

ALKBH5 maintains CD4+ T cell pathogenicity during autoimmune disease via regulating IFN-γ and CXCL2.
Abstract
N6-methyladenosine (m6A) modification is dynamically regulated by “writer” and “eraser” enzymes. m6A “writers” have been shown to ensure the homeostasis of CD4+ T cells, but the “erasers” functioning in T cells is poorly understood. Here, we reported that m6A eraser AlkB homolog 5 (ALKBH5), but not FTO, maintains the ability of naïve CD4+ T cells to induce adoptive transfer colitis. In addition, T cell–specific ablation of ALKBH5 confers protection against experimental autoimmune encephalomyelitis. During the induced neuroinflammation, ALKBH5 deficiency increased m6A modification on interferon-γ and C-X-C motif chemokine ligand 2 messenger RNA (mRNA), thus decreasing their mRNA stability and protein expression in CD4+ T cells. These modifications resulted in attenuated CD4+ T cell responses and diminished recruitment of neutrophils into the central nervous system. Our findings reveal an unexpected specific role of ALKBH5 as an m6A eraser in controlling the pathogenicity of CD4+ T cells during autoimmunity.
INTRODUCTION
N6-methyladenosine (m6A) is one of the most abundant modifications in mRNA and primarily distributed in 3′ untranslated regions, long internal exons, and around the stop codons of mRNA (1, 2). This modification is mainly controlled by three types of proteins: methyltransferases as “writers,” demethylases as “erasers,” and specific m6A-binding proteins as “readers” (3). By affecting adenosine methylation, these proteins regulate mRNA metabolism, including decay, splicing, and translation (4–6). Although the presence of m6A is associated with numerous physiological and pathological processes, the understanding of the impact of this modification on immune cell development and functionality remains limited.
The m6A “writer” complex consists of three core proteins: methyltransferase-like 3 (METTL3), METTL14, and Wilms tumor 1–associated protein (3, 7). Our previous results demonstrated that METTL3 governs the homeostasis and differentiation of T cells by targeting the interleukin-17 (IL-7)/signal transducer and activator of transcription 5 (STAT5)/suppressor of cytokine signaling (SOCS) pathway (8) and controls the function and stability of regulatory T cells (Tregs) by targeting the IL-2/STAT5/ suppressor of cytokine signaling (SOCS) signaling pathway (9). A recent study indicated that METTL3-mediated mRNA m6A methylation promotes the function of dendritic cells (DCs), up-regulating the translation of target genes responsible for activating T cells and enhancing cytokine production induced by the Toll-like receptor 4/nuclear factor κB signaling (10). These results not only suggest that the writer proteins enhance the degradation of target mRNAs to exert physiological functions in immune cells (3, 11) but also raise the possibility that m6A “eraser” enzymes may be involved in regulating immune cell homeostasis.
To date, two m6A eraser enzymes have been identified: the fat mass and obesity-associated protein (FTO) (12) and the alkylated DNA repair protein AlkB homolog 5 (ALKBH5) (13). FTO modulates alternative splicing by removing m6A in the vicinity of splice sites and preventing the binding of serine- and arginine-rich splicing factor 2 (14). ALKBH5 regulates the stability of target mRNA (15) and decreases the export of mRNA to the cytoplasm (13). Previous research demonstrated that ALKBH5-deficient macrophages restrict viral infection more effectively than wild-type (WT) cells by increasing mRNA decay and reducing the expression of α-ketoglutarate dehydrogenase (OGDH) protein, thus conferring host resistance against viral infection (16). Given the importance of the writer proteins in T cell function, it remains to be established whether, and, if so, to what extent, the m6A eraser proteins contribute to the regulation of T cell homeostasis and function.
Here, we found that, in comparison with FTO, ALKBH5 mRNA expression was specifically up-regulated upon T cell activation. Therefore, we constructed lineage-specific deletion of ALKBH5 in T cells (Alkbh5flox/floxCd4Cre) and documented that the transfer of naïve ALKBH5-deficient CD4+ T cells into lymphopenic Rag2−/− recipients failed to induce colitis. In addition, Alkbh5flox/floxCd4Cre mice with experimental autoimmune encephalomyelitis (EAE) exhibited a less severe course of the disease than WT littermates. Moreover, the ablation of ALKBH5 increased m6A modification on interferon-γ (IFN-γ) and C-X-C motif chemokine ligand 2 (CXCL2) mRNA in CD4+ T cells, thereby reducing mRNA stability and corresponding protein expression. These modifications resulted in impaired responses of CD4+ T cells and a decreased recruitment of neutrophils into the central nervous system (CNS) during neuroinflammation. These findings reveal that m6A eraser protein ALKBH5 exerts an unexpected biological function during T cell–mediated inflammation and autoimmunity.
RESULTS
The expression of ALKBH5 is up-regulated specifically upon T cell activation
Our previous results demonstrated that the deletion of m6A writer enzyme METTL3 in CD4+ T cells leads to the disruption of their homeostatic proliferation and differentiation into effector cells (8). Moreover, the lack of METTL3 in Tregs results in severe autoimmune disease because of the absence of suppressive activity of these cells (9). However, the effect of m6A eraser enzymes on the regulation of T cell development and function remains unclear. The analysis of the Immunological Genome Project database revealed that different T cell populations have an abundant expression of ALKBH5 and low expression of FTO (fig. S1A). We further validated that naïve CD4+ T cells predominately expressed ALKBH5 compared with FTO when activated with anti-CD3/CD28 for 0, 12, and 72 hours (fig. S1B). Given the significance of T helper (TH) cells in regulating immune responses (17), we cultured distinct TH cell subsets differentiated from naïve CD4+ T cells in vitro and found an increased expression of ALKBH5 mRNA in TH1, TH2, TH17, and Treg cells compared with naïve CD4+ T cells (Fig. 1A). Also, ALKBH5 mRNA expression was up-regulated in naïve CD4+ T cells upon in vitro activation with anti-CD3/CD28 antibodies at different time points (Fig. 1C). However, no evident change in the expression of FTO mRNA was observed in different subsets of TH cells or after activation of T cell receptor (TCR) signaling (Fig. 1, B and D). These findings suggest that ALKBH5, as a major m6A eraser enzyme, may have a role in regulating T cell function.
Fig. 1. ALKBH5 maintains the ability of naïve CD4+ T cells to induce adoptive transfer colitis.
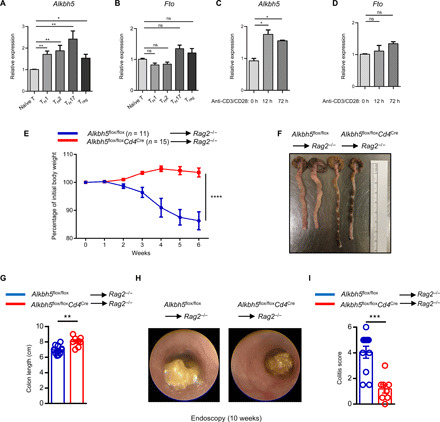
(A and B) Naïve CD4+ T cells isolated from WT mice were differentiated into TH1, TH2, TH17, and Treg cell subsets. The expression of Alkbh5 and Fto mRNA was measured by quantitative reverse transcription polymerase chain reaction (RT-qPCR). Data represent one of three independent experiments. (C and D) Naïve CD4+ T cells were activated with anti-CD3/CD28 for 0, 12, and 72 hours, and the expression of Alkbh5 and Fto mRNA was measured by qPCR. Data represent one of three independent experiments. (E) Naïve CD4+ T cells (5 × 105) from Alkbh5flox/floxCd4Cre mice or WT littermates were isolated, labeled with CellTrace, and transferred into Rag2−/− mice. The body weights of recipient mice were measured weekly (n = 11 to 15) and analyzed by two-way analysis of variance (ANOVA). Data represent three independent experiments. (F and G) The colons of mice from (E) were obtained at 12 weeks after the adoptive transfer colitis model, and representative photographs are shown in (F). (G) The length of the colon in each group was measured between the caecum and proximal rectum (n = 8 to 11) and analyzed by unpaired t test. Data represent three independent experiments. (H) Representative endoscopic views of the mouse colon after 10 weeks of adoptive transfer colitis are described under (E). (I) Colonoscopy severity score of mice in (E) after 10 weeks of adoptive transfer colitis (n = 8 to 11); results were analyzed by unpaired t test. Data represent three independent experiments. Data are shown as the means ± SEM. ns, not significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To determine the function of the eraser proteins in T cells, we used the CRISPR-Cas9 method to construct Alkbh5flox/flox mice (fig. S1C) and crossed the mice with Cd4Cre mice to obtain Alkbh5flox/floxCd4Cre offspring in which the expression of ALKBH5 mRNA and protein was specifically absent in CD4+ T cells (fig. S1, D and E). To further explore the role of ALKBH5 in the maintenance of T cell homeostasis in vivo, we compared the development of T cells in the thymus between WT littermates and Alkbh5flox/floxCd4Cre mice. The results demonstrated that the absence of ALKBH5 did not disrupt the late stages of T cell development in the thymus (fig. S2, A to C). After examining T cell homeostasis in peripheral lymphoid tissues, such as the spleen, inguinal lymph node (iLN), and mesenteric lymph node, we observed no differences between Alkbh5flox/floxCd4Cre and WT mice in T cell composition, activation, apoptosis, proliferation status, or cytokine secretion (figs. S2, A and D to G, S3, and S4). Moreover, there was no difference between these two genotypes in the composition of neutrophils, macrophages, DCs, and natural killer cells in the spleen and iLN during steady state (fig. S5), suggesting a dispensable role for T cell–specific ALKBH5 ablation in the development of other cell lineages. Collectively, these results demonstrate that the absence of ALKBH5 does not disrupt T cell development in the steady state.
