

Engineered red light-responsive OptoAAV system allows spatiotemporally controlled gene transfer at single-cell resolution.
Abstract
Methodologies for the controlled delivery of genetic information into target cells are of utmost importance for genetic engineering in both fundamental and applied research. However, available methods for efficient gene transfer into user-selected or even single cells suffer from low throughput, the need for complicated equipment, high invasiveness, or side effects by off-target viral uptake. Here, we engineer an adeno-associated viral (AAV) vector system that transfers genetic information into native target cells upon illumination with cell-compatible red light. This OptoAAV system allows adjustable and spatially resolved gene transfer down to single-cell resolution and is compatible with different cell lines and primary cells. Moreover, the sequential application of multiple OptoAAVs enables spatially resolved transduction with different transgenes. The approach presented is likely extendable to other classes of viral vectors and is expected to foster advances in basic and applied genetic research.
INTRODUCTION
Single-cell sequencing and, more recently, single-cell multimodal omics approaches are uncovering new biology, revolutionizing our understanding of cellular heterogeneity in multicellular systems at unprecedented resolution and scale (1–3). The insight gained at the single-cell level is rapidly increasing because of single-cell analytical techniques becoming more and more broadly accessible, easier to handle, and economically affordable (1). However, the development of technologies for engineering mammalian cells at the single-cell scale is lagging behind.
An ideal system for spatially controlled gene transfer at single-cell resolution would (i) control gene delivery at the level of cell entry to avoid side effects by off-target viral uptake; (ii) exhibit low invasiveness; (iii) allow repetitive application in the same culture for delivering different genes to different cells; (iv) rely on standard laboratory equipment; (v) allow high throughput; and (vi) be compatible with native (unmodified) and optimally also nondividing, primary cells. Spatially controlled nonviral and viral gene delivery is a highly active field of research, and multiple delivery technologies using microinjection, gene guns, magnetic fields, electric fields, sonication, and light have been developed (4, 5). In terms of spatial control, light-responsive delivery systems are the gold standard as light can easily be applied with unmatched spatiotemporal precision. Previously developed light-controlled gene delivery technologies involve optical transfection (6, 7), photochemical internalization (8), and light-controlled viral transduction (9–20). Although optical transfection based on laser-induced cell membrane perforation allows high spatial control, it is accompanied by high cell toxicity and restriction to small irradiation areas (4, 7). Photochemical internalization relies on endocytosis of nucleic acid or viral vectors and the subsequent light-induced permeabilization of the endosomal membrane by reactive oxygen species (ROS) produced by photosensitizing drugs (8). This method is inherently prone to off-target effects, as endocytosis occurs also in off-target cells and endosomal nucleic acid or viral vectors are also spontaneously released. Furthermore, intracellular generation of ROS may cause cell damage (8, 21).
Viral gene delivery systems have strong advantages compared to nonviral delivery systems (4, 5) such as high efficacy, prolonged transgene expression, and, depending on the vector system, the ability to also efficiently transduce nondividing and primary cells. To date, several light-controlled viral transduction approaches relying on different mechanisms have been developed as detailed in table S1. However, their widespread adoption is limited because these systems either need cytotoxic ultraviolet (UV) light for induction (9–14), require viral uptake to both target and off-target cells before optical control (9–11, 14–20), or rely on the pre-engineering of target cells to express the photoreceptor (20).
Here, we overcome the limitations described above by the development of OptoAAV to optically guide the selective transfer of genetic information into single target cells. OptoAAV is based on adeno-associated viral (AAV) vectors that play a key role as gene delivery vehicles in fundamental research and clinically licensed gene therapies (22–24). AAV vectors are nonenveloped, single-stranded DNA vectors that transduce both dividing and nondividing cells and provide an excellent safety profile due to the absence of AAV-associated pathologies and episomal persistence (22, 23). The OptoAAV technology (i) controls transduction at the level of viral cell entry, thus minimizing the impact on off-target neighboring cells; (ii) uses noninvasive, low-intensity, and tissue-penetrating red and far-red light; (iii) can sequentially be applied to transduce different cells with different genes within one culture; (iv) relies on standard hardware for optical stimulation [e.g., light-emitting diodes (LEDs) or a conventional confocal microscope]; (v) does not require the previous genetic engineering of target cells; and (vi) is compatible with nondividing immortalized and primary cells. As OptoAAV requires only brief periods of illumination (within the seconds range) with low-intensity red light applied by standard hardware, it should be compatible with high-throughput applications.
RESULTS AND DISCUSSION
Design of the OptoAAV system
The OptoAAV technology comprises an engineered AAV-2 and a light-responsive adapter protein that mediates selective interaction of the AAV with the target cell (Fig. 1). The viral vector is genetically modified to be blind to its natural cellular receptor [heparan sulfate proteoglycan (HSPG)] and to expose the phytochrome-interacting factor 6 (PIF6; amino acids 1 to 100) from Arabidopsis thaliana on the capsid surface (OptoAAV). The adapter protein consists of phytochrome B (PhyB; amino acids 1 to 651) of A. thaliana and a designed ankyrin repeat protein (DARPin) specific for a cell surface protein of the target cell (PhyB-DARPin). Upon illumination with red (~660 nm) light, PhyB of the adapter protein interacts with PIF6 on the viral vector, thus recruiting OptoAAV to the cell surface, which results in transduction of the target cell. In contrast, illumination with far-red (~740 nm) light dissociates the interaction between PhyB and PIF6 and consequently prevents transduction (Fig. 1).
Fig. 1. Design and mode of function of the light-controlled viral transduction system.
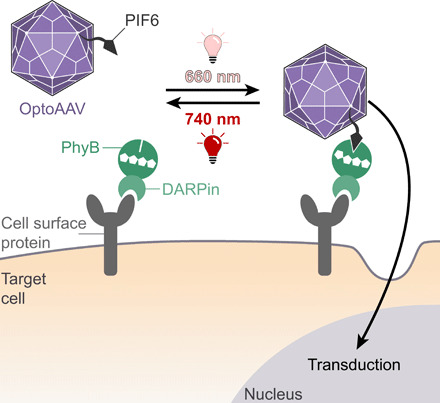
The OptoAAV system comprises (i) an engineered AAV-2 vector displaying PIF6 on its surface and containing mutations that ablate its natural tropism for HSPG (OptoAAV) and (ii) an adapter protein consisting of PhyB and a DARPin selectively binding to a specific cell surface protein. Illumination with 660-nm light induces the interaction of PhyB with PIF6 and thus the recruitment of the engineered OptoAAV to the cell surface resulting in transduction of the target cell.
Implementation of the OptoAAV system
To develop and characterize the OptoAAV system, we selected the DARPin E_01 showing high affinity [dissociation constant (KD) = 0.5 nM] to the human epidermal growth factor receptor (EGFR) that is overexpressed by many tumor cells (25, 26). This and other DARPins have previously been used for the retargeting of AAV (27–29), adenoviral (30) and lentiviral (31) vectors, and measles virus (32) either by exposing them on the viral surface or by using them as adapter mediating the interaction between the cell and the viral vector (33). We produced the fusion protein of DARPinEGFR and the photosensory domain of PhyB in Escherichia coli and purified it via its hexahistidine tag by immobilized metal affinity chromatography (IMAC; PhyB-DARPinEGFR; fig. S1). We verified PhyB photoswitching by acquiring the absorbance spectra upon 660- and 740-nm illumination (fig. S2) and validated the light-dependent interaction of PhyB-DARPinEGFR with PIF6 by size exclusion chromatography (fig. S3). Flow cytometry experiments revealed that PhyB-DARPinEGFR bound specifically via the DARPinEGFR to A-431 cells overexpressing EGFR (fig. S4). To demonstrate that PhyB-DARPinEGFR can recruit PIF6-tagged molecules to cells in a light-dependent manner, we analyzed light-induced recruitment of an mVenus-PIF6 fusion protein to A-431 cells by flow cytometry (Fig. 2A) and confocal microscopy (fig. S5). In addition, the microscopy experiment revealed that mVenus-PIF6 was massively internalized after 20-min incubation at 37°C, whereas it remained mainly membrane-localized when cells were incubated on ice. This observation is in line with internalization reported for EGFR (34).
Fig. 2. Characterization of OptoAAV system components.
