

Metabolic isotope labeling and imaging mass spectrometry delineate early events of precipitating Aβ plaque pathology.
Abstract
β-Amyloid (Aβ) plaque formation is the major pathological hallmark of Alzheimer’s disease (AD) and constitutes a potentially critical, early inducer driving AD pathogenesis as it precedes other pathological events and cognitive symptoms by decades. It is therefore critical to understand how Aβ pathology is initiated and where and when distinct Aβ species aggregate. Here, we used metabolic isotope labeling in APPNL-G-F knock-in mice together with mass spectrometry imaging to monitor the earliest seeds of Aβ deposition through ongoing plaque development. This allowed visualizing Aβ aggregation dynamics within single plaques across different brain regions. We show that formation of structurally distinct plaques is associated with differential Aβ peptide deposition. Specifically, Aβ1-42 is forming an initial core structure followed by radial outgrowth and late secretion and deposition of Aβ1-38. These data describe a detailed picture of the earliest events of precipitating amyloid pathology at scales not previously possible.
INTRODUCTION
Alzheimer’s disease (AD) poses an immense societal challenge, particularly as there are still no disease-modifying treatments (1–3). The major hallmarks of AD are the progressive accumulation of β-amyloid (Aβ) peptide derived from amyloid precursor protein (APP) and intracellular deposition of hyperphosphorylated tau protein (4–6). Although the importance of progressive Aβ plaque deposition in AD is long recognized, exactly how plaques develop over time and their relation to toxicity or homeostatic response of the surrounding neuronal network is not clear (2, 4, 7, 8). Therefore, it is of great relevance to delineate the fate of different amyloid aggregates in evolving Aβ pathology.
A major challenge in investigating Aβ pathology at submicron levels is the need for appropriate imaging technologies that combine the necessary spatial resolution, sensitivity, and specificity to probe these processes. The limited understanding of amyloidogenic protein aggregation in AD pathogenesis relates directly to the lack of effective imaging tools with high chemical, spatial, and longitudinal precision (9, 10).
Recent advances in using metabolic, in vivo labeling with stable isotopes (SIL), followed by mass spectrometry (MS) were demonstrated to be powerful tools to measure SIL kinetics (SILK) and protein turnover such as SILAC (SIL by amino acids in cell cultures) (11, 12), SIL of mammals (13), and SILK (14). In particular, in the context of AD, pioneering studies have been demonstrating the potential of SIL for both mechanistic studies in AD mouse models (15, 16) as well as for clinical, in vivo applications in patients with AD (14, 17). In these SILK studies, intravenous infusions of 13C6-Leu allowed Aβ and tau turnover dynamics to be quantified in continuously collected cerebrospinal fluid (CSF) from patients with AD, revealing highly relevant insights into protein clearance impairment associated with plaque pathology (14, 18–22). However, while these tools are very useful, they provide no or only very limited information about spatial isotope incorporation.
Emerging chemical imaging technologies such as imaging MS (IMS) (23) greatly increase the resolution of these events. The combination of isotope labeling of proteins with nanoscale secondary ion imaging (NanoSIMS) of these isotopes, termed multi-isotope imaging (24), has made it possible for measuring spatial protein turnover kinetics in cells and tissues and most relevantly demonstrated isotope incorporation in plaques in a hospice study of patients with AD and in transgenic APP mice, also referred to as SILK-SIMS (24, 25).
While NanoSIMS imaging provides outstanding data on nanoscopic isotope enrichment, one challenge of the technique is the limited molecular information retrievable, permitting analysis of intact peptides and proteins. This is something that can be achieved with matrix-assisted laser desorption/ionization MS–based IMS (MALDI-IMS), which has been successfully demonstrated for monitoring spatial enrichment and metabolization of intraperitoneally injected, stable isotope–labeled drug candidates (26) or intranasally instilled and intraperitoneally injected stable isotope–labeled phospholipids (27). Most importantly, MALDI-IMS allows for comprehensive, chemically specific Aβ peptide imaging of Aβ pathology in AD mouse models (10, 28) and in postmortem human AD brain tissue (29, 30).
We here set out to take advantage of these isotope labeling and imaging methods to follow Aβ peptide–specific aggregation dynamics in a recently developed knock-in mouse model of AD with familial APP mutations (31). We performed metabolic SILs of these mice combined with multimodal IMS, for spatial delineation of differential, stable isotope label incorporation in situ. Given that this imaging paradigm was implemented and applied in the context of AD pathology, this approach is herein, hence, referred to as imaging SILK (iSILK). The here described iSILK experiments allowed visualizing aggregation dynamics of different Aβ peptides within single plaques and across different brain regions in evolving plaque pathology from early deposition to later plaque growth. Specifically, using a comprehensive setup of different labeling schemes, we found that Aβ pathology in APPNL-G-F mice precipitates in the cortex by forming small dense core deposits consisting of Aβ1-42. Later events in early plaque pathology involve plaque growth upon homogenous Aβ1-42 deposition, deposition in the hippocampus, and secretion and deposition of Aβ1-38.
RESULTS
Stable isotopes can be metabolically incorporated in specific Aβ peptides and accumulate into extracellular plaques upon amyloid pathology onset
A central issue in delineating AD pathogenesis is that amyloid plaque pathology starts to develop long before any cognitive symptoms occur. It is therefore of great relevance to delineate the fate of different Aβ species in evolving plaque pathology, from early accumulation and aggregation to deposition and maturation of extracellular plaques. However, this is challenging as early Aβ aggregation, plaque formation, and plaque growth are constantly changing over time (32, 33). We therefore set out to examine the Aβ aggregation dynamics in APPNL-G-F mice. We aimed to delineate the initial events and sites of plaque onset in the brain by following amyloid aggregation. For this, we used an iSILK paradigm, based on metabolic isotope labeling, followed by spatial delineation of isotope-resolved aggregation kinetics using IMS.
We provided 15N-labeled protein diet to APPNL-G-F mice from ages 7 to 17 weeks (11-week PULSE period; Fig. 1A) to ensure that all APP is labeled prior and during initial Aβ secretion, Aβ deposition, and plaque onset, which, in the APPNL-G-F mouse model, takes place at around week 8 (31). Consequently, all the Aβ that was secreted and accumulated during rising amyloid just before deposition should contain 15N label. A CHASE period was omitted to avoid interference of heterogeneously labeled Aβ produced during the washout period.
Fig. 1. iSILK strategy to probe evolving plaque pathology using multimodal chemical imaging.
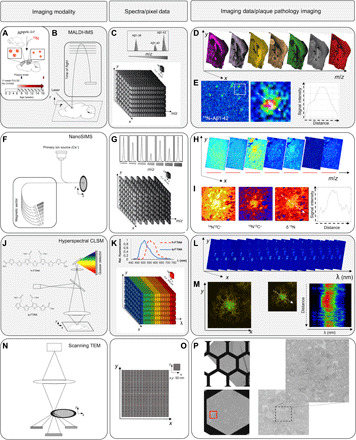
(A) APPNL-G-F mice were fed 15N diet, leading to metabolic isotope incorporation into APP and release and deposition of labeled Aβ peptide that is delineated with IMS. (B) MALDI-IMS uses a laser for analyte desorption and ionization and allows detection of intact Aβ peptides at 10 μm (C). (D) MALDI single ion images outline different Aβ isotopologues across the brain at a single plaque level (E). Here, total 15N–Aβ1-42 [Σ mass/charge ratio (m/z) 4450 to 4465] localizes predominantly to the plaque core. (F) NanoSIMS uses a highly focused, high-energy, primary ion beam for analyte desorption and ionization, allowing imaging of polyatomic species at 50 nm with high sensitivity with up to 7 m/z channels in parallel (G and H). NanoSIMS allows quantification of stable isotope enrichment (15N/14N above natural 15N abundance) at nanoscopic structures such as single plaques, although without intact molecular information. (I) Here, predominant 15N enrichment is detected at the core. (J to P) IMS is complemented with light and electron microscopy (EM) for additional structural information. (J to M) Structural amyloid imaging with confocal laser scanning microscopy (CLSM) and hyperspectral detection. (J) Double LCO staining with tetrameric formyl thiophene acid (q-FTAA) and heptameric formyl thiophene acid (h-FTAA). q-FTAA binds preferentially to mature fibrils. h-FTAA recognizes both mature and immature, prefibrillar Aβ. (K) The LCO probes have different emission properties that are delineated by CLSM using a spectral detector providing a continuous emission spectrum (32+2 channels) within each pixel (K), generating a lambda stack (L). Linear unmixing generates normalized emission profiles across single plaque and structural annotation based on the LCO emission profile (M). A characteristic blue shift is observed at the core indicating q-FTAA binding and mature, amyloid fibril content. (N) Scanning transmission EM (STEM) imaging on embedded tissues samples before NanoSIMS. STEM provides ultrahigh resolution at low nanometers (O), allowing visualization of fibrillar content in single plaques (P).
We analyzed the stable isotope enrichment in brain tissue and plaques in these mice using a multimodal chemical imaging paradigm (fig. S1), including MALDI-IMS (Fig. 1, B to E) and NanoSIMS (Fig. 1, F to I). The IMS experiments were complemented with fluorescent, confocal microscopy using conformation-sensitive amyloid dyes [luminescent conjugated oligothiophenes (LCOs) (34); Fig. 1, J to M] and scanning transmission electron microscopy (STEM; Fig. 1, N to P). The Aβ identity of deposits detected with MALDI-IMS and LCO microscopy was further validated using immunohistochemistry (fig. S2, A to G) and by off-tissue analysis using laser microdissection together with immunoprecipitation and MS (fig. S2, H to J) (29). This further allowed Aβ sequence validation of the intact peptide signals detected in MALDI-IMS (fig. S3).
The results show that 15N-labeled amino acids are incorporated into APP and Aβ and deposit within Aβ plaques upon plaque formation (Fig. 1E). In detail, MALDI-IMS detected solely labeled 15N–Aβ1-42, but no unlabeled 14N–Aβ1-42, demonstrating that PULSE feeding began early enough to capture all of the initial secretion and deposition of Aβ (Fig. 2, A to F). Here, 15N–Aβ1-42 showed distinct localization to amyloid plaques with characteristic accumulation at the plaque core (Fig. 1E). Similarly, NanoSIMS of these plaques revealed distinct plaque structures that showed characteristic enrichment of 15N label (δ15N; Fig. 1I and fig. S4, A to D). In contrast, no label was detected in control animals (fig. S4, E to H). NanoSIMS and complemental STEM show that isotope enrichment was highest at the compact, core structure in the plaques (Fig. 1, I and P, and fig. S4). Hyperspectral microscopy of these plaques using LCO-based, conformation-sensitive, amyloid staining (Fig. 1, J to M) (35) indicated that the core structure consisted of mature, Aβ fibrils, while the periphery was characterized by immature, prefibrillar Aβ aggregation intermediates (fig. S5).