ALKBH5 controls CD4+ T cell ability to induce autoimmune colitis
To establish whether ALKBH5 might maintain naïve T cell homeostasis in vivo, we isolated naïve CD4+ T cells from Alkbh5flox/floxCd4Cre mice and WT littermates, injected them separately into Rag2−/− mice to induce adoptive transfer colitis, and measured the body weight of recipient mice weekly. Adoptive transfer colitis is a well-established autoimmune colitis model to evaluate the homeostatic expansion of naïve T cells (8). As expected, mice that had received WT naïve CD4+ T cells began to lose weight at 2 weeks after transfer (Fig. 1E). In contrast, mice that had received ALKBH5-deficient naïve CD4+ T cells continued to gain weight, displayed milder colitis by endoscopy examination, and had a longer colon, a measure of colonic health and integrity, than recipients of WT CD4+ T cells (Fig. 1, E to I). These results imply that ALKBH5 controls the ability of naïve CD4+ T cells to induce adoptive transfer colitis.
We then analyzed the composition of donor CD4+ T cells at 12 weeks after the induction of adoptive transfer colitis. The infiltration of CD4+ T cells derived from Alkbh5flox/floxCd4Cre mice in the recipient’s colon was lower in comparison with cells derived from WT mice (Fig. 2, A to C). Moreover, the in vivo activation state and proliferation ability of naïve CD4+ T cells derived from WT and Alkbh5flox/floxCd4Cre mice were comparable (Fig. 2, D to G). Together, these results indicate that the failure of ALKBH5-deficient naïve CD4+ T cells to infiltrate into colon tissue after the induction of adoptive transfer colitis results in their reduced ability to promote colitis.
Fig. 2. Loss of ALKBH5 results in the failure of CD4+ T cells to accumulate in colon tissue during adoptive transfer colitis.
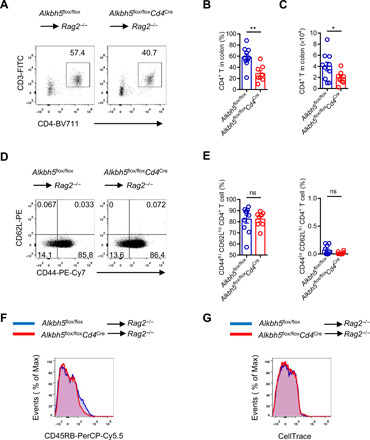
(A) Representative dot plots of the composition of colonic CD4+ T cells derived from Alkbh5flox/floxCd4Cre mice and WT littermates after 12 weeks of adoptive transfer colitis. (B and C) Ratio (B) and absolute number (C) of colonic CD4+ T cells described under (A) (n = 8 to 10); results were analyzed by unpaired t test. (D) Representative dot plots show the expression of CD44 and CD62L in colonic CD4+ T cells derived from Alkbh5flox/floxCd4Cre mice and WT littermates after 12 weeks of adoptive transfer colitis. (E) Ratio of colonic CD44hi CD62Llo CD4+ T and CD44lo CD62Lhi CD4+ T cells described under (D) (n = 8 to 10); results were analyzed by unpaired t test. (F) Representative histograms show the distribution of CD45RB staining intensity on colonic CD4+ T cells from Alkbh5flox/floxCd4Cre mice and WT littermates after 12 weeks of adoptive transfer colitis. (G) Representative histograms show the distribution of CellTrace staining intensity on colonic CD4+ T cells from Alkbh5flox/floxCd4Cre mice and WT littermates after 12 weeks of adoptive transfer colitis. Data represent three independent experiments and are shown as the means ± SEM. *P < 0.05 and **P < 0.01.
T cell–specific deletion of ALKBH5 confers protection against EAE
To further identify the role of ALKBH5 in regulating T cell function in vivo, we generated an EAE model. EAE is a T cell–driven autoimmune disease and is the most commonly used experimental model of the human inflammatory demyelinating disease-multiple sclerosis (18, 19). During the development of EAE, CD4+ T cells specific for antigens expressed in the CNS myelin initiate a localized inflammation that leads to demyelination, axonal transection, and clinical deficits (18). We found that immunizing WT mice with a myelin-specific autoantigen, myelin oligodendrocyte glycoprotein (MOG) peptide MOG35–55, led to overt EAE clinical scores (Fig. 3A). However, Alkbh5flox/floxCd4Cre mice were resistant to EAE induction (Fig. 3A). Histological staining revealed reduced lymphocyte infiltration and demyelination in the spinal cord of Alkbh5flox/floxCd4Cre mice than in WT littermates (Fig. 3B). These results demonstrate that the deletion of ALKBH5 in T cells mediates protection against experimental neuroinflammation.
Fig. 3. The deletion of ALKBH5 in T cells confers protection against EAE.
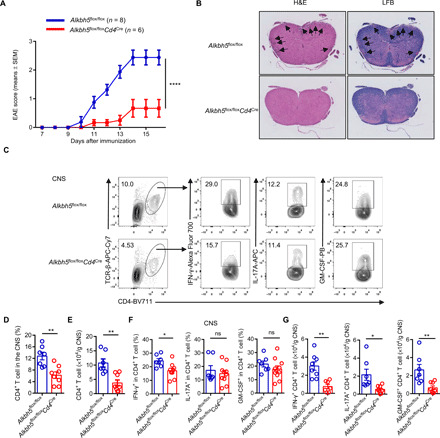
(A) Alkbh5flox/floxCd4Cre mice and WT littermates were immunized with the MOG35–55 peptide emulsified in a complete Freund’s adjuvant, clinical EAE scores obtained daily are shown (n = 6 to 8); results were analyzed by two-way ANOVA. (B) Representative images of hematoxylin and eosin (H&E) staining (left) and luxol fast blue (LFB) staining (right) of spinal cords of mice described under (A) after 30 days of EAE are shown. The black arrows indicate lymphocyte infiltration in H&E-stained sections and demyelinated lesions in LFB-stained sections. (C) Representative dot plots show the composition of CD4+ T cells and the expression of IFN-γ, IL-17A, and GM-CSF in CD4+ T cells from the CNS of Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE. APC, Allophycocyanin; PB, Pacific Blue. (D and E) Percentages (D) and absolute numbers (E) of CD4+ T cells described under (C) are shown (n = 7 to 10); results were analyzed by unpaired t test. (F and G) Percentages (F) and quantification (G) of IFN-γ–, IL-17A–, and GM-CSF–positive CD4+ T cells described under (C) are shown (n = 7 to 10); results were analyzed by unpaired t test. Data represent one of four independent experiments and are shown as the means ± SEM. *P < 0.05, **P < 0.01, and ****P < 0.0001.
