

Genetically engineered cell membrane–coated nanoparticles enable targeted drug delivery to sites of inflammation.
Abstract
As numerous diseases are associated with increased local inflammation, directing drugs to the inflamed sites can be a powerful therapeutic strategy. One of the common characteristics of inflamed endothelial cells is the up-regulation of vascular cell adhesion molecule–1 (VCAM-1). Here, the specific affinity between very late antigen–4 (VLA-4) and VCAM-1 is exploited to produce a biomimetic nanoparticle formulation capable of targeting inflammation. The plasma membrane from cells genetically modified to constitutively express VLA-4 is coated onto polymeric nanoparticle cores, and the resulting cell membrane–coated nanoparticles exhibit enhanced affinity to target cells that overexpress VCAM-1 in vitro. A model anti-inflammatory drug, dexamethasone, is encapsulated into the nanoformulation, enabling improved delivery of the payload to inflamed lungs and significant therapeutic efficacy in vivo. Overall, this work leverages the unique advantages of biological membrane coatings to engineer additional targeting specificities using naturally occurring target-ligand interactions.
INTRODUCTION
The chemical and physiological changes associated with inflammation are an important part of the innate immune system (1). Proinflammatory processes can lead to the release of cytokines such as interleukin-6 (IL-6) and tumor necrosis factor, which are capable of effecting vascular changes to improve immune responses at a site of stress or injury (2). These may include vasodilation and an increase in vascular permeability, which can promote more efficient immune cell recruitment (3, 4). On the cellular level, proinflammatory cytokines cause the up-regulation of specific surface markers, including vascular cell adhesion molecule–1 (VCAM-1) or intercellular adhesion molecule–1 (ICAM-1), which allow for immune cell adhesion at the site of inflammation (5, 6). Although inflammation is an integral process that is required for survival, a dysregulated immune system is implicated in a wide range of disease states (7, 8). The disease relevance of inflammation is further supported by the fact that inflammatory markers such as cellular adhesion molecules are often implicated in pathogenesis (9, 10), and these have been explored as therapeutic and diagnostic targets.
Nanoparticle-based platforms, especially those functionalized with active targeting ligands, have the potential to serve as powerful tools for managing a wide range of diseases associated with inflammation (11). Along these lines, the targeted delivery of anti-inflammatory agents to the vasculature of affected sites via cell adhesion molecules represents a promising strategy (12–14). Using inflammation as the cue, a diverse range of nanodelivery systems have been designed to target up-regulated markers such as VCAM-1 and ICAM-1 (15–20), and this approach has been leveraged to treat conditions such as cancer and cardiovascular diseases (21–23). More recently, cell membrane coating technology has garnered considerable attention in the field of nanomedicine (24, 25). From erythrocytes to cancer cells, virtually any type of cell membrane can be coated onto the surface of nanoparticles, resulting in nanoformulations with enhanced functionality that can be custom-tailored to specific applications (26, 27). In particular, cell membrane–coated nanoparticles have proven to be effective drug delivery systems owing to their extended circulation times and disease-homing capabilities (26–28). The targeting ability of these biomimetic nanoparticles is often mediated by proteins that are expressed on the source cells, and this bestows the nanoparticles with the ability to specifically interact with various disease substrates. For example, nanoparticles coated with the membrane derived from platelets were shown to specifically target bacteria as well as the exposed subendothelium in damaged vasculature (29). A similar platform was shown to target the lungs in a murine model of cancer metastasis (30). On top of the natural biointerfacing capabilities of cell membrane–coated nanoparticles, their traits can be further enhanced by introducing exogenous moieties onto the membrane surface. One way to achieve this is to tether targeting ligands via a lipid anchor, which can then be inserted into the cell membrane (31, 32). Red blood cell membrane–coated nanoparticles, which exhibit prolonged blood circulation, have been functionalized in this manner to enhance their cancer targeting ability.
Instead of relying on post-fabrication methods to introduce additional functionality, cell membrane–coated nanoparticles can be developed using the membrane from genetically engineered source cells (33). A wide range of tools are available to introduce or up-regulate the expression of specific surface markers (34, 35), and this approach enables researchers to augment the functionality of cell membrane–based nanodelivery platforms based on application-specific needs (36, 37). In this study, we genetically engineered cell membrane–coated nanoparticles to specifically target sites of inflammation (Fig. 1). Inflamed endothelial cells are known to up-regulate the expression of VCAM-1 to recruit immune cells such as leukocytes that express its cognate ligand, very late antigen–4 (VLA-4) (38). To exploit this interaction, we genetically modified a source cell line to stably express VLA-4 and harvested the engineered membrane to coat polymeric nanoparticle cores. A potent anti-inflammatory drug, dexamethasone (DEX), was used as a model payload to be loaded for the treatment of inflammation. The ability of the final nanoformulation to target inflamed cells without compromising the activity of DEX was first tested in vitro. Then, therapeutic efficacy was evaluated in vivo using a murine model of endotoxin-induced lung inflammation.
Fig. 1. Schematic illustration of genetically engineered cell membrane–coated nanoparticles for targeted drug delivery to inflamed lungs.
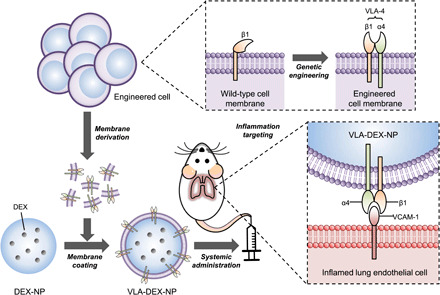
Wild-type cells were genetically engineered to express VLA-4, which is composed of integrins α4 and β1. Then, the plasma membrane from the genetically engineered cells was collected and coated onto dexamethasone-loaded nanoparticle cores (DEX-NP). The resulting VLA-4–expressing cell membrane–coated DEX-NP (VLA-DEX-NP) can target VCAM-1 on inflamed lung endothelial cells for enhanced drug delivery.