Fig. 2. SILK imaging of intraplaque heterogeneity identifies core formation to precede Aβ plaque growth.
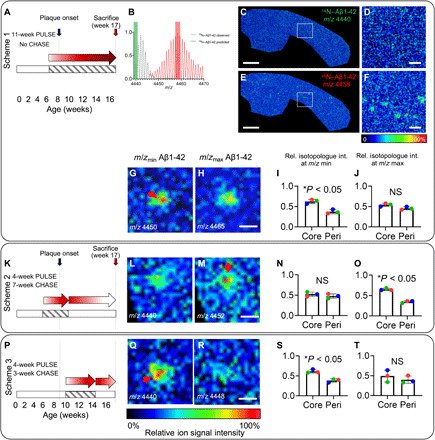
(A to J) Labeling Scheme 1 with 11 weeks of 15N diet, starting week 7 without CHASE (A) to establish 15N incorporation before plaque pathology onset. (B) MALDI MS spectrum–microdissected plaques showing isotopologue pattern of Aβ1-42 and 15N incorporation by comparison of the observed trace (red) and the isotope pattern predicted for unlabeled Aβ1-42 (gray). (C to F) MALDI-IMS ion maps of Aβ1-42 show no unlabeled Aβ1-42 signal (C and D), while solely 15N-labeled Aβ1-42 was observed with distinct plaque localization (E and F). Scale bars, 1000 μm (C and E) and 75 μm (D and F). (G and H) Ion maps of cortical plaques for least labeled Aβ1-42 isotopologue (m/z 4450) (G) and maximally labeled Aβ1-42 (m/z 4465) (H). (I and J) Bar plots for ion intensity of (G) (I) and (H) (J) showing significantly higher intensity of less-labeled Aβ1-42 (G) in the core (I), while the maximally labeled Aβ1-42 (H) was not different between core and periphery (J). (K) Scheme 2. (L and M) Ion maps of cortical plaques for least labeled Aβ1-42 (unlabeled; m/z 4440) (L) and maximally labeled Aβ1-42 (m/z 4452) (M). (N and O) Bar plots for ion intensity of (L) (N) and (M) (O). Statistical analysis validates higher intensity of maximally labeled Aβ1-42 (M) in the core (O), while the least labeled (in this case, nonlabeled) Aβ1-42 (L) was not different between core and periphery (N). (P) Scheme 3. (Q and R) Ion maps of cortical plaques for least labeled Aβ1-42 (unlabeled; m/z 4440) (Q) and maximally labeled Aβ1-42 (m/z 4448) (R). Bar plots for ion intensity of (Q) (S) and (R) (T). Statistical analysis validates higher intensity of less-labeled Aβ1-42 (Q) in the core (S), while the maximally labeled Aβ1-42 (R) was not different between core and periphery (T). Bar plots (I, J, N, O, S, and T) indicate means ± SD. n = 3 animals, N = 10 plaques per animal; MALDI intensity scale (G, H, L, M, Q, and R), 0 to 100% relative (rel.) ion intensity (int.). Scale bars, 30 μm (H, M, and R). NS, not significant.
MALDI-IMS peptide data show that plaques in APPNL-G-F mice at 17 weeks contain predominantly Aβ1-42 along with Aβ1-38. The plaque-associated Aβ species observed at the early age, 17 weeks, showed all 15N incorporation with varying degrees of 15N and 14N, while no unlabeled Aβ was observed in 14N (Fig. 2, B to F). The absence of unlabeled Aβ peptides in plaques of these labeled animals verifies that starting labeling at week <7 allows for in time, metabolic introduction of 15N to cover the plaque onset period from before Aβ plaque deposition.
These results show that the general iSILK labeling and analysis paradigm proved suitable to generate metabolically labeled Aβ that deposited in extracellular Aβ plaques. The data show that initiation of 15N feeding right before the onset of Aβ plaque pathology is sufficient to achieve 15N labeling of all the Aβ peptides present in extracellular Aβ plaques. Moreover, complementary structural imaging identified morphological heterogenous amyloid structures within the Aβ plaques, i.e., plaque core and periphery (fig. S5). This allows correlation of individual, differentially labeled Aβ peptides to different plaque structures such as core and periphery and which of these intrafeature structures evolve in what order during plaque formation.
Initial plaque formation in APPNL-G-F mice starts with formation of a dense core
AD pathology presents itself with a wide phenotypic heterogeneity among patients with AD, which, among others, manifests itself in the formation of morphological heterogeneous plaques, including diffuse and cored deposits (5, 36, 37). As diffuse plaques are also observed in amyloid-positive individuals without cognitive defects (29, 38), Aβ deposition into cored plaques has been linked with neurotoxic pathology in a wide variety of familial AD (fAD) cases, as well as in sporadic AD (sAD) (36, 39, 40). The actual chain of events underlying heterogenous plaque pathology and formation of diffuse and cored plaques, respectively, are still not fully understood. This can be addressed using iSILK, where spatial mapping of relative isotope accumulation across histological features allows delineation of the degree of Aβ deposition over time within different plaque structures.
A major observation for the first experiments showed differences in the degree of labeling of the detected Aβ peptides across the plaque structure. MALDI-IMS showed that the relative intensity of total 15N–Aβ1-42 signal was highest in the center of Aβ plaques [Fig. 1E, ion image: Σ mass/charge ratio (m/z) 4450 to 4465). Similarly, the corresponding NanoSIMS data showed pronounced 15N enrichment (δ15N) to be associated with these core structures (Fig. 1I and fig. S4, A and D).
Assuming that 15N isotope enrichment signal at the plaque, as observed with NanoSIMS, originates almost exclusively from Aβ and that the degree of enrichment corresponds to the degree of deposition, higher 15N enrichment at the core could be explained in two ways. On the one hand, more isotope enrichment at the core results from longer Aβ deposition and, hence, earlier deposition at the core. On the other hand, more isotope enrichment at the core could indicate merely a more core-specific peptide deposition in general that can be different over time and take place at any time during the labeling period, something that cannot be differentiated by looking solely at the total 15N signal, detected by NanoSIMS. This highlights the need for using complementary techniques with specificity toward the distinct Aβ peptides, as well as the differentially labeled species of the same Aβ peptide (isotopologues), something that is both warranted by MALDI-IMS.
Inspection of the MALDI-IMS peptide spectra for single-plaque region of interest (ROI) shows a characteristic isotope envelope for 15N-labeled Aβ species reflecting the degree of stable isotope incorporation and deposition (Fig. 2B and fig. S6, A to D). The generation of single ion images for various m/z species within the isotope envelope for Aβ peptides allows spatial delineation of deposition patterns of differentially labeled isoforms of the same Aβ peptide (fig. S6, D to G).
By monitoring Aβ1-42 deposition in the first labeling experiment (Fig. 2, A to J, and fig. S6, C to G), we observed that less-labeled 15N–Aβ1-42 species was (m/z 4450) localized to the core as compared to the more peripheral parts of the plaque (Fig. 2, G and I, and fig. S6F), while more label containing 15N–Aβ1-42 species (m/z 4465) showed no significant localization to either the core or periphery and deposited evenly over the plaque region (Fig. 2, H and J, and fig. S6G). Since the less-labeled 15N–Aβ1-42 must originate from earlier-secreted and likely earlier-deposited Aβ1-42, deposition at the core suggests that core structure formation is an initial event in seeding plaque pathology in these mice. In contrast, the deposition pattern of later-synthesized Aβ1-42, as reflected in increased content of 15N, was more homogenous across the plaque.
Together, these data point toward plaque formation in APPNL-G-F being initiated by seeding as dense core and outward growth as a consequence of later, plaque-wide deposition across this initial seeded plaque structures. This consequently results in morphological heterogenous cored deposits with a diffuse, prefibrillar periphery.
The data from the first 11-week full-stop labeling scheme (Scheme 1 and Fig. 2, A to J), without CHASE period, suggested that increased isotope deposition at the core of the plaque is a consequence of more extended deposition. We therefore hypothesized that plaque formation in APPNL-G-F mice is initiated by seeding as a dense core and plaque growth is a consequence of later, more homogenous deposition across seeded plaque structures.
To address this question, we performed more PULSE/CHASE labeling experiments to test and validate the findings on plaque formation observed for Scheme 1. Specifically, we designed two schemes with a 4-week 15N PULSE period starting at either week 6, before plaque deposition (Scheme 2 and Fig. 2, K to O), or at week 10, during plaque deposition (Scheme 3 and Fig. 2, P to T). Both schemes included a CHASE (i.e., washout) period allowing for unlabeled Aβ to be introduced to further delineate consequential events within a shorter time scale upon a certain stimulus.
For Scheme 2, we supplied a 15N diet from weeks 6 to 10 followed by a 7-week CHASE (Fig. 2, K to O). The aim was to ensure 15N labeling of APP at an even earlier stage before plaque pathology onset, as determined in Scheme 1. Using a shorter PULSE and a CHASE period allowed us to delineate the fate of early synthesized Aβ as compared to Aβ secreted and deposited during the washout. This can be achieved by mapping spatial intensity patterns of highly labeled Aβ species in comparison to their less and unlabeled isoforms as encoded in their isotope envelope pattern (fig. S6, H to L).
Our results show that the maximally labeled Aβ 15N-1-42 species (m/z 4452) was localized to the plaque core (Fig. 2, M and O, and fig. S6L). Conversely, unlabeled Aβ1-42 (m/z 4440), which was consequently produced after washout, was found to show an even distribution pattern across the plaque area (Fig. 2, L and N, and fig. S6K). This is in line with the results obtained for Scheme 1, where early, less-labeled Aβ 15N-1-42 was localized to the core, while later-secreted, more-labeled Aβ 15N-1-42 localized homogenously across the plaque (Fig. 2, G to J).
Together, this further strongly suggests that dense core formation represents the earliest seeding event in extracellular plaque deposition in the APPNL-G-F mice used in this study. This can be attributed to the arctic mutation of APP increasing the aggregation propensity of full-length Aβ1-42arc (41). This is well in line with immunohistochemical observations in APPNL-G-F mice (fig. S2, E to G) (31) and for transgenic APPArcSwe mice (42).