The analysis of T cell composition by flow cytometry documented that Alkbh5flox/floxCd4Cre mice had decreased infiltration of CD4+ T cells in the CNS but an increased CD4+ T cell number in the draining lymphoid nodes (dLNs) than WT mice (Figs. 3, C to E, and 4, A to D). Recently, CD8+ T cells were shown to be involved in EAE pathogenesis (20, 21); however, we did not found differences in the composition of CD8+ T cells in the CNS or dLN between WT and Alkbh5flox/floxCd4Cre mice (fig. S6, F and G). Considering the importance of pathogenic CD4+ T cells in triggering inflammatory cascades, we sought to measure the expression of critical proinflammatory cytokines derived from CD4+ T cells, such as IFN-γ, IL-17A, and granulocyte-macrophage colony-stimulating factor (GM-CSF) in EAE (22, 23). We found that ALKBH5-deficient CD4+ T cells displayed less robust IFN-γ secretion in the CNS than the cells from WT mice, but the expression of IL-17A and GM-CSF during neuroinflammation remained unaffected (Fig. 3, C and F). Given the decreased number of CD4+ T cells in the CNS of ALKBH5-deficient mice, the number of different effector T cell subsets was also lower than in WT mice (Fig. 3, E and G). However, despite the increased CD4+ T cell number in the dLN of Alkbh5flox/floxCd4Cre mice, the percentage of effector T cells expressing IFN-γ, IL-17A, or GM-CSF within the population of CD4+ T cells in the dLN of Alkbh5flox/floxCd4Cre mice was similar to that in WT littermates (Fig. 4, B, D to F). The comparison of the activation, proliferation, and apoptosis of CD4+ T cells from Alkbh5flox/floxCd4Cre and WT mice did not reveal any differences between these mice in EAE (fig. S6, A to E). Collectively, these data demonstrate that ALKBH5 deficiency inhibits CD4+ T cell trafficking into the CNS during EAE and decreases the secretion of IFN-γ in the CNS.
Fig. 4. ALKBH5 deficiency promotes CD4+ T cell retention in draining lymph nodes during EAE.
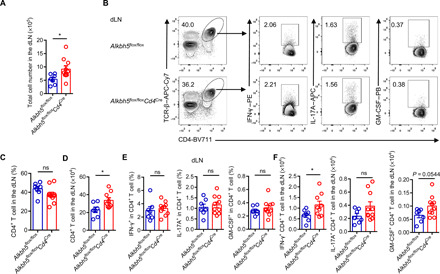
(A) Alkbh5flox/floxCd4Cre mice and WT littermates were immunized with the MOG35–55 peptide emulsified in a complete Freund’s adjuvant, and total cell numbers in draining lymph nodes (dLNs) were counted after 14 days (n = 7 to 10); results were analyzed by unpaired t test. (B) Representative dot plots show the composition of CD4+ T cells and expression of IFN-γ, IL-17A, and GM-CSF in CD4+ T cells in the dLN from Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE. PE, Phycoerythrin. (C and D) Percentages (C) and absolute numbers (D) of CD4+ T cells in the dLN from Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE (n = 7 to 10); results were analyzed by unpaired t test. (E and F) Percentages (E) and quantification (F) of IFN-γ–, IL-17A–, and GM-CSF–positive CD4+ T cells in the dLN from Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE (n = 7 to 10); results were analyzed by unpaired t test. Data represent one of four independent experiments and are shown as the means ± SEM. *P < 0.05.
Our previous work indicated that Tregs require the writer enzyme METTL3 to initiate its suppressive functions (9), whether the less EAE pathogenesis observed in Alkbh5flox/floxCd4Cre mice was due to the increased Tregs inhibitory abilities originating from eraser enzyme ALKBH5 remains unknown. Therefore, we bred Alkbh5flox/flox mice with Foxp3Cre mice to obtain Alkbh5flox/floxFoxp3Cre offspring in which the expression of ALKBH5 was specifically deleted in Tregs. However, the deficiency of ALKBH5 in Tregs did not affect EAE development (fig. S7), which rules out the possibility of Tregs in mediating the present phenotypes we observed in Alkbh5flox/floxCd4Cre mice.
Since T cells instruct myeloid cells to induce inflammation and trigger pathologic changes during experimental neuroinflammation (24), we assessed the composition of different myeloid cells in the CNS, dLN, and spleen in Alkbh5flox/floxCd4Cre and WT mice during the course of EAE. There was less accumulation of neutrophils in the CNS and increased retention in the dLN and spleen in Alkbh5flox/floxCd4Cre mice (Fig. 5, A and D). However, both groups of mice displayed comparable composition of macrophages and DCs in different organs (Fig. 5, B, C, E, and F). Thus, the lack of ALKBH5 in T cells specifically inhibits the ability of T cells to recruit neutrophils into the CNS during neuroinflammation.
Fig. 5. T cell–specific ALKBH5 ablation inhibits neutrophil recruitment in the CNS during EAE.
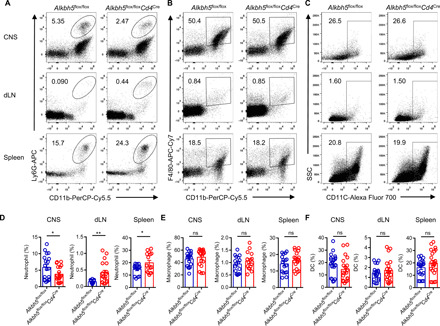
(A to C) Representative dot plots show the composition of (A) neutrophils (CD45+ CD11b+ Ly6G+), (B) macrophages (CD45+ CD11b+ F4/80+), and (C) DCs (CD45+ CD11c+) in the CNS, dLN, and spleen of Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE. (D to F) Frequency of (D) neutrophil, (E) macrophage, and (F) DC in the CNS, dLN, and spleen of Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE (n = 15 to 20); results were analyzed by unpaired t test. Data represent four to five independent experiments and are shown as the means ± SEM. *P < 0.05 and **P < 0.01.
Lack of ALKBH5 suppresses the IL-17 signaling pathway in CD4+ T cells during EAE
To investigate the mechanisms by which ALKBH5 regulates encephalitogenic CD4+ T cell function, we performed RNA sequencing (RNA-seq) analysis of CD4+ T cells isolated from the CNS of Alkbh5flox/floxCd4Cre and WT control mice subjected to EAE. RNA-seq analysis showed that 103 genes were up-regulated and 66 genes were down-regulated in Alkbh5flox/floxCd4Cre-derived CD4+ T cells compared to WT cells (Fig. 6A). On the basis of the enrichment pathway analysis of the RNA-seq data, IL-17 signaling was the most down-regulated pathway in Alkbh5flox/floxCd4Cre-derived CD4+ T cells compared with WT cells (Fig. 6, B and C). Cxcl2, Cxcl10, and Ifng, which play important roles in IL-17 signaling pathways (25–27), showed the significant down-regulation in CD4+ T cells from Alkbh5flox/floxCd4Cre mice with EAE when compared with cells from WT mice (Fig. 6A). These genes were demonstrated to participate in pathogenic function and chemotaxis of T cells and myeloid cells during EAE (25). A previous study indicated that TH17 cells in the CNS lose the ability to secrete IL-17A and begin to produce IFN-γ, representing an almost exclusive source of this cytokine during EAE; they are usually called “ex-TH17 cells” (27). Consistent with these findings, we noticed that the lack of ALKBH5 decreased the ratio of IFN-γ+ IL-17A− CD4+ T cells but did not affect IFN-γ− IL-17A+ CD4+ T cells in the CNS (Fig. 6, D and E), indicating that the absence of IFN-γ in ALKBH5-deficient ex-TH17 cells might partially explain the less pronounced pathologic changes induced by EAE in Alkbh5flox/floxCd4Cre mice than in WT littermates.
Fig. 6. Lack of ALKBH5 results in impaired IL-17 signaling pathway in CD4+ T cells during EAE.
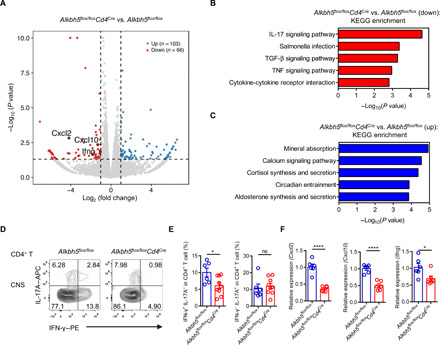
(A) Volcano plot exhibiting the differentially expressed genes (|fold change| > 2; P < 0.05; up-regulated genes, blue; down-regulated genes, red) in Alkbh5flox/floxCd4Cre-derived CD4+ T cells compared to WT-derived CD4+ T cells isolated from the CNS during EAE. Cxcl2, Cxcl10, and Ifng belong to IL-17 signaling pathways, highlighted by a circle. (B) According to the P value in RNA-seq data for CD4+ T cells obtained from Alkbh5flox/floxCd4Cre mice and WT littermates during EAE, the IL-17 signaling pathway ranks as the top down-regulated Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway in Alkbh5flox/floxCd4Cre-derived CD4+ T cells when compared with WT-derived CD4+ T cells. Two biological replicates were used for analysis. (C) The enriched KEGG pathways of significantly down-regulated genes in CD4+ T cells derived from Alkbh5flox/floxCd4Cre mice and WT littermates during EAE. RNA-seq data obtained in two biological replicates were used for analysis. (D) Representative dot plots show the expression of IL-17A and IFN-γ in CNS CD4+ T cells from Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE. (E) Frequency of IFN-γ+ IL-17A− and IFN-γ− IL-17A+ CNS CD4+ T cell from Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE (n = 6 to 9) was analyzed by unpaired t test. Data represent one of five independent experiments. (F) qPCR measurements of the expression of Cxcl2, Cxcl10, and Ifng mRNA in CD4+ T cells isolated from Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE (n = 6) were analyzed by unpaired t test. Data represent one of three independent experiments. Data are shown as the means ± SEM. *P < 0.05 and ****P < 0.0001.
