

The structure of CCR5 in complex with [6P4]CCL5 and the Gi protein reveals its activation mechanism.
Abstract
The human CC chemokine receptor 5 (CCR5) is a G protein–coupled receptor (GPCR) that plays a major role in inflammation and is involved in cancer, HIV, and COVID-19. Despite its importance as a drug target, the molecular activation mechanism of CCR5, i.e., how chemokine agonists transduce the activation signal through the receptor, is yet unknown. Here, we report the cryo-EM structure of wild-type CCR5 in an active conformation bound to the chemokine super-agonist [6P4]CCL5 and the heterotrimeric Gi protein. The structure provides the rationale for the sequence-activity relation of agonist and antagonist chemokines. The N terminus of agonist chemokines pushes onto specific structural motifs at the bottom of the orthosteric pocket that activate the canonical GPCR microswitch network. This activation mechanism differs substantially from other CC chemokine receptors that bind chemokines with shorter N termini in a shallow binding mode involving unique sequence signatures and a specialized activation mechanism.
INTRODUCTION
The human CC chemokine receptor 5 (CCR5) is a G protein–coupled receptor (GPCR) that plays a major role in inflammation by recruiting and activating leukocytes (1). CCR5 is also the principal HIV coreceptor (2), is involved in the pathology of both cancer (3) and neuroinflammation (4), and has been implicated in the inflammatory complications of coronavirus disease 2019 (COVID-19) (5, 6). Soon after the discovery of CCR5, it became evident that its natural chemokine ligands inhibit HIV entry (7), with CCL5 (RANTES) being most efficient, acting both by blocking the binding site for the viral glycoprotein gp120 and by promoting CCR5 endocytosis (8). Modifications of the N-terminal region of CCL5 preceding residue C10 yielded HIV entry inhibitors with significantly higher potency (9–11). These analogs belong to a group of over 100 engineered CCL5 N-terminal variants that show notable differences in their anti-HIV, endocytotic, affinity, and signaling properties ranging, e.g., from super-agonist to strong antagonist behavior (10, 11). The molecular basis of these N-terminal structure–related activity differences is currently unclear.
Whereas a good structural understanding has been reached of the activation mechanisms of class A GPCRs by small-molecule ligands (12), the activation mechanism of the chemokine receptor subclass is not yet well understood. Inactive structures of a number of chemokine receptors have been solved, including complexes of CCR5 with the engineered chemokine antagonist [5P7]CCL5 (13), the viral gp120•human CD4 complex (14), the HIV inhibitor maraviroc (15), and other small-molecule antagonists (16). In contrast, only two active-state human chemokine receptor complex structures are currently available: CCL20•CCR6•Go (17) and CXCL8•CXCR2•Gi (18). In these structures, CCL20 and CXCL8 adopt a shallow binding mode in which the chemokine N terminus is not deeply inserted into the orthosteric pocket and activation apparently involves transmission of forces directly from the extracellular domain of the receptor. There are also two inverse-agonist–bound (19), one agonist-bound, and one apo (20) structures available of the viral chemokine receptor US28. All these structures are in active conformation. However, US28 is constitutively active and can engage thousands of distinct chemokine sequences, some of them leading to a moderate increase in activity (20). Thus, chemokine-induced activation of US28 is thought to result from a rather sequence-insensitive mechanism, in which the steric bulk of the ligand is more important than specific interactions between the chemokine and the receptor (20).
In contrast to CCR6 and CXCR2, many native human chemokine receptors such as CCR5 have chemokine ligands with longer N termini, which likely insert more deeply into the orthosteric pocket of the receptor. As many agonist and antagonist CCL5 ligand variants have been identified for CCR5 that differ only in the composition of the first ~10 residues but not in their length (11). The decisive contacts for CCR5 signaling interactions must be located at the bottom of the orthosteric pocket rather than at the extracellular surface of the receptor.
With the aim of elucidating the apparently different activation mechanisms of CC chemokine receptors and to provide a general structural explanation for the variable pharmacology of CCL5 N-terminal variants, we solved the structure of wild-type human CCR5 in complex with the super-agonist [6P4]CCL5 and the Gi heterotrimer.
RESULTS
Overall structure of the [6P4]CCL5•CCR5•Gi complex
A stable [6P4]CCL5•CCR5•Gi complex was obtained by incubating detergent-solubilized human wild-type full-length CCR5 with the Gi heterotrimer and [6P4]CCL5 (fig. S1). The complex was treated with apyrase to hydrolyze guanosine diphosphate and was further stabilized by addition of the Fab fragment Fab16 (21, 22), which recognizes an interface between the Gα and Gβγ subunits of the Gi heterotrimer (fig. S2). Single-particle cryo–electron microscopy (cryo-EM) analysis with extensive particle classification yielded a three-dimensional (3D) density map with a nominal global resolution of 3.15 Å (Fig. 1A, fig. S3, and table S1). The map is well resolved for most of parts of CCR5, the [6P4]CCL5 N terminus, the Gi heterotrimer (fig. S4), and Fab16. The density of the globular core of [6P4]CCL5 and the adjacent CCR5 N terminus and extracellular parts of the receptor have less defined density, indicating relative flexibility in these parts of the structure. A 3D variability analysis of the cryo-EM data (movie S1) and molecular dynamics (MD) simulations of the atomic model (fig. S5A) reveal a certain degree of mobility of the [6P4]CCL5 core, the receptor N terminus, the extra- and intracellular loops, and transmembrane (TM) helices 5, 6, and 7. Still, the MD simulations indicate persistent interactions mediated by the N-terminal residues 0 to 8, the β1/β3-strands, and the 30s loop of the chemokine (fig. S5B). Apart from a small ~5° difference in the orientation, the position of the [6P4]CCL5 core is very similar to that of [5P7]CCL5 in the inactive [5P7]CCL5•CCR5 complex (Figs. 1C and 2A). Nevertheless, this minor change in orientation also leads to small (1 to 2 Å) but noticeable movements at the extracellular ends of TM1 and TM7 (Fig. 2B).
Fig. 1. Cryo-EM structure of the [6P4]CCL5•CCR5•Fab16 complex.
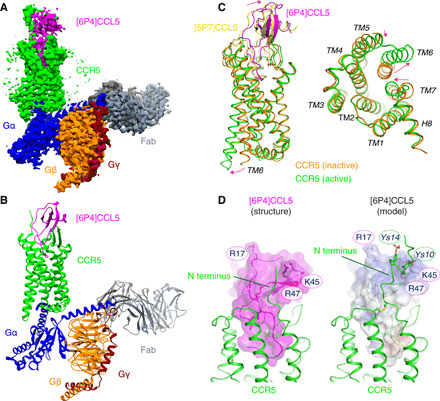
(A) Cryo-EM map of the [6P4]CCL5•CCR5•Gi•Fab16 complex colored by subunits ([6P4]CCL5, magenta; CCR5, green; Gαi, blue; Gβ, orange; Gγ, maroon; and Fab16, gray). (B) Atomic model of the [6P4]CCL5•CCR5•Gi•Fab16 complex in the same view and color scheme as shown in (A). (C) Side and cytoplasmic views of the structural overlay of active CCR5 (green) in complex [6P4]CCL5 (magenta) and inactive CCR5 [orange; Protein Data Bank (PDB) ID: 5UIW] in complex with [5P7]CCL5 (yellow). Substantial structural changes between two conformations are indicated by red arrows. The C1013.25-C178ECL2, C20N-term-C2697.25 disulfide bridges conserved in chemokine receptors are shown in dark yellow. (D) Interactions between the [6P4]CCL5 core and the CCR5 N terminus at the CRS1 site (left, cryo-EM structure; right, cryo-EM/NMR–based model). In the model, sulfo-tyrosines sY10 and sY14 are depicted as sticks, and the [6P4]CCL5 surface is colored according to its electrostatic potential (negative, red; positive, blue).
Fig. 2. Deep insertion of CCL5 chemokines into the orthosteric CCR5 ligand pocket.