RESULTS AND DISCUSSION
VLA-4 is a heterodimer that is formed by the association of integrin α4 with integrin β1 (39). To generate a cell line constitutively displaying the full complex, we elected to modify wild-type C1498 cells (C1498-WT), which were confirmed to express high levels of integrin β1 but lack integrin α4 (Fig. 2A). Following viral transduction of C1498-WT to introduce the integrin α4 gene, a subpopulation of the resulting engineered cells (referred to as C1498-VLA) was found to express both VLA-4 components (Fig. 2B). After successfully establishing C1498-VLA, the cells were harvested and their membrane was derived by a process involving cell lysis and differential centrifugation. The cell membrane was then coated onto poly(lactic-co-glycolic acid) (PLGA) nanoparticle cores that were prepared by a single emulsion method. Membrane-coated nanoparticles prepared with the membrane from C1498-WT and C1498-VLA (referred to as WT-NP and VLA-NP, respectively) both had an average diameter of approximately 175 nm, which was slightly larger than the uncoated PLGA cores (Fig. 2C). In terms of zeta potential, the membrane-coated nanoparticles exhibited a surface charge of approximately −20 mV, which was less negative than the PLGA cores (Fig. 2D). Both the size and zeta potential data suggested proper membrane coating, which was further verified for VLA-NP by transmission electron microscopy, which clearly showed a membrane layer surrounding the core (Fig. 2E). Western blotting analysis was used to probe for the two components of VLA-4 on the nanoformulations (Fig. 2F). As expected, both integrins α4 and β1 were found on VLA-NP, whereas only integrin β1 was present on WT-NP. To evaluate long-term stability of the membrane-coated nanoparticles, they were suspended in 10% sucrose solution at 4°C, and their size was monitored over the course of 8 weeks (Fig. 2G). Neither nanoparticle sample exhibited a significant increase in size during this period.
Fig. 2. Development and characterization of inflammation-targeting nanoparticles.
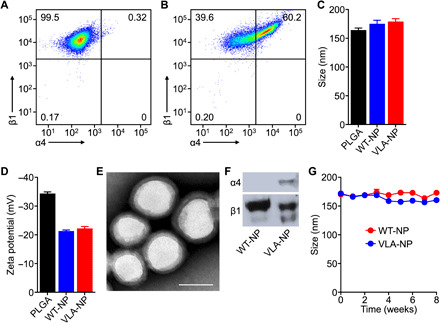
(A and B) Expression of integrins α4 and β1 on C1498-WT (A) and C1498-VLA (B) cells was confirmed by flow cytometry. (C and D) The average diameter (C) and surface zeta potential (D) of PLGA cores, WT-NP, and VLA-NP were confirmed by dynamic light scattering (n = 3, mean + SD). (E) Representative transmission electron microscopy image of VLA-NP (scale bar, 100 nm). (F) Western blots for integrins α4 and β1 on WT-NP and VLA-NP. (G) Size of WT-NP and VLA-NP when stored in solution over a period of 8 weeks (n = 3, mean ± SD).
The binding of VLA-NP was assessed in two different in vitro experiments. First, C1498-WT transduced to constitutively express high amounts of VCAM-1 (referred to as C1498-VCAM) was used as a model target cell. The expression of VCAM-1 on C1498-VCAM was confirmed via flow cytometry (Fig. 3A). Whereas the C1498-WT cells did not show any expression, the C1498-VCAM cells yielded a signal that was over an order of magnitude higher than the isotype control. To evaluate binding, fluorescent dye–labeled WT-NP or VLA-NP were incubated with either C1498-WT or C1498-VCAM (Fig. 3, B and C). For each pairing, the incubation was performed either with or without anti–VCAM-1 to block the specific interaction between VLA-4 and VCAM-1. For the samples with blocking, cells were first incubated with the antibody for 30 min before nanoparticle treatment. After incubating with the nanoparticles for 30 min, the cells were washed twice and were analyzed by flow cytometry. The data revealed that there was significant nanoparticle binding only when VLA-NP were paired with C1498-VCAM. The level of binding was reduced back to baseline levels in the presence of anti–VCAM-1, thus confirming the specificity of the interaction. In contrast, there was no evidence of specific binding when VLA-NP were paired with C1498-WT, which does not express the cognate receptor for VLA-4. The same held true for the WT-NP paired with either cell type, where antibody blocking had no impact on the relative nanoparticle binding.
Fig. 3. In vitro binding.
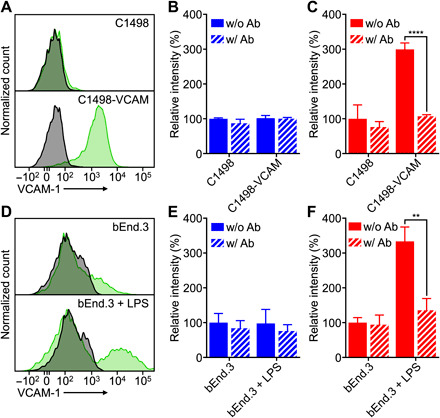
(A) Expression of VCAM-1 on C1498-WT and C1498-VCAM cells (gray, isotype antibody; green, anti–VCAM-1). (B and C) Binding of WT-NP (B) or VLA-NP (C) to C1498-WT or C1498-VCAM cells; blocking was performed by preincubating cells with anti–VCAM-1 (n = 3, mean + SD). ****P < 0.0001, Student’s t test. (D) Expression of VCAM-1 on untreated or LPS-treated bEnd.3 cells (gray, isotype antibody; green, anti–VCAM-1). (E and F) Binding of WT-NP (E) or VLA-NP (F) to untreated or LPS-treated bEnd.3 cells; blocking was performed by preincubating cells with anti–VCAM-1 (n = 3, mean + SD). **P < 0.01, Student’s t test.
Next, we elected to study the nanoparticle binding to endothelial cells, which represent a more biologically relevant target compared to the artificially engineered C1498-VCAM cells. For this purpose, we used a murine brain endothelial cell line, bEnd.3, whose VCAM-1 expression can be up-regulated in the presence of proinflammatory signals (40). To induce an inflamed state, bEnd.3 cells were treated with bacterial lipopolysaccharide (LPS), and the level of VCAM-1 expression was evaluated using flow cytometry (Fig. 3D). Whereas expression of VCAM-1 was near baseline levels for the untreated bEnd.3 cells, those that were treated with LPS exhibited a distinct population with elevated VCAM-1. As we observed in the previous experiment with C1498-VCAM cells, enhanced nanoparticle binding was only observed when VLA-NP were paired with inflamed bEnd.3 cells, and antibody blocking reduced the levels back to baseline (Fig. 3, E and F). When incubating with noninflamed bEnd.3 cells, there was no evidence of specific binding interactions, and the same held true for the control WT-NP paired with bEnd.3 cells regardless of their inflammatory status. The data in these two studies confirmed the successful engineering of membrane-coated nanoparticles with the ability to target inflammation based on the interaction between VLA-4 and VCAM-1.