IFN-γ not only promotes the pathogenic potential of CD4+ T cells (23) but also disrupts the blood-brain barrier and enhances their transendothelial migration into the brain (28). These findings are consistent with the lower infiltration of CD4+ T cells into the CNS in Alkbh5flox/floxCd4Cre mice subjected to EAE than in WT littermates (Fig. 3, C to E). Moreover, both Cxcl2 and Cxcl10 can be induced by IL-17 during EAE (25), raising the possibility that these genes are involved in the IL-17 signaling pathway and may be modulated by ALKBH5. Therefore, we validated the potential m6A target genes by real-time polymerase chain reaction (PCR) and confirmed that the expression of Cxcl2, Cxcl10, and Ifng mRNA was indeed reduced in ALKBH5-deficient CD4+ T cells relative to WT cells during neuroinflammation (Fig. 6F). These findings show that Cxcl2, Cxcl10, and Ifng involved in the IL-17 signaling pathway represent potential m6A targets.
ALKBH5 ablation decreases IFN-γ and CXCL2 mRNA stability
On the basis of our previous m6A RNA immunoprecipitation (RIP)–sequencing (RIP-seq) data obtained from WT CD4+ T cells (8), m6A peaks were enriched on Cxcl2, Cxcl10, and Ifng mRNAs (Fig. 7A), we hypothesized that these mRNAs are m6A targets directly modulated by ALKBH5. To explore how the loss of ALKBH5 affects the expression of Cxcl2, Cxcl10, and Ifng mRNA during neuroinflammation, we evaluated the m6A modification on these genes using m6A RIP-quantitative reverse transcription PCR (RT-qPCR). This approach was based on the essential function of ALKBH5 as an m6A eraser (13). In comparison with WT CD4+ T cells, only Cxcl2 and Ifng m6A enrichment was found to be specifically increased in ALKBH5-deficient CD4+ T cells (Fig. 7B). However, the m6A level of Cxcl10 mRNA remained unchanged (Fig. 7B), implying that Cxcl10 mRNA is not the target of m6A modification by ALKBH5 in CD4+ T cells during EAE. The m6A methylation of mRNA primarily affects its stability by promoting RNA decay (4, 5). To further validate whether the degradation of Cxcl2 and Ifng mRNA was modulated by m6A, we performed RNA decay assays and found that the levels of both Cxcl2 and Ifng mRNA decreased faster in ALKBH5-deficient CD4+ T cells than in WT CD4+ T cells after actinomycin-D treatment for different hours (Fig. 7, C and D). These results suggest that ALKBH5 ablation specifically increases m6A modification on Cxcl2 and Ifng mRNA, decreasing their stability in CD4+ T cells during EAE.
Fig. 7. ALKBH5 ablation promotes m6A RNA modification on Cxcl2 and Ifng mRNAs, decreasing their stability and expression.
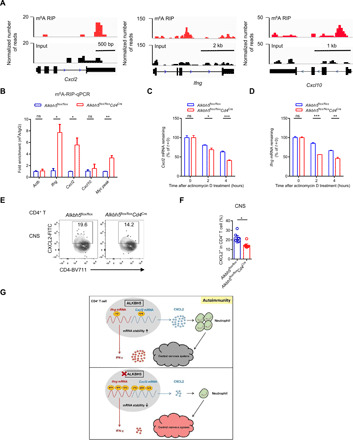
(A) m6A RIP-seq results from our published database (accession number GSE100048) (8) were obtained from CD4+ T cells isolated from WT mice, and peaks on Cxcl2, Ifng, and Cxcl10 mRNAs were then visualized using IGV. bp, base pair. (B) m6A RIP-qPCR assay was applied to detect m6A enrichment on Cxcl2, Cxcl10, and Ifng mRNA in CD4+ T cells isolated from the CNS of Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE. Results were presented as fold increase versus immunoglobulin G and analyzed by unpaired t test. Actb, m6A negative control; Myc peak, m6A-positive control. (C) Degradation of Cxcl2 mRNA was detected in CD4+ T cells isolated from the CNS of Alkbh5flox/floxCd4Cre mice and WT littermates at 14 days of EAE after treating with actinomycin D for 0, 2, and 4 hours (n = 3); the values of residual RNAs were normalized to 0 hours. (D) Degradation of Ifng mRNA was detected in CD4+ T cells isolated from the CNS of Alkbh5flox/floxCd4Cre mice and WT littermates at 14 days of EAE after treating with actinomycin D for 0, 2, and 4 hours (n = 3); the values of residual RNAs were normalized to 0 hours. (E) Representative dot plots show the expression of CXCL2 on CD4+ T cells in the CNS of Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE. FITC, fluorescein isothiocyanate. (F) Percentages of CXCL2-expressing CD4+ T cells in the CNS of Alkbh5flox/floxCd4Cre mice and WT littermates after 14 days of EAE (n = 5 to 7); results were analyzed by unpaired t test. (G) Working model summarize how ALKBH5 and m6A RNA modification regulate CD4+ T cell pathogenicity and promote autoimmunity during EAE. Data represent one of three independent experiments and are shown as the means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
Since we have demonstrated that the expression of IFN-γ protein in ALKBH5-deficient CD4+ T cells in the CNS during EAE was lower than in WT CD4+ T cells (Fig. 3, C and F), we measured the expression of CXCL2 protein in the CNS CD4+ T cells during the experimental neuroinflammation by flow cytometry. We found that the expression of CXCL2 protein was lower in ALKBH5-deficient CD4+ T cells than in WT CD4+ T cells (Fig. 7, E and F). CXCL2 is a potent chemokine that can recruit different myeloid cells, particularly neutrophils, to promote robust inflammation in the meninges in EAE (29, 30). We also observed that the deficiency of ALKBH5 in CD4+ T cells specifically inhibited neutrophil recruitment into the CNS during EAE (Fig. 5). Thus, these data indicate that the enhancement of m6A modification in ALKBH5-deficient CD4+ T cells leads to a decreased stability of Cxcl2 and Ifng mRNA and protein, suppressing CD4+ T cell function and neutrophil recruitment during the CNS autoimmunity (Fig. 7G).
DISCUSSION
m6A modification regulates many aspects of mRNA metabolism, including the splicing, stability, and translation of mRNA (31, 32). Several identified m6A machines, such as the writers, erasers, and readers, participate in the control of numerous fundamental biological processes, such as cell differentiation (33, 34), tumorigenesis (35, 36), DNA damage repair (37), and immune responses (10, 16, 38, 39). Our previous work also suggested that T cell homeostasis can be regulated by m6A writer proteins METTL3 and METTL14 (8, 9). However, whether m6A eraser proteins contribute to T cell development and function remains unknown. In the present study, we investigated the involvement of the eraser protein ALKBH5 in CD4+ T cell–mediated pathogenesis and identified target mRNAs regulated by ALKBH5 during autoimmunity.
ALKBH5, the second identified demethylase, has been reported to control immune responses in some recent studies. For example, the synthesis of type I IFN (IFN-I), initiated by double-stranded DNA or human cytomegalovirus, is regulated by ALKBH5 (40). RNA helicase DEAD-box 46 recruits ALKBH5 to restrain antiviral innate immune responses by erasing m6A modification of antiviral transcripts entrapped in the nucleus (41). Moreover, ALKBH5-deficient macrophages display IFN-I–independent resistance to viral exposure by increasing mRNA decay and reducing the expression of OGDH protein (16). However, whether ALKBH5 is involved in governing T cell immune responses has not been demonstrated. By using lineage-specific deletion of ALKBH5 in T cells, we documented that ALKBH5 does not affect the development and function of T cell in vivo at a steady state, but it controls the ability of CD4+ T cells to induce adoptive transfer colitis and EAE pathogenesis. These results imply the potential and importance of ALKBH5 in promoting CD4+ T cell function during pathologic processes and indicate the functional similarity between ALKBH5 and m6A writer enzyme METTL3 in directing CD4+ T cell function (8). Besides, although our previous research demonstrated that METTL3 controls the global suppressive functions of Tregs (9), the lack of ALKBH5 in Tregs does not affect their inhibitory roles against EAE development in our current work, which further indicates the selectivity and asymmetry in the actions of the m6A writers and erasers in controlling CD4+ T cells function. These results depict the complexity of this epigenetic regulation and raise the possibility that other unidentified erasers may play opposite roles in modulating CD4+ T cell function compared with the writers.