To further validate these results, we performed an additional labeling scheme (Scheme 3, Fig. 2, P to T, and fig. S6, M to Q), where 15N diet was supplied at a later time point at week 10, than in Schemes 1 and 2. The aim was to dissect the initial plaque formation phase (weeks 8 to 10) from the following phase of early plaque growth (week 10 onward). We hypothesized that early-produced Aβ will not contain any label and will deposit specifically at the core during pathology onset, similar to the results obtained for Schemes 1 and 2.
The MALDI-IMS data for Scheme 3 shows a distinct, predominant localization of unlabeled, early-secreted Aβ1-42 (m/z 4440) to the core in cortical deposits (Fig. 2, Q and S, and fig. S6P). Conversely, later-synthesized, more-labeled 15N–Aβ1-42 (m/z 4448) shows a homogenous distribution pattern across the plaque ROI (Fig. 2, R and T, and fig. S6Q). This further supports the findings of the previous schemes and verifies that Aβ pathology in APPNL-G-F mice appears to be initiated through formation of small core seeds, followed by homogenous plaque-wide deposition during plaque growth (31).
Plaque pathology in APPNL-G-F evolves through initial deposition of Aβ1-42 in the cortex, followed by deposition in the hippocampus
Along with intraplaque-specific, structural details on plaque growth in APPNL-G-F mice, iSILK allows us to identify deposition dynamics across different brain areas. This is of relevance to identify how plaque pathology spreads across the brain and, ultimately, to resolve the chain of mechanisms in how plaque formation affects vulnerable brain regions leading up to cognitive defects.
The aim was therefore to quantify the relative amounts of plaque-associated Aβ1-42 across cortical and hippocampal deposits. We hypothesized that the deposited Aβ1-42 species that constitute the earliest isoforms for plaques in specific brain regions allow for relative comparison of where across these brain regions plaque pathology is precipitating first. For this, we isolated plaque ROI spectral data of hippocampal and cortical deposits (fig. S6, R to V). We compared the isotopologue signature of plaque-associated Aβ1-42 that encodes the degree of labeling, as indicated by the shift in m/z value (fig. S6, W to Z). For unbiased, quantitative comparison of label incorporation, we performed curve analytics of the isotope envelope for plaque-associated Aβ1-42 signal that, in turn, encodes the degree of label incorporation. Here, to estimate the shift in m/z value as caused by the 15N label and hence difference in label content, we compared the centroid of the isotope envelope (fig. S6, W to Z). On the basis of the labeling scheme, this shift in the isotopologue signature indicates earlier or later deposition.
For Scheme 1, Aβ1-42 that is no difference in label incorporation as compared to hippocampal plaques (Fig. 3, A and B) attributed to the large overlap of the two isotope distributions as further highlighted in the single ion images that do not allow to distinguish plaques with earlier- and later-labeled Aβ1-42 (Fig. 3, C to F). NanoSIMS imaging of cortical and hippocampal plaques showed a significantly higher degree of total 15N isotope enrichment for plaques in the cortex as compared to plaques in the hippocampus (fig. S7). This would suggest that plaques in the cortex are formed earlier as they can thereby accumulate more 15N over time.
Fig. 3. Single Aβ plaque quantification identifies primary brain regions for plaque deposition.
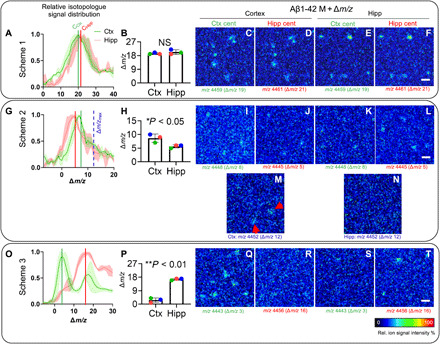
Comparative analysis of MALDI-IMS spectral data of whole plaque ROI from cortex and hippocampus for Aβ1-42 isotopologue distributions. (A) The Δm/z value corresponds to the shift of the isotopologue signals compared to the unlabeled peptide signal. Centroid values (average Δm/z value) are calculated for the corresponding Aβ1-42 isotopologue distribution curves of whole plaques in the cortex (Ctx) versus hippocampus (Hipp) that serve as a measure of average label incorporation into Aβ1-42 in those regions. (B) For Scheme 1, statistical analysis of the centroid data shows no difference between cortical and hippocampal plaques. (C to F) Single ion images of the Aβ1-42 peptide isotopologue signal m/z at MAβ1-42 + Δm/z for each region (MAβ1-42 = 4440; m/z = 4440 + Δm/z). Because of the overlap of isotopologue distributions, no difference is observed for Scheme 1. (G to N) For Scheme 2, comparison of signal from individual plaques in cortex and hippocampus revealed higher 15N–Aβ1-42 signal being present in cortical plaques (H). (I to L) Single ion maps for Scheme 2 show that although better separated, both isotope distributions are still overlapping at their respective centroids. When comparing isotopologue signal at the maximally labeled Aβ1-42 detected in Scheme 2 [m/z 4452; Δm/z = 12; blue, dashed line (G)], plaques are detected in the cortex [(M), arrows] but not hippocampus (N). (O and P) For Scheme 3, cortical plaques showed significantly less isotope incorporation (75% less) as compared to hippocampal plaques (P). (Q to T) Single ion images verify that low-labeled (early) Aβ1-42 (m/z 4443) is detected in the cortex (Q) but not in the hippocampus (S). Conversely, no higher labeled Aβ1-42 is detected in the cortex (R) but solely detected in hippocampal deposits (T). Bar plots (B, H, and P) indicate means ± SD. n = 3 animals, N = 10 (Ctx) and N = 5 (Hipp) plaques per animal; MALDI intensity scale, 0 to 100% relative ion intensity. Scale bars, 75 μm (F, L, and T).
Initial deposition of plaques in the cortex is further supported by the results obtained for Scheme 2. Here, using a short PULSE and inclusion of a CHASE period showed even more pronounced differences in region-specific plaque label content. In this experiment, Aβ1-42 in cortical plaques showed a 1.75-fold increase in label incorporation (175%, P < 0.05) as compared to hippocampal plaques (Fig. 3, G and H). This is in line with the assumption that in this labeling scheme, which started before plaque onset, early-deposited Aβ1-42 contains more 15N label as compared to later Aβ1-42 deposited produced during the washout phase. This is further illustrated in the single ion maps generated of isotopologue signals (m/z) that correspond to the different centroid values (m/zcentr. = MAβ1-42 + Δm; Fig. 3, I to L), although discrimination is complicated as those ion maps show distribution of isotopologues that represent average incorporation values and show a large overlap at the corresponding m/z value (Fig. 3, G and I to L). This is due to the isotope distribution pattern of different regional plaque types showing too much overlap. However, as the statistical analysis provide an unbiased overview of differences in average label incorporation, visualizing the maximal m/z values detected for the corresponding isotope distributions can illustrate specific deposition patterns of those peptide species that are secreted and deposited very early. Single ion maps for the maximally labeled Aβ1-42 isotopologues (m/zmax 4452; Fig. 3G, blue dashed line) show solely deposition in the cortex but not hippocampus (Fig. 3, M and N), indicating that the cortex is the primary site of early plaque deposition in this mouse model.
Last, using a complimentary labeling Scheme 3 (Fig. 3, O to T), starting after plaque onset, we aimed to further verify these results and even achieve a pronounced separation of the differentially labeled species. In this scheme, early-formed plaques should contain greater amounts of early and, hence, unlabeled Aβ1-42, leading to more distinct isotopologue patterns in between the regions that can be better discriminated. For Scheme 3, Aβ1-42 in cortical plaques showed fourfold less label incorporation (25%, P < 0.01) as compared to hippocampal plaques (Fig. 3, O and P). Aβ1-42 single ion maps for the corresponding centroid values (m/zcentr. = MAβ1-42 + Δm), representing average isotope incorporation of the different plaque populations, show distinct differences in spatial deposition pattern (Fig. 3, Q to T). Here, low-labeled, early, Aβ1-42 (m/zcentr. 4443) peptides are solely observed in the cortex (Fig. 3Q), while later-secreted more-labeled Aβ1-42 peptides (m/zcentr. 4456) were exclusively detected in the hippocampus (Fig. 3T), which further suggests that cortical plaques form earlier than hippocampal plaques.
To further validate these observations, we evaluated regional plaque load in a short time- course series of mice between 9 and 11 weeks using fluorescent amyloid staining (fig. S8, A to C). At 9 weeks, initial plaque deposition is observed primarily in the cortex, covering twice the area covered by the very few deposits observed in the hippocampus (fig. S8, D to F). Similarly, more pronounced deposition in cortical areas as compared to the hippocampus was observed with evolving pathology at 10 and 11 weeks (fig. S8, B and C).
Together, these data show that in these mice, plaque pathology precipitates primarily in cortical areas. The isotope labeling data further suggest that cortical plaques form earlier than hippocampal plaques. This is well in agreement with previous data described for APPNL-G-F mice and in transgenic mouse models carrying the Arctic and Swedish APP mutation (31, 41–43).
Formation and deposition of C-terminally truncated Aβ1-38 occur after initial plaque formation
The initiation and progression of amyloid plaque pathology have been suggested to be associated with secretion and aggregation of distinct Aβ peptide truncations, where both C- and N-terminal truncations have been reported (39, 40, 44, 45). Given the young age of the mice used in this study, this posed an opportunity to investigate what other terminally truncated species are associated with the initial Aβ pathology in the APPNL-G-F mice. The MALDI-IMS data for all labeling schemes revealed the presence of Aβ1-38 in plaques, however, at lower amounts than Aβ1-42. This suggests that C-terminal processing and deposition are later events in early, developing plaque pathology. However, it is still under debate where and when formation and deposition of different Aβ processing products take place. We hypothesized that iSILK can be used to delineate the chain of events during sequential Aβ production and deposition, i.e., whether Aβ1-38 production occurs either through processing of early-secreted, full-length Aβ1-42 but through delayed deposition or whether this Aβ1-38 secretion and deposition are time-linked processes that take place later.
