome annotation of the reference chromosome (GenBank accession no. CP046396) and applying snpEff (version 4.3 t) [33]. The EnteroBase core-genome MLST and whole-genome MLST schemes were used to distinguish core and accessory genes [34].
Recombination analysis
Gubbins version 2.4.1. (settings: -f 30) was used to detect recombination regions with coreSNP alignment and tree as input [35].
Pyseer analysis
To compare our homoplasy-associated analysis method to an alternative method, we used Pyseer (version 1.3.6) [36]. In short, variant calling files (VCFs) were obtained from snippy, and bcftools (version 1.11) was used to merge and filter the VCFs from all samples to a single VCF [37]. A phylogenetic distance file was calculated by using the phylogeny_distance.py included in Pyseer on the corrected for recombination phylogeny of Gubbins. Finally, the distance, trait and VCF file was used to run Pyseer (default fixed effects) with settings --min-af 0.01, --max-af 0.99.
Visualization of data
The interactive tree of life web-based tool (iTOL) version 5.3 was used to visualize the phylogenetic tree [38]. Information about CTX resistance, presence of the pampC gene, campC hyperproduction as defined, MLST and phylogroup, as well as alignments of promoter and attenuator region, were incorporated into the visualization. The sequence logo of the promoter and the attenuator alignment were generated using the web-based application WebLogo, version 3.7 [39] (http://weblogo.threeplusone.com). A chromosome ideogram of the E. coli isolate ampC_0069 reference chromosome was visualized using the circos software package, version 0.69-8 [40]. Consistency index scores and significant mutations associated with CTX resistance were plotted in the ideogram. Gubbins results were displayed by using Phandango [41].
Overview of the method
A workflow graph of the method is visualized in Fig. 1, using the web-based application yEd Live version 4.4.2 (https://www.yworks.com/yed-live/).
Fig. 1.

Schematic of the workflow used to perform the homoplasy-based association analysis. Starting from the top, (a) the de novo assembly of the NextSeq/MiSeq reads and (b) the hybrid assembly of the reference chromosome ampC_069. On the left side, (c) the alignment of promoter/attenuator region. In the middle, (d) the coreSNP analysis for the phylogeny used in (e) the homoplasy analysis combined with (f) the fullSNP data, on the right, which was also used for (g) the statistics (Fisher's exact test and FDR) to relate CTX resistance to SNP positions. (h) Inferring recombination events using Gubbins.
Results
E. coli collection
To study genetic homoplasy events in suspected AmpC-producing E. coli, FOX-resistant ESBL phenotype negative E. coli isolates (n=172) were selected, as previously described by Coolen et al. [16] (see Table S1). The entire collection was subjected to WGS, followed by de novo assembly of the sequence reads to obtain contigs.
MLST and phylogroup variants
To access the genetic diversity of our E. coli collection, we identified both MLST and phylogroup variants of each of the 172 E. coli isolates. A total of 75 different STs were identified, of which ST131 (8.1 %, n=14), ST38 (7.0 %, n=12) and ST73 (7.0 %, n=12) were the most prevalent. The STs of 13 isolates are unknown. Phylogroup stratification revealed that the isolates belonged to eight different phylogroups (Table 1). Phylogroup B1, B2 and D were the most prevalent. One isolate belonged to E . coli clade IV (strain no. ampC_0128).
Table 1.
Table of the distribution of the different AmpC promoter and attenuator variants, as well as the numbers of different MLST STs and phylogroups per grouped genotype (pampC, putative hyperproducers and putative low-level AmpC producers)
|
Genotype |
Isolate |
Promoter variant |
Attenuator variant |
MLST |
Phylogroup |
|---|---|---|---|---|---|
|
pampC |
n=90 |
3 variants |
6 variants |
44 STs and 4 unknown |
|
|
Putative hyperproducers |
n=61 |
13 variants |
14 variants |
30 STs and 5 unknown |
|
|
Putative low-level AmpC producers |
n=21 |
8 variants |
5 variants |
14 STs and 4 unknown |
|
|
Total |
n=172 |
21 variants |
18 variants |
75 STs and 13 unknown |
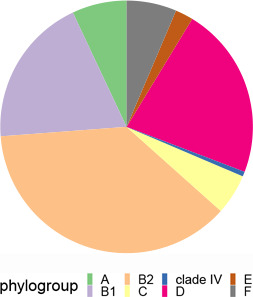
|
ampC promoter and attenuator variants
We examined the whole E. coli genome. However, we firstly focused on mutations in the ampC promoter and attenuator region. Previously described mutations in the ampC promoter region that according to described literature lead to ‘hyperproduction’ of AmpC were detected in 61 (35.5 %) of the isolates [6, 24, 25] (see Tables S1 and S2). These isolates were, therefore, labelled as putative hyperproducers. Analysis of the promotor area (−42 to −8) resulted in 20 different variants and the wild-type (see Table 1). In the attenuator region (+17 to +37), 18 different variants were identified (see Table 1). One isolate (ampC_0128) showed an unusual promoter variant, a four nucleotide deletion (−45_−42delATCC). Moreover, an insertion (21_22insG) of unknown function was detected in the attenuator of this isolate (ampC_0128) as well (see Tables S1 and S2).
pampC variants
As we aimed to associate chromosomal mutations with CTX resistance, differentiation of pampC-harbouring isolates from non-pampC-harbouring isolates was required. Genomic analysis showed that 90 (52.3 %) of the isolates harboured a pampC gene of which bla CMY-2 was the most prevalent (n=78). One isolate harboured two different pampC genes (bla CMY-4 and bla DHA-1) (ampC_0119). One isolate contained a bla CTX-M-27 gene combined with a bla CMY-2 gene (ampC_0114), but was ESBL disc test negative (see Table S1). In 21 (12.2 %) of the isolates, neither pampC nor described mutations related to AmpC hyperproduction were detected and are noted as putative low-level AmpC producers.