To fully understand how ALKBH5 regulates CD4+ T cell responses in vivo, we used the EAE model in which CD4+ T cells are critical to initiate a localized inflammatory process (18). We documented that ALKBH5 deficiency in CD4+ T cells renders mice resistant to EAE. During the onset of the disease, myelin-specific CD4+ T cells are activated, migrate through the blood-CNS barrier, and home to the CNS where they secrete a large amount of proinflammatory cytokines and chemokines. As a result, more pathogenic cells are attracted, initiating an inflammatory cascade (18). The analysis of the immune cells from ALKBH5-deficient mice after the EAE challenge identified a reduction in the infiltration of CD4+ T cells into the CNS, with more retention in the dLN. This finding indicated that ALKBH5 is essential for the recruitment of CD4+ T cells into target organs. Mechanistically, although our previous published CD4+ T cell m6A RIP-seq database (8) suggested that m6A peaks are enriched on Cxcl2, Cxcl10, and Ifng mRNAs, the m6A RIP-qPCR results further demonstrated that only the m6A levels on Cxcl2 and Ifng mRNAs were substantially increased in ALKBH5-deficient CD4+ T cells, without affecting m6A enrichments on Cxcl10 mRNA. These data jointly reveal that Cxcl2 and Ifng are m6A targets specifically regulated by ALKBH5 and showed the selectivity of writers and erasers in choosing the m6A sites. However, we still cannot exclude the presence of other potential m6A targets that may be modulated by ALKBH5, for we have limited access to obtain enough CD4+ T cells from the CNS during EAE to carry out the m6A RIP-seq. The combination of m6A RIP-seq and RNA-seq with the T cells isolated from the CNS before and during EAE could further illuminate additional drivers that may contribute to the observed phenotypes in our study, which will depend on the future technological advances in m6A RIP-seq.
We demonstrated that ALKBH5 controls CD4+ T cell function during EAE by erasing the m6A modification on CXCL2 and IFN-γ mRNA, both of the genes involved in the IL-17 signaling pathway. Considering the selectively decreased IFN-γ secretion by ALKBH5-deficient CD4+ T cells in the CNS demonstrated in our current study, the IL-17 signaling pathway indicates that a specific T cell subset, i.e., ex-TH17 cells, which are unstable and cease to express IL-17A but begin to produce IFN-γ in EAE (27), may represent the target cells regulated by ALKBH5. Besides, IFN-γ can enhance the pathogenicity and transendothelial migration of CD4+ T cells during EAE (23, 28), this is consistent with the observation that the deficiency of ALKBH5 dampened the infiltration of CD4+ T cells into the CNS. But whether the more retention of ALKBH5-deficient CD4+ T cells in the dLN is due to attenuated CNS inflammation or a cell-intrinsic deficiency remains to be further determined. Moreover, tissue damage in the CNS requires neutrophil infiltration in which CXCL2 activated by IL-17A plays a major promoting role (29, 42). The present study also provided evidence that ALKBH5 enhances the expression of CXCL2 protein in CD4+ T cells, which can explain the increased migration of neutrophils into the CNS during EAE in WT littermates in comparison with Alkbh5flox/floxCd4Cre mice. Collectively, these data document the previously unrecognized role of ALKBH5 in regulating CD4+ T cell function in autoimmune reactions.
Although both ALKBH5 and FTO are important eraser proteins, only the expression of ALKBH5 mRNA increased upon T cell activation in our study. To exclude entirely the potential of FTO in regulating T cell homeostasis or pathology in autoimmunity, we also constructed T cell–specific FTO knockout mice. Unexpectedly, we found that the ablation of FTO in T cells does not impair T cell development or EAE pathogenesis (fig. S8), which contrasts with the ability of ALKBH5 to regulate CD4+ T cell function during neuroinflammation. It will be interesting to elucidate different functions between these two erasers in T cells. One possibility can be advanced that different cell types or pathology processes can be regulated by distinct erasers. Besides, unlike the specificity of ALKBH5 toward m6A methylation, FTO also functions as the demethylase for other RNA modification, including N6-2’-O-dimethyladenosine (m6Am) and N1-methyladenosine (m1A) (43). The complex cross-talk among different epigenetic regulations resulting from the lack of FTO in CD4+ T cells may lead to genetic compensation response. Thus, our study shows that, generally, the regulation of RNA methylation and, specifically, the removal of m6A from key mRNAs are important regulatory steps in controlling the differentiation of T cells into effector cells and the pathogenicity of CD4+ T cells during autoimmunity. Given that ALKBH5 is critical in controlling the pathologic effects of CD4+ T cells in vivo, whether ALKBH5 may also govern CD8+ T cell function upon confrontation with internal or external stimuli needs to be further studied.
Together, our work reveals an important role of ALKBH5 in modulating CD4+ T cell function during EAE. Specifically, ALKBH5 decreases m6A modification in CXCL2 and IFN-γ mRNA, increasing transcript stability and protein expression, thus leading to enhanced responses of CD4+ T cells and more infiltration of neutrophils into the CNS during neuroinflammation. Considering the importance of m6A modification as a critical regulator on CD4+ T cell function, blocking or inhibiting ALKBH5 may provide previously unidentified strategies for therapeutic interventions in autoimmune diseases.
MATERIALS AND METHODS
Mice
Alkbh5 flox/flox mice were constructed using the CRISPR-Cas9–based genome-editing system by inserting two loxP sites into the loci flanking the first exon, as previously described (44). The guide RNA (gRNA) and donor oligos used for the Alkbh5 left side loxP were caggctcagcagccacttaa ggg and tt*g*c*ccgaattttcgggttgacatacacctctagctctctcctgctcaggctcagcagccacttaa ataacttcgtataatgtatgctatacgaagttat gggaatcgtctctgagtggttggagacctgagaggactgtattcacactccacttgtttgctc*a*t*t and for the Alkbh5 right side loxP ttgtgtctgaaactcatagc agg and gg*t*t*ctgactcgccttttttctttttgcgtgcaaaccataggaagcttgtgtctgaaactcatagc ataacttcgtataatgtatgctatacgaagttat aggataagactaatgtggaattgtgtacctgcaggcaggtttgaagtggccatagtagcttat*c*t*g. The correct integration of the lox sequences into the genome loci was verified by genotyping the offspring and sequencing the PCR products.
Fto flox/flox mice were also constructed using the CRISPR-Cas9–based genome-editing system as previously described (44). The gRNA and donor oligos used for the Fto left side loxP were gcgctcactggagagtgtct ggg and tt*t*g*tttgtgttgaagattattagtagttagagcttcaacctgacagcgctcactggagagtgtct ataacttcgtataatgtatgctatacgaagttat gggcgttggttttgtataaggtttgttttttagactggcaaggtaagaaccctgctggggaaa*a*g*t and for the Fto right side loxP ggctccgaagagtacttgat tgg and ct*t*a*actgaatgtggcagaggtcgttgggtggacagtttattgtagggctccgaagagtacttgat ataacttcgtataatgtatgctatacgaagttat tggaaactaacttgtatctaattcgactttacaccttatatacttatatttttggctctgggt*t*g*t. The correct integration of the lox sequences into the genome loci was verified by genotyping the offspring and sequencing the PCR products.
Cd4Cre mice were purchased from the Jackson laboratory, and Foxp3Cre mice were constructed before in R.A.F.’s laboratory (45) and have been fully backcrossed with C57BL/6 mice for more than 10 generations. We crossed Alkbh5flox/flox mice with Cd4Cre mice to obtain Alkbh5flox/floxCd4Cre offspring in which the expression of ALKBH5 is specifically absent in CD4+ T cells; the Alkbh5flox/flox offsprings were considered as WT littermate controls in our experiments. Besides, Alkbh5flox/flox mice were bred with Foxp3Cre mice to obtain Alkbh5flox/floxFoxp3Cre offspring in which the expression of ALKBH5 is specifically absent in Tregs. Because the Foxp3Cre transgene is on the X chromosome, we only used male mice to carry out experiments in our study in comparison to WT littermate controls. We also crossed Ftoflox/flox mice with Cd4Cre mice to obtain Ftoflox/floxCd4Cre offspring in which the expression of FTO is specifically absent in CD4+ T cells.
All mice were bred and maintained under specific pathogen–free conditions at the animal facility of Yale University School of Medicine. Mice were used at 8 to 12 weeks of age to carry out experiments. Animals were randomly divided into experimental groups, and each cage housed mice from different experimental groups. Animal procedures were approved by the Institutional Animal Care and Use Committee of Yale University.