We therefore compared the degree of labeling and the spatial deposition pattern of differentially labeled Aβ1-38 isoforms across the different labeling schemes. For Scheme 1, we observed that the degree of 15N labeling for Aβ1-38 deduced from the centroid values of the relative, isotope distribution curves was not statistically different to the degree of isotope incorporation observed for Aβ1-42 (Fig. 4, A and B). Ion maps for different labeled Aβ1-38 corresponding to the centroid values (average isotope incorporation) detected for both Aβ1-38 and Aβ1-42 displayed a similar spatial distribution pattern across the plaque (Fig. 4, C and D). While the isotope patterns of Aβ1-38 and Aβ1-42 could not be discriminated using the long labeling approach in Scheme 1, results for Scheme 2 showed that Aβ1-38 contained 43% less isotope as compared to Aβ1-42 (Fig. 4, E and F). This suggests that all Aβ1-38 has been secreted and deposited, later than Aβ1-42, during the washout period after initial plaque formation. However, similar to Scheme 1, no difference in spatial pattern was observed for the different Aβ1-38 isotopologues with the different centroid values, as a consequence of too extensive overlap of the isotopologue curves (Fig. 4, G and H). The hypothesis that Aβ1-38 secretion and deposition succeed that of Aβ1-42 is however further supported by single ion images generated for m/z shifts corresponding to the maximal Aβ1-42 label incorporation detected (Fig. 4, I and J). Here, only the corresponding Aβ1-42 isotopologue is detected and localized to plaques (Fig. 4J), while no correspondingly labeled Aβ1-38 species was detected (Fig. 4I).
Fig. 4. Deposition of Aβ1-38 peptide occurs after Aβ1-42, as indicated by differences in 15N incorporation.
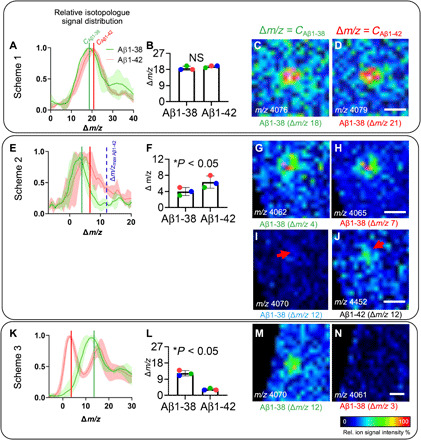
(A) MALDI-IMS spectral data of cortical plaques show relative isotopologue distributions for Aβ1-38 and Aβ1-42. Δm/z indicates the shift of isotopologue signals compared to the m/z of the monoisotopic, unlabeled peptide. Centroid values for the curves fitted to the isotopologue distributions of Aβ1-38 (green, CAβ1-38) and Aβ1-42 (red line, CAβ1-42), indicating average label incorporation. (A to D) For Scheme 1, no significant difference in average isotope incorporation in between Aβ1-38 and Aβ1-42 was observed because of overlap of the relative isotopologue curves (B). (C and D) Single ion images of Aβ1-38 (m/z 4058) + Δm/z for the different centroid values (Δm/z = CAβ1-38 and Δm/z = CAβ1-42), representing average, relative isotope incorporation observed for Aβ1-38 and Aβ1-42. No difference in ion distribution was observed for the different Aβ1-38 isotopologues at Δm/z = CAβ1-42 and CAβ1-38. (E to J) For Scheme 2, Aβ1-42 showed significantly higher label incorporation (+12) as compared to Aβ1-38 (+4) (F). (G and H) Single ion images of Aβ1-38 + Δm/z for the different centroid values show no difference in ion distribution due to broad overlap of the distribution curves (E). (I and J) Ion maps for the Aβ1-38 isotopologue at maximum label incorporation as observed for Aβ1-42 (Δm/z = 12) (E). The ion signal of maximally labeled Aβ1-42 (m/z 4458) was found to localize to plaques (J), while no signal is detected for the corresponding Aβ1-38 isotopologue (m/z 4070) (I). (K to N) In Scheme 3, Aβ1-38 showed significantly higher incorporation (+12) compared to Aβ1-42 (+3) (K and L). (M and N) Single ion images of Aβ1-38 + Δm/z for the different centroid values. For Δm/z = CAβ1-38, Aβ1-38 was found to localize to plaques (M), while no signal is detected for the Aβ1-38 isotopologue at Δm/z = CAβ1-42 (K and N). Bar plots (B and F) indicate means ± SD. n = 3 animals, N = 10 plaques per animal; MALDI intensity scale, 0 to 100% relative ion intensity. Scale bars, 30 μm (D, H, J, and N).
For further validation, we performed two additional labeling experiments deduced from Scheme 2 with modified CHASE periods: Scheme 2b, 4 weeks, and Scheme 2c, 10 weeks (fig. S9). These data confirm differential degrees of label incorporation in Aβ1-42 and Aβ1-38, with Aβ1-42 containing relatively more label. Further, similar rates of label washout were observed for both peptides (fig. S9). Together, these additional experiments further support that Aβ1-42 is secreted and deposited before Aβ1-38. Last, this was even more prominent in Scheme 3, with labeling starting at week 10 (Fig. 4, K to N). Here, Aβ1-38 showed 400% more average label incorporation compared to Aβ1-42 (Fig. 4L). This was further supported by the single ion maps of Aβ1-38 isotopologues with isotope incorporation degrees that correspond to average labeling values observed for both Aβ1-38 and Aβ1-42 (Fig. 4, M and N). Here, the Aβ1-38 signal of the isotopologue species corresponding to average label incorporation observed for Aβ1-38 showed characteristic plaque localization (Fig. 4M). In contrast, no signal was observed for the Aβ1-38 isotopologue corresponding to the label incorporation observed for Aβ1-42 (Fig. 4N). This further suggests that Aβ1-38 secretion and deposition is taking place at a later point of time as compared to Aβ1-42, which can be delineated with well-defined PULSE/CHASE experiments. Together, this validates that both production and deposition of Aβ1-38 represent a downstream event that occurs only after the initial seeding and plaque formation through deposition of Aβ1-42.
DISCUSSION
We used a novel chemical imaging paradigm, iSILK, based on metabolic SILs followed by multimodal IMS, for delineating the early events in developing plaque pathology in APPNL-G-F mice. We show that metabolically labeled Aβ accumulate in plaques upon pathology onset and can be traced with multimodal IMS at cellular length scales.
To follow dynamic biochemical events in vivo is inherently challenging. Other approaches using fluorescent dyes and in vivo two-photon imaging (TPI) provide intriguing data on in vivo plaque aggregation dynamics (32, 46, 47). Similarly, clinical imaging methods such as positron emission tomography (PET) allow amyloid pathology to be followed in vivo (48, 49). Moreover, elegant in vivo labeling techniques that allow ex vivo imaging have been presented (33). The main limitation of those approaches is the restricted molecular information about what amyloid peptides are involved, as well as where and when those species aggregate and deposit. A further limiting factor is that those techniques rely on indirect detection, using distinct chemical probes that require acute, in vivo amyloid staining, which can first occur after the aggregates are formed. Further limitations of those tools are with respect to spatial resolution (PET) or restrictions to superficial cortical layers (TPI). Similarly, ex vivo time series that can be performed with more specific and better resolved techniques such as immunohistochemistry or IMS are limited to postmortem tissues. Further, this requires distinct, predefined time schemes that provide limited longitudinal information. This makes ex vivo time series studies inherently difficult to design to capture varied pathology onsets, such as genetically triggered plaque pathology, and, in addition, require extensive amounts of animals. In the context of studying plaque pathology, this would be even more challenging for other AD mouse models, such as APPNL-F knock-in mice or various transgenic models, with delayed plaque pathology onset, starting between 6 and 10 months (31, 42, 50, 51).
The introduction of SIL greatly increases the longitudinal resolution for the study of biochemical events both in appropriate model systems (11, 13, 15) and in patients, although only in proximal fluids such as blood and CSF (14). This allows following pathogenic events over a long period of time that are otherwise hard to capture. While SIL-based protein analyses approaches provide comprehensive insight in protein regulation, processing, and interaction dynamics, those tools provide limited spatial information (13, 52). In turn, NanoSIMS imaging methods for monitoring stable isotope enrichment in cells and tissues have only limited molecular information (24, 25, 53). The main advantage of the here used MALDI-IMS approach is maintained molecular specificity that facilitates to comprehensively follow intact peptides that further constitute the actual pathogenic player, in this case, Aβ peptides.
The iSILK approach allowed us to visualize aggregation dynamics of all individual Aβ species within single plaques and across different brain regions. Using a comprehensive PULSE/CHASE labeling scheme setup, we found that Aβ pathology in APPNL-G-F mice precipitates first in the cortex by formation as a small dense core, with deposits consisting of Aβ1-42. Later events in early plaque pathology involve homogenous Aβ1-42 deposition and plaque growth in the cortex followed by deposition in the hippocampus, and, eventually, secretion and deposition of Aβ1-38. The data provide a longitudinal picture for the biochemical basis underlying development of heterogenic, intraplaque morphology.
A limitation of MALDI-IMS, while providing intact peptide labeling data, is the restricted spatial resolution (10 μm). This poses challenges for accurate correlation between MALDI-IMS–iSILK data and structural aspects of amyloid aggregation and plaque formation. For further information about what amyloid aggregates are detected and can be correlated with labeled Aβ peptide localization, we investigated adjacent tissue sections with conformation sensitive amyloid probes (LCO) (34, 35, 54). We show that in APPNL-G-F mouse model, plaque formation involves an initial deposition of a small, tetrameric formyl thiophene acid (q-FTAA)–positive core that consists exclusively of Aβ1-42. In contrast, later deposited Aβ is localized homogenously across the plaque to both plaque core and periphery interspersed with immature, prefibrillar Aβ aggregation, as revealed by heptameric formyl thiophene acid (h-FTAA) staining.
While seeding of Aβ pathology is considered a key event in early amyloid plaque formation, the mechanism of subsequent plaque growth and maturation is not well understood. fAD/sAD display structurally distinct plaque pathology, including both formation of diffuse and dense core deposits (29, 55). This is of relevance, as dense, cored plaques are specific to AD plaque pathology. Although cored plaques are observed in all patients with sAD and fAD, structural differences within the core fibrils have been identified that, in turn, were specific to the etiological subtypes of AD (55). Morphological plaque heterogeneity has been suggested to depend on underlying Aβ peptide composition, where the presence of the core has been associated with deposition of shorter Aβ peptides and to represent a secondary mechanism in plaque maturation (29).