As a model anti-inflammatory payload, we selected DEX, which was loaded into the PLGA core by a single emulsion method before coating with either C1498-WT or C1498-VLA membrane to yield DEX-loaded WT-NP or VLA-NP (referred to as WT-DEX-NP or VLA-DEX-NP, respectively). When the drug content was measured by high-performance liquid chromatography (HPLC), it was determined that the encapsulation efficiency and drug loading yield were approximately 11 and 2 weight % (wt %), respectively (Fig. 4A). To evaluate drug release, VLA-DEX-NP was dialyzed against a large volume of phosphate-buffered saline (PBS), and the amount of drug retained within the nanoparticles was quantified over time (Fig. 4B). The results revealed an initial burst, where approximately 80% of the drug payload was released in the first hour, followed by a sustained release. The release profile was in agreement with previous reports on DEX-loaded PLGA formulations (41, 42), and the data showed a good fit with the Peppas-Sahlin model with a regression coefficient of 0.978 (43). To evaluate the biological activity of the DEX loaded within the nanoparticles, we used an in vitro assay based on the LPS treatment of DC2.4 dendritic cells, which causes an elevation in the levels of proinflammatory cytokines such as IL-6 (Fig. 4C). DC2.4 cells were first treated with either free DEX or VLA-DEX-NP for 2 hours, followed by incubation with LPS overnight. The supernatant was then collected to measure the concentration of IL-6 by an enzyme-linked immunosorbent assay (ELISA). It was shown that both free DEX and VLA-DEX-NP were able to attenuate IL-6 secretion in a drug concentration–dependent manner (Fig. 4D). Although free DEX more efficiently lowered IL-6 levels at drug concentrations of 0.01 and 0.1 μM, the level of inflammation was reduced to levels near baseline for both free DEX and VLA-DEX-NP at 1 μM of drug. The data indicated that the activity of the drug payload was retained after being loaded inside of VLA-NP. It was confirmed that neither PLGA cores nor VLA-NP without DEX loading had an impact on the level of IL-6 production by the DC2.4 cells (Fig. 4E).
Fig. 4. Drug loading and in vitro activity.
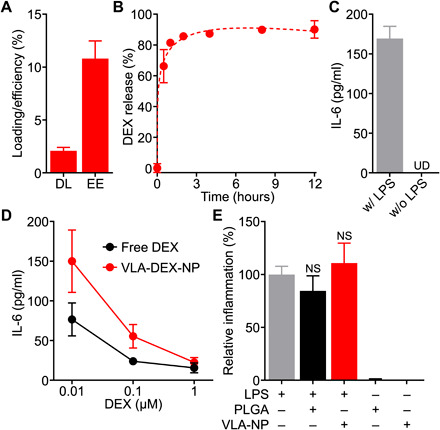
(A) Drug loading (DL) and encapsulation efficiency (EE) of dexamethasone (DEX) into VLA-NP (n = 3, mean + SD). (B) Drug release profile of VLA-DEX-NP (n = 3, mean ± SD). The data were fitted using the Peppas-Sahlin equation (dashed line). (C) Secretion of IL-6 by LPS-treated DC2.4 cells (n = 3, mean + SD). UD, undetectable. (D) Secretion of IL-6 by LPS-treated DC2.4 cells preincubated with DEX in free form or loaded into VLA-NP (n = 3, mean ± SD). (E) Relative inflammatory response, as measured by IL-6 secretion, of DC2.4 cells treated with LPS only, LPS and PLGA nanoparticles, LPS and VLA-NP, PLGA nanoparticles only, or VLA-NP only; all of the nanoparticles were empty without DEX loading (n = 3, mean + SD). NS, not significant (compared to the LPS-only group), one-way analysis of variance (ANOVA).
After confirming the biological activity of the VLA-DEX-NP formulation in vitro, we next sought to evaluate the formulation in vivo using a murine model of lung inflammation. The model was established by intratracheal injection of LPS directly into the lungs of BALB/c mice. To evaluate targeting ability, fluorescently labeled WT-NP or VLA-NP were injected intravenously after the induction of lung inflammation. After 6 hours, major organs, including the heart, lungs, liver, spleen, kidneys, and blood, were collected to assess nanoparticle biodistribution (Fig. 5A). The majority of the nanoparticles accumulated in the liver and spleen. Notably, a significant increase in accumulation of VLA-NP was observed in the lungs compared to WT-NP. This in vivo targeting result was in agreement with the in vitro findings where VLA-NP were able to specifically bind to inflamed cells. The safety of the formulation was assessed by monitoring the plasma levels of creatinine, a marker of kidney toxicity that was previously studied in the context of DEX nanodelivery (44). After 9 days of repeated daily administrations of free DEX or VLA-DEX-NP into healthy mice, it was shown that the creatinine concentration in mice receiving VLA-DEX-NP remained consistent with baseline levels, whereas it was significantly elevated in mice administered with free DEX (Fig. 5B).
Fig. 5. In vivo targeting, safety, and therapeutic efficacy.
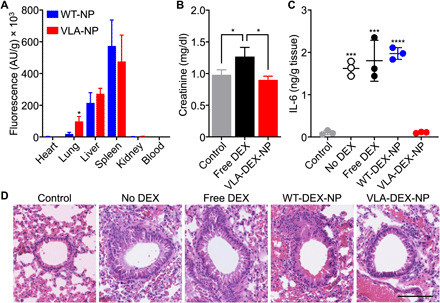
(A) Biodistribution of WT-NP or VLA-NP in a lung inflammation model 6 hours after intravenous administration (n = 3, mean + SD). *P < 0.05, Student’s t test. AU, arbitrary units. (B) Creatinine levels in the plasma of mice after repeated daily administrations for 9 days with free DEX or VLA-DEX-NP (n = 3, mean + SD). *P < 0.05, one-way ANOVA. (C) IL-6 levels in the lung tissue of mice intratracheally challenged with LPS and then treated intravenously with vehicle solution, free DEX, WT-DEX-NP, or VLA-DEX-NP (n = 3, mean ± SD). ***P < 0.001, ****P < 0.0001 (compared to VLA-DEX-NP), one-way ANOVA. (D) Representative hematoxylin and eosin–stained lung histology sections of mice intratracheally challenged with LPS and then treated intravenously with vehicle solution, free DEX, WT-DEX-NP, or VLA-DEX-NP (scale bar, 100 μm).