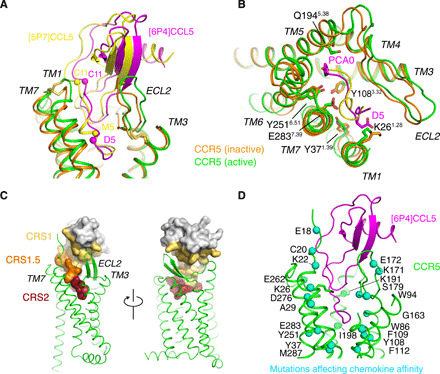
(A and B) Comparison between the insertion of the agonist [6P4]CCL5 (magenta) into active CCR5 (green) and the antagonist [5P7]CCL5 (yellow) into inactive CCR5 (orange; PDB ID: 5UIW) (A, side view; B, top view). Only the CCL5 N-terminal residues are shown in (B). Important residues participating in the CCL5-CCR5 interaction are marked. (C) Location of CRS1, CRS1.5, and CRS2 chemokine recognition sites in the [6P4]CCL5•CCR5•Gi complex. (D) Locations of previously identified CCR5 point mutants [as reviewed by (13)] that affect chemokine affinity within CCR5. Respective residues are shown in the [6P4]CCL5•CCR5•Gi complex as blue spheres. [6P4]CCL5 (magenta) and CCR5 (green) are shown in cartoon.
The open conformation of the intracellular part of the active CCR5 differs from all inactive CCR5 structures, thereby enabling binding of the G protein (Fig. 1C): TM6 is moved outward from the heptahelical bundle accompanied by further rearrangements of TM5, TM7, and intracellular loop 4 (ICL4). The moderate outward movement of TM6 and the arrangement of Gi relative to CCR5 (Fig. 1, A to C) agree with previous GPCR•Gi complexes (23).
CRS1 interactions
The interactions between [6P4]CCL5 and CCR5 can be separated into the three canonical chemokine recognition sites (CRS): CRS 1, 1.5, and 2 (Fig. 2C) (13, 24). CRS1 consists of the contacts of the chemokine core with the extracellular side of the receptor and is dominated by electrostatic interactions. The core of [6P4]CCL5 sits on top of a wide opening in the extracellular part of the CCR5 TM bundle, which is shaped by two disulfide bridges [C1013.25-C178ECL2, conserved in class A GPCRs, and C20N-term-C2697.25, specific to chemokine receptors; superscripts indicate the GPCRdb numbering scheme (25)] (Figs. 1C and 2A). The [6P4]CCL5 strand β1 makes extensive contacts with polar residues in extracellular loop (ECL) 2, while the CCR5 N terminus directs toward a shallow groove between the chemokine N-loop and 40s loop forming further extensive ionic and polar interactions. Interactions between CCR5 residues S17 and E18 and the chemokine residues R47 and Q48 are visible in the density. However, poorly defined density prevented building a model of the CCR5 N-terminal residues 1 to 15 with confidence. To gain insights into this region, these CCR5 residues were modeled (Fig. 1D) on the basis of the nuclear magnetic resonance (NMR) structure of CCL5 in complex with an N-terminal fragment (residues 1 to 27) of CCR5 sulfated at residues Y10 and Y14 (26). Sulfation at Y10 and Y14 of CCR5 is important for chemokine affinity (26–29) and is expected to be present also in the insect cell–expressed CCR5 used in the current study (30). The stability of the modeled interactions between the sulfated CCR5 N terminus and [6P4]CCL5 was assessed by MD (movie S2). The simulations reveal persistent interactions between sY10 and sY14 of CCR5 and residues in the N-loop, 40s loop, and β3-strand (fig. S6), including K45, R47, and R17 of [6P4]CCL5, in complete agreement with the NMR-observed nuclear Overhauser effect (NOE) contacts (29). In addition, comparison of MD trajectories between sulfated and nonsulfated CCR5 indicates that sulfation induces a higher number of contacts between the chemokine and the receptor N terminus (fig. S6), consistent with the higher affinity of the sulfated form (28).
CRS2 interactions and activation
The N terminus of [6P4]CCL5 reaches deep into the orthosteric pocket (CRS2) between the CCR5 7TM bundle (Fig. 2). In complete agreement with this deep binding mode, point mutations of many CCR5 residues lining the CRS2 site, the CRS1.5 site at the rim of the orthosteric pocket, and the extracellular N-terminal CRS1 site have been shown previously to affect the affinity of CCR5 for chemokines (Fig. 2D). This deep insertion contrasts with the shallow binding modes observed for the chemokines in the CCL20•CCR6•Go (17) and CXCL8•CXCR2•Gi (18) complexes (see below). Of note, the N-terminal residues preceding C10 and C11 of monomeric CCL5 in solution undergo large amplitude motions on the nanosecond time scale, as revealed by 15N relaxation data (31). However, they adopt a fixed conformation in the CCR5 complex.
The [6P4]CCL5 residues 0 to 3 form the distal N terminus, which is located at the bottom of CRS2 (Figs. 2, A and C, and 3, A and B). As compared to [5P7]CCL5, the deeper binding pose of [6P4]CCL5 slightly relocates the N-terminal pyroglutamate (PCA) group (Fig. 3A). The packing of CCR5 against the PCA group is not very tight, and our MD simulations of the [6P4]CCL5•CCR5 complex show that the PCA group interacts through dynamic water-mediated hydrogen bonds with nearby residues Q1945.38 and Y2516.51 (fig. S7A). An inspection of the electron density of the [5P7]CCL5•CCR5 crystal structure (13) allowed us to also model additional water molecules in the vicinity of the [5P7]CCL5-PCA, which similarly connect to surrounding residues K1915.35, Q1945.38, Y2516.51, N2586.58, and T2596.59 (fig. S7B). Thus, the PCA group of the chemokine does not appear to have well-defined contacts to CCR5. Rather, the surrounding CCR5 cavity may accommodate even larger moieties such as the alkyl chains of AOP- (9) or PSC-CCL5 (32), thereby increasing the potency of these ligands.
Fig. 3. Activation mechanism of CCR5 by [6P4]CCL5 at CRS2.
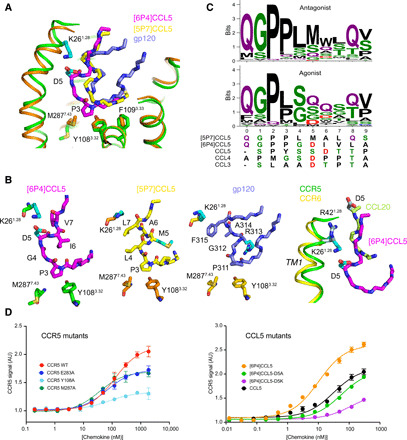
(A) Comparison of insertion depths of agonist [6P4]CCL5 (magenta), antagonist [5P7]CCL5 (yellow), and the antagonist V3 loop of gp120 (slate; PDB ID: 6MEO) into active CCR5 (green) and inactive CCR5 (orange). Important interacting residues are shown as sticks. (B) Detailed view of [6P4]CCL5 N terminus inserted into active CCR5, [5P7]CCL5 N terminus, and gp120 V3 loop inserted into inactive CCR5; and comparison of the insertion of [6P4]CCL5 N terminus into active CCR5 and of CCL20 N terminus into active CCR6 (PDB ID: 6WWZ). (C) Sequence composition of the N termini of CCL5 natural amino acid variants (table S2) with low (≤20%, N = 83, top) and high (≥50%, N = 34, bottom) calcium signaling. The N termini of [5P7]CCL5, [6P4]CCL5, and wild-type CCL3 to CCL5 (all agonists) are shown below. (D) Effect of CCR5 (left) and CCL5 (right) mutations on CCR5 signaling activity as monitored by Ca2+ flux measurements in human embryonic kidney (HEK) cells. Data points represent mean peak height ± SD (n = 3). Data shown are representative of two (left) or three (right) independent experiments. Fits to data points by a three-parameter response model are shown as solid lines. Respective fit values for Emax and EC50 are listed in table S3. AU, arbitrary units.
[6P4]CCL5 residues 4 to 9 form the proximal N terminus, which acts as a hinge between the chemokine core and the distal N terminus (Figs. 2, A and C, and 3, A and B). Conspicuously, residues P3 to D5, which constitute the turn between the proximal and the distal [6P4]CCL5 N terminus, insert several angstroms deeper into the CCR5 orthosteric pocket than the corresponding residues of [5P7]CCL5 or the V3 loop of gp120 in the respective inactive complexes with CCR5 (Fig. 3A). Because of this deeper insertion, P3 of [6P4]CCL5 can displace CCR5 M2877.43 and Y1083.32 (Fig. 3, A and B), thereby apparently activating the canonical GPCR microswitch network (see below), which remains in the inactive conformation in the [5P7]CCL5 or gp120 complexes. A similar CCR5 activation mechanism involving M2877.43 has been suggested on the basis of a computational model of the CCR5/CCL5 complex (13).