Reference chromosome
To be able to reconstruct an accurate phylogeny, we selected E. coli isolate ampC_0069, one of the strains of the study, to use as self-constructed reference chromosome for SNP calling. The reference chromosome was constructed through a hybrid assembly of n=4 4 231 09 2×150 bp Illumina NextSeq 500 paired-end reads together with n=218 475 PacBio Sequel SMRT subreads (median 5640 bp). This resulted in a high-quality full circular chromosome of E. coli isolate ampC_0069, with a size of 5 056 572 bp. This isolate belongs to ST648 and contains a plasmid-encoded bla CMY-42. The full circular chromosome was uploaded to GenBank under accession number CP046396 and was used for further analysis. Genome annotation with the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) identified 4720 coding sequences.
SNP calling
The mapping of the reads of all isolates to the reference chromosome E. coli ampC_0069 (accession no. CP046396) resulted in a coreSNP alignment containing 314 200 variable core SNP positions. For further details per isolate see Table S3. To validate our SNP calling method, we compared the ampC_0069 Illumina NextSeq 500 paired-end reads to the reference chromosome of ampC_0069, resulting in 0 SNPs detected, supporting that the SNP calling data and method produce no false positives.
Phylogenetic tree based on coreSNP
The coreSNP alignment was used for further analysis. Fig. 2 illustrates the approximately maximum-likelihood phylogenetic tree of all 172 isolates based on the coreSNP alignment. The tree has a robust topology as indicated by the SH-like (Shimodaira–Hasegawa like) branch support, only three positions have a value ≤60 % [29]. When focusing on the ampC promoter mutations, they were most prevalent in phylogroups B1, B2 and C, although they were present in all phylogroups except phylogroup E that lacked mutations in either the promoter or attenuator region. Interestingly, two positions previously highlighted by Tracz et al., −42 and −32, are only mutated in the absence of a pampC gene, even in isolates with a similar MLST ST (ST12, ST88 and ST131). The −42C>T mutation, which results in an alternate displaced promoter box and, therefore, leads to increased resistance [6], is present in 24 isolates in five distinct phylogroups and in 17 separate phylogenetic branches, indicating that this mutation is homoplastic. Additionally, the −32T>A mutation in the promoter, previously also associated with resistance [6], is present in 20 isolates in three distinct phylogroups and in 14 separate phylogenetic branches. To quantify the level of homoplasy, we calculated the consistency index.
Fig. 2.

Approximately maximum-likelihood phylogenetic tree of all 172 E. coli isolates based on the coreSNP alignment with the resistance for CTX, pampC gene presence, MLST STs, phylogroups, and the alignments of the promoter and the attenuator region. Positions with a SH-like (Shimodaira–Hasegawa like) branch support ≤60 % are indicated as red dots. Scale bar indicates branch length calculated by FastTree.
Genomic homoplastic mutations
We calculated the consistency index for all SNP positions on the E. coli reference chromosome. A low consistency index value for a position indicates a high degree of inconsistency with the chromosomal phylogeny and can be calculated by HomoplasyFinder as described in earlier studies [10, 42, 43]. As can be observed in Fig. 3, results clearly indicate position −42 (4 470 140) and −32 (4 470 150) are the lowest scoring positions on the consistency index concerning the promoter and attenuator region, respectively, 0.05882353 and 0.07142857 (see also Fig. S1). To access how extreme these values are, we calculated the consistency index for all SNP positions in the chromosome (see Fig. S2). All consistency indexes <1.0 are plotted in the outer ring (ring A) of Fig. 4. Results show that only 9640 out of 5 056 572 positions (0.19 %) had a consistency index ≤0.07142857 (see Fig. 4, ring a, cut-off is indicated by a black circle). This clearly indicates that positions with low consistency indexes are rare, but not unique. Although these 9640 positions have a low consistency index, we do not yet know their relation to CTX resistance.
Fig. 3.

WebLogo sequence logo with both bits and probability score for the promoter and the attenuator region. Consistency index and the minimum number of changes on the tree per position are represented in the bar charts below the sequence logos. Orange bars are positions containing SNPs that are tested for homoplasy. Blue bars are positions containing insertions that are tested for homoplasy.
Fig. 4.

circos plot for the full chromosome of ampC_0069 (accession no. CP046396) with, per position, the various metrics used. (a) The blue coloured ring represents the consistency index results per genomic position. The two red dots indicate the −42 and −32 position on the promoter. The black circle line indicates the 0.07142857 consistency index value. (b) The ring with a red background shows all positions that were significantly associated with CTX resistance in all non-pampC-harbouring E. coli isolates. Larger bars pointing outwards indicate multiple significant associated positions in a small genomic region. (c) The ring with the green background shows all 24 positions that have a low consistency index of ≤0.05882353 and are significantly associated with CTX resistance in all non-pampC-harbouring E. coli isolates.
CTX-resistance measurements
CTX MIC measurements from Coolen et al. [16] in relation to the genotype of the E. coli isolates are shown in Table S1. Eighty-four of ninety (93.3 %) pampC-harbouring E. coli were CTX resistant (MIC >2 mg l−1) based on EUCAST clinical breakpoints. Twenty-two of sixty-one (36.1 %) isolates categorized as putative hyperproducers based on Tracz et al. were CTX resistant, primarily isolates with the −42 (n=15, 62.5 % of isolates with −42 mutation) or −32 mutation (n=2, 10.0 % of isolates with −32 mutation). The pampC genes never occurred simultaneously with the −42 or −32 mutations in any of these isolates. As depicted in Fig. 2, the non-pampC strains with a phenotype of CTX >2 mg l−1 were present in all phylogroups, although CTX-resistant isolates with the −42 or −32 mutation were predominantly present in phylogroups B1, B2 and C.
Genotype to phenotype
To be able to link E. coli chromosomal mutations to CTX resistance, we excluded all E. coli isolates with a plasmid containing an ampC β-lactamase gene. The association of SNPs with the CTX-resistance phenotype (MIC >2 mg l−1) was tested in the remaining 82 isolates using Fisher’s exact test. After FDR correction to 0.05, 45 998 significant positions were found (see Fig. 4, ring b). Mutation C>T on position −42 of the ampC promoter was found to be significantly associated with CTX resistance (FDR=0.034). However, position −32 A>T was not significantly associated with CTX resistance (FDR=1).