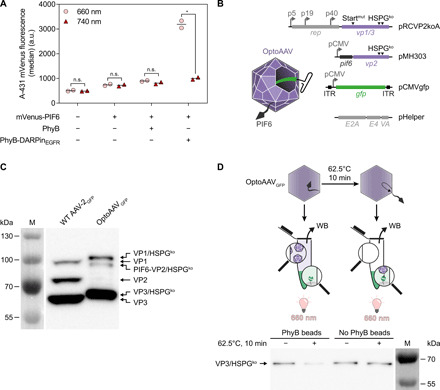
(A) Light-controlled recruitment of mVenus-PIF6 to cells. A-431 cells were incubated with 100 nM PhyB or PhyB-DARPinEGFR and/or 200 nM mVenus-PIF6 for 20 min under 740- or pulsed 660-nm light. Afterward, cellular mVenus fluorescence was analyzed by flow cytometry. n > 2200 cells per sample. Mean is indicated. n.s. (not significant), P ≥ 0.05; *P < 0.05. a.u., arbitrary units. (B) Construction of OptoAAVs. OptoAAVs were produced in HEK-293T cells by cotransfection of four plasmids. pRCVP2koA encodes nonstructural proteins required for replication (rep) and the viral capsid proteins VP1 and VP3. Expression of vp2 was prevented by a silent mutation within the vp2 start codon T138 (Startmut), and the natural HSPG tropism was ablated by two mutations (R585A and R588A; HSPGko). pMH303 encodes PIF6 fused to VP2 (HSPGko) under the CMV promoter. pCMVgfp contains a self-complementary AAV genome encoding GFP. pHelper provides adenoviral genes for AAV production. ITR, inverted terminal repeat. (C) Western blot (B1 antibody) against capsid proteins of wild-type (WT) AAV-2GFP and OptoAAVGFP. Representative data of n = 3 batches. M, protein size marker. (D) Binding of OptoAAVGFP to PhyB. OptoAAVsGFP (optionally preincubated at 62.5°C for 10 min) were incubated with or without PhyB-functionalized beads under 660-nm light for 1 hour. After pelleting beads, supernatants were analyzed for unbound OptoAAVGFP by Western blot (WB; B1 antibody) against VP3. Representative data of n = 4 experiments.
To display PIF6 on the capsid of AAV-2, we genetically fused the active phytochrome binding domain of PIF6 to the N terminus of the viral capsid protein VP2 (20). We placed the coding sequence of the PIF6-VP2 fusion protein under control of a cytomegalovirus (CMV) promoter (plasmid pMH303) and prevented expression of native VP2 by deletion of its start codon in the plasmid pRCVP2koA (Fig. 2B) (27). To prevent transduction of cells in a light-independent manner, these plasmids additionally contained the mutations R585A and R588A in the cap genes, ablating the natural tropism of AAV-2 for its natural receptor HSPG (Fig. 2B) (35). As model transgene, we selected the green fluorescent protein (GFP) or the red fluorescent protein mScarlet under control of the constitutive CMV promoter encoded on a self-complementary genome. Self-complementary AAV vectors are characterized by a higher transduction efficiency at the expense of only half of the loading capacity (36). We produced our OptoAAVs using the helper-free packaging system in human embryonic kidney (HEK)–293T cells and purified assembled AAV capsids by iodixanol gradient ultracentrifugation (37). We confirmed the incorporation of PIF6-VP2 in OptoAAVGFP and the ablation of native VP2 by Western blotting against the viral capsid proteins VP1, VP2, and VP3 (Fig. 2C). From one OptoAAVGFP production (ten 15-cm dishes), we determined an average genomic titer of (1.6 ± 0.6) × 1011 vector genomes (vg). We next analyzed the light-controlled interaction of OptoAAVGFP with PhyB. To this aim, we incubated OptoAAVGFP with PhyB-functionalized agarose beads under 660-nm light and subsequently analyzed the supernatant for unbound viral particles by Western blotting against the viral capsid proteins (Fig. 2D). OptoAAVGFP did not bind to the PhyB beads, suggesting that PIF6 was not accessible or exposed on the capsid surface. As previous studies showed that the N terminus of VP2 becomes exposed upon limited heat shock (38, 39), we incubated our OptoAAVsGFP for 10 min at 62.5°C and repeated the binding experiment to the PhyB beads. Upon heat treatment, we observed binding of OptoAAVGFP to PhyB (Fig. 2D).
Red light-controlled transduction
After the characterization of the individual components, we tested the ability of the system to transduce A-431 cells in a light-dependent manner. For this, we used OptoAAVsGFP that had been previously incubated for different periods of time at different temperatures (Fig. 3A and figs. S6 and S7A). We observed a 5.6- to 81-fold increase in the percentage of transduced cells at 660- compared to 740-nm illumination (low light intensity, both 20 μmol m−2 s−1, equals 0.36 and 0.32 mW cm−2, respectively). In agreement with the above-described in vitro binding studies (Fig. 2D), heat treatment of OptoAAVGFP resulted in a strong increase of transduced cells (from ~2 to ~40%). On the basis of these data, we exposed PIF6 on OptoAAVs by a 10-min incubation step at 62.5°C in all further experiments. Transduction experiments with untreated and heated unmodified [wild-type (WT)] AAV-2 vectors revealed that this heat treatment reduced the infectious titer 5.9-fold (fig. S7B). For OptoAAV, a likely similar loss of infectivity is outcompeted by the gain in light-controlled infectivity caused by the heat-induced exposure of PIF6. Control experiments verified that the adapter protein and OptoAAVGFP are both required for transduction and that the transduction efficiency of unmodified AAV-2 vectors in the presence of the adapter protein is not affected by 660- and 740-nm illumination (Fig. 3B). The percentage of transduced A-431 cells reached up to 65% with increasing OptoAAVGFP titer, while the 660-nm light-induced 37-fold change in transduced cells was not affected by the viral titer. We further demonstrated that the percentage of transduced target cells can precisely be fine-tuned by adjusting the illumination intensity and/or period (Fig. 3C). We observed light-controlled viral transduction over a broad range of adapter protein concentration (0.5 to 500 nM) with the highest percentage of transduced cells at 50 nM (fig. S8A). The system was functional in phosphate-buffered saline (PBS) supplemented with bovine serum albumin (BSA) or fetal calf serum (FCS) and in different commonly used cell culture media (fig. S8B). Furthermore, the transduction efficiency increased with the incubation time of the cells with OptoAAVGFP and the adapter protein (fig. S8C). Although cellular binding of the used DARPinEGFR does not trigger EGFR signaling (26), multiple AAV-bound PhyB-DARPinEGFR adapter proteins may cross-link and activate EGFR under 660-nm illumination. However, analysis of EGFR and extracellular signal–regulated kinases 1/2 (Erk1/2) phosphorylation by Western blotting did not show any receptor activation by the OptoAAVEGFR system (fig. S9). Using the PhyB-DARPinEGFR adapter protein, we could further transduce the EGFR-expressing tumor cell lines A549, HeLa, and MDA-MB-231 in a light-dependent manner, but not CHO-K1 cells lacking EGFR (fig. S10) (40).
Fig. 3. Characterization of the OptoAAV system.
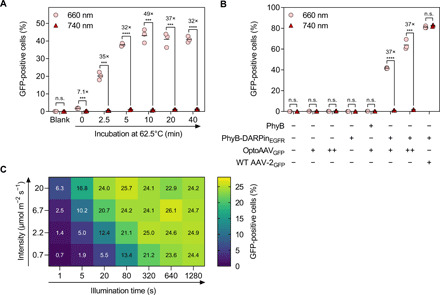
(A) Heating of OptoAAV. A-431 cells were incubated with heat-treated OptoAAVsGFP [multiplicity of infection (MOI), genomic particles per cell: 3.4 × 104] and 50 nM PhyB-DARPinEGFR in PBS supplemented with 1% BSA for 2 hours under 740- or pulsed 660-nm illumination. Afterward, cells were washed and incubated in medium under continued illumination for 46 hours before analysis of transduced (GFP-positive) cells by flow cytometry. n > 5600 cells per sample. (B) Influence of OptoAAV components. A-431 cells were incubated for 2 hours in PBS supplemented with 1% BSA and 50 nM PhyB or PhyB-DARPinEGFR, OptoAAVGFP [MOI(+): 4.5 × 104; MOI(++): 1.4 × 105], or WT AAV-2GFP (MOI: 9.3 × 103). The experiment was performed as in (A) with OptoAAVGFP preincubated at 62.5°C for 10 min. n > 300 cells per sample. (C) Dose dependency. A-431 cells incubated with 50 nM PhyB-DARPinEGFR and OptoAAVGFP (MOI: 2.8 × 104) in PBS supplemented with 10% FCS were illuminated as indicated with 630-nm light and incubated afterward in the dark. Two hours after illumination started, cells were washed and incubated in medium for 46 hours under 740-nm light before flow cytometry analysis. Samples illuminated with 780-nm light within the first 2 hours showed 0.21% GFP-positive cells. Values obtained from biological duplicates. n > 3800 cells per sample. Mean is indicated. n.s., P ≥ 0.05; ***P < 0.001; ****P < 0.0001.