The deeper pose of the [6P4]CCL5 N terminus partially overlaps with that of the antagonist maraviroc (fig. S8) (15). However, maraviroc inserts its phenyl ring between Y1083.32 and F1093.33 of the “aromatic connector” (see below), thereby apparently blocking the conformational rearrangement necessary for activation. A comparison of the chemokine N termini in the [6P4]CCL5•CCR5 versus the CCL20•CCR6 complexes (Fig. 3B) also shows the much reduced insertion depth of the latter, which prevents it from reaching sites corresponding to the activation switches identified at the bottom of the orthosteric pocket of CCR5.
The different insertion depth of the [6P4]CCL5 and [5P7]CCL5 N termini into CRS2 is caused by a markedly different structure of their proximal N-terminal residues 5 to 8, a short helix in [5P7]CCL5 and an extended coil in [6P4]CCL5 (Fig. 2, A and B, and 3, A and B). The hinge function of this structure is presumably key to the control of receptor activation. In [6P4]CCL5, D5 of the extended hinge forms an ionic interaction with K261.28 in TM1, whereas the side chain of the equivalent M5 of [5P7]CCL5 points in the opposite direction forming a helical turn (Fig. 3B). Apparently, this helical turn is also pushed sideways by unfavorable interactions between [5P7]CCL5 L7 and K261.28. Very similar interactions and conformations are present in the inactive gp120•CCR5 complex, with F315, R313, and P311 taking the roles of [5P7]CCL5 L7, M5, and P3, respectively. Besides the ionic D5-K261.28 interactions, the extended backbone at residues 2, 4, and 5 of the active [6P4]CCL5 is further stabilized by contacts to E2837.39.
The structural finding that the CCL5 the N-terminal hinge conformation controls the insertion depth of its residues 3 to 5 and thereby the activation state of CCR5 is corroborated by a statistical analysis of the pharmacological properties of CCL5 N-terminal amino acid variants. Currently, ~140 of these have been characterized for G protein signaling (table S2), CCR5 internalization, and anti-HIV activity (11). Sequence analysis shows that residues 0 to 3 (highest abundance: QGPL, distal N terminus) and 8 and 9 (highest abundance: QV, proximal N terminus) are highly similar between N-terminal variants with low (N = 83) and high (N = 34) signaling activity (Fig. 3C). The latter is expected since the panel of tested variants was to some extent biased toward these residues (11). In contrast, strong differences are observed for residues 4 to 7 in the proximal N terminus: In agonist variants, the small, hydrophilic, or negatively charged amino acids S, Q, G, and D dominate, whereas antagonist variants contain mostly the large hydrophobic amino acids L, M, and W. Apparently, the small hydrophilic residues direct the hinge toward K261.28 in TM1, whereas the large hydrophobic residues make the hinge collapse to a helical turn. In agreement with their agonist pharmacology, both [6P4]CCL5 and wild-type CCL5 as well as the other major CCR5 agonist chemokines CCL3 (MIP1α) and CCL4 (MIP1β) contain an aspartic acid residue at positions 5 or 6 (Fig. 3C), which presumably stabilizes the extended hinge by forming a salt bridge to K261.28.
Essential parts of the proposed CCR5 activation mechanism were tested by CCR5 and [6P4]CCL5 point mutants using cellular Ca2+ flux activation assays in human embryonic kidney (HEK) cells (Fig. 3D and table S3). Consistent with our model, the CCR5 M2877.43A, Y1083.32A, and E2837.39A mutations all reduced Emax for [6P4]CCL5 activation by ~40 to 70% without affecting the half-maximal effective concentration (EC50). This indicates that these mutations reduce signaling without modifying chemokine affinity. In contrast, the [6P4]CCL5 D5A and D5K mutations decreased Emax by 30 and 60%, respectively and increased EC50 ~5- to 10-fold. Similar observations were made in Chinese hamster ovary (CHO) cells expressing CCR5 (table S3). The fact that these [6P4]CCL5 mutations reduce both signaling and binding affinity agrees with our proposed mechanism, since the unfavorable interactions of the CCR5 residue K261.28 with an alanine or lysine at position 5 in [6P4]CCL5 are expected to alter the hinge structure and reduce the binding enthalpy.
Signal transmission from CRS2 to the microswitch network
As described above, the straight conformation of the [6P4]CCL5 proximal N terminus pushes the region around residue P3 toward the bottom of CRS2, with the backbone of residues 2, 4, and 5 of [6P4]CCL5 interacting with E2837.39 (Fig. 4A). The straight hinge appears to be further stabilized by contacts of [6P4]CCL5 residues P3 to D5 to a cluster of the hydrophobic CCR5 residues F852.59, W862.60, Y892.63, and L1043.28. These contacts may act as “counter bearing” to promote the force of the [6P4]CCL5 N terminus toward the bottom of CRS2.
Fig. 4. Transmission of the chemokine agonist signal to the receptor activation switches.
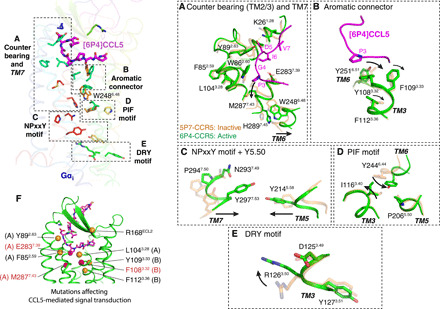
Top left: Residue groups connecting the N-terminal region of [6P4]CCL5 (magenta) to key CCR5 activation switches. Relevant residues are shown as sticks. For clarity, only part of the receptor structure is shown. (A) Counter bearing in TM2/3 and transmission through TM7. (B) Transmission through the aromatic connector. (C) Activation of the NPxxY motif and Y2145.58 in TM5. (D) Activation of the PIF motif. (E) Activation of the DRY motif. (F) Point mutations shown to affect CCR5 activation but not chemokine affinity [orange, mutations from literature (see main text); red, mutations performed in this work]. The letters next to the residue numbers direct to the panel where these residues are shown in more detail.
The deeper placement of residue P3 of [6P4]CCL5 as compared to [5P7]CCL5 forces a relocation of M2877.43 in the receptor (Figs. 3A and 4A), which is accompanied by noticeable local changes in the backbone of TM7 (fig. S9) that bring the intracellular half of this helix toward the receptor core. This movement allows H2897.45 to push onto W2486.48, possibly assisting the relocation of TM6 (Fig. 4A). P3 also lies on top of an aromatic connector formed by CCR5 residues Y1083.32, F1093.33, and F1123.36 (Fig. 4B), forcing the movement of Y1083.32 and resulting in a cascade of aromatic side chain relocations that transmit the activation signal to the receptor core. This apparently switches the PIF motif (P2065.50, I1163.40, and Y2446.44) to an active conformation (Fig. 4D) and induces the large-scale movement of TM6. The relocation of TM6 and TM7 coincides with local structural changes in the NPxxY motif (Fig. 4C), leading to the formation of the conserved water-mediated interaction between Y2977.53 and Y2145.58 (33) and the opening of the binding pocket for H5 of Gi, which includes R1263.50 in the open conformation of the intrahelical ionic lock of the DRY motif (Fig. 4E). The MD simulations show that all these mentioned residues maintain stable contacts corresponding to the active conformation of the receptor (fig. S10).
An overview of all CCR5 point mutations (either previously described or generated in this study) that hinder signaling but do not reduce chemokine binding (Fig. 4F) confirms the essential aspects of the global signal transmission from the chemokine binding site to the canonical GPCR microswitch network. Such mutations comprise (i) F852.59, Y892.63, and L1043.28 (34, 35), suggesting that shaping and clasping of the hinge by the TM2/TM3 counter bearing is necessary for agonist efficacy; (ii) Y1083.32, F1093.33, and F1123.36 [(35) and this study], proving the importance of the aromatic connector; and (iii) E2837.39 and M2877.43 (this study), showing the involvement of residues in TM7 in the shaping of the agonist conformation of the chemokine and as a possible route to the rearrangement of W2486.48.