Along that line, protofibrillar and oligomeric Aβ aggregates are considered the neurotoxic species that drive Aβ pathology, and our observations that mature fibrils, as observed in the core structure, precede protofibrillar Aβ species appears counter to both hypotheses on amyloid gain of toxicity and plaque formation dynamics proposed for AD pathology in humans. There are two possible explanations for this discrepancy. First, the limited spatial resolution and sensitivity of MALDI-IMS may prevent detection of small, prefibrillar aggregates that might precede the detected core structures, captured by this method in humans. However, on this point, it is noteworthy that the hyperspectral, LCO microscopy experiments did not show any h-FTAA–positive, immature amyloid structures that were not correlated with the MALDI-IMS signal. While this suggest that the earliest seeds detected by conformation-sensitive LCO microscopy are also captured by MALDI-IMS, extensive ex situ structural characterization of the various aggregation intermediates at the nanoscale poses an exciting challenge that can be subject for follow-up studies. A second more straightforward explanation for the orthogonal observations on heterogenic plaque formation is that the genetic background of the APPNL-G-F model used in the present study may result in a different outcome from that seen in humans. Multiple mutations have previously been described to have an effect on plaque morphology (55). In APPNL-G-F mice, the arctic APP mutation leads to early formation of a pronounced plaque core. This is likely a consequence of rapid aggregation due to increased aggregation propensity of Aβ, which is in line with observations from transgenic mice (41, 42). In addition, the Swedish mutation leads to increased production of mainly Aβ1-40 but also Aβ1-42 (41, 42). The shift toward predominant production of Aβ1-42 as observed here is attributed to the Iberian APP mutation (I716F) that lies outside the Aβ sequence near the γ-secretase cleavage site (31, 56). While the Aβarc plaque phenotypics of the APPNL-G-F model are not representative for human AD pathology, these mice provide a suitable model system for studying Aβ plaque pathology–associated mechanisms by showing exacerbated neuroinflammation and accelerated age-related memory impairment as previously noted (31). We make use of the accelerated plaque pathology in this mouse model as this allows us to follow plaque pathology onset in a reasonable time scheme, while still providing relevant pathobiochemical information about cored plaque morphotypes specific to both human sAD and fAD pathology. It has been reported that distinct structural features within the dense core fibrils can be specific for heterogenous plaque pathology among patients across different etiologies (55). Our iSILK data described here can therefore serve as a primer on how to study the effects of genetic background on plaque polymorphism and heterogenous plaque pathology, respectively.
A further advantage of the iSILK approach includes the possibility to study deposition dynamics across brain regions and across different Aβ species. Our experiments identified the cortex as primary site of deposition followed by deposition in subcortical areas, mainly the hippocampus, which is in line with previous observation in this mouse model (31). Further, our data indicate that the Aβ1-38 is the second main peptide present in the APPNL-G-F mice. Recently, MALDI-based three-dimensional reconstruction of amyloid plaques in aged APPNL-G-F mice suggested the presence of Aβ1-42 in cores that were surrounded by Aβ1-38 in most but not all plaques (28). SILK imaging provides strong evidence for a sequence of events that can explain the chemical phenotype of these plaques. This is further in line with enzyme-linked immunosorbent assay results for these mice (31), where the other main peptide detected was the Aβ1-38. Presence of shorter C- and N-terminally truncated peptides within single plaques has been suggested to represent intraplaque metabolism of Aβ1-42 toward shorter fragments, a process facilitated by enzymatic activity (57, 58). While this might still be the case for the N-terminally truncated fragments as shown by the similar distribution pattern of the Aβx-40 and Aβx-42 detected in human brain tissue (29), this is likely not the case for the C-terminally truncated peptides. Given that throughout Schemes 2 and 3, Aβ1-38 showed different degrees of stable isotope incorporation as compared to Aβ1-42, our data suggest that the generation of Aβ1-38 represents an independent event, probably through γ-secretase–mediated APP processing.
Together, the data presented here demonstrate that iSILK is an innovative approach to studying the Aβ plaque formation dynamics over time in experimental AD. The refined methods for imaging metabolically labeled, endogenous peptides and proteins hold further great potential for investigations on studying other genetic mouse models of AD pathology and delineate the effect of different mutations on developing amyloid pathology as well as genetic and pharmacological efforts to modify Aβ aggregation propensity. This is relevant to developing strategies to target AD pathogenesis in time particular with respect to ongoing antibody-based drug trials that target Aβ aggregation intermediates, where previous efforts have not been successful. The iSILK principles and technologies demonstrated here can furthermore be of great potential for studying other disease mechanisms such as for studying tumor development in experimental cancer research.
MATERIALS AND METHODS
Chemicals
MouseExpress (15N, 98%) mouse feed kit (15N/14N) was obtained from Cambridge Isotope Laboratories (Andover, MA, USA). All solvents used in the study were of high-performance liquid chromatography–MS grade. Acetone (Ac), acetonitrile (ACN), absolute ethanol (EtOH), and methanol (MeOH) were obtained from Thermo Fisher Scientific (Hampton, NH, USA). Glacial acetic acid was purchased from VWR Chemicals (Radnor, PA, USA). Paraformaldehyde (PFA), glutaraldehyde, sodium cacodylate buffer, osmium tetroxide, and Agar 100 resin were obtained from Agar Scientific (Stansted, Essex, UK). Formic acid (FA), trifluoroacetic acid (TFA), α-cyano-4-hydroxycinnamic acid (CHCA), and 2,5-dihydroxyacetophenone (2,5-DHAP) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Tissue-Tek optimal cutting temperature compound was purchased from Sakura Finetek (AJ Alphen aan den Rijn, Netherlands). Indium tin oxide (ITO)–coated conductive glass slides and peptide calibration standard I were obtained from Bruker Daltonics (Bremen, Germany). PELCO copper grids were obtained from Ted Pella (Redding, CA, USA). The double-distilled H2O (ddH2O) was obtained from a Milli-Q purification system (Merck Millipore, Darmstadt, Germany). The 0.17 polyethylene naphthalate (PEN) membrane slides and Adhesive Cap 500 opaque tubes were purchased from Zeiss/P.A.L.M. Microlaser Technologies (Bernsried, Germany). Dako fluorescence mounting medium was obtained from Agilent (Santa Clara, CA, USA).
Animal experiment
All procedures and experiments on APP knock-in mice (NL-G-F) were performed at University College London with local ethical approval 6 May 2016 in agreement with guidelines of the Institutional Animal Care and Use Committee and the Animals (Scientific Procedures) Act 1986. Male APP knock-in mice (APPNL-G-F) carrying humanized Aβ sequence, along with Swedish mutation (KM670/671NL) on exon 16, as well as Arctic (E693G) and the Beyreuther/Iberian mutations (I716F) on exon 17, were used in the study. To study spatiotemporal Aβ plaque metabolism, mice were fed MouseExpress (15N, 98%) mouse feed (PULSE) based on 15N following the different labeling schemes:
Scheme 1. PULSE, 10 weeks (weeks 7 to 17), no CHASE (n = 3);
Scheme 2. PULSE, 4 weeks (weeks 6 to 10), CHASE, 7 weeks (n = 3);
Scheme 2b. PULSE, 4 weeks (weeks 6 to 10), CHASE, 4 weeks, (n = 3);
Scheme 2c. PULSE, 4 weeks (weeks 6 to 10), CHASE, 10 weeks (n = 3);
Scheme 3. PULSE, 4 weeks (weeks 10 to 14), CHASE, 4 weeks (n = 3).
Control animals (n = 3) for each scheme received 14N spirulina diet. The mice were euthanized after the CHASE period. Following brain isolation, one-half was snap-frozen in liquid N2-cooled isopentane, while the other was immediately dissected into anatomical regions and immersion fixed in 4% PFA, and thereafter processed for EM.
In addition to the iSILK mouse experiments, a small, time-course study on 8- to 10-week-old male, APPNL-G-F mice was performed (n = 3 per time point; N = 2 tissue sections per mouse). These animals were euthanized via decapitation, the brain was rapidly extracted, and one hemisphere was drop-fixed into 4% PFA and was left overnight at 4°C. Hemispheres were then transferred into 30% sucrose and 0.03% sodium azide in 0.01 M phosphate-buffered saline (PBS) for storage.
Tissue preparation
Frozen and PFA-fixed tissue preparation
One hemisphere was snap-frozen directly after isolation using liquid nitrogen (−150°C)–cooled isopentane (fig. S1). For MALDI-IMS (and LCO analysis) 12-μm cryosections were collected from fresh frozen brain tissue on a cryostat microtome (Leica CM 1520, Leica Biosystems, Nussloch, Germany) at −18°C. For MALDI-IMS, the sections were thaw mounted on conductive ITO glass slides (Bruker Daltonics, Bremen, Germany). For LMPC (laser microdissection and pressure catapulting) , 12-μm-thick fresh frozen sections were cut on and mounted on 0.17 PEN membrane slides. All tissues were stored at −80°C.
PFA-fixed tissue collected for time course plaque staining was sectioned transverse to the long axis of the hippocampus at 30 μm using a frozen sledge microtome (Leica). Free-floating sections were collected and stored in 0.03% sodium azide in PBS at 4°C.
EM tissue preparation of PFA fixed tissue
For EM and NanoSIMS, following harvesting one brain hemisphere was immediately dissected into anatomical regions and immersion fixed in 4% PFA at 4°C (fig. S1). Tissue was then transferred to modify Karnovsky fixative, containing 2% PFA, and 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, for 4 hours [1 hour at room temperature (RT), 3 hours at 4°C, on a rotor]. Further tissue processing was performed using Automatic Microwave Tissue Processor (Leica EM AMW, Leica Microsystems, Wetzlar, Germany). Here, tissue was washed four times with 0.1 M sodium cacodylate buffer. Tissue was postfixed with 1% OsO4 and 1% KFe in 0.1 M sodium cacodylate buffer for 45 min at RT. For additional contrasting, en bloc staining with 0.5% uranyl acetate in ddH2O was performed for 45 min. Dehydration was performed with rising concentrations of EtOH (50, 70, 85, and 95% and two times with absolute EtOH) and two times with 100% Ac at RT, 10 min each. Samples were embedded in Agar 100 resin, and the resin was polymerized for 24 hours at 60°C. All samples within an isotopic labeling scheme were embedded using the same batch of resin, under the same conditions. Semithin tissue sections (350 nm, diamond knife) were obtained with an ultramicrotome (Leica EM UC6, Leica Microsystems, Wetzlar, Germany). Here, first 350-μm sections were collected on glass microscope slides and stained with toluidine blue for light microscopy, to identify Aβ plaque–rich regions of the resin block and guide EM analysis. The block was trimmed, and 350-nm-thick serial sections were cut and collected onto formvar-coated PELCO copper grids (fig. S4, I to N).
EM analysis
EM observations were carried out on a focused ion beam combined with a scanning electron microscope, FIB-SEM workstation through annular bright-field STEM analysis (GAIA3, Tescan, Brno-Kohoutovice, Czech Republic). The instrument was operated at 30.0 kV, with working distance of 5.3 mm, yielding a spot size of approximately 7 nm, and effective pixel size of approximately 20 nm at ×12,000 magnification used for collection of single plaque images. Image treatment and analysis were performed in Fiji and Adobe Photoshop.