While EGFR is known to be internalized by endocytosis (34), we next asked whether active internalization of the cellular target receptor is a prerequisite for the functionality of the OptoAAV system. To this aim, we displayed SpyCatcher on the surface of HEK-293T cells by fusing it to the secretion signal and transmembrane domain of major histocompatibility complex I (MHC I). Afterward, we covalently coupled purified PhyB-mCherry-SpyTag to SpyCatcher (41). The MHC I transmembrane domain has previously been used to anchor proteins stably on the cell membrane (42) and does, to our knowledge, not contain an endocytosis function. We observed only minimal internalization of PhyB-mCherry-SpyTag on the engineered HEK-293T by confocal microscopy in comparison to the massive internalization of EGFR on A-431 cells (fig. S11A). As we were able to transduce the engineered PhyB-displaying HEK-293T cells with OptoAAVGFP in a light-dependent manner with a similar efficiency as for the A-431 cells (fig. S11B), we suggest that active internalization of the cellular target receptor is not required for the OptoAAVGFP system. We hypothesize that viral uptake is induced upon cell attachment by binding of the OptoAAV to co-receptors such as the AAV receptor (43).
To demonstrate that the OptoAAV system can be expanded to other cell lines with minimal or absent EGFR expression, we modularly exchanged the DARPinEGFR with DARPins specific for EpCAM (DARPin Ec1) (44), Her2/ErbB2 (DARPin 9_29) (25), and CD4 (DARPin D55.2) (45) (figs. S1 and S2). Using these adapter proteins, we were able to selectively transduce further cell lines (Fig. 4A) and primary human CD4-positive T lymphocytes (Fig. 4B) in a light-dependent manner.
Fig. 4. Modular adaptation of the OptoAAV system to different cell types.
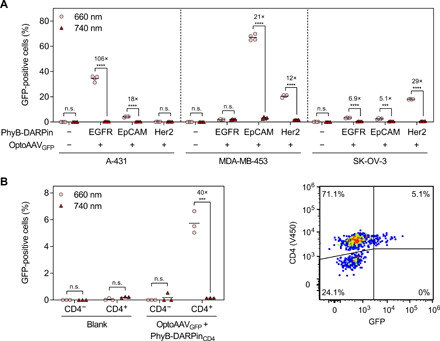
(A) Transduction of different cell lines with PhyB-DARPin adapter proteins specific for the cell surface receptors EGFR, EpCAM, and Her2. The cell lines were incubated as indicated with 50 nM PhyB-DARPin and OptoAAVGFP (MOI: 4.1 × 104) in PBS supplemented with 10% FCS for 2 hours under 740- or pulsed 660-nm illumination. Afterward, cells were washed and incubated in medium under continued illumination for 46 hours before analysis of transduced (GFP-positive) cells by flow cytometry. n > 1200 cells per sample. (B) Light-controlled transduction of primary human CD4+ T cells. T cells were incubated with 50 nM PhyB-DARPinCD4 and OptoAAVGFP (MOI: 4.7 × 104) in PBS supplemented with 10% FCS for 6 hours under 740- or pulsed 660-nm illumination. Afterward, cells were incubated in medium under continued illumination for 42 hours. Following staining of the cells with a V450-labeled anti-CD4 antibody, the number of transduced (GFP-positive) CD4-positive and CD4-negative cells was analyzed by flow cytometry. n > 900 cells per sample. Mean is indicated. n.s., P ≥ 0.05; ***P < 0.001; ****P < 0.0001.
Spatiotemporal control of transduction
Next, we aimed at transducing cells in a spatially resolved manner. To this end, we incubated A-431 cells for 10 min with the adapter protein under 740-nm illumination (Fig. 5A). After a washing step to remove unbound adapter protein, we added OptoAAVGFP or OptoAAVmScarlet and illuminated the cells spatially resolved with 660-nm light for 9 s using a photomask. Following incubation for 2 hours in the dark, we washed the cells, incubated them for another 46 hours under 740-nm light, and visualized the transduced cells by microscopy (Fig. 5A and fig. S12A). We observed spatially resolved transduction (i.e., expression of the fluorescent proteins) with a spatial resolution in the 100-μm range, while nontransduced cells were distributed over the whole wells as seen in the 4′,6-diamidino-2-phenylindole (DAPI) images. We next tested whether OptoAAVs encoding different transgenes can be used to sequentially transduce cells in a spatially resolved manner. To this aim, we performed the just described experiment with OptoAAVGFP and photomask 1. Following 2-hour incubation in the dark and a subsequent washing step, we repeated the same procedure with OptoAAVmScarlet and photomask 2 (Fig. 5B and fig. S12, B and C). This approach enabled the spatially resolved transduction of cells with two different transgenes and can likely be expanded to additional transgenes. In areas illuminated with both photomasks, we observed cells expressing both transgenes, indicating that the OptoAAV system can deliver several transgenes into one target cell (fig. S12B).
Fig. 5. Spatiotemporally controlled transduction.
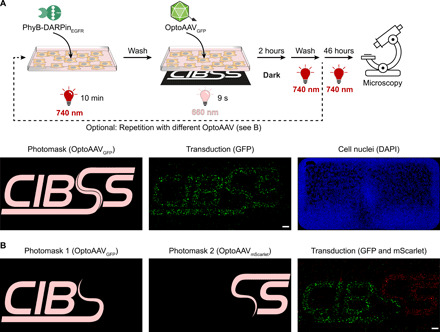
(A) Transduction with one transgene. A-431 cells were incubated with 50 nM PhyB-DARPinEGFR in PBS supplemented with 10% FCS under 740-nm light for 10 min. After washing, OptoAAVsGFP (MOI: 1.7 × 104) in PBS supplemented with 10% FCS were added, and cells were illuminated for 9 s from the bottom with 660-nm light (15 μmol m−2 s−1) using the photomask with simultaneous global 740-nm illumination (3 μmol m−2 s−1) from the top. Following 2-hour incubation in the dark, cells were washed and incubated for 46 hours in medium under 740-nm light. Last, cells were fixed, DAPI-stained, and imaged by confocal microscopy. Representative images from n = 4 experiments. (B) Transduction with two transgenes. A-431 cells were spatially resolved transduced as in (A) using photomask 1 and OptoAAVGFP (MOI: 1.5 × 104). Following incubation in the dark, the procedure was repeated using photomask 2 and OptoAAVmScarlet (MOI: 1.7 × 104). After the second incubation step in the dark, cells were washed and incubated for 44 hours in medium under 740-nm light. Last, cells were fixed, DAPI-stained, and imaged. The DAPI image is shown in fig. S12C. Representative images from n = 2 experiments. Scale bars, 1 mm.
We next extended the spatial resolution to the single-cell level by illumination of one selected cell using a conventional confocal microscope equipped with a 633-nm laser. To track the illuminated cell over the course of the experiment (48 hours), we seeded A-431 cells, of which 1% were fluorescently labeled (eFluor670), on a coverslip with a labeled grid (Fig. 6A). Following incubation with the adapter protein and addition of OptoAAVGFP under 740-nm light, we illuminated a single isolated eFluor670-labeled cell with low-intensity 633-nm laser light (~120 ms; Fig. 6A). After 48 hours, we analyzed the transduction by microscopy. In 4 of 17 experiments (24%), the single illuminated eFluor670-labeled cell showed GFP expression (for a representative successful experiment, see fig. S13A). As the success rate was lower than transduction of eFluor670-labeled cells at 660 nm in comparable flow cytometry experiments (38%; fig. S13B), we hypothesized that absorption of the 633-nm laser by the eFluor670 dye might have reduced photoactivation of PhyB. Therefore, we repeated the experiment with carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled cells, which have no absorbance at 633 nm and OptoAAVmScarlet. In 9 of 15 experiments (60%), we were now able to transduce the single illuminated CFSE-labeled cell (Fig. 6B and fig. S14A), which is in agreement with the transduction rate at 660 nm of comparable flow cytometry experiments (58%; fig. S14C). From the nonilluminated cells, 1.0% showed mScarlet fluorescence, although mainly with a much lower intensity than the illuminated cell (fig. S14A). This off-target transduction rate is similar with the one under constant 740-nm illumination in comparable flow cytometry experiments (0.6%; fig. S14B).
Fig. 6. Spatially resolved transduction of single cells using a conventional confocal microscope.
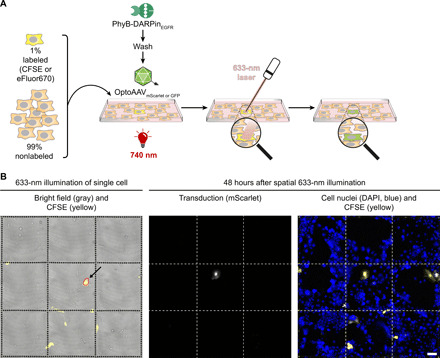
(A) Experimental workflow. A-431 cells containing 1% fluorescently labeled (CFSE or eFluor670) cells were seeded on a gridded coverslip. Forty-eight hours after seeding, cells were incubated with 50 nM PhyB-DARPinEGFR in PBS supplemented with 10% FCS under 740-nm light for 10 min. After washing, OptoAAVsGFP or OptoAAVsmScarlet in PBS supplemented with 10% FCS were added and a bright field and CFSE or eFluor670 image was acquired under 740-nm illumination (200 μmol m−2 s−1). After switching the 740-nm light off, a single CFSE or eFluor670-positive cell was illuminated with the 633-nm laser of the confocal microscope. Following 2-hour incubation in the dark, cells were washed and incubated for 46 hours in medium under 740-nm light. Last, cells were fixed, DAPI-stained, and imaged by confocal microscopy. (B) Light-controlled transduction of a single CFSE-stained A-431 cell. The experiment was performed as in (A) with OptoAAVmScarlet (MOI: 4.9 × 104). The area illuminated with the 633-nm laser is encircled in red. Representative images from n = 9 successful experiments (out of 15). All experiments are shown in fig. S14A. Scale bar, 100 μm.