Gi interactions
The binding interface of Gi to CCR5 is mediated exclusively by the Gα subunit (fig. S11) and can be divided into two main regions: the rim and the core (Fig. 5A and fig. S12). The rim contains two clearly separated parts (Fig. 5B): a proximal side formed by the end of the αN helix (αNβ1) and nearby β strands (β2β3) in Gα and ICL2 in the receptor and a distal side formed by β strands (α4β6) in Gα and ICL3 in the receptor. In addition, the rim also includes interactions between α5 in Gi and ICL2/3 of the receptor. The core of the CCR5•Gi complex interface is formed exclusively by interactions of α5 in Gi (Fig. 5A). Here, the Gi α5 helix interacts with the cytoplasmic sides of TM2, TM3, and TM5 in one side of the core binding pocket, while the C-terminal hook of α5 (residues 352 to 354) leans toward TM6 and ICL4.
Fig. 5. Binding interfaces between CCR5 and Gαi.
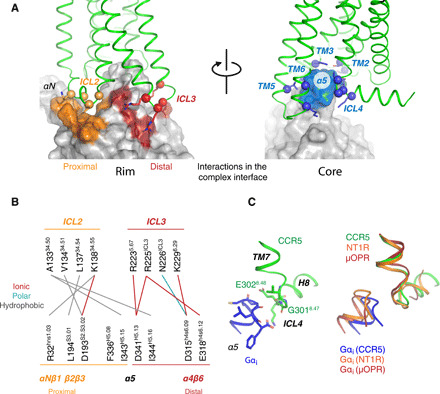
(A) Binding interfaces at the rim (left) and core (right) of the complex. Each interface is colored on the surface of the G protein and interacting residues in the receptor are shown as spheres (Cα) and sticks (side chains). (B) Residue-residue interactions at the rim of the binding interface. (C) Left: Structure of the α5 hook and ICL4 in the [6P4]CCL5•CCR5 complex. Key residues are shown as sticks. Right: Comparison between the α5 hook and ICL4 in the Gi-bound receptors CCR5, neurotensin type 1 (NT1R), and μ-opioid (μOPR).
These interfaces are common to all GPCR/G protein complexes, as they arise from the common overall relative orientation of the bound components. However, analysis of the currently available complexes reveals that the precise location and nature of the individual interface contacts vary to a certain degree (fig. S13). At the proximal rim of the interface, contacts are mostly hydrophobic and consistent with other Gi complexes. At the distal rim, we observe several ionic interactions absent in other structures. However, the most noticeable differences lie in the core region of the binding interface, where we observe different contacts between the hook of α5 (the last three C-terminal residues of Gi) and ICL4 of CCR5. This is due to a distinct conformation of ICL4 of CCR5 in which G3018.47 and E3028.48 slightly relocate compared to, e.g., the Gi complexes of the neurotensin type 1 (NT1R) or μ-opioid (μOPR) receptors, resulting in a different set of interactions between E3028.48 and the hook of α5 (Fig. 5C). A 3D variability analysis of the cryo-EM density reveals structural heterogeneity around ICL4 that allows to model a main conformation as shown in Fig. 5C and a second minor conformation that is similar to the NTR1 and μOPR complexes (fig. S14, A to C). The MD simulations indicate that ICL4 reverts to a preferred conformation in the absence of Gαi (fig. S14D). An analysis of further solved GPCR•Gi structures also highlights the structural plasticity of ICL4 (fig. S15).
Structure-activity relationship of CCR5 chemokine ligands
The comparison between our structure and the inactive [5P7]CCL5•CCR5 complex (13) allows us to precisely pinpoint the activation mechanism of CCR5 by a chemokine agonist (Fig. 6A). The overall binding poses of the [5P7]CCL5 antagonist (13) and the [6P4]CCL5 agonist are similar, with the globular core of the chemokine held by the receptor N terminus and ECL2 and the chemokine N terminus reaching deep into the receptor TM bundle. However, despite having the same 10-residue length, the N termini of the two CCL5 derivatives differ in their amino acid sequences. This results in different chemokine/receptor interactions in this region: small, hydrophilic, or negatively charged residues in sequence positions 4 and 5 of [6P4]CCL5 lead to a straight conformation of the proximal N terminus that pushes residue P3 against the bottom of CRS2. Thus, P3 exerts a force that is bolstered on the counter bearing hydrophobic residues W862.60 and Y892.63 onto the aromatic connector and residue M2877.43. This triggers the canonical GPCR activation switches resulting in the relocation of TM5/6/7 and the stabilization of the receptor active conformation. In contrast, the large hydrophobic residues at positions 4 and 5 in [5P7]CCL5 force the proximal N-terminal hinge into a turn structure making P3 recede (Figs. 6A and 3B) and thereby leaving the receptor in the inactive state. The highly conserved (~70% in nonolfactory human class A GPCRs) residue W2486.48 lies at the center of these activating conformational changes, connecting the rearrangements at H2897.45 and Y2446.44 and, thus, the large-scale relocation of TM7 and TM6.
Fig. 6. Activation mechanism of CC chemokine receptors.
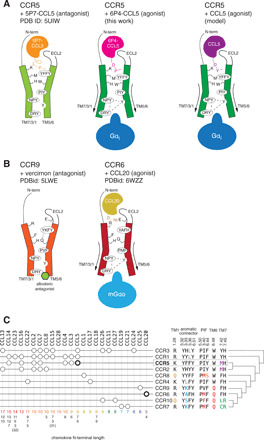
(A) CCR5 bound to the antagonist [5P7]CCL5 (left, orange), the super-agonist [6P4]CCL5 (center, magenta), and the natural agonist CCL5 (right, purple). Key residues in the activation mechanism of CCR5 are shown. (B) Proposed activation mechanism for CCR6 by CCL20 (yellow, right) by comparing with the structure of CCR9 (left). (C) Pairing between CC chemokine receptors and CCL chemokines (59). At the right, the sequence composition of key positions is shown, together with the phylogenetic relationship between the receptors. The lengths of the CCL chemokine N termini according to UniProt (60) are shown at the bottom. The available active CCR/CCL complex structures are shown in bold.
On the basis of their N-terminal sequence, we expect that other identified CCR5 agonist or antagonist chemokines feature respective similar deeper (6P4[CCL5]-active-like) or less deep (5P7[CCL5]-inactive-like) positions of their N-terminal turns within CRS2. Using our structure as a template, we modeled the wild-type agonist CCL5 bound to CCR5 (fig. S16 and movie S3). CCL5, as the [6P4]CCL5 agonist, features an aspartate in its N terminus (D6) able to interact with K261.28. The MD simulations reveal a similar deep binding pose of the CCL5 N terminus with a straight-hinge conformation of residues 5 to 8 where Y3 could be playing the role of P3 in [6P4]CCL5 to engage the aromatic connector and M2877.43 (Fig. 6A). As expected, a previous model of the active CCL5•CCR5 complex based on the inactive [5P7]CCL5•CCR5 structure (13) does not show this straight-hinge conformation but rather the inactive helical turn. The CCR5 chemokine agonist ligands CCL3 and CCL4 are closely related to CCL5 having similar N-terminal sequence lengths and compositions (Fig. 3C). We therefore expect that these chemokines also adopt the straight-hinge conformation and use the same activation mechanism as [6P4]CCL5 or CCL5 with the aspartates at position 5 and the bulky residues at position 2 carrying out analogous functions.
DISCUSSION
The activation mechanism in CCL5/CCR5, in which the N terminus of the chemokine reaches deep into the TM bundle, differs substantially from that of CCL20/CCR6, where a much shorter CCL20 adopts a shallower binding pose and engages a noncanonical activation mechanism (Figs. 3B and 6B) (17). Thus, CC chemokine receptors can apparently be activated through two very different mechanisms by “long” and “short” chemokines. But what are the molecular features in the receptor that determine the type of activation? A phylogenetic analysis of CC chemokine receptors (fig. S17) puts CCR5 and CCR6 into distinct subgroups. A more detailed sequence comparison of key residues in the activation mechanism shows that CC chemokine receptors can be divided into two main groups according to the nature of the residue at position 6.48 (W versus Q) and, to some extent, of the aromatic connector (Fig. 6C). CC chemokine receptors featuring the conserved W6.48 (CCR1, CCR2, CCR3, CCR4, CCR5, and CCR8) tend to be more promiscuous and preferentially recognize chemokines with longer N termini (9 to 14 residues) (Fig. 6C). On the other hand, CC chemokine receptors featuring Q6.48 (CCR6, CCR9, CCR7, and CCR10) bind to only a few (1 to 2) chemokines with short N termini (4 to 9 residues). Although position 6.48 allows for a certain degree of variability in human class A GPCRs (70% W, 15% F, 5% Y, and 10% other), a Q at this position is exclusive of this subgroup of chemokine human receptors, supporting the uniqueness of this “shallow” activation mechanism.