NanoSIMS data acquisition
Following STEM analysis of fixed samples, ion microprobe (NanoSIMS 50L, Cameca, Gennevilliers, France) measurements were performed on the same copper grids. Sections were gold-coated before being transferred to the NanoSIMS.
NanoSIMS images were obtained by rasterizing a 16-keV Cs+ primary ion beam across the sample surface to generate negative secondary ions that were extracted and separated by a magnetic sector. For our study, the NanoSIMS was set up to measure the following secondary ions species: 12C15N−, 12C14N−, 12C13C−, 12C12C−, 19F−, and 32S−. The instrument was tuned to a minimum mass resolving power of >8000 (Cameca definition), enough to resolve isobaric interferences.
Before acquisition, the area of analysis was implanted with a high-current (~22 pA), defocused beam to remove the gold-coating, implant Cs+ ion into the surface of the sample and reach a sufficient amount of sputtered secondary ions. Images of surfaces (35 by 35 μm or 40 by 40 μm) were then acquired using constant settings (current of 0.7 to 0.8 pA, spot size of about 150 nm, dwell time of 5 ms, number of pixels of 256 by 256, and 10 layers).
To provide the isotopic composition of an unlabeled reference tissue, images were taken of isotopically unlabeled mice Aβ plaques (prepared in an identical manner) at the start of each day of analysis. The mean 12C15N−/12C14N− ratio for the control images was 0.00367794 ± 0.000028 2σ (N = 5). A total of three Aβ plaques (N = 3) per region (cortex/hippocampus) per labeled animal (n = 3) were analyzed. Similarly, three cortical Aβ plaques (N = 3) were analyzed in each 14N spirulina-fed APPNL-G-F control mice (n = 3).
All the obtained data were dead time corrected and aligned before stacking, using the L’IMAGE software (L. Nittler, Carnegie Institution of Washington, DC, USA). ROIs were drawn around individual Aβ plaques, using the contour lines on the 12C14N− image. These Aβ plaque ROIs were used to quantify the mean enrichment of 15N in each region. The nitrogen isotope ratio images were obtained by taking the ratio between the cumulated 12C15N− and 12C14N− images. The deviation (i.e., enrichment) from the natural abundant 15N determined in unlabeled control mice is reported in per mil using the following parameter
Univariate comparisons between the groups were performed using paired, two-tailed t test (P < 0.05) (fig. S7).
Matrix deposition for MALDI-IMS
Before MALDI-IMS sections, fresh frozen tissue sections mounted on ITO glasses were thawed and dried in a desiccator for 15 min. A series of sequential washes of 100% EtOH (60 s), 70% EtOH (30 s), Carnoy’s fluid (6:3:1 EtOH/CHCl3/acetic acid) (110 s), 100% EtOH (15 s), H2O with 0.2% TFA (60 s), and 100% EtOH (15 s) was carried out. For enhanced plaque extraction, tissues were subjected to FA vapor for 20 min. 2,5-DHAP was used as matrix compound and applied using an HTX TM-Sprayer (HTX Technologies LLC, Carrboro, NC, USA). A matrix solution of 2,5-DHAP (15 mg/ml) in 70% ACN/2% CH3COOH/2% TFA was sprayed onto the tissue sections using the following instrumental parameters: nitrogen flow (10 pounds per square inch), spray temperature (75°C), nozzle height (40 mm), eight passes with offsets and rotations, and spray velocity (1000 mm/min), and isocratic flow of 100 μl/min using 70% ACN as pushing solvent.
MALDI-IMS data acquisition
MALDI-IMS experiments were performed on a rapifleX MALDI–time-of-flight (TOF) instrument (Bruker Daltonics). Measurements were performed at 5-μm spatial resolution, with the laser operating at a frequency of 10 kHz, a laser power of 90%, and 200 shots per pixel. Data were acquired in linear positive mode in the mass range of 2000 to 20,000 m/z (mass resolution of 500 at 4000 m/z). Preacquisition calibration of the system was performed using a combination of peptide calibration standard I and synthetic Aβ peptides (Aβ1-38, Aβ1-39, Aβ1-40, Aβ1-42, Aβ1-43, Aβ1-44, Aβ1-45, Aβ1-46, Aβ1-47, and Aβ1-48), to ensure calibration over the entire range of potential Aβ species. Acquisition and subsequent processing were performed in flexImaging (version 5.0, Bruker Daltonics).
LCO staining and immunohistochemistry
Fluorescent plaque staining was performed using LCO to further investigate plaque pathology. For the time series in young APPNL-G-F mice, plaque staining was performed using the commercially available LCO derivative Amytracker 480 (Ebba Biotech AB, Stockholm, Sweden) to follow plaque pathology over time. PFA-fixed tissue sections (N = 2) from mice at 9 to 11 weeks (n = 3 animals) were initially washed once with PBS for 10 min, followed by labeling using for 30 min at RT in the dark and a subsequent wash in PBS for 10 min. Sections nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; 1:10,000 in PBS) for 5 min, followed by a final wash in PBS for 10 min, and stored in the dark at 4°C overnight. Free-floating sections were mounted onto Superfrost Plus glass slides (Thermo Fisher Scientific) using Fluoromount-G medium (SouthernBiotech, Birmingham, AL, USA) with coverslips placed over them.
For plaque feature validation of the MALDI-IMS data, as well as for structural amyloid analysis, a double LCO stain strategy with two LCO, q-FTAA and h-FTAA, was used. Here, 12-μm-thick fresh frozen tissue sections, adjacent to those used for MALDI-IMS, were analyzed. Before staining, the sections were thawed in a desiccator and fixed using absolute EtOH, 70% EtOH, and PBS for 10 min each. Tissues were double-stained with q-FTAA and h-FTAA (2.4 μM q-FTAA and 0.77 μM h-FTAA in PBS) similar to a previously described protocol (58). Sections were incubated for 30 min at RT in the dark, rinsed with PBS, desiccated, mounted with Dako fluorescence mounting medium, and stored in dark at 4°C for a minimum of 24 hours before microscopy. For Aβ immunolabeling, 6E10 antibody (1:500; Invitrogen, Life Technologies) was incubated overnight at 4°C, followed by secondary antibody staining with Alexa Fluor 488 (1:1000; Invitrogen).
Fluorescent microscopy
Bright-field images of the toluidine blue–stained tissue and epifluorescence LCO plaque images were acquired using a wide-field microscope (Axio Observer Z1, Zeiss, Jena, Germany). The images were acquired with a Plan-Apochromat 10×/0.3 differential interference contrast objective.
The hyperspectral imaging of double LCO–stained tissues was performed using an inverted laser scanning confocal microscope (ELYRA PS.1 SIM/PAL-M LSM780, Zeiss, Jena, Germany), equipped with a 32-channel GaAsP spectral detector, in parallel spectral detection design, enabling simultaneous 32+2-channel spectral readout in lambda mode. The acquisition was performed using a 35-nW, 458-nm argon laser, with the Plan-Apochromat 20×/0.8 objective. The continuous emission was acquired in the range of 405 to 750 nm (58). Linear unmixing, a function within the Zen 2011 (Zeiss) software, was used to differentiate between the q-FTAA and h-FTAA fluorescent signals in the double-stained samples and distinguish between true LCO fluorescence spectrum and unwanted autofluorescence, from, for instance, lipofuscin.
For time series plaque staining, whole hippocampal and cortical regions were imaged using a 20× objective on an epifluorescence EVOS FL auto cell imaging system microscope (Life Technologies, Carlsbad, CA, USA). LCO were detected at a wavelength of 488 nm and DAPI at a wavelength of 356 nm.
LMPC-based Aβ plaques isolation and MS
Before laser microdissection, the tissue was double-stained with q-FTAA and h-FTAA similarly to double staining for LCO analysis (fig. S5), using ddH2O instead of PBS. The tissue was desiccated but not mounted. Microdissection was performed using an LMPC microscope (PALM MicroBeam LMPC microscope, Zeiss/P.A.L.M. Microlaser Technologies, Bernsried, Germany) equipped with a 355-nm pulsed ultraviolet laser. The individual Aβ plaques as visualized with LCO double-stain strategy were collected in Adhesive Cap 500 opaque tubes and stored at −20°C before extraction. In total, an area of 50,000 μm2 of amyloid-positive plaques was collected.
Aβ immunoprecipitation
To the isolated amyloid plaques, 50 μl of 70% FA, with 5 mM EDTA, was added. The samples were sonicated for 5 min and incubated for 1 hour at 24°C. The samples were then neutralized to pH 7 using 0.5 M tris buffer. Aβ peptides were than purified through immunoprecipitation using Aβ-specific antibodies (antibodies 6E10 and 4G8, Signet Laboratories), coupled to magnetic Dynabeads M-280 Sheep Anti-Mouse (Invitrogen), as described previously (59). The supernatant was collected and dried through lyophilization.
MS analysis of immunoprecipitation plaque extracts
For MS, the samples were reconstituted in 5 μl of 20% ACN/0.1% FA. For MALDI MS of peptide extracts, the samples were deposited onto a MALDI target using the seed layer preparation. Here, a matrix seed layer [CHCA (20 mg/ml), 90% Ac, 10% MeOH, and 0.005% TFA] was prespotted onto the target, followed by subsequent coapplication of 1 μl of sample mixed with 1 μl of second matrix solution [CHCA (15 mg/ml) and 50% ACN/0.1% TFA]. MALDI MS of plaque extracts analysis was performed on a ultrafleXtreme MALDI-TOF/TOF instrument (Bruker Daltonics) in reflector-positive mode. Data were collected in the mass range of 500 to 5500 m/z (5000 shots, laser frequency of 1000 Hz, laser focus: medium). External calibration was performed by the deposition of a peptide calibrant mix 1 (Bruker Daltonics) spotted adjacent to the sample spots on the target. Further, to verify the identity of the observed peptides, a liquid chromatography–tandem MS (MS/MS) analysis, using alkaline mobile phase, of the brain was carried out using a Q Exactive quadrupole-orbitrap hybrid mass spectrometer equipped with a heated electrospray ionization source (HESI-II) (Thermo Scientific) and UltiMate 3000 binary pump, column oven, and autosampler (Thermo Scientific), as previously described (60), but with the Q Exactive operated in data-dependent mode. Briefly, the resolution settings were 70,000, and target values were 1 × 106 both for MS and MS/MS acquisitions. Acquisitions were performed with one microscan per acquisition. Precursor isolation width was 3 m/z units, and ions were fragmented by so-called higher-energy collision-induced dissociation at a normalized collision energy of 25.