Outlook
In summary, we demonstrated that the OptoAAV technology enables the spatiotemporally resolved and light dose-dependent selective transduction of native cell lines and primary cells using low-intensity red light. Because of its modular design, the system can be customized to target cell types of choice by switching to an adapter protein with the desired specificity. OptoAAV intrinsically features a two-factor control for specificity. Successful transduction requires both recognition of the target cell type by the adapter protein and the light stimulus, leading to vector binding and internalization. Such AND-type control was shown in previous studies to substantially increase target specificity (46). Using a conventional confocal microscope, OptoAAV allowed the selective transduction of single cells by local illumination. These experiments could allow perturbing biological processes at the single-cell level to reconstruct and understand the implications of cell heterogeneity. In addition, the sequential application of different OptoAAVs enabled the selective transduction of cells with different transgenes within the same culture. This feature may be of particular interest when applying OptoAAV for the delivery of genes encoding differentiation factors in tissue engineering for regenerative medicine. Although we only tested the in vitro functionality of OptoAAV so far, it may also be applied for the site-specific in vivo gene delivery in fundamental research or (cancer) gene therapy. Another promising area of application would be in neurosciences. Light-responsive ion channels such as channelrhodopsin (ChR) are routinely used for the light-responsive induction of action potentials in neurons, both in tissue culture and in vivo (47). In living animals, ChRs are often introduced via AAV-based vectors, and the injection of viral vectors can lead to substantial spread of opsin expression, e.g., caused by the backflow of the vector along the injection tract. While this might sometimes be a desired feature, it is often a detrimental complication, hampering conclusions about the role of an activated or deactivated brain area (48). Using OptoAAV, gene delivery could be targeted with high spatial resolution and selectivity to the cells of interest by (i) choosing an appropriate adapter protein and (ii) by local illumination with red light. For transduction, the same waveguide-based illumination hardware as used for activation of ChR could be used. In an in vivo setting, the spatial resolution and tissue penetration may be further increased by two-photon activation of the OptoAAV system as recently shown for a bacterial phytochrome (49). Moreover, OptoAAV may be used to obtain further insights into viral cell entry, e.g., by determining required binding times (50). Because of its modular design, we suggest that the OptoAAV approach could serve as blueprint for rendering further classes of viral vectors light-responsive.
MATERIALS AND METHODS
Cloning of plasmids
The nucleic acid sequences of all plasmids generated in this study are depicted in table S2. The plasmids were assembled by Gibson (51) or AQUA (52) cloning, and the used templates for polymerase chain reaction (PCR) are mentioned in table S2. The codon-optimized (for E. coli) sequences for the DARPins Ec1 (EpCAM) (44), 9_29 (Her2/ErbB2) (25), and D55.2 (CD4) (45) and the codon-optimized (for human and E. coli) sequence for SpyCatcher003 (41) were ordered as gBlocks from Integrated DNA Technologies (Coralville, IA). All other sequences or mutations were introduced with oligonucleotides and PCR.
Illumination
If not indicated otherwise, samples were illuminated with microcontroller-regulated illumination panels containing LEDs with ~660-nm (LED660N-03, Roithner Lasertechnik, Vienna, Austria; peak wavelength: 660 nm; LH W5AM, Osram Opto Semiconductors, Regensburg, Germany; peak wavelength: 660 nm; LST1-01F06-PRD1-00, Opulent Americas, Raleigh, NC; peak wavelength: 655 nm) or 740-nm (LED740-01AU, SMB1N-740D, both Roithner Lasertechnik; LZ4-00R308, LED Engin, San Jose, CA) peak wavelengths. For spatially resolved illumination, custom laser photoplot films (4000 DPI, JD Photo Data, Herts, UK) were used as photomask. The experiment depicted in Fig. 3C was performed with optoPlate-96 (53) equipped with 630-nm (150141RB73100, Würth Elektronik, Niedernhall, Germany) and 780-nm (SMT780-27, Marubeni, Tokyo, Japan) LEDs, and illumination protocols were defined with optoConfig-96 (54). For illumination at the confocal microscope, a pE-4000 LED light source (CoolLED, Andover, UK) with 740 nm was used. After illumination of the samples with the indicated wavelengths, they were only handled under dim green safe light until the end of the experiment to prevent photoswitching of PhyB. Light intensities were measured with an AvaSpec-ULS2048 fiber-optic spectrometer (Avantes BV, Apeldoorn, The Netherlands). Where indicated, pulsed 660-nm illumination (5 min ON, 55 min OFF) was used to prevent continuous cycling of PhyB between the binding and nonbinding state as occurring under continuous 660-nm light (50). If not indicated otherwise, samples were illuminated at an intensity of 20 μmol m−2 s−1.
Protein production and purification
For production of the different PhyB-DARPin proteins and PhyB-mCherry-SpyTag, the corresponding plasmid (see table S2) encoding the fusion protein and the biosynthesis enzymes for the chromophore phycocyanobilin (PCB) was transformed into E. coli BL21 Star (DE3) (Thermo Fisher Scientific, Waltham, MA, catalog no. C601003), and transformed bacteria were selected in LB medium supplemented with streptomycin (100 μg ml−1) (55). Bacteria were grown at 30°C and 150 rpm to an OD600 (optical density at 600 nm) of 0.8, and expression was induced by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside. After incubation for 20 hours at 18°C and 150 rpm in the dark, bacteria were harvested by centrifugation at 6500g, resuspended in lysis buffer [50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP) (pH 8.0)], shock-frozen in liquid nitrogen, and stored at −80°C. For IMAC-based protein purification, resuspended bacteria were thawed and disrupted using a French Press (APV 2000, APV Manufacturing, Bydgoszcz, Poland) at 1000 bar. Following clarification of the lysate by centrifugation at 30,000g for 1 hour, the supernatant was loaded onto an equilibrated Ni-NTA column (Qiagen, Hilden, Germany, catalog no. 30430). Next, the column was washed with 15 column volumes (CVs) of wash buffer [50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, and 0.5 mM TCEP (pH 8.0)], and the purified protein was eluted in 4 CVs of elution buffer [50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, and 0.5 mM TCEP (pH 8.0)]. Afterward, the buffer was exchanged to PBS [2.7 mM KCl, 1.5 mM KH2PO4, 8.1 mM Na2HPO4, and 137 mM NaCl (pH 7.4)] supplemented with 0.5 mM TCEP by ultrafiltration (polyethersulfone membrane, 10-kDa molecular weight cut-off), and the protein (~10 mg ml−1) was shock-frozen in aliquots in liquid nitrogen and stored at −80°C. Before the cell experiments, proteins were thawed, diluted 1:10 in PBS supplemented with 0.5 mM TCEP, and used on the same day. The protein mVenus-PIF6 was produced from plasmid pMH302 in E. coli BL21 Star (DE3)pLysS (Thermo Fisher Scientific, catalog no. C602003) at 30°C for 5 hours and purified by IMAC using buffers without TCEP followed by a buffer exchange to PBS as described above. PhyB was produced from plasmid pMH1105 in E. coli and purified by IMAC as described previously (56). FRB-mCherry-DARPin was produced from plasmid pMH212 in E. coli and purified by IMAC as described previously (28). Afterward, the buffer of PhyB and FRB-mCherry-DARPin was exchanged to PBS supplemented with 0.5 mM TCEP as described above.
Protein characterization
The identity and purity of the proteins was analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) followed by Zn2+ staining of the chromophore PCB [incubation in 1 mM zinc acetate for 10 min followed by imaging of fluorescence under UV light (312 nm) using an agarose gel documentation system (Intas, Göttingen, Germany)] and by Coomassie staining of proteins. As protein size standard, a PageRuler prestained protein ladder (Thermo Fisher Scientific, catalog no. 26616) or a Pierce prestained protein molecular weight marker (Thermo Fisher Scientific, catalog no. 26612) was used. Protein concentration was determined by Bradford assay (Bio-Rad, Hercules, CA, catalog no. 500-0006) using BSA (Sigma-Aldrich, St. Louis, MO, catalog no. 05479) as standard. The absorbance spectra were acquired with an Infinite M200 Pro microplate reader (Tecan, Männedorf, Switzerland). The interaction of PhyB-DARPinEGFR and mVenus-PIF6 was analyzed by size exclusion chromatography on a light-protected Superdex 200 10/300 GL column (GE Healthcare, Freiburg, Germany, catalog no. 17-5175-01) connected to the Äkta Explorer Fast Protein Liquid Chromatography System (GE Healthcare) using PBS as running buffer at a flow rate of 0.5 ml min−1. The column was calibrated with a gel filtration standard (Bio-Rad, catalog no. 151-1901).