It is interesting to observe that many chemokines undergo posttranslational proteolytic processing leading to different N-terminal lengths, which may constitute a layer of regulation (36, 37). Thus, a CCL5 variant lacking the first two N-terminal residues (CCL53–68) behaves as a natural chemotaxis inhibitor, and a 10-fold higher concentration compared to wild-type CCL5 is required to induce a significant calcium response (37). Similarly, CCL54–68 has an about 10-fold lower affinity for CCR5 compared to CCL53–68 or wild-type CCL5 and is less potent in stimulating lymphocyte chemotaxis or inhibiting HIV infection (36). These findings are in complete agreement with the lack of contacts at the bottom of the CCR5 CRS2 region expected for such CCL5 truncations.
The structure of CCR5 in an active conformation allows us to elucidate a novel activation pathway of CC chemokine receptors by a chemokine agonist. In CCR5 and related receptors (CCR1, CCR2, CCR3, and CCR4), the respective cognate chemokines have long N termini and bind deep into the orthosteric pocket (CRS2), thereby triggering the rearrangement of an aromatic connector in TM3 and TM6 and of the TM7 backbone. The activating force exerted by the deep binding [6P4]CCL5 N terminus appears to be stabilized by a cluster of hydrophobic CCR5 residues in TM2 and TM3 that line the extended N-terminal hinge of this agonist chemokine. Understanding this force balance may help in the design of small-molecule agonists, which could activate the connector region at the bottom of CRS2 by pushing against this counter bearing.
W6.48 lies at the center of these conformational changes connecting the receptor activation pathways through TM7 and TM6. In contrast, a subgroup of CC chemokine receptors (CCR6, CCR7, CCR9, and CCR10) harbors a Q residue at this position, a unique feature in human class A GPCRs. The cognate chemokines of these receptors have shorter N termini featuring a shallow binding mode and a specialized mode of activation. We expect that our findings will help to rationalize the relationship between sequence, structure, and activity of chemokines and their receptors and aid drug discovery.
METHODS
Protein expression and purification
The wild-type human CCR5 gene containing a C-terminal 3C cleavage site followed by a FLAG-tag was cloned into the pFastBac1 vector and expressed in Spodoptera frugiperda Sf9 insect cells using the baculoviral infection system. CCR5 expression and membrane preparation were performed as described (15). Membranes from a 1-liter culture of Sf9 cells were resuspended in 10 ml of lysis buffer containing iodoacetamide (2 mg/ml), and EDTA-free complete protease inhibitor cocktail tablets, and incubated at 4°C for 1 hour. Then, membranes were solubilized by supplementing 0.5% lauryl maltose neopentyl glycol (LMNG) at 4°C for 3 hours. The soluble fraction was isolated by centrifugation at 140,000g and incubated with 1 ml of M2 anti-FLAG affinity resin overnight at 4°C. The latter column was washed with 10 column volumes (CV) of washing buffer 1 [25 mM Hepes, 400 mM NaCl, 10% glycerol, and 0.1% LMNG (w/v), pH 7.5], followed by 10 CV of washing buffer 2 (25 mM Hepes, 400 mM NaCl, 2 mM adenosine 5′-triphosphate, 5 mM MgCl2, 10% glycerol, and 0.1% LMNG, pH 7.5) and subsequently washed with another 6 CV of washing buffer 1. The receptor was eluted with 3 CV of elution buffer consisting of 25 mM Hepes, 400 mM NaCl, 0.01% LMNG, and FLAG peptide (200 μg/ml; DYKDDDDK; pH 7.5).
A DNA construct of [5P14]CCL5 cloned into a pET32a vector was a gift of P. LiWang. The DNA sequence of [6P4]CCL5 was obtained by mutating this [5P14]CCL5 construct using standard QuickChange polymerase chain reaction. [6P4]CCL5 with enterokinase-cleavable N-terminal thioredoxin fusion and hexa-histidine tags was expressed in the Escherichia coli BL21 (DE3) strain cultured in Lysogeny broth media. Protein production was induced with 1 mM isopropyl β-d-thiogalactopyranoside when the optical density at 600 nm reached 0.7 to 0.8. After induction, cells were grown for 20 hours at 22°C and then harvested by centrifugation. Ten grams of the cell pellet was resuspended in 50 ml of resuspension buffer (50 mM tris, 6 M guanidinium HCl, and 200 mM NaCl, pH 8.0) and lysed using a French press. The supernatant was isolated by centrifugation at 27,000g for 1 hour and applied to a 5-ml HisTrap column. The column was washed with 10 CV of resuspension buffer and eluted with 3 CV of 60 mM NaOAc, 200 mM NaCl, and 6 M guanidinium HCl. β-Mercaptoethanol (20 mM) was added to the elution fraction and incubated for 1 hour. The denatured protein was added dropwise into 250 ml of folding buffer (550 mM l-arginine hydrochloride, 20 mM tris, 200 mM NaCl, 1 mM EDTA, 1 mM reduced glutathione, and 0.1 oxidized glutathione, pH 8.0) and incubated overnight at 4°C. The solution was concentrated [molecular weight cutoff (MWCO), 10 kDa] and dialyzed in 20 mM tris, 200 mM NaCl, and 2 mM CaCl2 (pH 8.0). To cleave the fusion tags, enterokinase (New England Biolabs) was added, and the solution was incubated for 24 hours at room temperature. The protein was separated from the fusion tag using an acetonitrile gradient on a C4 reversed-phase chromatography column (Vydac, Hesperia, CA) and then lyophilized. The lyophilizate was resuspended in 25 mM phosphate buffer (pH 4). The N-terminal amino acid of [6P4]CCL5 glutamine (Q0) was cyclized at 37°C for 48 hours.
The human Gαi subunit (Gαi1) with an N-terminal tobacco etch virus (TEV) protease–cleavable deca-histidine tag was expressed in the E. coli BL21 (DE3) strain and purified as described (22).
The transducin heterotrimer was isolated from the rod outer segment of bovine retina (W L Lawson Company) and Gβ1γ1 was separated from Gαt with Blue Sepharose 6 Fast Flow (GE Healthcare) as described (22). The Gαi1β1γ1 heterotrimer (Gi) was prepared by mixing equimolar amounts of Gαi1 and Gβ1γ1 and incubated at 4°C for 1 hour shortly before use for CCR5-Gi complex formation. Fab16 was produced by papain digestion of immunoglobulin G16 as described (22).
Formation of the [6P4]CCL5•CCR5•Gi•Fab16 complex
Pooled fractions of CCR5 eluted from the anti-FLAG resin and a molar excess of Gi heterotrimer were mixed together and incubated for 30 min. Then, an equimolar amount of [6P4]CCL5, together with apyrase (25 mU/ml), was added and incubated for another 2 hours. The complex was mixed with molar excess (1:1.4) of Fab16 and further incubated for at least 1 hour. The mixture of [6P4]CCL5•CCR5•Gi and Fab16 was concentrated using an Amicon Ultra concentrator (MWCO, 100 kDa) and loaded onto a Superdex 200 Increase 10/300 GL column for size exclusion chromatography (SEC) with buffer containing 25 mM Hepes, 150 mM NaCl, and 0.01% LMNG (pH 7.5). The protein quality of each fraction was evaluated by SDS–polyacrylamide gel electrophoresis (fig. S1, A and B). Fractions showing good purity and complex integrity were pooled together and concentrated for EM grid preparation.