The therapeutic efficacy of VLA-DEX-NP was then evaluated following the same experimental design as the targeting study. After 6 hours, the lungs were collected and homogenized, and the homogenate was then clarified by centrifugation and filtered through a 0.22-μm porous membrane before measuring the concentration of IL-6 by ELISA. As shown in Fig. 5C, the VLA-DEX-NP formulation was able to completely abrogate lung inflammation, while both free DEX and WT-DEX-NP did not have any discernable effect. The fact that WT-DEX-NP were not able to significantly reduce lung IL-6 levels suggested that systemic exposure to DEX was not a major contributor to the efficacy observed with VLA-DEX-NP. The efficacy of the formulation against lung inflammation was further confirmed by analyzing lung sections stained with hematoxylin and eosin (Fig. 5D). Leukocyte recruitment and peribronchial thickening, which are hallmarks of lung inflammation (45, 46), were prominent in the lungs of mice receiving no treatment, free DEX, or WT-DEX-NP. In contrast, minimal leukocyte recruitment and no peribronchial thickening were observed for the group treated with VLA-DEX-NP, and there were no other signs of toxicity present in these lung sections. Overall, the results from the in vivo studies confirmed the benefit of targeted delivery to inflamed lungs using VLA-NP as a drug nanocarrier.
In conclusion, we have engineered cell membrane–coated nanoparticles that can be used to specifically target and treat localized lung inflammation via systemic administration. A host cell positive for integrin β1 was modified to express integrin α4. Together, the two protein markers formed VLA-4, which specifically interacts with VCAM-1, a common marker for inflammation found on vascular endothelia. Nanoparticles fabricated using the membrane from these genetically engineered cells were able to leverage this natural affinity to target inflamed sites, including in a murine model of LPS-induced lung inflammation. When the nanoparticles were loaded with DEX, an anti-inflammatory drug, significant therapeutic efficacy was achieved in vivo. Future studies will comprehensively evaluate the safety profile of the VLA-DEX-NP formulation, obtain additional lung-specific efficacy readouts, elucidate the optimal time window for treatment, and assess clinical relevance using additional animal models of severe inflammatory disease. As pathological inflammation is heavily implicated in a number of important disease conditions (7, 47), the reported biomimetic platform could be leveraged to improve the in vivo activity of various therapeutic payloads through enhanced targeting. Notably, VCAM-1 up-regulation has been observed in renal pathologies as well as in inflamed cerebral vasculature (48, 49). In addition, DEX has been shown to be effective at managing the inflammation associated with COVID-19 (50), and a targeted formulation capable of localizing the drug to the lungs may help to further boost its therapeutic profile. In this work, we specifically engineered the nanoparticles to display VLA-4, which is a complex, multicomponent membrane–bound ligand that would otherwise be infeasible to incorporate using traditional synthetic strategies. This highlights the advantages of using genetic engineering techniques to expand the wide-ranging utility of cell membrane coating technology. In particular, the generalized application of this approach would enable researchers to streamline the development of new targeted nanoformulations by using target-ligand interactions that occur in nature. Combined with the biocompatibility and biointerfacing characteristics that are inherent to cell membrane coatings, the work presented here could initiate a new wave of biomimetic nanomedicine with finely crafted functionalities.
MATERIALS AND METHODS
Cell culture
Wild-type C1498 mouse leukemia cells (TIB-49, American Type Culture Collection) were cultured at 37°C in 5% CO2 with Dulbecco’s modified Eagle’s medium [DMEM; with l-glutamine, glucose (4.5 g/liter), and sodium pyruvate; Corning] supplemented with 10% bovine growth serum (BGS; Hyclone) and 1% penicillin-streptomycin (Pen-Strep; Gibco). Engineered C1498-VCAM cells were cultured with DMEM supplemented with 10% U.S. Department of Agriculture (USDA) fetal bovine serum (FBS; Omega Scientific), 1% Pen-Strep, and hygromycin B (400 μg/ml; InvivoGen). Engineered C1498-VLA cells were cultured with DMEM supplemented with 10% USDA FBS, 1% Pen-Strep, and puromycin (1 μg/ml; InvivoGen). bEnd.3 mouse brain endothelial cells (CRL-2299, American Type Culture Collection) were cultured with DMEM supplemented with 10% BGS and 1% Pen-Strep. AmphoPhoenix cells (obtained from the National Gene Vector Biorepository) were cultured with DMEM supplemented with 10% BGS and 1% Pen-Strep. DC2.4 mouse dendritic cells (SCC142, Sigma-Aldrich) were cultured with DMEM supplemented with 10% BGS and 1% Pen-Strep.
Genetic engineering
Engineered C1498-VLA and C1498-VCAM cells were created by transducing C1498-WT. Briefly, the genes for integrin α4 (MG50049-M, Sino Biological) and VCAM-1 (MG50163-UT, Sino Biological) gene were cloned into pQCXIP and pQCXIH plasmids (Clontech), respectively, using an In-Fusion HD cloning kit (Clontech) following the manufacturer’s protocol, yielding pQCXIP-α4 and pQCXIH-VCAM-1. AmphoPhoenix cells were plated onto 100-mm tissue culture dishes containing 10 ml of medium at 3 × 105 cells/ml and cultured overnight. The cells were transfected with pQCXIP-α4 or pQCXIH-VCAM-1 using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. The supernatant of the transfected AmphoPhoenix was collected and used to resuspend C1498-WT cells, which were then centrifuged at 800g for 90 min. After the spin, the transduced cells were incubated for 4 hours before the media were changed with fresh media. Fluorescently labeled antibodies, including FITC (fluorescein isothiocyanate) anti-mouse CD49d (R1-2, BioLegend), Alexa647 anti-mouse/rat CD29 (HMβ1-1, BioLegend), or PE (phycoerythrin) anti-mouse CD106 (STA, BioLegend), were used to assess the expression levels of VLA-4 or VCAM-1. Data were collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo software. All of the engineered cells were sorted using a Becton Dickinson FACSAria-II flow cytometer to select for cells expressing high levels of VLA-4 or VCAM-1.