Homoplasy-based association analysis
Combining the outcome of the homoplasy analysis with the significant CTX-resistance-associated positions results in genomic positions associated with CTX resistance that have evolved multiple times independently. After selecting the lowest scoring consistency index positions, ≤0.05882353, 24 relevant genomic positions were identified that had both a low consistency index and a significant association with CTX resistance. Most notably, 1 of these 24 positions is position −42. Only 2 mutations of those 24 that were located in genes were non-synonymous: a (conservative) missense mutation in the type II secretion system protein L (gspL) gene leading to a Ser330Thr alteration, and a mutation in the hydroxyethylthiazole kinase (thiM) gene resulting in a Thr122Ala alteration according to the annotation of E. coli strain ampC_0069 (accession no. CP046396). In addition to the non-synonymous mutation found on the gspL gene, eight synonymous mutations are also located in genes annotated as being part of the type II secretion system. A complete overview is presented in Tables 2 and S4.
Table 2.
Details of the 24 positions with a significant association with CTX resistance (FDR ≤0.05) and with a consistency index ≤0.05882353
|
Homoplasy-based association analysis |
Pyseer |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Genomic position |
Mutation |
ACGT counts |
Min changes |
CI |
Crosstab |
AF |
Fisher P value |
FDR |
Gene name |
Genomic position |
Mutation |
AF |
Filter P value |
lrt P value |
|
810 581 |
A>G |
41 : 0 : 122 : 0 |
18 |
0.056 |
0 : 22 : 26 : 34 |
6.83E−01 |
8.11E−05 |
0.039 |
glcE |
810 581 |
A>G |
0.683 |
1.87E−04 |
2.09E−02 |
|
810 791 |
G>T |
0 : 0 : 54 : 109 |
17 |
0.059 |
0 : 22 : 28 : 32 |
6.59E−01 |
1.68E−05 |
0.035 |
glcE |
810 791* |
GGGT>TGGC |
0.646 |
4.08E−04 |
1.94E−02 |
|
815 680 |
C>T |
0 : 69 : 0 : 93 |
18 |
0.056 |
2 : 20 : 34 : 26 |
5.61E−01 |
1.06E−04 |
0.042 |
glcA |
815 680 |
C>T |
0.561 |
1.20E−04 |
2.07E−02 |
|
824 522 |
T>C |
0 : 82 : 0 : 73 |
18 |
0.056 |
2 : 20 : 36 : 24 |
5.37E−01 |
3.58E−05 |
0.036 |
gspC |
824 522* |
T>C |
0.378 |
8.08E−06 |
1.00E+00 |
|
828 695 |
A>G |
79 : 0 : 74 : 0 |
18 |
0.056 |
3 : 19 : 41 : 19 |
4.63E−01 |
1.14E−05 |
0.034 |
gspF |
828 695 |
A>G |
0.463 |
1.08E−05 |
3.26E−02 |
|
830 684 |
T>C |
0 : 80 : 0 : 73 |
18 |
0.056 |
2 : 20 : 37 : 23 |
5.24E−01 |
1.61E−05 |
0.034 |
gspI |
830 684* |
T>C |
0.488 |
3.74E−05 |
1.06E−01 |
|
830 708 |
A>G |
68 : 0 : 85 : 0 |
19 |
0.053 |
2 : 20 : 36 : 24 |
5.37E−01 |
3.58E−05 |
0.036 |
gspI |
830 708* |
A>G |
0.11 |
6.41E−01 |
5.02E−01 |
|
830 732 |
T>C |
0 : 82 : 0 : 71 |
17 |
0.059 |
2 : 20 : 36 : 24 |
5.37E−01 |
3.58E−05 |
0.036 |
gspI |
830 732 |
T>C |
0.537 |
4.20E−05 |
4.19E−02 |
|
831 564 |
G>A |
71 : 0 : 83 : 0 |
17 |
0.059 |
5 : 17 : 45 : 15 |
3.90E−01 |
2.76E−05 |
0.036 |
gspK |
831 564 |
G>A |
0.39 |
1.71E−05 |
2.37E−01 |
|
832 152 |
T>C |
0 : 86 : 0 : 72 |
18 |
0.056 |
2 : 20 : 28 : 22 |
5.83E−01 |
1.08E−05 |
0.034 |
gspK |
832 152* |
CGTCG>TGTCA |
0.195 |
6.94E−03 |
2.29E−01 |
|
832 287 |
A>T |
81 : 0 : 0 : 79 |
17 |
0.059 |
2 : 20 : 37 : 23 |
5.24E−01 |
1.61E−05 |
0.034 |
gspK |
832 287* |
T>A |
0.232 |
6.73E−02 |
5.73E−01 |
|
833 451 |
A>T |
64 : 0 : 0 : 98 |
20 |
0.05 |
1 : 21 : 34 : 26 |
5.73E−01 |
1.03E−05 |
0.034 |
gspL |
833 451* |
T>A |
0.378 |
1.69E−04 |
9.04E−02 |
|
843 887 |
G>A |
64 : 0 : 40 : 0 |
19 |
0.053 |
21 : 1 : 29 : 31 |
3.90E−01 |
7.00E−05 |
0.036 |
Unnamed |
843 884* |
GTTGCGC>TCGACGT |
0.354 |
4.08E−04 |
1.08E−01 |
|
1 863 004 |
A>G |
126 : 0 : 46 : 0 |
18 |
0.056 |
9 : 13 : 53 : 7 |
2.44E−01 |
3.37E−05 |
0.036 |
thiM |
1 862 989* |
CTGTCTCATAAGCCAACCGCC > TTATCTTTTAAACCGGCAGCG |
0.232 |
4.55E−05 |
3.30E−02 |
|
1 911 551 |
T>C |
0 : 116 : 0 : 44 |
17 |
0.059 |
16 : 6 : 14 : 46 |
6.34E−01 |
7.02E−05 |
0.036 |
Unnamed |
1 911 551* |
C>T* |
0.28 |
1.22E−03 |
5.28E−01 |
|
1 946 016 |
A>G |
58 : 0 : 91 : 0 |
23 |
0.043 |
17 : 5 : 17 : 43 |
5.85E−01 |
1.03E−04 |
0.041 |
ugd |
1 946 016* |
A>G* |
0.402 |
5.02E−02 |
4.92E−01 |
|
1 946 067 |
G>T |
0 : 0 : 65 : 84 |
22 |
0.045 |
18 : 4 : 20 : 40 |
5.37E−01 |