MALDI-IMS data analysis
Plaque ROIs (whole plaque, core, and periphery) were annotated on the basis of the coaligned LCO fluorescent images acquired on consecutive sections. Total ion current normalized average spectra of the annotated ROIs were exported as *.csv file.
Isotope pattern analysis was performed in GraphPad Prism (version 7). Here, MALDI-IMS ROI spectral data were loaded into Prism, and average distribution curves were fitted to the individual peptide isotope signal pattern. The centroid was used as a measure for average isotope incorporation, allowing either signal comparisons of the corresponding isotopologue (m/z value) and for comparative analysis in between distinct ROI (plaque cortex versus plaque hippocampus) and peptides (Aβ1-42 and Aβ1-38). Univariate comparisons between the groups were performed using unpaired, two-tailed t test (P < 0.05) (fig. S6).
For individual peak statistics, ROI spectra data files were imported into Origin (version 8.1, OriginLab, Northampton, MA, USA) for peak detection and peak width determination using the implemented peak analyzer function. The determined peak widths that serve as bin borders for peak integration were exported as tab delimited text file. The bin borders were used for area under curve peak integration within each bin (peak-bin) of all individual ROI average spectra using an in-house developed R script (61). Data were log-transformed. Univariate comparisons of distinct isotopologue signals (m/z) between the intraplaque regions (core versus periphery) were performed using unpaired, two-tailed t test (P < 0.05).
Electrospray ionization–MS/MS peptide identification
For electrospray ionization–MS/MS data analysis, MS and MS/MS spectra were deconvoluted using Mascot Distiller before submission to database search using the Mascot search engine (both Matrix Science), as described previously (62). The MS/MS spectra were searched toward a local database containing mutant human APP sequences using the following search parameters: precursor mass ± 15 parts per million; fragment mass ± 0.05 Da; no enzyme; no fixed modifications; variable modifications including deamidated (NQ), Glu- > pyro-Glu (N-term E), oxidation (M); instrument default.
Acknowledgments
We thank J. Lundgren at the Pathology Division at Sahlgrenska University Hospital and the staff at Centre for Cellular Imaging (CCI), Core Facilities, The Sahlgrenska Academy at the University of Gothenburg for assistance with the EM preparations and the hyperspectral microscopy data acquisition. P. Hammarström and P. Nilsson at Linköping University are acknowledged for donating the q-FTAA and h-FTAA compounds. Funding: The Swedish Research Council VR (no. 2018-02181 to J.H. and no. 2018-02532 to H.Z.), the European Research Council (no. 681712 to H.Z.), Alzheimerfonden (to J.H., K.B., and K.M.S.), Demensfonden (to W.M.), Ahlén Stiftelsen (to J.H.), Stiftelsen Gamla Tjänarinnor (to J.H., K.B., and W.M.), Stohnes Stiftelse (to J.H. and W.M.), Torsten Söderberg Foundation (to K.B.), Federal Ministry of Education and Research (BMBF, FH-Impuls Partnerschaft M2Aind, project M2OGA, and Förderkennzeichen 13FH8I02IA to C.H.), Alzheimer Research UK (ARUK-PPG2018B-025 to F.A.E. and J.H.), Medical Research Council (MR/N013867/1 to K.V.), and The Cure Alzheimer’s Fund (to F.A.E.) are acknowledged for financial support. H.Z. is a Wallenberg Scholar. Author contributions: J.H. and F.A.E. conceived and designed the study. W.M., K.M.S., T.E., M.K.P., S.E., K.V., and J.W. performed experiments. W.M., K.M.S., T.E., M.K.P., S.E., K.V., J.W., H.Z., K.B., A.M., C.H., F.A.E., and J.H. analyzed and discussed the data. J.H. and W.M. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg4855/DC1
REFERENCES AND NOTES
- 1.Blennow K., de Leon M. J., Zetterberg H., Alzheimer’s disease. Lancet 368, 387–403 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Serrano-Pozo A., Frosch M. P., Masliah E., Hyman B. T., Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1, a006189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheltens P., Blennow K., Breteler M. M. B., de Strooper B., Frisoni G. B., Salloway S., van der Flier W. M., Alzheimer’s disease. Lancet 388, 505–517 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Braak H., Braak E., Neuropathological staging of Alzheimer related changes. Acta Neuropathol. 82, 239–259 (1991). [DOI] [PubMed] [Google Scholar]
- 5.Thal D. R., Capetillo-Zarate E., Del Tredici K., Braak H., The development of amyloid beta protein deposits in the aged brain. Sci. Aging Knowledge Environ. 2006, re1 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Thal D. R., Rüb U., Orantes M., Braak H., Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Armstrong R. A., β-Amyloid plaques: Stages in life history or independent origin? Dement. Geriatr. Cogn. Disord. 9, 227–238 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Cummings D. M., Liu W., Portelius E., Bayram S., Yasvoina M., Ho S. H., Smits H., Ali S. S., Steinberg R., Pegasiou C. M., James O. T., Matarin M., Richardson J. C., Zetterberg H., Blennow K., Hardy J. A., Salih D. A., Edwards F. A., First effects of rising amyloid-β in transgenic mouse brain: Synaptic transmission and gene expression. Brain 138, 1992–2004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westermark G. T., Johnson K. H., Westermark P., Staining methods for identification of amyloid in tissue. Methods Enzymol. 309, 3–25 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Carlred L., Michno W., Kaya I., Sjövall P., Syvänen S., Hanrieder J., Probing amyloid-β pathology in transgenic Alzheimer’s disease (tgArcSwe) mice using MALDI imaging mass spectrometry. J. Neurochem. 138, 469–478 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M., Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Westman-Brinkmalm A., Abramsson A., Pannee J., Gang C., Gustavsson M. K., von Otter M., Blennow K., Brinkmalm G., Heumann H., Zetterberg H., SILAC zebrafish for quantitative analysis of protein turnover and tissue regeneration. J. Proteomics 75, 425–434 (2011). [DOI] [PubMed] [Google Scholar]
- 13.McClatchy D. B., Dong M. Q., Wu C. C., Venable J. D., Yates J. R. III, 15N metabolic labeling of mammalian tissue with slow protein turnover. J. Proteome Res. 6, 2005–2010 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bateman R. J., Munsell L. Y., Morris J. C., Swarm R., Yarasheski K. E., Holtzman D. M., Human amyloid-β synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12, 856–861 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hark T. J., Rao N. R., Castillon C., Basta T., Smukowski S., Bao H., Upadhyay A., Bomba-Warczak E., Nomura T., O’Toole E. T., Morgan G. P., Ali L., Saito T., Guillermier C., Saido T. C., Steinhauser M. L., Stowell M. H. B., Chapman E. R., Contractor A., Savas J. N., Pulse-chase proteomics of the App knockin mouse models of Alzheimer’s disease reveals that synaptic dysfunction originates in presynaptic terminals. Cell Syst. 12, 141–158.e9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savas J. N., Wang Y. Z., DeNardo L. A., Martinez-Bartolome S., McClatchy D. B., Hark T. J., Shanks N. F., Cozzolino K. A., Lavallée-Adam M., Smukowski S. N., Park S. K., Kelly J. W., Koo E. H., Nakagawa T., Masliah E., Ghosh A., Yates J. R. III, Amyloid accumulation drives proteome-wide alterations in mouse models of Alzheimer’s disease-like pathology. Cell Rep. 21, 2614–2627 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paterson R. W., Gabelle A., Lucey B. P., Barthélemy N. R., Leckey C. A., Hirtz C., Lehmann S., Sato C., Patterson B. W., West T., Yarasheski K., Rohrer J. D., Wildburger N. C., Schott J. M., Karch C. M., Wray S., Miller T. M., Elbert D. L., Zetterberg H., Fox N. C., Bateman R. J., SILK studies—Capturing the turnover of proteins linked to neurodegenerative diseases. Nat. Rev. Neurol. 15, 419–427 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bateman R. J., Siemers E. R., Mawuenyega K. G., Wen G., Browning K. R., Sigurdson W. C., Yarasheski K. E., Friedrich S. W., DeMattos R. B., May P. C., Paul S. M., Holtzman D. M., A γ-secretase inhibitor decreases amyloid-β production in the central nervous system. Ann. Neurol. 66, 48–54 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mawuenyega K. G., Sigurdson W., Ovod V., Munsell L., Kasten T., Morris J. C., Yarasheski K. E., Bateman R. J., Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 330, 1774 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Y., Potter R., Sigurdson W., Santacruz A., Shih S., Ju Y. E., Kasten T., Morris J. C., Mintun M., Duntley S., Bateman R. J., Effects of age and amyloid deposition on Aβ dynamics in the human central nervous system. Arch. Neurol. 69, 51–58 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potter R., Patterson B. W., Elbert D. L., Ovod V., Kasten T., Sigurdson W., Mawuenyega K., Blazey T., Goate A., Chott R., Yarasheski K. E., Holtzman D. M., Morris J. C., Benzinger T. L. S., Bateman R. J., Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers. Sci. Transl. Med. 5, 189ra77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patterson B. W., Elbert D. L., Mawuenyega K. G., Kasten T., Ovod V., Ma S., Xiong C., Chott R., Yarasheski K., Sigurdson W., Zhang L., Goate A., Benzinger T., Morris J. C., Holtzman D., Bateman R. J., Age and amyloid effects on human central nervous system amyloid-beta kinetics. Ann. Neurol. 78, 439–453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanrieder J., Phan N. T. N., Kurczy M. E., Ewing A. G., Imaging mass spectrometry in neuroscience. ACS Chem. Nerosci. 4, 666–679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinhauser M. L., Bailey A. P., Senyo S. E., Guillermier C., Perlstein T. S., Gould A. P., Lee R. T., Lechene C. P., Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature 481, 516–519 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wildburger N. C., Gyngard F., Guillermier C., Patterson B. W., Elbert D., Mawuenyega K. G., Schneider T., Green K., Roth R., Schmidt R. E., Cairns N. J., Benzinger T. L. S., Steinhauser M. L., Bateman R. J., Amyloid-β plaques in clinical Alzheimer’s disease brain incorporate stable isotope tracer in vivo and exhibit nanoscale heterogeneity. Front. Neurol. 