Cell membrane derivation
The membranes from C1498-WT and engineered C1498-VLA cells were derived using a previously described method with some modifications (51). First, the cells were harvested and washed in a starting buffer containing 30 mM tris-HCl (pH 7.0) (Quality Biological) with 0.0759 M sucrose (Sigma-Aldrich) and 0.225 M d-mannitol (Sigma-Aldrich). The washed cells were resuspended in an isolation buffer containing 0.5 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (Sigma-Aldrich), a phosphatase inhibitor cocktail (Sigma-Aldrich), and a protease inhibitor cocktail (Sigma-Aldrich). Then, the cells were homogenized using a Kinematica Polytron PT 10/35 probe homogenizer at 70% power for 15 passes. The homogenate was first centrifuged at 10,000g in a Beckman Coulter Optima XPN-80 ultracentrifuge for 25 min. The supernatant was then collected and centrifuged at 150,000g for 35 min. The resulting pellet of cell membrane was washed and stored in a solution containing 0.2 mM ethylenediaminetetraacetic acid (USB Corporation) in UltraPure DNase-free/RNase-free distilled water (Invitrogen). Total membrane protein content was quantified by a BCA protein assay kit (Pierce).
Synthesis of membrane-coated nanoparticles
Polymeric cores were prepared by a single emulsion process using carboxyl-terminated 50:50 PLGA (0.66 dl/g; LACTEL absorbable polymers). For DEX-loaded PLGA cores, 500 μl of PLGA (50 mg/ml) in dichloromethane (DCM; Sigma-Aldrich) was mixed with 500 μl of DEX (10 mg/ml) in acetone. This mixture was added to 5 ml of 10 mM tris-HCl (pH 8) and sonicated using a Thermo Fisher Scientific 150E Sonic Dismembrator at 70% power for 2 min. The sonicated mixture was added to 10 ml of 10 mM tris-HCl (pH 8) and was magnetically stirred at 700 rpm overnight. For 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DiD, ex/em = 644/663 nm; Biotium) labeling, 500 μl of PLGA (50 mg/ml) in DCM was mixed with 500 μl of DiD (20 μg/ml) in DCM. This mixture was added to 5 ml of 10 mM tris-HCl (pH 8) and sonicated using a Thermo Fisher Scientific 150E Sonic Dismembrator at 70% power for 2 min. The sonicated mixture was added to 10 ml of 10 mM tris-HCl (pH 8) and was magnetically stirred at 700g for 3 hours. Empty PLGA core preparation followed the same procedure, except substituting the DiD solution for 500 μl of neat DCM. To coat the polymeric cores with cell membranes, the nanoparticle cores were first centrifuged at 21,100g for 8 min. The pellets were resuspended in solution containing membranes derived from C1498-WT or C1498-VLA. The mixture was sonicated in a 1.5-ml disposable sizing cuvette (BrandTech Scientific Inc.) using a Thermo Fisher Scientific FS30D bath sonicator at a frequency of 42 kHz and a power of 100 W for 3 min. For the in vitro studies, UltraPure water and sucrose were added to adjust the polymer concentration to 1 mg/ml and the sucrose concentration to 10%. For the in vivo studies, UltraPure water and sucrose were added to adjust the polymer concentration to 10 mg/ml and the sucrose concentration to 10%.
Nanoparticle characterization
The size and surface zeta potential of WT-NP and VLA-NP were measured by dynamic light scattering using a Malvern ZEN 3600 Zetasizer. For electron microscopy visualization, a VLA-NP sample was negatively stained with 1 wt % uranyl acetate (Electron Microscopy Sciences) on a carbon-coated 400-mesh copper grid (Electron Microscopy Sciences) and visualized using a JEOL 1200 EX II transmission electron microscope. The presence of VLA-4 on WT-NP and VLA-NP was determined using western blotting. First, the samples were adjusted to 1 mg/ml protein content, followed by the addition of NuPAGE 4× lithium dodecyl sulfate sample loading buffer (Novex) and heating at 70°C for 10 min. Then, 25 μl was loaded into the wells of 12-well Bolt 4 to 12% Bis-Tris gels (Invitrogen) and ran at 165 V for 45 min in MOPS running buffer (Novex). The proteins were transferred for 60 min at a voltage of 10 V onto 0.45-μm nitrocellulose membranes (Pierce) in Bolt transfer buffer (Novex). Nonspecific interactions were blocked using 5% milk (Genesee Scientific) in PBS (Thermo Fisher Scientific) with 0.05% Tween 20 (National Scientific). The blots were probed using anti-integrin α4 antibody (B-2, Santa Cruz Biotechnology) or anti-integrin β1 antibody (E-11, Santa Cruz Biotechnology). The secondary staining was done using the corresponding horseradish peroxidase–conjugated antibodies (BioLegend). Membranes with stained samples were developed in a dark room using ECL western blotting substrate (Pierce) and an ImageWorks Mini-Medical/90 Developer. Long-term stability of WT-NP and VLA-NP in 10% sucrose solution was tested by storing the particles at 4°C for 2 months with weekly size measurements.
Binding studies
The expression level of VCAM-1 on C1498-WT, C1498-VCAM, untreated bEnd.3 cells, and bEnd.3 cells treated overnight with LPS (1 μg/ml) from Escherichia coli K12 (LPS; InvivoGen) was evaluated as described above. For the first binding study, 5 × 104 cells, either C1498-WT or C1498-VCAM, were collected and resuspended in 160 μl of DMEM containing 0.5% USDA FBS, 1% bovine serum albumin (BSA; Sigma-Aldrich), and 1 mM MnCl2 (Sigma-Aldrich). For blocking, anti-mouse CD106 antibody was added to the cells, followed by incubation at 4°C for 30 min. Then, 40 μl of DiD (1 mg/ml)–labeled WT-NP or VLA-NP was added, and the mixture was incubated at 4°C for another 30 min. After washing the cells twice with PBS, the fluorescent signals from the cells were detected using flow cytometry. For the second study, 5 × 104 bEnd.3 cells were plated and then either left untreated or pretreated with LPS overnight. The media were then removed and replaced with 160 μl of DMEM containing 0.5% USDA FBS, 0.8% BSA, and 1 mM MnCl2. For blocking, anti-mouse CD106 antibody was added to the cells, followed by incubation at 4°C for 30 min. Then, 40 μl of DiD (1 mg/ml)–labeled WT-NP or VLA-NP was added, and the mixture was incubated at 4°C for another 30 min. After washing the cells twice with PBS, the cells were detached by scraping, and the fluorescent signals from the cells were detected using flow cytometry. All data were collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo software.