1.25E−04 |
0.05 |
Non-coding region |
1 946 067 |
G>T |
0.537 |
9.58E−05 |
2.26E−01 |
|
1 946 072 |
T>A |
84 : 0 : 0 : 65 |
22 |
0.045 |
18 : 4 : 20 : 40 |
5.37E−01 |
1.25E−04 |
0.05 |
Non-coding region |
1 946 072 |
T>A |
0.537 |
9.58E−05 |
2.26E−01 |
|
1 952 745 |
A>G |
131 : 0 : 40 : 0 |
28 |
0.036 |
9 : 13 : 52 : 8 |
2.56E−01 |
7.67E−05 |
0.037 |
hisD |
1 952 745 |
G>A |
0.732 |
6.54E−05 |
7.96E−03 |
|
2 051 214 |
T>A |
137 : 0 : 0 : 34 |
19 |
0.053 |
11 : 11 : 5 : 55 |
8.05E−01 |
1.00E−04 |
0.041 |
Unnamed |
2 051 211* |
CACT>TACA |
0.659 |
3.92E−03 |
4.87E−01 |
|
2 051 220 |
T>A |
137 : 0 : 0 : 34 |
19 |
0.053 |
11 : 11 : 5 : 55 |
8.05E−01 |
1.00E−04 |
0.041 |
Unnamed |
2 051 220* |
T>A |
0.793 |
4.79E−06 |
5.72E−02 |
|
2 057 518 |
A>G |
111 : 0 : 60 : 0 |
19 |
0.053 |
7 : 15 : 49 : 11 |
3.17E−01 |
3.89E−05 |
0.036 |
dcm |
2 057 506* |
ATGTTTCCCTGCGCAGCGAGT > CTGCTATCCGGCACAACGTATT |
0.0122 |
9.66E−02 |
1.00E+00 |
|
2 068 593 |
G>A |
45 : 0 : 123 : 0 |
17 |
0.059 |
9 : 13 : 52 : 8 |
2.56E−01 |
7.67E−05 |
0.037 |
fliM |
2 068 593 |
G>A |
0.256 |
2.60E−05 |
1.28E−01 |
|
4 470 140 |
C>T |
0 : 147 : 0 : 24 |
17 |
0.059 |
7 : 15 : 51 : 9 |
2.93E−01 |
8.35E−06 |
0.034 |
ampC promoter |
4 470 140 |
C>T |
0.293 |
2.74E−06 |
5.42E−03 |
*pyseeridentified multiple mutation variants, affecting theoutcome, only the position with the highest allelefrequency (AF) is reported which overlaps the result of the homoplasy-based association analysis. Sequences in the pyseer mutation of the pyseer outcome that are bold are the corresponding position with thetested genomic position. crosstab:R/other:R/mutation:S/other:S/mutation.
AF, allele frequency; CI, Consistency Index; FDR, false discovery rate; lrt, likelihood ratio test.
Recombination analysis
To verify whether the level of homoplasy could be a result of recombination, we used the Genealogies Unbiased By recomBinations In Nucleotide Sequences (Gubbins) algorithm to predict recombination events in our isolate collection [35]. This analysis showed frequent recombination events in our 172 E. coli isolates (see Fig. S3). Results illustrate that recombination blocks cover the region of the gspL and the thiM gene, and their high homoplasy levels could, thus, be due to recurrent recombination rather than independent mutations. Nonetheless, position −42 in the ampC promoter is not located in a region affected by recombination, as shown in Fig. S3. Moreover, when inferring the phylogenetic tree corrected for recombination events as obtained from the Gubbins analysis, the −42 C>T mutation actually occurred in 18 independent branches rather than the 17 branches in the uncorrected tree. This supports our previous result that this mutation is homoplastic, and not the result of a recurrent recombination event.
Pyseer analysis
To add additional support to our findings, we used Pyseer as an alternative method to compare the 24 positions identified with the homoplasy-based association analysis (see Table 2). Pyseer identified 65 501 unique significant mutations associated with CTX resistance with filter P value ≤0.05 and 1097 unique positions with filter and likelihood ratio test (lrt) P value ≤0.05. Of the 24 positions identified with the homoplasy-based association analyses, we identified 8 positions also reported by the Pyseer method (see Table 2). Furthermore, the Pyseer method identified 6 complex mutation variants and a total of 14 positions that have multiple mutation variants overlapping the same genomic positions as found by the homoplasy-based method. The most dominant position associated with CTX resistance is the −42 C>T promoter mutation as indicated by both methods. No further positions on the promoter or the attenuator were found significantly associated with CTX resistance.
Discussion
We present a genome-wide analysis in which homoplastic mutations are associated with antibiotic resistance in E. coli . By comparing WGS data of 172 E. coli isolates to a reference chromosome, we were able to reconstruct the evolution of the genomes and therewith map recurrent events, allowing us to detect homoplasy associated with CTX resistance.