Cell isolation
The spleen, thymus, and lymphoid node were pressed through a 200-gauge mesh. Splenic single-cell leukocyte suspensions were prepared by lysing the erythrocytes with red cell lysis buffer (Thermo Fisher Scientific). The CNS including the brain and spinal cord were removed and pressed through a 200-gauge mesh, and CNS mononuclear cells were then collected following 40% Percoll (GE Healthcare, no. 17089101) density gradient centrifugation.
For isolation of colon lymphocytes, the colon was cut and flushed with ice-cold phosphate-buffered saline (PBS) and then cut into 1-cm-long piece and incubated in extraction buffer (2% fetal bovine serum, 2 mM EDTA, and 1 mM dithiothreitol in PBS) with shaking (200 rpm) at 37°p for 30 min. After extensive washing to remove intraepithelial lymphocytes, the lamina propria was minced and incubated at 37° with shaking (200 rpm) for 60 min in digestion buffer [RPMI with 2% fetal bovine serum, type II collagenase (1 mg/ml), and dispase (0.5 mg/ml)]. The supernatant was collected, and lymphocytes were purified by Percoll gradient centrifugation.
Antibody staining and flow cytometry
Monoclonal antibodies (mAbs) against CD3 (145-2C11), CD4 (RM4-5), CD8 (53-6.7), CD11b (M1/70), CD11c (N418), CD44 (IM7), CD62L (MEL-14), CD45 (30-F11), CD45RB (C363-16A), F4/80 (BM8), GM-CSF (MP1-22E9), IFN-γ (XMG1.2), IL-17A (TC11-18H10.1), Ly6G (1A8), TCR-β (H57-597), and tumor necrosis factor–ɑ (MP6-XT22) were purchased from BioLegend (San Diego, CA, USA). The isolated cells were incubated with rat serum to block Fc receptors followed by labeling with fluorescently labeled antibodies. For intracellular cytokines and CXCL2 detection, cells were stimulated with phorbol 12-myristate 13-acetate (50 ng/ml; Sigma-Aldrich, St. Louis, MO, USA) and ionomycin (1 μg/ml; Sigma-Aldrich) in the presence of GolgiPlug (1 μl/ml; BD Biosciences, San Jose, CA, USA) for 4 hours. After surface staining, cells were fixed, permeabilized using a FoxP3/Transcription Factor Buffer Set (eBioscience), and then stained with mAbs against the intracellular molecules. For intracellular CXCL2 staining, cells were incubated with goat anti-mouse CXCL2 antibody (Invitrogen, no. PA5-47015) followed by Alexa Fluor 488–conjugated anti-goat immunoglobulin G (IgG) secondary antibody (Invitrogen, no. A-11055). All data were collected with BD LSR II and analyzed with FlowJo software (Tree Star, Ashland, OR, USA).
Adoptive transfer colitis
Naïve CD4+ T cells were purified from spleens by using the EasySep Mouse Naïve CD4+ T Cell Isolation Kit (STEMCELL) and labeled with CellTrace Violet (Thermo Fisher Scientific). A total of 5 × 105 naïve CD4+ T cells from Alkbh5flox/floxCd4Cre and WT littermate control mice were transferred into Rag2−/− mice respectively, and recipient mice were measured body weight weekly.
Endoscopic procedures
Colonoscopy was performed in a blinded fashion for colitis scoring using a high-resolution mouse video Coloview system (Karl Storz, Germany). Colitis scoring was based on stool consistency, vascularity, the granularity of mucosal surface, and translucency of the colon (46–48).
Induction and assessment of EAE
EAE was induced by subcutaneous injection with 200 μg of mouse MOG35–55 peptide (Prespec) emulsified in complete Freund’s adjuvant containing heat-killed Mycobacterium tuberculosis H37RA (BD Difco) on day 0. Pertussis toxin (200 ng; List Biological Laboratories) was intravenously injected on the day of immunization and 2 days after immunization. Mice were then monitored and scored every day using the following clinical score assessment standard: 0, no clinical signs; 1, limp tail; 2, paraparesis (weakness, incomplete paralysis of one or two hind limbs); 3, paraplegia (complete paralysis of two hind limbs); 4, paraplegia with forelimb weakness or paralysis; and 5, moribund or death, as described previously (49).
Apoptosis assay
7-amino-actinomycin D (7-AAD) was adopted to detect the apoptotic state of the cells, and annexin V+ or annexin V/7-AAD double-positive cells are apoptotic cells, the double-negative cells are viable.
RNA library preparation, sequencing, and differentially expressed genes analysis
Alkbh5flox/floxCd4Cre and Alkbh5flox/flox mice were induced with EAE and monitored until the Alkbh5flox/flox mice reached the EAE score 2-3. CD4+ T cells were sorted from the CNS of Alkbh5flox/floxCd4Cre and Alkbh5flox/flox mice. Total RNA was extracted using the TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. RNA purity and quantification were assessed using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA), and RNA integrity was evaluated using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). RNA libraries were prepared with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB) according to the manufacturer’s instructions.
The libraries were sequenced on an Illumina HiSeq X Ten platform (150–base pair paired-end reads) by OE Biotech Co. Ltd. (Shanghai, China). About 50 million raw reads for each sample were generated. Raw reads were processed using Trimmomatic (50) to get high-quality clean reads. The clean reads were mapped to the mouse genome (GRCm38.p6) using HISAT2 (51). FPKM (52) of each gene was calculated using Cufflinks (53), and the read counts of each gene were obtained by HTSeq-count (54). The DESeq2 (55) was used to normalize the raw counts and identify differentially expressed genes (DEGs; |fold change| > 2; P < 0.05). Volcano plots and bar plots were performed using the R package. Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of DEGs was performed using phyper function form R package “stats” based on the hypergeometric distribution. Two independent biological replicates were performed for RNA-seq.
Reverse transcription qPCR
Total RNA was isolated from CD4+ T cells with TRIzol reagent (Invitrogen) as described in the manufacturer’s instructions and reverse-transcribed using the Maxima H Minus Reverse Transcriptase Kit (Thermo Fisher Scientific, no. EP0753). All qPCRs were run on Bio-Rad CFX96 real-time system using iTaq Universal SYBR Green Supermix (Bio-Rad, no. 1725124), and β-actin was used as an internal control to normalize the data across different samples.
Primer sequences used for qPCR are as follows: Actb (forward, 5′-AGTGTGACGTTGACATCCGT-3′; reverse, 5′-GCAGCTCAGTAACAGTCCGC-3′), Alkbh5 (forward, 5′-CGCGGTCATCAACGACTACC-3′; reverse, 5′- ATGGGCTTGAACTGGAACTTG-3′), Fto (forward, 5′-GCCTCGGTTTAGTTCCACTCAC-3′; reverse, 5′-GTCGCCATCGTCTGAGTCATTG-3′), Ifng (forward, 5′-CAGCAACAGCAAGGCGAAA-3′; reverse, 5′-CTGGACCTGTGGGTTGTTGAC-3′), Cxcl2 (forward, 5′-CCCTGCCAAGGGTTGACTTC-3′; reverse, 5′-GCAAACTTTTTGACCGCCCT-3′), Cxcl10 (forward, 5′-CCTATGGCCCTCATTCTCAC-3′; reverse, 5′-CTCATCCTGCTGGGTCTGAG-3′), and Myc peak (forward, 5′-GCTTCGAAACTCTGGTGCAT-3′; reverse, 5′-AATTCCAGCGCATCAGTTCT-3′).
RNA degradation assay
RNA degradation assay was conducted as previously described (8, 56). CD4+ T cells were purified by the EasySep Mouse CD4+ T Cell Isolation Kit (STEMCELL Technologies) and plated on a 96-well plate with 5 × 105 cells per well. Actinomycin D (Sigma-Aldrich) was added in each well at a final concentration of 5 μM. CD4+ T cells were harvested at 0, 2, and 4 hours after adding actinomycin D. The cells were processed as described in the “Reverse transcription qPCR” section, and the data were normalized to the t = 0 time point.
m6A RIP-qPCR
m6A RIP-qPCR assay was performed as described previously (16). In brief, CD4+ T cells were purified by the EasySep Mouse CD4+ T Cell Isolation Kit (STEMCELL Technologies). Total RNA extracted from CD4+ T cells was purified with the Dynabeads mRNA Purification Kit (Invitrogen) according to the manufacturer’s instructions. The purified polyadenylated mRNA was incubated with anti-m6A antibody (Synaptic Systems) or rabbit IgG in immunoprecipitating (IP) wash buffer (10 mM tris-HCL, 0.1% NP-40, and 150 mM NaCl supplemented with ribonuclease inhibitor (pH 7.4) for 2 hours at 4°C. Protein A beads (Thermo Fisher Scientific, no. 21348) were added and incubated at 4°C for 2 hours. After incubation, the immunoprecipitated beads-m6A antibody-mRNA complex was extensively washed with IP wash buffer. TRIzol reagent (Invitrogen) was added to elute m6A nucleotide. The Maxima H Minus Reverse Transcriptase Kit (Thermo Fisher Scientific, no. EP0753) was used for cDNA synthesis. All qPCRs were run on Bio-Rad CFX96 real-time system using iTaq Universal SYBR Green Supermix (Bio-Rad, no. 1725124). Actb was used as m6A negative control, and Myc peak was set as m6A-positive control.
m6A-RIP-seq analysis
m6A RIP-seq results from our published database (accession number GSE100048) (8) were obtained from CD4+ T cells isolated from WT mice, and peaks were then visualized using IGV (57).