Cell culture
A-431 [human epidermoid carcinoma; German Collection of Microorganisms and Cell Cultures (DSMZ), Braunschweig, Germany, catalog no. ACC 91; cell line identity verified by short tandem repeat (STR) profiling], A549 (human lung carcinoma; CLS, Eppelheim, Germany, catalog no. 300114), HeLa [human cervix adenocarcinoma; American Type Culture Collection (ATCC), Manassas, VA, catalog no. CCL-2], MDA-MB-231 (human breast adenocarcinoma, obtained from Signalling Factory Core Facility, University of Freiburg, Germany; cell line identity verified by STR profiling), and HEK-293T (HEK, DSMZ, catalog no. ACC 635) cells were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) complete [DMEM (PAN Biotech, Aidenbach, Germany, catalog no. P04-03550) supplemented with 10% (v/v) FCS (PAN Biotech, catalog no. P30-3602), penicillin (100 U ml−1), and streptomycin (100 μg ml−1)]. SK-OV-3 cells (human ovary adenocarcinoma; ATCC, catalog no. HTB-77) were maintained in McCoy’s 5A medium (Sigma-Aldrich, catalog no. M8403) supplemented with 10% (v/v) FCS, 2 mM l-glutamine (Thermo Fisher Scientific, catalog no. 25030-024), penicillin (100 U ml−1), and streptomycin (100 μg ml−1). MDA-MB-453 cells (human breast metastatic carcinoma; ATCC, catalog no. HTB-131) were cultivated in RPMI 1640 medium (Thermo Fisher Scientific, catalog no. 61870-010) supplemented with 10% (v/v) FCS, penicillin (100 U ml−1), and streptomycin (100 μg ml−1). CHO-K1 (Chinese hamster ovary; DSMZ, catalog no. ACC 110) cells were maintained in HTS medium (Cell Culture Technologies, Gravesano, Switzerland, catalog no. CHTS) supplemented with 10% (v/v) FCS, 2 mM l-glutamine, penicillin (100 U ml−1), and streptomycin (100 μg ml−1). All cells were cultivated at 37°C in a humidified atmosphere containing 5% CO2 and passaged upon reaching a confluence of ~80%.
Generation of stable HEK-293T–SpyCatcher cell line
To generate HEK-293T cells stably expressing SpyCatcher-MHCI-moxBFP, we used lentiviral transduction as described previously (50). Briefly, HEK-293T cells were transfected with the lentiviral packaging plasmid pCMV dR8.74, the envelope plasmid pMD2 vsvG (both gifts from D. Trono), and the transfer plasmid pOSY115 by polyethylenimine (PEI)–mediated transfection. Forty-eight hours after transfection, lentiviral particle-containing HEK-293T supernatant was harvested, filtered using a 0.45-μm filter, and concentrated by centrifugation through a 20% (w/v) sucrose cushion in PBS supplemented with 1 mM EDTA at 10,000g at 4°C for 4 hours. After centrifugation, the supernatant was discarded and the viral particles were resuspended in medium using 1% of the original cell supernatant volume. HEK-293T cells were transduced with increasing amounts of concentrated lentiviral particles to obtain ~95% transduced cells. Transduction [blue fluorescent protein (BFP)] and surface expression (anti-hemagglutinin antibody) were analyzed after 48 hours by flow cytometry.
Analysis of protein binding to cells
To analyze the binding of PhyB and PhyB-DARPinEGFR to A-431 cells, A-431 cells were detached with trypsin/EDTA solution (PAN Biotech, catalog no. P10-023500) and washed once with DMEM complete. Afterward, 8 × 105 cells ml−1 were incubated with 1 μM PhyB or PhyB-DARPinEGFR in DMEM complete for 2 hours at 37°C in the dark. Next, cells were washed with PBS, resuspended in PBS supplemented with 2% (v/v) FCS, and analyzed for PhyB fluorescence with a Gallios flow cytometer (Beckman Coulter, Brea, CA) using a 638-nm laser for excitation and a 660/20-nm bandpass filter for emission.
To analyze the recruitment of mVenus-PIF6 to the cell surface by flow cytometry, 5 × 104 A-431 cells were seeded per well of a 24-well plate. After 24 hours, cells were incubated with the indicated concentrations of proteins in 500 μl of PBS supplemented with 1% (w/v) BSA under the indicated illumination condition. After washing with PBS, cells were trypsinized and mVenus fluorescence was analyzed with a Gallios flow cytometer using a 488-nm laser for excitation and a 525/40-nm bandpass filter for emission. To analyze the recruitment of mVenus-PIF6 to the cell surface and EGFR/PhyB-mCherry-SpyTag internalization by microscopy, 7 × 104 A-431 cells or 9 × 104 HEK-293T–SpyCatcher cells were seeded per well of a 24-well plate, each containing a coverslip [previously coated with collagen I (50 μg ml−1; Thermo Fisher Scientific, catalog no. A1048301) in 20 mM acetic acid for 3 hours]. After 24 hours, cells were incubated as indicated with the different proteins and, after two washing steps with PBS, fixed with 4% (w/v) paraformaldehyde (PFA) in PBS for 20 min. Next, nuclei were stained with TO-PRO-3 (Thermo Fisher Scientific, catalog no. T3605; 0.5 μM in PBS for 15 min), and cells were imaged on a Zeiss LSM 880 laser scanning confocal microscope (Zeiss, Oberkochen, Germany) using a 63× Plan Apochromat objective [numerical aperture (NA): 1.4]. mVenus, mCherry, and TO-PRO-3 were excited with a 514-, 561-, and 633-nm laser and detected between 517 to 543 nm, 570 to 597 nm, and 643 to 720 nm, respectively (pinhole was adjusted to image a 1.0-μm section for each channel).
AAV vector production and purification
AAV vectors were produced using the adenovirus helper-free packaging system (37). For production of OptoAAVs, HEK-293T cells were transfected with the plasmids pMH303, pRCVP2koA (27) (gift from H. Büning), pHelper (Cell Biolabs, San Diego, CA, catalog no. VPK-402) and the self-complementary vector plasmid pCMVgfp or pCMVmScarlet (both plasmids were gifts from D. Grimm) in an equimolar ratio. For production of WT AAV-2 vectors encoding GFP, cells were transfected with the plasmids pAAV-RC2 (Cell Biolabs, catalog no. VPK-402), pHelper and the self-complementary vector plasmid pCMVgfp in an equimolar ratio. Briefly, 8 × 106 HEK-293T cells were seeded per 15-cm cell culture dish, and after 24 hours, cells were transfected with 60 μg of total plasmid DNA mixed with 200 μg of PEI (MW, 25,000) in 3 ml Opti-MEM (Thermo Fisher Scientific, catalog no. 22600-134). After 72 hours, cells were scraped from the cell culture plates, pelleted by centrifugation (400g for 15 min), washed once with PBS, and resuspended in virus lysis solution [50 mM tris-HCl and 150 mM NaCl (pH 8.5); 500 μl per 15-cm dish]. After lysing the cells by five freeze-thaw cycles, the lysate was incubated with benzonase (50 U ml−1; Merck Millipore, Darmstadt, Germany, catalog no. 70664-3) at 37°C for 1 hour before cell debris were removed by centrifugation at 4000g for 15 min. Last, AAVs were purified from the supernatant by discontinuous iodixanol density gradient centrifugation (57). Centrifugation was performed for 2 hours at 171,000g and 4°C using the Optima L-90 K ultracentrifuge equipped with a Type 70.1 Ti fixed-angle rotor (Beckman Coulter). OptoAAVs from 10 plates were purified per Quick-Seal polypropylene tube (Beckman Coulter, catalog no. 342413). After centrifugation, the 40% iodixanol fraction containing OptoAAVs or WT AAVs was aliquoted, shock-frozen in liquid nitrogen, and stored at −80°C.
AAV characterization and quantification
Purified AAV vectors were analyzed by Western blotting against the viral capsid proteins. To this aim, iodixanol-purified AAVs were mixed with SDS loading buffer [final concentration: 62 mM tris-HCl (pH 6.8), 10% (v/v) glycerol, 2% (w/v) SDS, 2.5% (v/v) 2-mercaptoethanol, and 0.01% (w/v) bromophenol blue sodium salt] and incubated at 95°C for 5 min. After separating the samples by SDS-PAGE, the proteins were transferred onto a polyvinylidene difluoride membrane, and the membrane was blocked with blocking buffer [PBS containing 3% (w/v) milk powder] at 4°C overnight. Following incubation of the membrane for 2 hours with a hybridoma cell supernatant containing AAV-2–specific B1 antibody (58) (gift from D. Grimm) diluted 1:10 in blocking buffer, the membrane was washed with PBS-T [PBS supplemented with 0.05% (v/v) Tween 20] and incubated for 1 hour with secondary anti-mouse horseradish peroxidase (HRP)–conjugated antibody (GE Healthcare, catalog no. NA931) diluted 1:2000 in blocking buffer. After washing the membrane with PBS-T, ECL Prime Western blotting detection reagent (GE Healthcare, catalog no. RPN2232) was added and chemiluminescence was imaged with the ImageQuant LAS 4000 Mini System (GE Healthcare). In agreement with previous studies, the mutations R585A and R588A resulted in reduced mobility of the viral capsid proteins in SDS-PAGE (27).