Our foremost finding is the significant association of the −42C>T mutation, in the ampC promoter, with CTX resistance that evolved independently at least 17 times in five distinct phylogroups. The −42C>T mutation has been confirmed in former studies to result in AmpC hyperproduction in E. coli [6, 24, 44]. Nelson et al. demonstrated an 8 to 18 times increase in activity of AmpC when cloning the promoter upstream a lac operon [44]. Conversely, Caroff et al. found a decrease in expression of AmpC when cloning the promoter with a −42T>C mutation in a pKK232-8 reporter plasmid with a chloramphenicol acetyltransferase gene [24]. Tracz et al. confirmed that the −42C>T mutation has the strongest effect on the ampC promoter, resulting in a high expression of the ampC gene as detected by qRT-PCR [6]. Despite the fact that the −42C>T mutation has such a strong effect on AmpC production, the effect of the mutation on CTX MICs had not been confirmed. Moreover, the contribution of convergent evolution on this position relative to the role of the expansion of a clone with a beneficial mutation at this position has not been determined. That being the case, this study provides evidence that this −42 C>T mutation is not a result of a recombination event and most likely evolved many times independently.
In the current study, we see a strong correlation between the −42C>T mutation and CTX resistance, even though there were exceptions, as not all isolates with this mutation were considered resistant according to EUCAST guidelines. As described by Coolen et al., the MIC for CTX in putative AmpC hyperproducers was generally higher than in the putative low-level AmpC producers, though the range in CTX MICs overlapped [16]. Yet, the lowest MIC measured in the isolates with the −42C>T mutation was 0.75 mg l−1, which is at the higher end of the EUCAST epidemiological cut-off values (ECOFF) distribution. The variation in phenotypical testing could be an explanation, although an interplay of AmpC hyperproduction and other strain-specific factors as previously described by Tracz et al. may also be considered [6].
In order to avoid biasing towards a single method, in our case the homoplasy-based association method, we performed the analysis using Pyseer on the same data set. The outcome of the Pyseer analysis provided a similar number of CTX-resistance-associated mutations and also confirmed the strong association of the −42 C>T mutation. Nonetheless, when zooming in on the identified positions by the homoplasy-based method, not all 24 mutations were significantly associated with CTX by Pyseer. An explanation for this discrepancy is that the combination of snippy and Pyseer identifies various mutation variants on the same overlapping genomic region by stratifying it as complex, indels and/or SNP. This greatly affects the power of the test on certain positions. An example of the differences in mutation variants is illustrated in Table 2.
Remarkably, we observed that the −42C>T mutation never occurs in the presence of a pampC gene (0 out of 24 cases). This was even noticed in isolates with the same MLST, i.e. ST88 −42C>T (n=3) and pampC (n=1), suggesting preferred exclusivity for one of the resistance mechanisms. One study mentioned the co-occurrence of the −42C>T mutation and a pampC gene in only 1 out of 36 strains [45]. One could speculate that the exclusivity is a matter of what arrives first, the plasmid or the mutation, after which there is no selective advantage for the second mechanism, or that there is actually a fitness cost to having both the mutation and the plasmid relative to having only the mutation or the plasmid. This hypothesis might be a start for future studies to determine the relative fitness and resistance provided by the −42C>T mutation relative to isolates harbouring a pampC gene.
The study performed by Tracz et al. showed that position −32T>A on the promotor of ampC associates with AmpC hyperproduction that results in elevated MIC levels for FOX [6]. Surprisingly, in the current study, no significant association of −32T>A with CTX resistance was noticed despite its low consistency index. Only 2 out of 20 isolates with the −32T>A were CTX resistant, 4 out of 20 showed an intermediate elevated CTX MIC, and 14 were susceptible to CTX. Although we do not know under which conditions this mutation did arise, it can be speculated that the high level of homoplasy at the −32 position is associated with a different trait, e.g. resistance against another antibiotic.
Prior studies discovered the importance of mutations in the promoter elements. Although an existing promoter is often copied upstream to the gene, a de novo promoter can also evolve out of an existing sequence region. Random sequences can even evolve expression comparable to the wild-type promoter elements after only a single mutation [46]. This means that the de novo creation of a promoter region within the E. coli may be much more often the result of mutation rather than a rearrangement. Furthermore, these promotor elements evolve to only a few forms, indicating convergent evolution [47], as also observed in the present study. All encountered variants seem to result in a sequence that resembles the E. coli consensus σ70 promoter more than the wild-type sequence they are derived from [6].
Next to the −42C>T promoter mutation, we detected 23 other positions in our analysis that are associated with CTX resistance and have extremely high levels of homoplasy. Most of these are synonymous mutations, with only two missense mutations (thiM and gspL) found. It is remarkable that one missense mutation (p.Ser330Thr) is located in gspL that encodes a protein of the type II secretion system. The type II secretion system is used by many Gram-negative bacteria to translocate folded proteins from the periplasm, through the outer membrane, into the extracellular milieu [48]. The system is composed of 12–15 different general secretory pathway (Gsp) proteins and is related to virulence of various pathogenic E. coli , e.g. EHEC (enterohaemorrhagic E. coli ) and UPEC (uropathogenic E. coli ) [49–51]. It could be that in our selection of mainly clinical samples a certain predilection has occurred towards isolates with particular virulence traits and not based on mechanistic benefits. The gspL gene has been described as being part of the accessory genome of E. coli [52]. Our study supports this finding as some strains did not harbour this gene. Additionally, we found evidence that recombination events in the type II secretion system could be the underlying cause of the extreme homoplasy levels. Still, it is remarkable that missense mutation p.Ser330Thr in the gspL gene correlates with the CTX-resistance trait even though it is most likely caused by a recombination event. To the best of our knowledge, no relationship between the type II secretion system and CTX resistance has been observed before. One could hypothesize that the mutation is a secondary adaptation needed to cope with the elevated AmpC production, as the peptidoglycan (PG) layer is affected by AmpC hyperproduction and the type II secretion system contains proteins that are partly localized in the periplasm [53, 54].