Drug loading and release
Drug loading and encapsulation efficiency were measured using HPLC on an Agilent 1220 Infinity II gradient liquid chromatography system equipped with a C18 analytical column (Brownlee). VLA-DEX-NP samples were dissolved overnight in 80% acetonitrile (ACN; EMD Millipore) and then centrifuged at 21,100g for 8 min to collect the supernatant for analysis. The solutions were run through the column at a flow rate of 0.3 ml/min and DEX was detected at a wavelength of 242 nm. The DEX release profile was obtained by loading 200 μl of VLA-DEX-NP (1 mg/ml) into Slide-A-Lyzer MINI dialysis devices (10K molecular weight cutoff; Thermo Fisher Scientific) and floating them on 1 liter of PBS stirred at 150 rpm. At each time point, dialysis cups were retrieved, and their contents were centrifuged at 21,100g for 8 min. The pellets were dissolved in 80% ACN overnight and processed as described above for HPLC analysis.
In vitro activity of DEX
The biological activity of DEX was evaluated in vitro using a test system involving the LPS treatment of DC2.4 dendritic cells. To validate the system, DC2.4 cells were first plated onto a 24-well tissue culture plate at 5 × 104 cells per well and cultured overnight with or without LPS at a concentration of 1 μg/ml. Then, supernatant was collected, and the concentration of IL-6 was measured using a mouse IL-6 ELISA kit (BioLegend) according to the manufacturer’s protocol. To compare free DEX and VLA-DEX-NP, the two formulations were first added to the culture medium at final drug concentrations of 0.01, 0.1, and 1 μM, followed by 2 hours of incubation. For free DEX, 1000× stock solutions were prepared at 0.01, 0.1, and 1 mM in dimethyl sulfoxide. Then, the cells were treated with LPS overnight before measuring the concentration of IL-6 in the supernatant. To test the effect of empty nanoparticles, either PLGA cores or VLA-NP at a final concentration of 1 μg/ml were first incubated with the cells for 2 hours, followed by an overnight incubation either with or without LPS before measuring IL-6 levels.
In vivo inflammation targeting and biodistribution
All animal experiments were performed in accordance with the National Institutes of Health (NIH) guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California San Diego. To induce lung inflammation in mice, 30 μl of LPS (400 μg/ml) in PBS was injected intratracheally into male BALB/c mice (Charles River Laboratories). At 1 hour after LPS injection, 100 μl of DiD (10 mg/ml)–labeled WT-NP or VLA-NP was administered intravenously. After 6 hours, the heart, lungs, liver, spleen, kidneys, and blood were collected. All solid tissues were washed with PBS and suspended in 1 ml of PBS before being homogenized with a Biospec Mini-Beadbeater-16. The homogenates and blood were then diluted 4× with PBS and added to a 96-well plate, and fluorescence was measured using a BioTek Synergy Mx microplate reader. For each sample, the background signal measured from the corresponding organ or blood of control mice that did not receive any treatment was subtracted.
In vivo safety
Male BALB/c mice were intravenously injected with 100 μl of free DEX or VLA-DEX-NP, each at a drug concentration of 200 μg/ml, daily for the first 7 days. Then, for the next 2 days, the dosage was doubled by injecting 200 μl of each formulation at the same drug concentration. At 24 hours after the last injection, blood was collected by submandibular puncture and collected into tubes containing sodium heparin (Sigma-Aldrich). Plasma samples were obtained by taking the supernatant of the blood after centrifuging at 800g for 10 min. Creatinine levels were measured using a creatinine colorimetric assay kit (Cayman Chemical Company) according to the manufacturer’s protocol.
Inflammation treatment studies
To treat lung inflammation, male BALB/c mice were first intratracheally challenged with 30 μl of LPS (400 μg/ml) in PBS. At 1 hour after the challenge, 100 μl of free DEX, WT-DEX-NP, and VLA-DEX-NP, each at a drug concentration of 200 μg/ml, was injected intravenously. After 6 hours, the lungs were collected and homogenized as described above. The homogenates were centrifuged at 10,000g, and the supernatants were filtered through 0.22-μm polyvinylidene difluoride syringe filters (CELLTREAT). The concentration of IL-6 was measured using a mouse IL-6 ELISA kit according to the manufacturer’s protocol. For histology analysis, the lungs were collected after 6 hours and fixed in 10% phosphate-buffered formalin (Fisher Chemical) for 24 hours. The fixed lungs were sectioned, followed by hematoxylin and eosin (Sakura Finetek) staining. Histology slides were prepared by the Moores Cancer Center Tissue Technology Shared Resource (Cancer Center Support Grant P30CA23100). Images were obtained using a Hamamatsu NanoZoomer 2.0-HT slide scanner and analyzed using the NanoZoomer Digital Pathology software.
Acknowledgments
Funding: This work was supported by the National Institutes of Health under award no. R01CA200574 and the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense under grant no. HDTRA1-18-1-0014. J.H.P. was supported by a National Institutes of Health 5T32CA153915 training grant from the National Cancer Institute. Author contributions: J.H.P., Y.J., R.H.F., and L.Z. conceived and designed the experiments. J.H.P., Y.J., J.Z., H.G., A.M., and J.H. performed all experiments. All authors analyzed and discussed the data. J.H.P., A.M., R.H.F., and L.Z. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper.