Western blot
CD4+ T cells from lymphoid nodes were purified by the EasySep Mouse CD4+ T Cell Isolation Kit (STEMCELL Technologies), and total protein from CD4+ T cells was extracted with radioimmunoprecipitation assay lysis buffer (Beyotime, no. P0013E) supplemented with protease inhibitors (Thermo Fisher Scientific, no. 78443). Antibodies against ALKBH5 (Sigma-Aldrich, no. HPA007196) were diluted in 5% nonfat milk buffer at the concentration of 1:1000 and incubated at 4°C overnight. After extensively washing the membrane with 0.1% PBST buffer three times, the horseradish peroxidase–conjugated secondary antibody (Cell Signaling Technology, no. 7074) was added to the membrane and incubated at room temperature for 1 hour. The final signal was detected by enhanced chemiluminescence (ECL) with pico ECL using ChemiDoc MP (Bio-Rad), and glyceraldehyde-3-phosphate dehydrogenase (Cell Signaling Technology, no. 2118S) was used as the internal control.
T cell ex vivo differentiation
Naïve CD4+ T cells were purified from the spleens by using the EasySep Mouse Naïve CD4+ T Cell Isolation Kit (STEMCELL Technologies) and cultured with anti-CD3 mAb (10 μg/ml; 145-2C11) and anti-CD28 mAb (2 μg/ml; PV-1) in the presence of defined mouse recombinant cytokines and blocking antibodies. In brief, TH1 was induced with IL-12 (10 ng/ml) and anti–IL-4 mAb (10 μg/ml; 11B11); TH2 was induced with IL-4 (10 ng/ml) and anti–IFN-γ mAb (10 μg/ml; XMG1.2); TH17 was induced with IL-6 (20 ng/ml), IL-23 (20 ng/ml), anti–IL-4 mAb (10 μg/ml; 11B11) and anti–IFN-γ mAb (10 μg/ml; XMG1.2); Treg was induced with IL-2 (50 U/ml), IL-23 (20 ng/ml), transforming growth factor–β (2 ng/ml), anti–IL-4 mAb (10 μg/ml; 11B11), and anti–IFN-γ mAb (10 μg/ml; XMG1.2). All cytokines were purchased from R&D Systems. After culturing for 4 days, the cells were used for RT-qPCR.
Histology
At 30 days after the EAE model, mice were sacrificed and their spinal cords were fixed in 10% neutral-buffered formalin and embedded in paraffin. Serial paraffin sections (4 μm) were cut and stained with hematoxylin and eosin staining and luxol fast blue.
Statistics
Unpaired Student’s t tests and two-way analysis of variance (ANOVA) were used to compare pairs of groups, and all data are presented as the means ± SEM. P values of < 0.05 were considered statistically significant.
Acknowledgments
We would like to thank J. Alderman, C. Lieber, P. Ranney, L. Evangelisti, C. Hughes, E. Hughes-Picard, M. Yang, K. Mao, S. Hu, and Y. Zhou for technical and administrative assistance. We would like to thank all members of the R.A.F. laboratory and the H.-B.L. laboratory for discussion. Funding: This work was supported by the National Natural Science Foundation of China (32070917/82030042/91753141 to H.-B.L., 81901569 to J.Z., and 31900683 to X.Z.), Shanghai Science and Technology Commission (20JC1417400/201409005500/20JC1410100 to H.-B.L.), the Postdoctoral Innovation Talent Support Program (BX20190214 to J.Z.), the Shanghai Super Postdoctoral Program (J.Z. and X.Z.), China Postdoctoral Science Foundation (2020 M671149 to J.Z., 2019 M660089 to X.Z.), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (H.-B.L.), the startup fund from the Shanghai Jiao Tong University School of Medicine (H.-B.L. and R.A.F.), and the Howard Hughes Medical Institute (R.A.F.). Author contributions: J.Z. designed the research, performed experiments, and drafted the manuscript. X.Z. designed the research and performed experiments. J.H. contributed to the discussion and revised the manuscript. R.Q., H.C., and Y.Y. performed the bioinformatic analysis. Z.Y. and H.X. performed experiments. C.D. and L.Y. offered important experimental methods. Q.Z. and Z.W. contributed to the discussion. R.A.F. and H.-B.L. supervised the research, coordinated the projects, and revised the manuscript. Competing interests: R.A.F. is a consultant for GSK and Zai Lab Ltd. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Sequenced reads have been deposited in the SRA (accession number PRJNA673064). The floxed Alkbh5 and Fto mouse strains can be provided by H.-B. L. pending on scientific review and a completed material transfer agreement. Request for those mouse lines should be submitted to H.-B. L. (huabing.li@shsmu.edu.cn). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg0470/DC1
REFERENCES AND NOTES
- 1.Cao G., Li H. B., Yin Z., Flavell R. A., Recent advances in dynamic m6A RNA modification. Open Biol. 6, 160003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao B. S., Roundtree I. A., He C., Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tong J., Flavell R. A., Li H.-B., RNA m6A modification and its function in diseases. Front. Med. 12, 481–489 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Nachtergaele S., He C., Chemical modifications in the life of an mRNA transcript. Annu. Rev. Genet. 52, 349–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaccara S., Ries R. J., Jaffrey S. R., Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 20, 608–624 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Mapperley C., van de Lagemaat L. N., Lawson H., Tavosanis A., Paris J., Campos J., Wotherspoon D., Durko J., Sarapuu A., Choe J., Ivanova I., Krause D. S., von Kriegsheim A., Much C., Morgan M., Gregory R. I., Mead A. J., O'Carroll D., Kranc K. R., The mRNA m6A reader YTHDF2 suppresses proinflammatory pathways and sustains hematopoietic stem cell function. J. Exp. Med. 218, e20200829 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu C., Chen W., He J., Jin S., Liu Y., Yi Y., Gao Z., Yang J., Yang J., Cui J., Zhao W., Interplay of m6A and H3K27 trimethylation restrains inflammation during bacterial infection. Sci. Adv. 6, eaba0647 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H. B., Tong J., Zhu S., Batista P. J., Duffy E. E., Zhao J., Bailis W., Cao G., Kroehling L., Chen Y., Wang G., Broughton J. P., Chen Y. G., Kluger Y., Simon M. D., Chang H. Y., Yin Z., Flavell R. A., m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 548, 338–342 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong J., Cao G., Zhang T., Sefik E., Amezcua Vesely M. C., Broughton J. P., Zhu S., Li H., Li B., Chen L., Chang H. Y., Su B., Flavell R. A., Li H. B., m6A mRNA methylation sustains Treg suppressive functions. Cell Res. 28, 253–256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H., Hu X., Huang M., Liu J., Gu Y., Ma L., Zhou Q., Cao X., Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat. Commun. 10, 1898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi H., Wei J., He C., Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y. G., He C., N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng G., Dahl J. A., Niu Y., Fedorcsak P., Huang C. M., Li C. J., Vågbø C. B., Shi Y., Wang W. L., Song S. H., Lu Z., Bosmans R. P., Dai Q., Hao Y. J., Yang X., Zhao W. M., Tong W. M., Wang X. J., Bogdan F., Furu K., Fu Y., Jia G., Zhao X., Liu J., Krokan H. E., Klungland A., Yang Y. G., He C., ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao X., Yang Y., Sun B. F., Shi Y., Yang X., Xiao W., Hao Y. J., Ping X. L., Chen Y. S., Wang W. J., Jin K. X., Wang X., Huang C. M., Fu Y., Ge X. M., Song S. H., Jeong H. S., Yanagisawa H., Niu Y., Jia G. F., Wu W., Tong W. M., Okamoto A., He C., Rendtlew Danielsen J. M., Wang X. J., Yang Y. G., FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 24, 1403–1419 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X. C., Jin F., Wang B. Y., Yin X. J., Hong W., Tian F. J., The m6A demethylase ALKBH5 controls trophoblast invasion at the maternal-fetal interface by regulating the stability of CYR61 mRNA. Theranostics 9, 3853–3865 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y., You Y., Lu Z., Yang J., Li P., Liu L., Xu H., Niu Y., Cao X., N6-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 365, 1171–1176 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Zhu J., Yamane H., Paul W. E., Differentiation of effector CD4 T cell populations*. Annu. Rev. Immunol. 28, 445–489 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Constantinescu C. S., Farooqi N., O'Brien K., Gran B., Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 164, 1079–1106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mastorakos P., McGavern D., The anatomy and immunology of vasculature in the central nervous system. Sci. Immunol. 