The use of genomic data to detect homoplasy events is an accepted scientific technique [55–57]. In Mycobacterium tuberculosis , it is a well-known method for identifying advantageous mutations, as they are likely to be associated with phenotypes such as drug resistance, heightened transmissibility or host adaptation [12–15]. In other species, e.g. Staphylococcus aureus and Burkholderia pseudomallei , the method has been effectively applied to identify mutations associated with antibiotic resistance or virulence-associated genes [58, 59]. Homoplasy-based association analysis limits phylogenetic bias by correcting for genetic relatedness of strains with the same phenotype, thereby increasing statistical power to find true associations [14]. Taking this into account, the use of homoplasy-based association analysis seems viable to relate polymorphic sites to phenotypic traits in bacteria. Still, studies on other genera than mycobacteria are scarce. To our knowledge, no homoplasy studies have used this method on E. coli.
The increase of 3GC resistance imposes a clinical threat by restricting treatment options and it is essential to understand the underlying resistance mechanisms. To be able to explore these mechanisms, we selected primarily clinical E. coli strains. The current study is directed on exploring AmpC-mediated CTX resistance. Therefore, we included isolates that are already suspected for increased AmpC production based on elevated FOX resistance. Since a random sample of E. coli would limit finding homoplasy-based associated promoter mutations with CTX resistance. A downside of these selection criteria might be that we over-estimated certain genetic variants associated with the trait, as we do not know the frequency of these variants in the general population. Despite the fact that the spontaneous mutation rate in E. coli is relatively low [60], it is still likely that this particular mutation occurs often in the general population, given the vast amounts of E. coli in the environment [61], providing ample opportunities for adaptation to antibiotics and arguing for antibiotics of which genomic adaptation requires multiple mutations in order to develop resistance.
The findings of this study have a number of implications for future practice. This study not only grants insights into how chromosome-encoded antibiotic resistance evolves, but also provides potential strategies for future homoplasy-based association studies. Furthermore, the use of genome-wide homoplasy-based analysis could be applied to optimize outbreak analysis. Prior studies have optimized outbreak analysis by removing recombinant regions [62, 63]. Homoplasy events disturbs the true phylogeny; hence, removing genomic positions that are heavily affected by homoplasy could improve tree topology, thereby refining outbreak analysis, although this strategy is still under debate [64].
Conclusions
To conclude, our method demonstrates extreme levels of homoplasy in E. coli that are significantly associated with CTX resistance. Greater access to WGS data provides new opportunities to perform large-scale genome-wide analysis. Homoplasy-based methods can have a potential role in future studies as they constitute an effective strategy to relate phenotypic traits to variable genomic positions.
Supplementary Data
Funding information
The authors received no specific grant from any funding agency.
Acknowledgements
Special thanks to A. C. J. Soer (Department of Medical Microbiology and Radboudumc Center for Infectious Diseases, Radboudumc, Nijmegen, The Netherlands), B. A. Lamberts (Department of Medical Microbiology and Immunology, Rijnstate, Arnhem, The Netherlands) and C. Verhulst (Department of Infection Control, Amphia Ziekenhuis, Breda, The Netherlands and Laboratory for Microbiology, Microvida, Breda, The Netherlands) for handling the samples in the laboratory and creating the Illumina sequence libraries. Many thanks to M. P. Kwint and R. Derks (Department of Human Genetics, Radboudumc, Nijmegen, The Netherlands) for helping with SMRT sequencing on the PacBio Sequel I. Many thanks also to M. Janssens (Laboratory for Medical Microbiology and Immunology, Elisabeth-Tweesteden Hospital, Tilburg, The Netherlands), S. Van Leest (Laboratory for Microbiology, Microvida, Bravis Hospital, The Netherlands), K. T. Veldman and D. J. Mevius (Department of Bacteriology and Epidemiology, Wageningen Bioveterinary Research, Lelystad, The Netherlands), E.A. Reuland (Medical Microbiology and Infection Control, Amsterdam UMC VUmc, Amsterdam, The Netherlands), W. H. F. Goessens (Erasmus University Medical Center, Rotterdam, The Netherlands), R. W. Bosboom (Department of Medical Microbiology and Immunology, Rijnstate, Arnhem, The Netherlands) and P. Vos (Check-Points, Wageningen, The Netherlands) for providing samples of which some are included in this study.
Author contributions
H. F. L. W., J. A. J. W. K. and M. A. H. conceived and supervised the study. J. P. M. C., E. P. M. D., E. K., J. A. S., J. J. V., W. J. G. M. and K. N. performed the data acquisition. J. P. M. C. and E. P. M. D. performed the data analysis. J. P. M. C. performed bioinformatic analysis. J. P. M. C., E. P. M. D. and M. A. H. performed the data interpretation and wrote the manuscript. All authors read and approved the final manuscript.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Footnotes
Abbreviations: campC, chromosome-mediated ampC; CTX, cefotaxime; ESBL, extended-spectrum β-lactamase; EUCAST, European Committee on Antimicrobial Susceptibility Testing; FDR, false discovery rate; FOX, cefoxitin; 3GC, third-generation cephalosporin; MIC, minimum inhibitory concentration; MLST, multilocus sequence typing; NCBI, National Center for Biotechnology Information; pampC, plasmid-mediated ampC; qRT-PCR, quantitative reverse-transcriptase PCR; SMRT, single-molecule real-time; ST, sequence type; VCF, variant calling file; WGS, whole-genome sequencing.
All supporting data, code and protocols have been provided within the article or through supplementary data files. Four supplementary tables and three supplementary figures are available with the online version of this article.