REFERENCES AND NOTES
- 1.Netea M. G., Balkwill F., Chonchol M., Cominelli F., Donath M. Y., Giamarellos-Bourboulis E. J., Golenbock D., Gresnigt M. S., Heneka M. T., Hoffman H. M., Hotchkiss R., Joosten L. A. B., Kastner D. L., Korte M., Latz E., Libby P., Mandrup-Poulsen T., Mantovani A., Mills K. H. G., Nowak K. L., O’Neill L. A., Pickkers P., van der Poll T., Ridker P. M., Schalkwijk J., Schwartz D. A., Siegmund B., Steer C. J., Tilg H., van der Meer J. W. M., van de Veerdonk F. L., Dinarello C. A., A guiding map for inflammation. Nat. Immunol. 18, 826–831 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perlstein R. S., Whitnall M. H., Abrams J. S., Mougey E. H., Neta R., Synergistic roles of interleukin-6, interleukin-1, and tumor necrosis factor in the adrenocorticotropin response to bacterial lipopolysaccharide in vivo. Endocrinology 132, 946–952 (1993). [DOI] [PubMed] [Google Scholar]
- 3.Naldini A., Carraro F., Role of inflammatory mediators in angiogenesis. Curr. Drug Targets Inflamm. Allergy 4, 3–8 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Huang A. L., Vita J. A., Effects of systemic inflammation on endothelium-dependent vasodilation. Trends Cardiovasc. Med. 16, 15–20 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meager A., Cytokine regulation of cellular adhesion molecule expression in inflammation. Cytokine Growth Factor Rev. 10, 27–39 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Kelly M., Hwang J. M., Kubes P., Modulating leukocyte recruitment in inflammation. J. Allergy Clin. Immunol. 120, 3–10 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Kotas M. E., Medzhitov R., Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pawelec G., Goldeck D., Derhovanessian E., Inflammation, ageing and chronic disease. Curr. Opin. Immunol. 29, 23–28 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Cybulsky M. I., Iiyama K., Li H., Zhu S., Chen M., Iiyama M., Davis V., Gutierrez-Ramos J. C., Connelly P. W., Milstone D. S., A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Invest. 107, 1255–1262 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q., Massagué J., Molecular pathways: VCAM-1 as a potential therapeutic target in metastasis. Clin. Cancer Res. 18, 5520–5525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin K., Luo Z., Zhang B., Pang Z., Biomimetic nanoparticles for inflammation targeting. Acta. Pharm. Sin. B 8, 23–33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howard M. D., Hood E. D., Zern B., Shuvaev V. V., Grosser T., Muzykantov V. R., Nanocarriers for vascular delivery of anti-inflammatory agents. Annu. Rev. Pharmacol. Toxicol. 54, 205–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shuvaev V. V., Ilies M. A., Simone E., Zaitsev S., Kim Y., Cai S., Mahmud A., Dziubla T., Muro S., Discher D. E., Muzykantov V. R., Endothelial targeting of antibody-decorated polymeric filomicelles. ACS Nano 5, 6991–6999 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garnacho C., Albelda S. M., Muzykantov V. R., Muro S., Differential intra-endothelial delivery of polymer nanocarriers targeted to distinct PECAM-1 epitopes. J. Control. Release 130, 226–233 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visweswaran G. R. R., Gholizadeh S., Ruiters M. H. J., Molema G., Kok R. J., Kamps J. A. A. M., Targeting rapamycin to podocytes using a vascular cell adhesion molecule-1 (VCAM-1)-harnessed SAINT-based lipid carrier system. PLOS ONE 10, e0138870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R., Kowalski P. S., Morselt H. W. M., Schepel I., Jongman R. M., Aslan A., Ruiters M. H. J., Zijlstra J. G., Molema G., van Meurs M., Kamps J. A. A. M., Endothelium-targeted delivery of dexamethasone by anti-VCAM-1 SAINT-O-Somes in mouse endotoxemia. PLOS ONE 13, e0196976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leus N. G. J., Morselt H. W. M., Zwiers P. J., Kowalski P. S., Ruiters M. H. J., Molema G., Kamps J. A. A. M., VCAM-1 specific PEGylated SAINT-based lipoplexes deliver siRNA to activated endothelium in vivo but do not attenuate target gene expression. Int. J. Pharm. 469, 121–131 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Calderon A. J., Bhowmick T., Leferovich J., Burman B., Pichette B., Muzykantov V., Eckmann D. M., Muro S., Optimizing endothelial targeting by modulating the antibody density and particle concentration of anti-ICAM coated carriers. J. Control. Release 150, 37–44 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muro S., Gajewski C., Koval M., Muzykantov V. R., ICAM-1 recycling in endothelial cells: A novel pathway for sustained intracellular delivery and prolonged effects of drugs. Blood 105, 650–658 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Muro S., Cui X., Gajewski C., Murciano J. C., Muzykantov V. R., Koval M., Slow intracellular trafficking of catalase nanoparticles targeted to ICAM-1 protects endothelial cells from oxidative stress. Am. J. Physiol. Cell Physiol. 285, C1339–C1347 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Mlinar L. B., Chung E. J., Wonder E. A., Tirrell M., Active targeting of early and mid-stage atherosclerotic plaques using self-assembled peptide amphiphile micelles. Biomaterials 35, 8678–8686 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Rouleau L., Berti R., Ng V. W. K., Matteau-Pelletier C., Lam T., Saboural P., Kakkar A. K., Lesage F., Rhéaume E., Tardif J.-C., VCAM-1-targeting gold nanoshell probe for photoacoustic imaging of atherosclerotic plaque in mice. Contrast Media Mol. Imaging 8, 27–39 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Pan H., Myerson J. W., Hu L., Marsh J. N., Hou K., Scott M. J., Allen J. S., Hu G., San Roman S., Lanza G. M., Schreiber R. D., Schlesinger P. H., Wickline S. A., Programmable nanoparticle functionalization for in vivo targeting. FASEB J. 27, 255–264 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang R. H., Jiang Y., Fang J. C., Zhang L., Cell membrane-derived nanomaterials for biomedical applications. Biomaterials 128, 69–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang R. H., Kroll A. V., Gao W., Zhang L., Cell membrane coating nanotechnology. Adv. Mater. 30, 1706759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang R. H., Hu C.