Cryo-EM sample preparation and image acquisition
For cryo-EM, 3.5 μl sample (2.5 mg/ml) was directly applied to glow-discharged 200-mesh carbon grids (Quantifoil Cu R1.2/1.3, 200 mesh). Grids were immediately plunge-frozen in liquid ethane using an FEI Vitrobot Mark IV (Thermo Fisher Scientific) with a blotting time of 3 s. The grids were screened for ice thickness and particle distribution using a Glacios Cryo-TEM operated at 200 kV. Images were acquired from the selected grid using a Glacios Cryo-TEM (Thermo Fisher Scientific) operated at 200 kV equipped with a Gatan K3 Summit direct electron detector (Gatan Inc.). Automated data collection was carried out using SerialEM with a set of customized scripts enabling automated low-dose image acquisition (38, 39) and online prescreened during data collection using FOCUS (40). Movie stacks of 40 frames were obtained with a defocus range of −1.0 to −2.0 μm at a magnification of ×45,000 (nominally ×36,000) and the K3 detector operated in super-resolution mode (super-resolution pixel size, 0.556 Å). Each movie had a total accumulated dose exposure of ~49 e/Å2. A total of 2586 image stacks were collected for the [6P4]CCL5•CCR5•Gi•Fab16 complex.
Cryo-EM data processing
Contaminated micrographs were removed manually. Patch motion correction and patch contrast transfer function (CTF) parameter estimation were performed using algorithms implemented in cryoSPARC v2.15.0 (41). After sorting, micrographs with estimated resolution worse than 6.0 Å were discarded. The remaining motion-corrected images summed with dose weighting were used for all further image processing in cryoSPARC. Approximately 2.6 million particles were auto-picked and subjected to several rounds of reference-free 2D classification to remove false-positive particles. A total of 345,458 particles from 3D classes that demonstrated clear structural features were combined and subjected to 3D refinement, which led to a reconstruction at 3.6-Å resolution. Nonuniform refinement (42) with subsequent local refinements was performed in cryoSPARC v3.1.0 and improved the overall resolution to 3.15 Å [Fourier shell correlation (FSC) = 0.143].
The final set of homogeneous [6P4]CCL5•CCR5•Gi•Fab16 complex particles was subjected to 3D variability analysis implemented in CryoSPARC (43) using three variability components and a low-pass filter resolution of 4 Å after applying a soft mask to exclude solvent and micelle.
Reported resolutions calculated with a soft shape mask are based on the gold-standard FSC using the 0.143 criterion. The local resolution was determined using ResMap (44).
Model building and refinement
The crystal structures of the Gi heterotrimer [Protein Data Bank (PDB) ID: 5KDO], Fab16 (PDB ID: 6QNK), and the [5P7]CCL5•CCR5 complex (PDB ID: 5UIW) were used as initial templates for model building. The models were docked into the 3D map as rigid bodies in Chimera (45). The [6P4]CCL5 N terminus (up to the residue 9) was built ab initio. As compared to residues 1 to 9, a lower definition of the density was observed in the region of the N-terminal pyroglutamate (PCA0). The remaining part of [6P4]CCL5 was taken from the 5UIW structure. Several rounds of manual building were performed in Coot (46). The model was finalized by refinement in Phenix 1.18.2. (47) against the 3.15-Å cryo-EM map. Structural figures were prepared in Chimera and PyMOL (https://pymol.org/2/). The refinement statistics are summarized in table S1.
Amino acid sequence analysis
The analysis of N-terminal sequence similarity of the natural amino acid CCL5 variants (table S2) was carried using WebLogo (48).
Characterization of resistance to GTPγS
To assess the stability of the purified [6P4]CCL5•CCR5•Gi complexes with or without Fab16, they were incubated with 100 μM guanosine 5′-O-(3′-thiotriphosphate) (GTPγS) in 25 mM Hepes, 150 mM NaCl, and 0.01% LMNG (pH 7.5) for 1 hour at 4°C and analyzed by SEC with a Superdex 200 Increase 10/300 GL column monitoring the protein intrinsic tryptophan fluorescence (λex = 280 nm; λem = 350 nm). As standards, SEC analyses were also carried out on purified Fab16, Gβγ, Gαi, and mixtures thereof using identical buffer conditions. The respective chromatograms are shown in fig. S1C.
Cellular Ca2+ flux assays for receptor activation
Human CCL5 and reference standard [6P4]CCL5 were prepared by chemical synthesis as previously described (10, 11). [6P4]CCL5 variants (D5A and D5K), as well as a sample of unmodified [6P4]CCL5, were prepared using a previously described multiplex chemical synthesis approach (49).
For experiments involving CCR5 mutants, HEK cells were transiently transfected with expression vectors obtained by site-directed mutagenesis (Q5 Site-Directed Mutagenesis kit, New England Biolabs) of the parent FUGW-CCR5 vector, which was generated by Gibson Assembly (New England Biolabs) as previously described (50). HEK cells (1.25 × 106) were seeded overnight in 10-cm dishes and transfected with CCR5 expression vectors (jetPRIME, Polyplus Transfection) according to the manufacturer’s instructions. Cells were used in Ca2+ flux experiments 24 hours later. For experiments involving [6P4]CCL5 variants, HEK (10) and CHO (49) cell clones stably expressing CCR5 (HEK-CCR5 and CHO-CCR5, respectively) were used.
Ca2+ flux measurements were performed using a Functional Drug Screening System (FDSS) microcell device (HAMAMATSU). On the day of the experiment, cells were detached in phosphate-buffered saline (PBS) supplemented with 0.48 mM EDTA and added (20,000 cells per well) to wells of black-walled clear-bottom 384-well plates. Cells were then loaded with a calcium-sensitive fluorescent dye (Screen Quest Fluo-8 No Wash Calcium Assay Kit, AAT Bioquest) according to the manufacturer’s instructions. Fluorescence signals (excitation, 490 nm; emission, 525 nm) were recorded before and after addition of agonist (dissolved in PBS supplemented with 1% bovine serum albumin and 25 mM Hepes) at defined concentrations. Agonist responses were defined as the maximum Ca2+ flux fluorescence signal divided by that of a control well with cells treated with buffer only. Dose-response curves were fitted (GraphPad Prism) to the agonist responses R at each concentration using a three-parameter agonist versus response model, , where R0 presents the baseline and Rmax − R0 = Emax.
Modeling and MD simulations
CCR5 N-terminal residues 1 to 19 were built using as template residues 1 to 14 of the NMR solution structure of a doubly sulfated (at Y10 and Y14) N-terminal segment of CCR5 bound to CCL5 (PDB ID: 6FGP). The chemokine in the latter structure was then used as a guide for the structural alignment to our cryo-EM structure. Remaining residues 15 to 19 of CCR5 were then connected to the rest of the cryo-EM model using Modeller v9.16 (51). All models derived from Modeller were then subjected to 300 iterations of variable target function method optimization and MD and simulated annealing optimization (within Modeller), scored using the discrete optimized protein energy potential, and the best-scoring model was selected (Fig. 1D, right).
This model of CCR5 (residues 1 to 320) bound to [6P4]CCL5 was used for MD simulations of the nonsulfated and sulfated (Y10 and Y14) forms. Coordinates were first preprocessed using VMD1.9.3 (52). The receptor-ligand complex (i.e., CCR5-[6P4]CCL5 or CCR5-CCL5) was then embedded into a 90 Å × 90 Å lipid bilayer composed of 80% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and 20% cholesterol. The system was solvated with explicit water molecules, neutralized, and its ionic strength was adjusted using the CHARMM-GUI builder (53). Disulfide bridges were explicitly defined between C50-C11 and C34-C10 in CCL5 or [6P4]CCR5 and C1013.25-C178 and C20-C2697.25 in CCR5. Except for CCR5 residues D762.50, E2837.39, and E3028.48, which were protonated, all titratable residues of CCR5 and CCL5 were left in their dominant protonation state at pH 7.0. Before production runs, the geometry of the system was optimized by energy minimization and further relaxed by a sequence of equilibration steps where harmonic positional restraints were applied to all Cα atoms of the protein and gradually released throughout the equilibration. In the last equilibration step (i.e., before completely releasing all protein restraints), water, ion, and lipids were allowed to diffuse without restraints during 50 ns to allow for adequate equilibration of the lipid mixture. After equilibration was completed, five independent trajectories of each system were spawned from the last snapshot of the equilibrated trajectory using a random seed. Production simulations for each replica were run in the isothermal-isobaric (NPT) ensemble at 1013 bar and 310 K for 500 ns each. All simulations were run using Gromacs v2020 (54) with the CHARMM36m force field (55). Gromacs v2020 and VMD1.9.31 were used to postprocess and analyze all trajectories. MD simulation figures were rendered using VMD1.9.3 and the R ggplot2 library (56). Circular plots of residue contacts were generated using the mdciao library (57).