References
- 1.Gaynes R, Edwards JR, National Nosocomial Infections Surveillance System Overview of nosocomial infections caused by Gram-negative bacilli. Clin Infect Dis. 2005;41:848–854. doi: 10.1086/432803. [DOI] [PubMed] [Google Scholar]
- 2.Pitout JDD. Extraintestinal pathogenic Escherichia coli: a combination of virulence with antibiotic resistance. Front Microbiol. 2012;3:9. doi: 10.3389/fmicb.2012.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacoby GA. AmpC β-lactamases. Clin Microbiol Rev. 2009;22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez- L, Simonsen GS. EUCAST Detection of Resistance Mechanisms, 170711 ( https://aurosan.de/images/mediathek/servicematerial/EUCAST_detection_of_resistance_mechanisms.pdf) Växjö: European Committee on Antimicrobial Susceptibility Testing; 2017. [Google Scholar]
- 5.Tracz DM, Boyd DA, Bryden L, Hizon R, Giercke S, et al. Increase in ampC promoter strength due to mutations and deletion of the attenuator in a clinical isolate of cefoxitin-resistant Escherichia coli as determined by RT-PCR. J Antimicrob Chemother. 2005;55:768–772. doi: 10.1093/jac/dki074. [DOI] [PubMed] [Google Scholar]
- 6.Tracz DM, Boyd DA, Hizon R, Bryce E, McGeer A, et al. ampC gene expression in promoter mutants of cefoxitin-resistant Escherichia coli clinical isolates. FEMS Microbiol Lett. 2007;270:265–271. doi: 10.1111/j.1574-6968.2007.00672.x. [DOI] [PubMed] [Google Scholar]
- 7.De Smet AMGA, Kluytmans JAJW, Cooper BS, Mascini EM, Benus RFJ, et al. Decontamination of the digestive tract and oropharynx in ICU patients. N Engl J Med. 2009;360:20–31. doi: 10.1056/NEJMoa0800394. [DOI] [PubMed] [Google Scholar]
- 8.Aardema H, Bult W, Van Hateren K, Dieperink W, Touw DJ, et al. Continuous versus intermittent infusion of cefotaxime in critically ill patients: a randomized controlled trial comparing plasma concentrations. J Antimicrob Chemother. 2020;75:441–448. doi: 10.1093/jac/dkz463. [DOI] [PubMed] [Google Scholar]
- 9.Wake DB. Homoplasy: the result of natural selection, or evidence of design limitations? Am Nat. 1991;138:543–567. doi: 10.1086/285234. [DOI] [Google Scholar]
- 10.Crispell J, Balaz D, Gordon SV. HomoplasyFinder: a simple tool to identify homoplasies on a phylogeny. Microb Genom. 2019;5:000245. doi: 10.1099/mgen.0.000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kluge AG, Farris JS. Quantitative phyletics and the evolution of anurans. Syst Biol. 1969;18:1–32. doi: 10.1093/sysbio/18.1.1. [DOI] [Google Scholar]
- 12.Farhat MR, Shapiro BJ, Kieser KJ, Sultana R, Jacobson KR, et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis . Nat Genet. 2013;45:1183–1189. doi: 10.1038/ng.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mortimer TD, Weber AM, Pepperell CS. Signatures of selection at drug resistance loci in Mycobacterium tuberculosis . mSystems. 2018;3:e00108-17. doi: 10.1128/mSystems.00108-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruesen C, Chaidir L, van Laarhoven A, Dian S, Ganiem AR, et al. Large-scale genomic analysis shows association between homoplastic genetic variation in Mycobacterium tuberculosis genes and meningeal or pulmonary tuberculosis. BMC Genomics. 2018;19:122. doi: 10.1186/s12864-018-4498-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miotto P, Cabibbe AM, Feuerriegel S, Casali N, Drobniewski F, et al. Mycobacterium tuberculosis pyrazinamide resistance determinants: a multicenter study. mBio. 2014;5:e01819-14. doi: 10.1128/mBio.01819-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coolen JPM, Den Drijver EPM, Kluytmans JAJW, Verweij JJ, Lamberts BA, et al. Development of an algorithm to discriminate between plasmid- and chromosomal-mediated AmpC β-lactamase production in Escherichia coli by elaborate phenotypic and genotypic characterization. J Antimicrob Chemother. 2019;74:3481–3488. doi: 10.1093/jac/dkz362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beghain J, Bridier-Nahmias A, Le Nagard H, Denamur E, Clermont O. ClermonTyping: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb Genom. 2018;4:000192. doi: 10.1099/mgen.0.000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seemann T MLST. 2020 https://github.com/tseemann/mlst
- 20.Jolley KA, Maiden MCJ. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 22.Peter-Getzlaff S, Polsfuss S, Poledica M, Hombach M, Giger J, et al. Detection of AmpC beta-lactamase in Escherichia coli: comparison of three phenotypic confirmation assays and genetic analysis. J Clin Microbiol. 2011;49:2924–2932. doi: 10.1128/JCM.00091-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seemann T Abricate. 2020 https://github.com/tseemann/abricate
- 24.Caroff N, Espaze E, Gautreau D, Richet H, Reynaud A. Analysis of the effects of -42 and -32 ampC promoter mutations in clinical isolates of Escherichia coli hyperproducing AmpC. J Antimicrob Chemother. 2000;45:783–788. doi: 10.1093/jac/45.6.783. [DOI] [PubMed] [Google Scholar]
- 25.Siu LK, Lu P-L, Chen J-Y, Lin FM, Chang S-C. High-level expression of AmpC β-lactamase due to insertion of nucleotides between -10 and -35 promoter sequences in Escherichia coli clinical isolates: cases not responsive to extended-spectrum-cephalosporin treatment. Antimicrob Agents Chemother. 2003;47:2138–2144. doi: 10.1128/AAC.47.7.2138-2144.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haft DH, DiCuccio M, Badretdin A, Brover V, Chetvernin V, et al. RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018;46:D851–D860. doi: 10.1093/nar/gkx1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.EUCAST Breakpoint Tables for Interpretation of MICs and Zone Diameters, version 10.0 ( http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_10.0_Breakpoint_Tables.pdf) Växjö: European Committee on Antimicrobial Susceptibility Testing; 2020. [Google Scholar]
- 31.Mehta CR, Patel NR. A network algorithm for performing Fisher’s exact test in r × c contingency tables. J Am Stat Assoc. 1983;78:427–434. [Google Scholar]