-M., Luk B. T., Gao W., Copp J. A., Tai Y., O'Connor D. E., Zhang L., Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 14, 2181–2188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu C.-M., Zhang L., Aryal S., Cheung C., Fang R. H., Zhang L., Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. U.S.A. 108, 10980–10985 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dehaini D., Fang R. H., Zhang L., Biomimetic strategies for targeted nanoparticle delivery. Bioeng. Transl. Med. 1, 30–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu C.-M. J., Fang R. H., Wang K.-C., Luk B. T., Thamphiwatana S., Dehaini D., Nguyen P., Angsantikul P., Wen C. H., Kroll A. V., Carpenter C., Ramesh M., Qu V., Patel S. H., Zhu J., Shi W., Hofman F. M., Chen T. C., Gao W., Zhang K., Chien S., Zhang L., Nanoparticle biointerfacing by platelet membrane cloaking. Nature 526, 118–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J., Ai Y., Wang L., Bu P., Sharkey C. C., Wu Q., Wun B., Roy S., Shen X., King M. R., Targeted drug delivery to circulating tumor cells via platelet membrane-functionalized particles. Biomaterials 76, 52–65 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang R. H., Hu C.-M. J., Chen K. N. H., Luk B. T., Carpenter C. W., Gao W., Li S., Zhang D.-E., Lu W., Zhang L., Lipid-insertion enables targeting functionalization of erythrocyte membrane-cloaked nanoparticles. Nanoscale 5, 8884–8888 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei X., Zhan C., Shen Q., Fu W., Xie C., Gao J., Peng C., Zheng P., Lu W., A D-peptide ligand of nicotine acetylcholine receptors for brain-targeted drug delivery. Angew. Chem. Int. Ed. 54, 3023–3027 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Jiang Y., Krishnan N., Zhou J., Chekuri S., Wei X., Kroll A. V., Yu C. L., Duan Y., Gao W., Fang R. H., Zhang L., Engineered cell-membrane-coated nanoparticles directly present tumor antigens to promote anticancer immunity. Adv. Mater. 32, 2001808 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith H. O., Danner D. B., Deich R. A., Genetic transformation. Annu. Rev. Biochem. 50, 41–68 (1981). [DOI] [PubMed] [Google Scholar]
- 35.Knott G. J., Doudna J. A., CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lv P., Liu X., Chen X., Liu C., Zhang Y., Chu C., Wang J., Wang X., Chen X., Liu G., Genetically engineered cell membrane nanovesicles for oncolytic adenovirus delivery: A versatile platform for cancer virotherapy. Nano Lett. 19, 2993–3001 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Shi X., Cheng Q., Hou T., Han M., Smbatyan G., Lang J. E., Epstein A. L., Lenz H. J., Zhang Y., Genetically engineered cell-derived nanoparticles for targeted breast cancer immunotherapy. Mol. Ther. 28, 536–547 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nourshargh S., Alon R., Leukocyte migration into inflamed tissues. Immunity 41, 694–707 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Hemler M. E., Huang C., Takada Y., Schwarz L., Strominger J. L., Clabby M. L., Characterization of the cell surface heterodimer VLA-4 and related peptides. J. Biol. Chem. 262, 11478–11485 (1987). [PubMed] [Google Scholar]
- 40.Böggemeyer E., Stehle T., Schaible U. E., Hahne M., Vestweber D., Simon M. M., Borrelia burgdorferi upregulates the adhesion molecules E-selectin, P-selectin, ICAM-1 and VCAM-1 on mouse endothelioma cells in vitro. Cell Adhes. Commun. 2, 145–157 (1994). [DOI] [PubMed] [Google Scholar]
- 41.Kim D.-H., Martin D. C., Sustained release of dexamethasone from hydrophilic matrices using PLGA nanoparticles for neural drug delivery. Biomaterials 27, 3031–3037 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Gómez-Gaete C., Tsapis N., Besnard M., Bochot A., Fattal E., Encapsulation of dexamethasone into biodegradable polymeric nanoparticles. Int. J. Pharm. 331, 153–159 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Peppas N. A., Sahlin J. J., A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int. J. Pharm. 57, 169–172 (1989). [Google Scholar]
- 44.Lorscheider M., Tsapis N., Ur-Rehman M., Gaudin F., Stolfa I., Abreu S., Mura S., Chaminade P., Espeli M., Fattal E., Dexamethasone palmitate nanoparticles: An efficient treatment for rheumatoid arthritis. J. Control. Release 296, 179–189 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Ghosh S., Hoselton S. A., Asbach S. V., Steffan B. N., Wanjara S. B., Dorsam G. P., Schuh J. M., B lymphocytes regulate airway granulocytic inflammation and cytokine production in a murine model of fungal allergic asthma. Cell. Mol. Immunol. 12, 202–212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ren Y., Su X., Kong L., Li M., Zhao X., Yu N., Kang J., Therapeutic effects of histone deacetylase inhibitors in a murine asthma model. Inflamm. Res. 65, 995–1008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coussens L. M., Werb Z., Inflammation and cancer. Nature 420, 860–867 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marcos-Contreras O. A., Greineder C. F., Kiseleva R. Y., Parhiz H., Walsh L. R., Zuluaga-Ramirez V., Myerson J. W., Hood E. D., Villa C. H., Tombacz I., Pardi N., Seliga A., Mui B. L., Tam Y. K., Glassman P. M., Shuvaev V. V., Nong J., Brenner J. S., Khoshnejad M., Madden T., Weissmann D., Persidsky Y., Muzykantov V. R., Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood–brain barrier. Proc. Natl. Acad. Sci. U.S.A. 117, 3405–3414 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kowalski P. S., Zwiers P. J., Morselt H. W. M., Kuldo J. M., Leus N. G. J., Ruiters M. H. J., Molema G., Kamps J. A. A. M., Anti-VCAM-1 SAINT-O-Somes enable endothelial-specific delivery of siRNA and downregulation of inflammatory genes in activated endothelium in vivo. J. Control. Release 176, 64–75 (2014). [DOI] [PubMed] [Google Scholar]
- 50.The RECOVERY Collaborative Group , Dexamethasone in hospitalized patients with Covid-19. N. Engl. J. Med. 384, 693–704 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kroll A. V., Fang R. H., Jiang Y., Zhou J., Wei X., Yu C. L., Gao J., Luk B. T., Dehaini D., Gao W., Zhang L., Nanoparticulate delivery of cancer cell membrane elicits multiantigenic antitumor immunity. Adv. Mater. 29, 1703969 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]