The equilibrated model [6P4]CCL5 bound to CCR5 was used to model the binding pose of the wild-type CCL5. The sequence of CCL5 was threaded on [6P4]CCL5 (6P4: QGPPGDIVLACC/CCL5: SPYSSDTTP-CC) and steric clashes were relieved using the molecular graphics software PyMOL. Using this structure as a template, residues 1 to 9 of CCL5 and all residues within 8 Å around Y3 of CCL5 were remodeled with Modeller v9.16 using the protocol described above. The stability of the resulting binding pose was assessed by MD simulations using the protocol described above.
A list of simulations performed in this work is given in table S4. MD simulations were performed at the Paul Scherrer Institute computing cluster and at the Swiss National Supercomputing Centre (CSCS).
Electrostatic potentials were calculated using the Adaptive Poisson-Boltzmann Solver (APBS) method (58) as implemented in PyMOL using a concentration of 0.150 M for the +1 and 1 ion species. The biomolecular surface is colored from red (5 kT/e) to blue (+5 kT/e) according to the potential on the soluble accessible surface.
Acknowledgments
This work was supported by the Swiss National Science Foundation (grants 149927 and 173089 to S.G.; grant 192780 to X.D.; grant 184828 to O.H.; SNF R’EQUIP 177084 to T.M.; NCCR TransCure to H.S.; and SNF Sinergia 183563 to G.F.X.S.), the Synapsis Foundation (grant 2018-P104 to G.F.X.S.), and the European Union (grants FP6-EMPRO and FP7-CHAARM to S.G.). We acknowledge M. Chami and L. Kovacik (Biozentrum BioEM Lab) for help with cryo-EM data collection, P. LiWang (University of California) for a gift of the [5P14]CCL5 DNA construct, M. Rogowski (Biozentrum) for preparation of [6P4]CCL5, J. Muehle (Paul Scherrer Institute) for preparation of Fab16, M. Caubet (High Performance Computing and Emerging Technologies Group, Paul Scherrer Institute) for technical support with MD simulations, S. Isogai for helpful discussion, and the sciCORE facility of the University of Basel for the computer infrastructure. Author contributions: P.I., A.G., and S.G. conceived the study. P.I., C.-J.T., and F.P. expressed and purified proteins and developed the protocol for forming the [6P4]CCL5•CCR5•Gi•Fab16 complex. A.G. assisted with the expression and purification of CCR5. P.I. and C.-J.T. prepared cryo-EM grids. K.N.G. and P.I. collected the cryo-EM data. P.I. and N.D. processed the cryo-EM data and built and refined the model. X.D. and R.G.-G. designed and performed MD simulations. H.S. and T.M. provided guidance on EM sample preparation, data collection, and model refinement. C.B. generated the CCR5 mutants used in cellular Ca2+ flux assays. M.P.-B. synthesized chemokine analogs for use in the signaling assays, which were performed and analyzed by N.C. and F.C. P.I., X.D., O.H., and S.G. analyzed the structure and wrote the manuscript, with input from C.-J.T., A.G., and G.F.X.S. Competing interests: G.F.X.S. is a cofounder and scientific advisor of the companies leadXpro AG and InterAx Biotech AG. O.H. is the inventor of 6P4[CCL5] and is both a shareholder and an affiliate of Orion Biotechnology Switzerland, to whom the rights to the invention have been assigned. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The cryo-EM map of [6P4]CCL5•CCR5•Gi•Fab16 has been deposited in the Electron Microscopy Data Bank as entry EMD-12746 and the corresponding model in the Protein Data Bank as entry 7O7F.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg8685/DC1
REFERENCES AND NOTES
- 1.Scurci I., Martins E., Hartley O., CCR5: Established paradigms and new frontiers for a ‘celebrity’ chemokine receptor. Cytokine 109, 81–93 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Alkhatib G., The biology of CCR5 and CXCR4. Curr. Opin. HIV AIDS 4, 96–103 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aldinucci D., Casagrande N., Inhibition of the CCL5/CCR5 axis against the progression of gastric cancer. Int. J. Mol. Sci. 19, 1477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin-Blondel G., Brassat D., Bauer J., Lassmann H., Liblau R. S., CCR5 blockade for neuroinflammatory diseases — Beyond control of HIV. Nat. Rev. Neurol. 12, 95–105 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Chua R. L., Lukassen S., Trump S., Hennig B. P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M. T., Kazmierski J., Timmermann B., Twardziok S., Schneider S., Machleidt F., Müller-Redetzky H., Maier M., Krannich A., Schmidt S., Balzer F., Liebig J., Loske J., Suttorp N., Eils J., Ishaque N., Liebert U. G., von Kalle C., Hocke A., Witzenrath M., Goffinet C., Drosten C., Laudi S., Lehmann I., Conrad C., Sander L.-E., Eils R., COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Patterson B. K., Seethamraju H., Dhody K., Corley M. J., Kazempour K., Lalezari J. P., Pang A. P., Sugai C., Francisco E. B., Pise A., Rodrigues H., Ryou M., Wu H. L., Webb G. M., Park B. S., Kelly S., Pourhassan N., Lelic A., Kdouh L., Herrera M., Hall E., Aklin E., Ndhlovu L., Sacha J. B., Disruption of the CCL5/RANTES-CCR5 pathway restores immune homeostasis and reduces plasma viral load in critical COVID-19. medRxiv 2020.05.02.20084673 (2020). [Google Scholar]
- 7.Cocchi F., DeVico A., Garzinodemo A., Arya S., Gallo R., Lusso P., Identification of RANTES, MIP-1α, and MIP-1β as the major HIV-suppressive factors produced by Cd8+ T-cells. Science 270, 1811–1815 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Trkola A., Paxton W. A., Monard S. P., Hoxie J. A., Siani M. A., Thompson D. A., Wu L., Mackay C. R., Horuk R., Moore J. P., Genetic subtype-independent inhibition of human immunodeficiency virus type 1 replication by CC and CXC chemokines. J. Virol. 72, 396–404 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simmons G., Clapham P. R., Picard L., Offord R. E., Rosenkilde M. M., Schwartz T. W., Buser R., Wells T. N. C., Proudfoot A. E. I., Potent Inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 276, 276–279 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Hartley O., Gaertner H., Wilken J., Thompson D., Fish R., Ramos A., Pastore C., Dufour B., Cerini F., Melotti A., Heveker N., Picard L., Alizon M., Mosier D., Kent S., Offord R., Medicinal chemistry applied to a synthetic protein: Development of highly potent HIV entry inhibitors. Proc. Natl. Acad. Sci. U.S.A. 101, 16460–16465 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaertner H., Cerini F., Escola J. M., Kuenzi G., Melotti A., Offord R., Rossitto-Borlat I., Nedellec R., Salkowitz J., Gorochov G., Mosier D., Hartley O., Highly potent, fully recombinant anti-HIV chemokines: Reengineering a low-cost microbicide. Proc. Natl. Acad. Sci. U.S.A. 105, 17706–17711 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weis W. I., Kobilka B. K., The molecular basis of G protein-coupled receptor activation. Annu. Rev. Biochem. 87, 897–919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng Y., Han G. W., Abagyan R., Wu B., Stevens R. C., Cherezov V., Kufareva I., Handel T. M., Structure of CC chemokine receptor 5 with a potent chemokine antagonist reveals mechanisms of chemokine recognition and molecular mimicry by HIV. Immunity 46, 1005–1017.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaik M. M., Peng H., Lu J., Rits-Volloch S., Xu C., Liao M., Chen B., Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 565, 318–323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan Q., Zhu Y., Li J., Chen Z., Han G. W., Kufareva I., Li T., Ma L., Fenalti G., Li J., Zhang W., Xie X., Yang H., Jiang H., Cherezov V., Liu H., Stevens R. C., Zhao Q., Wu B., Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 341, 1387–1390 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng P., Chen H., Zhu Y., Wang Z., Li J., Luo R.-H., Wang J., Chen L., Yang L.-M., Jiang H., Xie X., Wu B., Zheng Y.-T., Liu H., Structure-based design of 1-heteroaryl-1,3-propanediamine derivatives as a novel series of CC-chemokine receptor 5 antagonists. J. Med. Chem. 61, 9621–9636 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Wasilko D. J., Johnson Z. L., Ammirati M., Che Y., Griffor M. C., Han S., Wu H., Structural basis for chemokine receptor CCR6 activation by the endogenous protein ligand CCL20. Nat. Commun. 11, 3031 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu K., Wu L., Yuan S., Wu M., Xu Y., Sun Q., Li S., Zhao S., Hua T., Liu Z.