- 32.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 33.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Z, Alikhan N-F, Mohamed K, Fan Y, Agama Study Group. et al. The EnteroBase user's guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020;30:138–152. doi: 10.1101/gr.251678.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lees JA, Galardini M, Bentley SD, Weiser JN, Corander J. pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics. 2018;34:4310–4312. doi: 10.1093/bioinformatics/bty539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Letunic I, Bork P. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hadfield J, Croucher NJ, Goater RJ, Abudahab K, Aanensen DM, et al. Phandango: an interactive viewer for bacterial population genomics. Bioinformatics. 2018;34:292–293. doi: 10.1093/bioinformatics/btx610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crispell J, Benton CH, Balaz D, De Maio N, Ahkmetova A, et al. Combining genomics and epidemiology to analyse bi-directional transmission of Mycobacterium bovis in a multi-host system. Elife. 2019;8:e45833. doi: 10.7554/eLife.45833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Dorp L, Gelabert P, Rieux A, De Manuel M, De-Dios T, et al. Plasmodium vivax malaria viewed through the lens of an eradicated European strain. Mol Biol Evol. 2020;37:773–785. doi: 10.1093/molbev/msz264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson EC, Elisha BG. Molecular basis of AmpC hyperproduction in clinical isolates of Escherichia coli . Antimicrob Agents Chemother. 1999;43:957–959. doi: 10.1128/AAC.43.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mulvey MR, Bryce E, Boyd DA, Ofner-Agostini M, Land AM, et al. Molecular characterization of cefoxitin-resistant Escherichia coli from Canadian hospitals. Antimicrob Agents Chemother. 2005;49:358–365. doi: 10.1128/AAC.49.1.358-365.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yona AH, Alm EJ, Gore J. Random sequences rapidly evolve into de novo promoters. Nat Commun. 2018;9:1530. doi: 10.1038/s41467-018-04026-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu S, Libchaber A. Some aspects of E. coli promoter evolution observed in a molecular evolution experiment. J Mol Evol. 2006;62:536–550. doi: 10.1007/s00239-005-0128-x. [DOI] [PubMed] [Google Scholar]
- 48.Korotkov KV, Sandkvist M, Hol WGJ. The type II secretion system: biogenesis, molecular architecture and mechanism. Nat Rev Microbiol. 2012;10:336–351. doi: 10.1038/nrmicro2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho TD, Davis BM, Ritchie JM, Waldor MK. Type 2 secretion promotes enterohemorrhagic Escherichia coli adherence and intestinal colonization. Infect Immun. 2008;76:1858–1865. doi: 10.1128/IAI.01688-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baldi DL, Higginson EE, Hocking DM, Praszkier J, Cavaliere R, et al. The type II secretion system and its ubiquitous lipoprotein substrate, SslE, are required for biofilm formation and virulence of enteropathogenic Escherichia coli . Infect Immun. 2012;80:2042–2052. doi: 10.1128/IAI.06160-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kulkarni R, Dhakal BK, Slechta ES, Kurtz Z, Mulvey MA, et al. Roles of putative type II secretion and type IV pilus systems in the virulence of uropathogenic Escherichia coli . PLoS One. 2009;4:e4752. doi: 10.1371/journal.pone.0004752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dunne KA, Chaudhuri RR, Rossiter AE, Beriotto I, Browning DF, et al. Sequencing a piece of history: complete genome sequence of the original Escherichia coli strain. Microb Genom. 2017;3:mgen000106. doi: 10.1099/mgen.0.000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vanderlinde EM, Strozen TG, Hernández SB, Cava F, Howard SP. Alterations in peptidoglycan cross-linking suppress the secretin assembly defect caused by mutation of GspA in the type II secretion system. J Bacteriol. 2017;199:e00617-16. doi: 10.1128/JB.00617-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Juan C, Torrens G, Barceló IM, Oliver A. Interplay between peptidoglycan biology and virulence in Gram-negative pathogens. Microbiol Mol Biol Rev. 2018;82:e00033-18. doi: 10.1128/MMBR.00033-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Read TD, Massey RC. Characterizing the genetic basis of bacterial phenotypes using genome-wide association studies: a new direction for bacteriology. Genome Med. 2014;6:109. doi: 10.1186/s13073-014-0109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen PE, Shapiro BJ. The advent of genome-wide association studies for bacteria. Curr Opin Microbiol. 2015;25:17–24. doi: 10.1016/j.mib.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 57.Shapiro BJ, David LA, Friedman J, Alm EJ. Looking for Darwin's footprints in the microbial world. Trends Microbiol. 2009;17:196–204. doi: 10.1016/j.tim.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 58.Alam MT, Petit RA, Crispell EK, Thornton TA, Conneely KN, et al. Dissecting vancomycin-intermediate resistance in Staphylococcus aureus using genome-wide association. Genome Biol Evol. 2014;6:1174–1185. doi: 10.1093/gbe/evu092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sahl JW, Allender CJ, Colman RE, Califf KJ, Schupp JM, et al. Genomic characterization of Burkholderia pseudomallei isolates selected for medical countermeasures testing: comparative genomics associated with differential virulence. PLoS One. 2015;10:e0121052. doi: 10.1371/journal.pone.0121052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee H, Popodi E, Tang H, Foster PL. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci USA. 2012;109:E2774–E2783. doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tenaillon O, Skurnik D, Picard B, Denamur E. The population genetics of commensal Escherichia coli . Nat Rev Microbiol. 2010;8:207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 62.Escobar-Páramo P, Sabbagh A, Darlu P, Pradillon O, Vaury C, et al. Decreasing the effects of horizontal gene transfer on bacterial phylogeny: the Escherichia coli case study. Mol Phylogenet Evol. 2004;30:243–250. doi: 10.1016/S1055-7903(03)00181-7. [DOI] [PubMed] [Google Scholar]
- 63.Price LB, Johnson JR, Aziz M, Clabots C, Johnston B, et al. The epidemic of extended-spectrum-β-lactamase-producing Escherichia coli ST131 is driven by a single highly pathogenic subclone, H30-Rx. mBio. 2013;4:e00377-13. doi: 10.1128/mBio.00377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hedge J, Wilson DJ. Bacterial phylogenetic reconstruction from whole genomes is robust to recombination but demographic inference is not. mBio. 2014;5:e02158. doi: 10.1128/mBio.02158-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.