The genomic titer of the purified AAVs was determined by quantitative real-time PCR (qPCR). Dilution series of the AAVs and of the vector plasmid pCMVgfp as standard were prepared, and a 134–base pair fragment was amplified from the CMV promoter using oligonucleotides 5′-TGCCCAGTACATGACCTTATGG-3′ and 5′-GAAATCCCCGTGAGTCAAACC-3′ and the ABsolute qPCR SYBR Green ROX mix (Thermo Fisher Scientific, catalog no. AB1163A). The qPCR was performed on a CFX384 thermocycler (Bio-Rad) using the following temperature protocol: 10 min at 95°C followed by 40 cycles of 15 s at 95°C, 5 s at 67°C, and 10 s at 72°C.
Binding of AAVs to PhyB beads
To analyze the accessibility and functionality of PIF6 on OptoAAVs, Ni-NTA agarose beads (Qiagen, catalog no. 30430) were washed with beads buffer [PBS supplemented with 0.01% (w/v) BSA] and incubated for 30 min with PhyB (3 mg ml−1; 40 μl of Ni-NTA agarose beads and 740 μg of PhyB per experiment). After washing with beads buffer, the PhyB beads were incubated with untreated or heated (62.5°C, 10 min) OptoAAVsGFP [10 μl of Opto-AAVGFP (1.7 × 1011 vg ml−1) and 40-μl PhyB beads per experiment] diluted in beads buffer for 1 hour under 660-nm light illumination. As control, OptoAAVsGFP were incubated at the same concentration without beads. After centrifugation (1600g, 1 min) to pellet the beads, the supernatants were analyzed for OptoAAVGFP by Western blotting using the B1 antibody.
Analysis of EGFR activation
To analyze EGFR activation by Western blotting, 6.5 × 104 A-431 cells were seeded per well of a 24-well plate. After 24 hours, the medium was exchanged to starvation medium (DMEM complete without FCS), and after cultivation for another 24 hours, the cells were incubated as indicated (recombinant human EGF was purchased from Sigma-Aldrich; catalog no. E9644). Following a washing step with PBS, cells were incubated with 100 μl of lysis buffer [20 mM tris-HCl (pH 7.5), 1 mM EDTA, 100 mM NaCl, 0.5% (v/v) Triton X-100, 0.1% (w/v) SDS, protease inhibitor (cOmplete protease inhibitor cocktail tablets, Roche, Basel, Switzerland, catalog no. 04693116001), 10 mM β-glycerophosphate, 50 mM sodium fluoride, 1 mM sodium orthovanadate, and 10 mM sodium pyrophosphate] for 10 min on ice. Next, the plate was incubated for 30 min at −20°C, and after subsequent thawing on ice, the lysed cells were transferred into microcentrifuge tubes and centrifuged at 10,000g at 4°C for 10 min. Afterward, the supernatants were mixed with SDS loading buffer and incubated at 95°C for 5 min. The samples were separated by SDS-PAGE and analyzed by Western blotting as described above for the AAV samples using the following buffers and antibodies: washing buffer: TBS-T [TBS (50 mM tris-HCl and 150 mM NaCl; pH 7.4) with 0.1% (v/v) Tween 20]; blocking buffer: TBS-T with 5% (w/v) BSA; primary antibodies (diluted 1:1000 in blocking buffer; incubation at 4°C overnight): EGFR [Cell Signaling Technology (CST), Danvers, MA, catalog no. 4267], pEGFR (Tyr1068; CST, catalog no. 3777), Erk1/2 (CST, catalog no. 9102), pErk1/2 (Thr202/Tyr204, CST, catalog no. 9101), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (CST, catalog no. 5174); secondary antibody (diluted 1:3000 in blocking buffer; 1-hour incubation): anti-rabbit immunoglobulin G, HRP-linked (CST, catalog no. 7074).
Light-controlled transduction
If not indicated otherwise, OptoAAVs were incubated at 62.5°C for 10 min in a heat block and stored afterward on ice (up to 6 hours) before use in the experiment. For the light-controlled transduction experiments that were analyzed by flow cytometry, 5000 cells were seeded per well of a 96-well plate in 100 μl of the corresponding medium. For experiments with the optoPlate-96, cells were seeded in a black 96-well plate with transparent bottom (Greiner, Frickenhausen, Germany, catalog no. 655090). After 24 hours, OptoAAVs were mixed with the indicated PhyB-DARPin protein in the indicated buffer/medium, illuminated for 5 min with 740-nm light, and added to the corresponding wells after washing cells once with PBS. After incubation for the indicated period under the indicated illumination condition, wells were washed with PBS and the cells were further incubated in their corresponding medium under the indicated illumination. Forty-eight hours after addition of the AAVs to the cells, the wells were washed with PBS and the cells were detached by the addition of 50 μl of trypsin/EDTA solution per well. Afterward, 200 μl of PBS supplemented with 5% (v/v) FCS was added to each well and the cells were analyzed for transgene expression using an Attune NxT flow cytometer (Thermo Fisher Scientific). BFP, GFP/CFSE, mScarlet, and eFluor670 were excited with a 405-, 488-, 561-, and 637-nm laser and detected using a 440/50-, 530/30-, 620/15-, and 670/14-nm emission filter, respectively. Autofluorescence of the cells was measured in the unused BFP or eFluor670 channel. Flow cytometry data were analyzed with FlowJo (v10.6.1, Becton, Dickinson and Company, Franklin Lakes, NJ), and the gating strategy used throughout this study is depicted in fig. S6. For experiments with the HEK-293T–SpyCatcher cell line, only BFP-positive cells (~95%) were used for the transduction analysis.
For spatially resolved transduction experiments using a photomask, 1 × 105 or 4 × 104 A-431 cells were seeded in 750 or 300 μl of medium per well of a μ-Slide four-well (ibidi, Gräfelfing, Germany, catalog no. 80426; Fig. 5) or μ-Slide eight-well (ibidi, catalog no. 80826; fig. S12, A and B) chambered coverslip, respectively. After 24 hours, cells were incubated as described sequentially with PhyB-DARPinEGFR and OptoAAV under the indicated illumination regime. Twenty-four hours after addition of OptoAAVs to the cells, cells were transferred from 37° to 30°C to reduce proliferation and were incubated for another 24 hours. Afterward, cells were fixed with 4% (w/v) PFA in PBS for 20 min, DAPI-stained (1 μg ml−1 in PBS), and imaged on a Zeiss LSM 880 laser scanning confocal microscope using a 10× EC Plan Neofluar objective (NA, 0.3). DAPI was excited with a 405-nm laser and detected between 417 and 470 nm. GFP and mScarlet were excited with 488- and 561-nm lasers and detected in lambda scanning mode (GFP: 499 to 695 nm, bin width: 8.9 nm; mScarlet: 570 to 695 nm, bin width: 8.9 nm), respectively. Afterward, GFP and mScarlet signals were separated from autofluorescence by linear unmixing using Zen Black (v2.3 SP1, Zeiss), tiles were stitched using Zen Blue (v3.1, Zeiss) and median-filtered in Fiji (59).
For spatially resolved transduction experiments using a confocal microscope, A-431 cells were stained with 1.5 or 5 μM cell proliferation dye CFSE (Thermo Fisher Scientific, catalog no. C34554) or eFluor670 (Thermo Fisher Scientific, catalog no. 65-0840-90) according to the manufacturer’s instructions, respectively. The stained cells were mixed with unstained cells in a ratio of 1:99, and 1.5 × 104 cells were seeded in 300 μl of medium per well of a μ-Slide eight-well grid-500 (ibidi, catalog no. 80826-G500) gridded (with lettered and numbered squares) and chambered coverslip. After 48 hours, cells were incubated as described sequentially with PhyB-DARPinEGFR and OptoAAVmScarlet or OptoAAVGFP within a stage-top incubator (Tokai Hit, Fujinomiya, Japan) installed on a Zeiss LSM 880 laser scanning confocal microscope. CFSE/transmission and eFluor670 images were acquired using a 25× LD LCI Plan Apochromat water objective (NA, 0.8) and a 488- and 633-nm laser, respectively. CFSE and eFluor670 fluorescence was detected at 490 to 570 nm and 651 to 740 nm, respectively. During imaging, cells were constantly illuminated with 740-nm light (200 μmol m−2 s−1). After selecting a single, isolated CFSE or eFluor670-positive cell for light-controlled transduction, the 740-nm light was switched off and the cell was illuminated spatially resolved with the 633-nm laser using the bleaching function of the microscope (pixel dwell time: 0.77 μs; pixel size: 0.554 μm; 633-nm laser intensity: 0.5%; 15 iterations). Afterward, the samples were processed and imaged as described above for the spatially resolved transduction experiments using photomasks. The grid was plotted into the fluorescence images based on its weak autofluorescence.