-J., Structural basis of CXC chemokine receptor 2 activation and signalling. Nature 585, 135–140 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Burg J. S., Ingram J. R., Venkatakrishnan A. J., Jude K. M., Dukkipati A., Feinberg E. N., Angelini A., Waghray D., Dror R. O., Ploegh H. L., Garcia K. C., Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miles T. F., Spiess K., Jude K. M., Tsutsumi N., Burg J. S., Ingram J. R., Waghray D., Hjorto G. M., Larsen O., Ploegh H. L., Rosenkilde M. M., Garcia K. C., Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. eLife 7, e35850 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maeda S., Koehl A., Matile H., Hu H., Hilger D., Schertler G. F. X., Manglik A., Skiniotis G., Dawson R. J. P., Kobilka B. K., Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat. Commun. 9, 3712 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai C.-J., Marino J., Adaixo R., Pamula F., Muehle J., Maeda S., Flock T., Taylor N. M., Mohammed I., Matile H., Dawson R. J., Deupi X., Stahlberg H., Schertler G., Cryo-EM structure of the rhodopsin-Gαi-βγ complex reveals binding of the rhodopsin C-terminal tail to the gβ subunit. eLife 8, e46041 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glukhova A., Draper-Joyce C. J., Sunahara R. K., Christopoulos A., Wootten D., Sexton P. M., Rules of engagement: GPCRs and G proteins. ACS Pharmacol. Transl. Sci. 1, 73–83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scholten D. J., Canals M., Maussang D., Roumen L., Smit M. J., Wijtmans M., de Graaf C., Vischer H. F., Leurs R., Pharmacological modulation of chemokine receptor function. Br. J. Pharmacol. 165, 1617–1643 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isberg V., de Graaf C., Bortolato A., Cherezov V., Katritch V., Marshall F. H., Mordalski S., Pin J.-P., Stevens R. C., Vriend G., Gloriam D. E., Generic GPCR residue numbers – Aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 36, 22–31 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abayev M., Rodrigues J. P. G. L. M., Srivastava G., Arshava B., Jaremko Ł., Jaremko M., Naider F., Levitt M., Anglister J., The solution structure of monomeric CCL5 in complex with a doubly sulfated N-terminal segment of CCR5. FEBS J. 285, 1988–2003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farzan M., Mirzabekov T., Kolchinsky P., Wyatt R., Cayabyab M., Gerard N. P., Gerard C., Sodroski J., Choe H., Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96, 667–676 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Duma L., Häussinger D., Rogowski M., Lusso P., Grzesiek S., Recognition of RANTES by extracellular parts of the CCR5 receptor. J. Mol. Biol. 365, 1063–1075 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Kessler N., Akabayov S. R., Moseri A., Cohen L. S., Sakhapov D., Bolton D., Fridman B., Kay L. E., Naider F., Anglister J., Allovalency observed by transferred NOE: Interactions of sulfated tyrosine residues in the N-terminal segment of CCR5 with the CCL5 chemokine. FEBS J. 288, 1648–1663 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Nisius L., Rogowski M., Vangelista L., Grzesiek S., Large-scale expression and purification of the major HIV-1 coreceptor CCR5 and characterization of its interaction with RANTES. Protein Expr. Purif. 61, 155–162 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Wiktor M., Hartley O., Grzesiek S., Characterization of structure, dynamics, and detergent interactions of the anti-hiv chemokine variant 5P12-RANTES. Biophys. J. 105, 2586–2597 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lederman M., Veazey R., Offord R., Mosier D., Dufour J., Mefford M., Piatak M., Lifson J., Salkowitz J., Rodriguez B., Blauvelt A., Hartley O., Prevention of vaginal SHIV transmission in rhesus macaques through inhibition of CCR5. Science 306, 485–487 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Grahl A., Abiko L. A., Isogai S., Sharpe T., Grzesiek S., A high-resolution description of β1-adrenergic receptor functional dynamics and allosteric coupling from backbone NMR. Nat. Commun. 11, 2216 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blanpain C., Doranz B., Bondue A., Govaerts C., De Leener A., Vassart G., Doms R., Proudfoot A., Parmentier M., The core domain of chemokines binds CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J. Biol. Chem. 278, 5179–5187 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Govaerts C., Bondue A., Springael J.-Y., Olivella M., Deupi X., Poul E. L., Wodak S. J., Parmentier M., Pardo L., Blanpain C., Activation of CCR5 by chemokines involves an aromatic cluster between transmembrane helices 2 and 3. J. Biol. Chem. 278, 1892–1903 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Lim J. K., Burns J. M., Lu W., DeVico A. L., Multiple pathways of amino terminal processing produce two truncated variants of RANTES/CCL5. J. Leukoc. Biol. 78, 442–452 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Proost P., Meester I. D., Schols D., Struyf S., Lambeir A.-M., Wuyts A., Opdenakker G., Clercq E. D., Scharpé S., Damme J. V., Amino-terminal truncation of chemokines by CD26/dipeptidyl-peptidase IV. Conversion of RANTES into a potent inhibitor of monocyte chemotaxis and HIV-1-infection. J. Biol. Chem. 273, 7222–7227 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Schorb M., Haberbosch I., Hagen W. J. H., Schwab Y., Mastronarde D. N., Software tools for automated transmission electron microscopy. Nat. Methods 16, 471–477 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biyani N., Righetto R. D., McLeod R., Caujolle-Bert D., Castano-Diez D., Goldie K. N., Stahlberg H., Focus: The interface between data collection and data processing in cryo-EM. J. Struct. Biol. 198, 124–133 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Punjani A., Zhang H., Fleet D. J., Non-uniform refinement: Adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Punjani A., Fleet D. J., 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle Cryo-EM. J. Struct. Biol. 213, 107702 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liebschner D., Afonine P. V., Baker M. L., Bunkóczi G., Chen V. B., Croll T. I., Hintze B., Hung L.-W., Jain S., McCoy A. J., Moriarty N. W., Oeffner R. D., Poon B. K., Prisant M. G., Read R. J., Richardson J. S., Richardson D. C., Sammito M. D., Sobolev O. V., Stockwell D. H., Terwilliger T. C., Urzhumtsev A. G., Videau L. L., Williams C. J., Adams P. D., Macromolecular structure determination using x-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Biol. Crystallogr. 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crooks G. E., Hon G., Chandonia J.-M., Brenner S. E., WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paolini-Bertrand M., Cerini F., Martins E., Scurci I., Hartley O., Rapid and low-cost multiplex synthesis of chemokine analogs. J. Biol. Chem. 293, 19092–19100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scurci I., Akondi K. B., Pinheiro I., Paolini-Bertrand M., Borgeat A., Cerini F., Hartley O., CCR5 tyrosine sulfation heterogeneity generates cell surface receptor subpopulations with different ligand binding properties. Biochim. Biophys. Acta Gen. Subj. 1865, 129753 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Webb B., Sali A., Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Humphrey W., Dalke A., Schulten K., VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Jo S., Kim T., Im W., Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLOS ONE 2, e880 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 55.Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B. L., Grubmüller H., MacKerell A. D. Jr., CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.H. Wickham, Ggplot2: Elegant Graphics for Data Analysis (Springer, New York, 2009). [Google Scholar]
- 57.G. Pérez-Hernández, gph82/mdciao (2021); https://github.com/gph82/mdciao.
- 58.Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A., Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bachelerie F., Ben-Baruch A., Burkhardt A. M., Combadiere C., Farber J. M., Graham G. J., Horuk R., Sparre-Ulrich A. H., Locati M., Luster A. D., Mantovani A., Matsushima K., Murphy P. M., Nibbs R., Nomiyama H., Power C. A., Proudfoot A. E. I., Rosenkilde M. M., Rot A., Sozzani S., Thelen M., Yoshie O., Zlotnik A., International union of basic and clinical pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 66, 1–79 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.UniProt Consortium , UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg8685/DC1