4, eaav0492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saligrama N., Zhao F., Sikora M. J., Serratelli W. S., Fernandes R. A., Louis D. M., Yao W., Ji X., Idoyaga J., Mahajan V. B., Steinmetz L. M., Chien Y. H., Hauser S. L., Oksenberg J. R., Garcia K. C., Davis M. M., Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature 572, 481–487 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner C. A., Roqué P. J., Mileur T. R., Liggitt D., Goverman J. M., Myelin-specific CD8+ T cells exacerbate brain inflammation in CNS autoimmunity. J. Clin. Invest. 130, 203–213 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Göbel K., Ruck T., Meuth S. G., Cytokine signaling in multiple sclerosis: Lost in translation. Mult. Scler. 24, 432–439 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Wagner C. A., Roqué P. J., Goverman J. M., Pathogenic T cell cytokines in multiple sclerosis. J. Exp. Med. 217, e20190460 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain A., Irizarry-Caro R. A., McDaniel M. M., Chawla A. S., Carroll K. R., Overcast G. R., Philip N. H., Oberst A., Chervonsky A. V., Katz J. D., Pasare C., T cells instruct myeloid cells to produce inflammasome-independent IL-1β and cause autoimmunity. Nat. Immunol. 21, 65–74 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karpus W. J., Cytokines and chemokines in the pathogenesis of experimental autoimmune encephalomyelitis. J. Immunol. 204, 316–326 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Onishi R. M., Gaffen S. L., Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 129, 311–321 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirota K., Duarte J. H., Veldhoen M., Hornsby E., Li Y., Cua D. J., Ahlfors H., Wilhelm C., Tolaini M., Menzel U., Garefalaki A., Potocnik A. J., Stockinger B., Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12, 255–263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonar S. A., Shaikh S., Joshi N., Atre A. N., Lal G., IFN-γ promotes transendothelial migration of CD4+T cells across the blood-brain barrier. Immunol. Cell Biol. 95, 843–853 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Simmons S. B., Liggitt D., Goverman J. M., Cytokine-regulated neutrophil recruitment is required for brain but not spinal cord inflammation during experimental autoimmune encephalomyelitis. J. Immunol. 193, 555–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Girbl T., Lenn T., Perez L., Rolas L., Barkaway A., Thiriot A., Del Fresno C., Lynam E., Hub E., Thelen M., Graham G., Alon R., Sancho D., von Andrian U. H., Voisin M. B., Rot A., Nourshargh S., Distinct compartmentalization of the chemokines CXCL1 and CXCL2 and the atypical receptor ACKR1 determine discrete stages of neutrophil diapedesis. Immunity 49, 1062–1076.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frye M., Harada B. T., Behm M., He C., RNA modifications modulate gene expression during development. Science 361, 1346–1349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roundtree I. A., Evans M. E., Pan T., He C., Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A. A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y. S., Ben-Haim M. S., Eyal E., Yunger S., Pinto Y., Jaitin D. A., Viukov S., Rais Y., Krupalnik V., Chomsky E., Zerbib M., Maza I., Rechavi Y., Massarwa R., Hanna S., Amit I., Levanon E. Y., Amariglio N., Stern-Ginossar N., Novershtern N., Rechavi G., Hanna J. H., Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347, 1002–1006 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Yoon K. J., Ringeling F. R., Vissers C., Jacob F., Pokrass M., Jimenez-Cyrus D., Su Y., Kim N. S., Zhu Y., Zheng L., Kim S., Wang X., Doré L. C., Jin P., Regot S., Zhuang X., Canzar S., He C., Ming G. L., Song H., Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell 171, 877–889.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen M., Wei L., Law C. T., Tsang F. H., Shen J., Cheng C. L., Tsang L. H., Ho D. W., Chiu D. K., Lee J. M., Wong C. C., Ng I. O., Wong C. M., RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 67, 2254–2270 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Weng H., Huang H., Wu H., Qin X., Zhao B. S., Dong L., Shi H., Skibbe J., Shen C., Hu C., Sheng Y., Wang Y., Wunderlich M., Zhang B., Dore L. C., Su R., Deng X., Ferchen K., Li C., Sun M., Lu Z., Jiang X., Marcucci G., Mulloy J. C., Yang J., Qian Z., Wei M., He C., Chen J., METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell 22, 191–205.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiang Y., Laurent B., Hsu C. H., Nachtergaele S., Lu Z., Sheng W., Xu C., Chen H., Ouyang J., Wang S., Ling D., Hsu P. H., Zou L., Jambhekar A., He C., Shi Y., RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkler R., Gillis E., Lasman L., Safra M., Geula S., Soyris C., Nachshon A., Tai-Schmiedel J., Friedman N., Le-Trilling V. T. K., Trilling M., Mandelboim M., Hanna J. H., Schwartz S., Stern-Ginossar N., m6A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 20, 173–182 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Du H., Zhao Y., He J., Zhang Y., Xi H., Liu M., Ma J., Wu L., YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 7, 12626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubio R. M., Depledge D. P., Bianco C., Thompson L., Mohr I., RNA m6A modification enzymes shape innate responses to DNA by regulating interferon β. Genes Dev. 32, 1472–1484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng Q., Hou J., Zhou Y., Li Z., Cao X., The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat. Immunol. 18, 1094–1103 (2017). [DOI] [PubMed] [Google Scholar]
- 42.McGinley A. M., Sutton C. E., Edwards S. C., Leane C. M., DeCourcey J., Teijeiro A., Hamilton J. A., Boon L., Djouder N., Mills K. H. G., Interleukin-17A serves a priming role in autoimmunity by recruiting IL-1β-producing myeloid cells that promote pathogenic T cells. Immunity 52, 342–356.e6 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Wei J., Liu F., Lu Z., Fei Q., Ai Y., He P. C., Shi H., Cui X., Su R., Klungland A., Jia G., Chen J., He C., Differential m6A, m6Am, and m1A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol. Cell 71, 973–985.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henao-Mejia J., Williams A., Rongvaux A., Stein J., Hughes C., Flavell R. A., Generation of genetically modified mice using the CRISPR-Cas9 genome-editing system. Cold Spring Harb Protoc 2016, pdb.prot090704, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan Y. Y., Flavell R. A., Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc. Natl. Acad. Sci. U.S.A. 102, 5126–5131 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Becker C., Fantini M. C., Neurath M. F., High resolution colonoscopy in live mice. Nat. Protoc. 1, 2900–2904 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Gagliani N., Amezcua Vesely M. C., Iseppon A., Brockmann L., Xu H., Palm N. W., de Zoete M. R., Licona-Limón P., Paiva R. S., Ching T., Weaver C., Zi X., Pan X., Fan R., Garmire L. X., Cotton M. J., Drier Y., Bernstein B., Geginat J., Stockinger B., Esplugues E., Huber S., Flavell R. A., Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523, 221–225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nowarski R., Jackson R., Gagliani N., de Zoete M. R., Palm N. W., Bailis W., Low J. S., Harman C. C., Graham M., Elinav E., Flavell R. A., Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 163, 1444–1456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wheeler M. A., Jaronen M., Covacu R., Zandee S. E. J., Scalisi G., Rothhammer V., Tjon E. C., Chao C. C., Kenison J. E., Blain M., Rao V. T. S., Hewson P., Barroso A., Gutiérrez-Vázquez C., Prat A., Antel J. P., Hauser R., Quintana F. J., Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 176, 581–596.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roberts A., Trapnell C., Donaghey J., Rinn J. L., Pachter L., Improving RNA-seq expression estimates by correcting for fragment bias. Genome Biol. 12, R22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., Pachter L., Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anders S., Pyl P. T., Huber W., HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang S. S., Lim J., Yu Z., Kong P., Sefik E., Xu H., Harman C. C. D., Kim L. K., Lee G. R., Li H. B., Flavell R. A., mRNA destabilization by BTG1 and BTG2 maintains T cell quiescence. Science 367, 1255–1260 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., Mesirov J. P., Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg0470/DC1