Transduction experiment with T cells
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy human donors by density gradient centrifugation (Ficoll-Paque). Afterward, PBMCs were resuspended in RPMI complete medium [RPMI 1640 medium supplemented with 10% (v/v) FCS, 10 mM Hepes (Thermo Fisher Scientific, catalog no. 15630-080), 10 μM sodium pyruvate (Thermo Fisher Scientific, catalog no. 11360-039), 1× minimum essential medium nonessential amino acids (PAN Biotech, catalog no. P08-32100), penicillin (50 U ml−1), and streptomycin (50 μg ml−1)] supplemented with interleukin-2 (IL-2; 500 U ml−1; PeproTech, Hamburg, Germany, catalog no. 200-02) and activated with anti-CD3/CD28 (1 μg ml−1) antibodies. After 72 hours, the remaining PBMCs were mostly T cells (60), which were used for light-controlled transduction using PhyB-DARPinCD4 and OptoAAVGFP as indicated. Six hours after addition of OptoAAVsGFP, the cells were incubated in RPMI complete medium supplemented with IL-2 (100 U ml−1) under the indicated illumination. After 42 hours, the cells were stained for CD4 expression by incubation with CD4-V450 antibody (BD Biosciences, San Jose, CA, catalog no. 560345) diluted 1:200 in PBS supplemented with 2% (v/v) FCS at 4°C for 15 min. After washing, the cells were resuspended in PBS supplemented with 2% (v/v) FCS and analyzed using an Attune NxT flow cytometer as described above. V450 fluorescence and autofluorescence were measured in the BFP and mScarlet channel, respectively. Autofluorescent cells (<1.7%) were excluded from the analysis.
Statistical analysis
Statistical significance was tested with unpaired two-sided t tests (no assumption of consistent standard deviations) with correction for multiple comparisons (Holm-Sidak method) using GraphPad Prism (v8.4.3, GraphPad Software, San Diego, CA).
Acknowledgments
We are grateful to M. R. Russ (University of Freiburg) for generation of the HEK-SpyCatcher cell line and providing PhyB-mCherry-SpyTag. We would like to thank R. M. Velasco Cárdenas, S. Brandl, K. Raute, and S. Minguet (University of Freiburg) for providing the primary human T cells. We thank the technical workshop of the Faculty of Biology for the design and construction of the illumination devices. We acknowledge the excellent scientific and technical assistance of the Signalling Factory Core Facility staff of the University of Freiburg for help on flow cytometry and providing cell lines. We thank the staff of the Life Imaging Center (LIC) in the Center for Biological Systems Analysis (ZBSA) of the University of Freiburg for help with microscopy resources, and the excellent support in image recording and analysis. We are grateful to A. Hartley, E. Wiedtke, and D. Grimm (Heidelberg University, Germany) for providing plasmids pCMVgfp and pCMVmScarlet, B1 antibody, and helpful advice. We would like to thank H. Büning (Hannover Medical School, Germany) for providing plasmid pRCVP2koA and for helpful advice. We thank J. Suh (Rice University, TX) for providing plasmid pVP2A-PIF6 (Addgene plasmid # 73369). Funding: This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy CIBSS, EXC-2189, Project ID: 390939984 and under the Excellence Initiative of the German Federal and State Governments EXC-294 and GSC-4, and in part by the Ministry for Science, Research, and Arts of the State of Baden-Württemberg. Author contributions: M.H. and W.W. conceived the project. M.H., C.J.-L., A.H., S.H., O.S.Y., W.W.A.S., C.H., M.D.Z., H.Y., H.J.W., and W.W. designed, analyzed, and interpreted experiments. M.H., C.J.-L., A.H., S.H., and H.J.W. performed experiments. M.H. and W.W. wrote the manuscript with support from all other authors. M.H., H.J.W., and W.W. supervised the project. Competing interests: The University of Freiburg has filed a patent application covering the technology described here, of which M.H. and W.W. are inventors. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabf0797/DC1
REFERENCES AND NOTES
- 1.Schier A. F., Single-cell biology: Beyond the sum of its parts. Nat. Methods 17, 17–20 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Wan Y., Wei Z., Looger L. L., Koyama M., Druckmann S., Keller P. J., Single-cell reconstruction of emerging population activity in an entire developing circuit. Cell 179, 355–372.e23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandberg R., Entering the era of single-cell transcriptomics in biology and medicine. Nat. Methods 11, 22–24 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Bono N., Ponti F., Mantovani D., Candiani G., Non-viral in vitro gene delivery: It is now time to set the bar! Pharmaceutics 12, 183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y., Bruggeman K. F., Franks S., Gautam V., Hodgetts S. I., Harvey A. R., Williams R. J., Nisbet D. R., Is viral vector gene delivery more effective using biomaterials? Adv. Healthc. Mater. 10, e2001238 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Stevenson D. J., Gunn-Moore F. J., Campbell P., Dholakia K., Single cell optical transfection. J. R. Soc. Interface 7, 863–871 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antkowiak M., Torres-Mapa M. L., Stevenson D. J., Dholakia K., Gunn-Moore F. J., Femtosecond optical transfection of individual mammalian cells. Nat. Protoc. 8, 1216–1233 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Jerjes W., Theodossiou T. A., Hirschberg H., Høgset A., Weyergang A., Selbo P. K., Hamdoon Z., Hopper C., Berg K., Photochemical internalization for intracellular drug delivery. From basic mechanisms to clinical research. J. Clin. Med. 9, 528 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goater J., Müller R., Kollias G., Firestein G. S., Sanz I., O'Keefe R. J., Schwarz E. M., Empirical advantages of adeno associated viral vectors in vivo gene therapy for arthritis. J. Rheumatol. 27, 983–989 (2000). [PubMed] [Google Scholar]
- 10.Ulrich-Vinther M., Maloney M. D., Goater J. J., Soballe K., Goldring M. B., O'Keefe R. J., Schwarz E. M., Light-activated gene transduction enhances adeno-associated virus vector-mediated gene expression in human articular chondrocytes. Arthritis Rheum. 46, 2095–2104 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Ito H., Goater J. J., Tiyapatanaputi P., Rubery P. T., O'Keefe R. J., Schwarz E. M., Light-activated gene transduction of recombinant adeno-associated virus in human mesenchymal stem cells. Gene Ther. 11, 34–41 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Pandori M. W., Sano T., Photoactivatable retroviral vectors: A strategy for targeted gene delivery. Gene Ther. 7, 1999–2006 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Pandori M. W., Hobson D. A., Olejnik J., Krzymanska-Olejnik E., Rothschild K. J., Palmer A. A., Phillips T. J., Sano T., Photochemical control of the infectivity of adenoviral vectors using a novel photocleavable biotinylation reagent. Chem. Biol. 9, 567–573 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Wang Y., Li S., Tian Z., Sun J., Liang S., Zhang B., Bai L., Zhang Y., Zhou X., Xiao S., Zhang Q., Zhang L., Zhang C., Zhou D., Generation of a caged lentiviral vector through an unnatural amino acid for photo-switchable transduction. Nucleic Acids Res. 47, e114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tahara M., Takishima Y., Miyamoto S., Nakatsu Y., Someya K., Sato M., Tani K., Takeda M., Photocontrollable mononegaviruses. Proc. Natl. Acad. Sci. U.S.A. 116, 11587–11589 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagihara Y., Sakamoto A., Tokuda T., Yamashita T., Ikemoto S., Kimura A., Haruta M., Sasagawa K., Ohta J., Takayama K., Mizuguchi H., Photoactivatable oncolytic adenovirus for optogenetic cancer therapy. Cell Death Dis. 11, 570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Høgset A., Engesaeter B. Ø., Prasmickaite L., Berg K., Fodstad Ø., Maelandsmo G. M., Light-induced adenovirus gene transfer, an efficient and specific gene delivery technology for cancer gene therapy. Cancer Gene Ther. 9, 365–371 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Bonsted A., Hogset A., Hoover F., Berg K., Photochemical enhancement of gene delivery to glioblastoma cells is dependent on the vector applied. Anticancer Res. 25, 291–297 (2005). [PubMed] [Google Scholar]
- 19.Bonsted A., Engesaeter B. O., Hogset A., Maelandsmo G. M., Prasmickaite L., D'Oliveira C., Hennink W. E., van Steenis J. H., Berg K., Photochemically enhanced transduction of polymer-complexed adenovirus targeted to the epidermal growth factor receptor. J. Gene Med. 8, 286–297 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Gomez E. J., Gerhardt K., Judd J., Tabor J. J., Suh J., Light-activated nuclear translocation of adeno-associated virus nanoparticles using phytochrome B for enhanced, tunable, and spatially programmable gene delivery. ACS Nano 10, 225–237 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Perillo B., Di Donato M., Pezone A., Di Zazzo E., Giovannelli P., Galasso G., Castoria G., Migliaccio A., ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 52, 192–203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang D., Tai P. W. L., Gao G., Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ravindra Kumar S., Miles T. F., Chen X., Brown D., Dobreva T., Huang Q., Ding X., Luo Y., Einarsson P. H., Greenbaum A., Jang M. J., Deverman B. E., Gradinaru V., Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nat. Methods 17, 541–550 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner H. J., Weber W., Fussenegger M., Synthetic biology: Emerging concepts to design and advance adeno-associated viral vectors for gene therapy. Adv. Sci. 8, 2004018 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steiner D., Forrer P., Plückthun A., Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J. Mol. Biol. 382, 1211–1227 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Boersma Y. L., Chao G., Steiner D., Wittrup K. D., Plückthun A., Bispecific designed ankyrin repeat proteins (DARPins) targeting epidermal growth factor receptor inhibit A431 cell proliferation and receptor recycling. J. Biol. Chem. 286, 41273–41285 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Münch R. C., Janicki H., Volker I., Rasbach A., Hallek M., Büning H., Buchholz C. J., Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 21, 109–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hörner M., Kaufmann B., Cotugno G., Wiedtke E., Büning H., Grimm D., Weber W., A chemical switch for controlling viral infectivity. Chem. Commun. 50, 10319–10322 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Hagen S., Baumann T., Wagner H. J., Morath V., Kaufmann B., Fischer A., Bergmann S., Schindler P., Arndt K. M., Müller K. M., Modular adeno-associated virus (rAAV) vectors used for cellular virus-directed enzyme prodrug therapy. Sci. Rep. 4, 3759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dreier B., Mikheeva G., Belousova N., Parizek P., Boczek E., Jelesarov I., Forrer P., Plückthun A., Krasnykh V., Her2-specific multivalent adapters confer designed tropism to adenovirus for gene targeting. J. Mol. Biol. 405, 410–426 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Münch R. C., Mühlebach M. D., Schaser T., Kneissl S., Jost C., Plückthun A., Cichutek K., Buchholz C. J., DARPins: An efficient targeting domain for lentiviral vectors. Mol. Ther. 19, 686–693 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friedrich K., Hanauer J. R., Prüfer S., Münch R. C., Völker I., Filippis C., Jost C., Hanschmann K. M., Cattaneo R., Peng K. W., Plückthun A., Buchholz C. J., Cichutek K., Mühlebach M. D., DARPin-targeting of measles virus: Unique bispecificity, effective oncolysis, and enhanced safety. Mol. Ther. 21, 849–859 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plückthun A., Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 55, 489–511 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Wiley H. S., Herbst J. J., Walsh B. J., Lauffenburger D. A., Rosenfeld M. G., Gill G. N., The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 266, 11083–11094 (1991). [PubMed] [Google Scholar]
- 35.Opie S. R., Warrington K. H. Jr., Agbandje-McKenna M., Zolotukhin S., Muzyczka N., Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 77, 6995–7006 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCarty D. M., Self-complementary AAV vectors; advances and applications. Mol. Ther. 16, 1648–1656 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Xiao X., Li J., Samulski R. J., Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 72, 2224–2232 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kronenberg S., Böttcher B., von der Lieth C. W., Bleker S., Kleinschmidt J. A., A conformational change in the adeno-associated virus type 2 capsid leads to the exposure of hidden VP1 N termini. J. Virol. 79, 5296–5303 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bleker S., Sonntag F., Kleinschmidt J. A., Mutational analysis of narrow pores at the fivefold symmetry axes of adeno-associated virus type 2 capsids reveals a dual role in genome packaging and activation of phospholipase A2 activity. J. Virol. 79, 2528–2540 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu X., Nagarajan H., Lewis N. E., Pan S., Cai Z., Liu X., Chen W., Xie M., Wang W., Hammond S., Andersen M. R., Neff N., Passarelli B., Koh W., Fan H. C., Wang J., Gui Y., Lee K. H., Betenbaugh M. J., Quake S. R., Famili I., Palsson B. O., Wang J., The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat. Biotechnol. 29, 735–741 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keeble A. H., Turkki P., Stokes S., Khairil Anuar I. N. A., Rahikainen R., Hytonen V. P., Howarth M., Approaching infinite affinity through engineering of peptide-protein interaction. Proc. Natl. Acad. Sci. U.S.A. 116, 26523–26533 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schamel W. W., Kuppig S., Becker B., Gimborn K., Hauri H. P., Reth M., A high-molecular-weight complex of membrane proteins BAP29/BAP31 is involved in the retention of membrane-bound IgD in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 100, 9861–9866 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pillay S., Meyer N. L., Puschnik A. S., Davulcu O., Diep J., Ishikawa Y., Jae L. T., Wosen J. E., Nagamine C. M., Chapman M. S., Carette J. E., An essential receptor for adeno-associated virus infection. Nature 530, 108–112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stefan N., Martin-Killias P., Wyss-Stoeckle S., Honegger A., Zangemeister-Wittke U., Plückthun A., DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J. Mol. Biol. 413, 826–843 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Schweizer A., Rusert P., Berlinger L., Ruprecht C. R., Mann A., Corthésy S., Turville S. G., Aravantinou M., Fischer M., Robbiani M., Amstutz P., Trkola A., CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics. PLOS Pathog. 4, e1000109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nissim L., Bar-Ziv R. H., A tunable dual-promoter integrator for targeting of cancer cells. Mol. Syst. Biol. 6, 444 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deisseroth K., Hegemann P., The form and function of channelrhodopsin. Science 357, eaan5544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De La Crompe B., Coulon P., Diester I., Functional interrogation of neural circuits with virally transmitted optogenetic tools. J. Neurosci. Methods 345, 108905 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Sokolovski S. G., Zherebtsov E. A., Kar R. K., Golonka D., Stabel R., Chichkov N. B., Gorodetsky A., Schapiro I., Möglich A., Rafailov E. U., Two-photon conversion of a bacterial phytochrome. Biophys. J. 120, 964–974 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yousefi O. S., Günther M., Hörner M., Chalupsky J., Wess M., Brandl S. M., Smith R. W., Fleck C., Kunkel T., Zurbriggen M. D., Höfer T., Weber W., Schamel W. W., Optogenetic control shows that kinetic proofreading regulates the activity of the T cell receptor. eLife 8, e42475 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gibson D. G., Young L., Chuang R.-Y., Venter J. C., Hutchison C. A. III, Smith H. O., Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Beyer H. M., Gonschorek P., Samodelov S. L., Meier M., Weber W., Zurbriggen M. D., AQUA cloning: A versatile and simple enzyme-free cloning approach. PLOS ONE 10, e0137652 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bugaj L. J., Lim W. A., High-throughput multicolor optogenetics in microwell plates. Nat. Protoc. 14, 2205–2228 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Thomas O. S., Hörner M., Weber W., A graphical user interface to design high-throughput optogenetic experiments with the optoPlate-96. Nat. Protoc. 15, 2785–2787 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Hörner M., Gerhardt K., Salavei P., Hoess P., Härrer D., Kaiser J., Tabor J. J., Weber W., Production of phytochromes by high-cell-density E. coli fermentation. ACS Synth. Biol. 8, 2442–2450 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Hörner M., Yousefi O. S., Schamel W. W. A., Weber W., Production, purification and characterization of recombinant biotinylated phytochrome B for extracellular optogenetics. Bio Protoc. 10, e3541 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zolotukhin S., Byrne B. J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R. J., Muzyczka N., Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 6, 973–985 (1999). [DOI] [PubMed] [Google Scholar]
- 58.Wistuba A., Kern A., Weger S., Grimm D., Kleinschmidt J. A., Subcellular compartmentalization of adeno-associated virus type 2 assembly. J. Virol. 71, 1341–1352 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hartl F. A., Beck-García E., Woessner N. M., Flachsmann L. J., Cárdenas R. M. V., Brandl S. M., Taromi S., Fiala G. J., Morath A., Mishra P., Yousefi O. S., Zimmermann J., Hoefflin N., Köhn M., Wöhrl B. M., Zeiser R., Schweimer K., Günther S., Schamel W. W., Minguet S., Noncanonical binding of Lck to CD3ε promotes TCR signaling and CAR function. Nat. Immunol. 21, 902–913 (2020). [DOI] [PubMed] [Google Scholar]
- 61.Beyer H. M., Thomas O. S., Riegel N., Zurbriggen M. D., Weber W., Hörner M., Generic and reversible opto-trapping of biomolecules. Acta Biomater. 79, 276–282 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Costantini L. M., Baloban M., Markwardt M. L., Rizzo M., Guo F., Verkhusha V. V., Snapp E. L., A palette of fluorescent proteins optimized for diverse cellular environments. Nat. Commun. 6, 7670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabf0797/DC1