9, 169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arts M., Soons Z., Ellis S. R., Pierzchalski K. A., Balluff B., Eijkel G. B., Dubois L. J., Lieuwes N. G., Agten S. M., Hackeng T. M., van Loon L. J. C., Heeren R. M. A., Olde Damink S. W. M., Detection of localized hepatocellular amino acid kinetics by using mass spectrometry imaging of stable isotopes. Angew. Chem. Int. Ed. Engl. 56, 7146–7150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellis S. R., Hall E., Panchal M., Flinders B., Madsen J., Koster G., Heeren R. M. A., Clark H. W., Postle A. D., Mass spectrometry imaging of phosphatidylcholine metabolism in lungs administered with therapeutic surfactants and isotopic tracers. J. Lipid Res. 62, 100023 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enzlein T., Cordes J., Munteanu B., Michno W., Serneels L., de Strooper B., Hanrieder J., Wolf I., Chávez-Gutiérrez L., Hopf C., Computational analysis of Alzheimer amyloid plaque composition in 2D- and elastically reconstructed 3D-MALDI MS images. Anal. Chem. 92, 14484–14493 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Michno W., Nyström S., Wehrli P., Lashley T., Brinkmalm G., Guerard L., Syvänen S., Sehlin D., Kaya I., Brinet D., Nilsson K. P. R., Hammarström P., Blennow K., Zetterberg H., Hanrieder J., Pyroglutamation of amyloid-βx-42 (Aβx-42) followed by Aβ1–40 deposition underlies plaque polymorphism in progressing Alzheimer’s disease pathology. J. Biol. Chem. 294, 6719–6732 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michno W., Wehrli P. M., Blennow K., Zetterberg H., Hanrieder J., Molecular imaging mass spectrometry for probing protein dynamics in neurodegenerative disease pathology. J. Neurochem. 151, 488–506 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Saito T., Matsuba Y., Mihira N., Takano J., Nilsson P., Itohara S., Iwata N., Saido T. C., Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 17, 661–663 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Burgold S., Bittner T., Dorostkar M. M., Kieser D., Fuhrmann M., Mitteregger G., Kretzschmar H., Schmidt B., Herms J., In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 121, 327–335 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Condello C., Schain A., Grutzendler J., Multicolor time-stamp reveals the dynamics and toxicity of amyloid deposition. Sci. Rep. 1, 19 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klingstedt T., Åslund A., Simon R. A., Johansson L. B. G., Mason J. J., Nyström S., Hammarström P., Nilsson K. P. R., Synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aggregates. Org. Biomol. Chem. 9, 8356–8370 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aslund A., Sigurdson C. J., Klingstedt T., Grathwohl S., Bolmont T., Dickstein D. L., Glimsdal E., Prokop S., Lindgren M., Konradsson P., Holtzman D. M., Hof P. R., Heppner F. L., Gandy S., Jucker M., Aguzzi A., Hammarström P., Nilsson K. P. R., Novel pentameric thiophene derivatives for in vitro and in vivo optical imaging of a plethora of protein aggregates in cerebral amyloidoses. ACS Chem. Biol. 4, 673–684 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickson T. C., Vickers J. C., The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer’s disease. Neuroscience 105, 99–107 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Ikeda S., Allsop D., Glenner G. G., Morphology and distribution of plaque and related deposits in the brains of Alzheimer’s disease and control cases. An immunohistochemical study using amyloid beta-protein antibody. Lab. Invest. 60, 113–122 (1989). [PubMed] [Google Scholar]
- 38.Dickson D. W., Crystal H. A., Mattiace L. A., Masur D. M., Blau A. D., Davies P., Yen S. H., Aronson M. K., Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol. Aging 13, 179–189 (1992). [DOI] [PubMed] [Google Scholar]
- 39.Iwatsubo T., Saido T. C., Mann D. M., Lee V. M., Trojanowski J. Q., Full-length amyloid-beta (1-42(43)) and amino-terminally modified and truncated amyloid-beta 42(43) deposit in diffuse plaques. Am. J. Pathol. 149, 1823–1830 (1996). [PMC free article] [PubMed] [Google Scholar]
- 40.Rijal Upadhaya A., Kosterin I., Kumar S., von Arnim C. A. F., Yamaguchi H., Fändrich M., Walter J., Thal D. R., Biochemical stages of amyloid-β peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain 137, 887–903 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Lord A., Kalimo H., Eckman C., Zhang X.-Q., Lannfelt L., Nilsson L. N. G., The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging 27, 67–77 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Lord A., Philipson O., Klingstedt T., Westermark G., Hammarström P., Nilsson K. P. R., Nilsson L. N. G., Observations in APP bitransgenic mice suggest that diffuse and compact plaques form via independent processes in Alzheimer’s disease. Am. J. Pathol. 178, 2286–2298 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Latif-Hernandez A., Sabanov V., Ahmed T., Craessaerts K., Saito T., Saido T., Balschun D., The two faces of synaptic failure in AppNL-G-F knock-in mice. Alzheimers Res. Ther. 12, 100 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iwatsubo T., Odaka A., Suzuki N., Mizusawa H., Nukina N., Ihara Y., Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ 42(43). Neuron 13, 45–53 (1994). [DOI] [PubMed] [Google Scholar]
- 45.Lemere C. A., Blusztajn J. K., Yamaguchi H., Wisniewski T., Saido T. C., Selkoe D. J., Sequence of deposition of heterogeneous amyloid β-peptides and APO E in Down syndrome: Implications for initial events in amyloid plaque formation. Neurobiol. Dis. 3, 16–32 (1996). [DOI] [PubMed] [Google Scholar]
- 46.Meyer-Luehmann M., Spires-Jones T. L., Prada C., Garcia-Alloza M., de Calignon A., Rozkalne A., Koenigsknecht-Talboo J., Holtzman D. M., Bacskai B. J., Hyman B. T., Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bolmont T., Haiss F., Eicke D., Radde R., Mathis C. A., Klunk W. E., Kohsaka S., Jucker M., Calhoun M. E., Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 28, 4283–4292 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klunk W. E., Engler H., Nordberg A., Wang Y., Blomqvist G., Holt D. P., Bergström M., Savitcheva I., Huang G. F., Estrada S., Ausén B., Debnath M. L., Barletta J., Price J. C., Sandell J., Lopresti B. J., Wall A., Koivisto P., Antoni G., Mathis C. A., Långström B., Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 55, 306–319 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Ikonomovic M. D., Klunk W. E., Abrahamson E. E., Mathis C. A., Price J. C., Tsopelas N. D., Lopresti B. J., Ziolko S., Bi W., Paljug W. R., Debnath M. L., Hope C. E., Isanski B. A., Hamilton R. L., DeKosky S. T., Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 131, 1630–1645 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee J.-E., Han P.-L., An update of animal models of Alzheimer disease with a reevaluation of plaque depositions. Exp. Neurobiol. 22, 84–95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Philipson O., Lord A., Gumucio A., O’Callaghan P., Lannfelt L., Nilsson L. N. G., Animal models of amyloid-β-related pathologies in Alzheimer’s disease. FEBS J. 277, 1389–1409 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Liao L., McClatchy D. B., Park S. K., Xu T., Lu B., Yates J. R. III, Quantitative analysis of brain nuclear phosphoproteins identifies developmentally regulated phosphorylation events. J. Proteome Res. 7, 4743–4755 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Contat C., Ancey P. B., Zangger N., Sabatino S., Pascual J., Escrig S., Jensen L., Goepfert C., Lanz B., Lepore M., Gruetter R., Rossier A., Berezowska S., Neppl C., Zlobec I., Clerc-Rosset S., Knott G. W., Rathmell J. C., Abel E. D., Meibom A., Meylan E., Combined deletion of Glut1 and Glut3 impairs lung adenocarcinoma growth. eLife 9, e53618 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wegenast-Braun B. M., Skodras A., Bayraktar G., Mahler J., Fritschi S. K., Klingstedt T., Mason J. J., Hammarström P., Nilsson K. P. R., Liebig C., Jucker M., Spectral discrimination of cerebral amyloid lesions after peripheral application of luminescent conjugated oligothiophenes. Am. J. Pathol. 181, 1953–1960 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Rasmussen J., Mahler J., Beschorner N., Kaeser S. A., Häsler L. M., Baumann F., Nyström S., Portelius E., Blennow K., Lashley T., Fox N. C., Sepulveda-Falla D., Glatzel M., Oblak A. L., Ghetti B., Nilsson K. P. R., Hammarström P., Staufenbiel M., Walker L. C., Jucker M., Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, 13018–13023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guardia-Laguarta C., Pera M., Clarimón J., Molinuevo J. L., Sànchez-Valle R., Lladó A., Coma M., Gómez-Isla T., Blesa R., Ferrer I., Lleó A., Clinical, neuropathologic, and biochemical profile of the amyloid precursor protein I716F mutation. J. Neuropathol. Exp. Neurol. 69, 53–59 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Portelius E., Bogdanovic N., Gustavsson M. K., Volkmann I., Brinkmalm G., Zetterberg H., Winblad B., Blennow K., Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 120, 185–193 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nyström S., Psonka-Antonczyk K. M., Ellingsen P. G., Johansson L. B. G., Reitan N., Handrick S., Prokop S., Heppner F. L., Wegenast-Braun B. M., Jucker M., Lindgren M., Stokke B. T., Hammarström P., Nilsson K. P. R., Evidence for age-dependent in vivo conformational rearrangement within Aβ amyloid deposits. ACS Chem. Biol. 8, 1128–1133 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Portelius E., Tran A. J., Andreasson U., Persson R., Brinkmalm G., Zetterberg H., Blennow K., Westman-Brinkmalm A., Characterization of amyloid β peptides in cerebrospinal fluid by an automated immunoprecipitation procedure followed by mass spectrometry. J. Proteome Res. 6, 4433–4439 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Pannee J., Portelius E., Minthon L., Gobom J., Andreasson U., Zetterberg H., Hansson O., Blennow K., Reference measurement procedure for CSF amyloid beta (Aβ)1–42 and the CSF Aβ1–42/Aβ1–40 ratio - a cross-validation study against amyloid PET. J. Neurochem. 139, 651–658 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Hanrieder J., Ljungdahl A., Fälth M., Mammo S. E., Bergquist J., Andersson M., L-DOPA-induced dyskinesia is associated with regional increase of striatal dynorphin peptides as elucidated by imaging mass spectrometry. Mol. Cell. Proteomics 10, M111.009308 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brinkmalm G., Portelius E., Öhrfelt A., Mattsson N., Persson R., Gustavsson M. K., Vite C. H., Gobom J., Månsson J. E., Nilsson J., Halim A., Larson G., Rüetschi U., Zetterberg H., Blennow K., Brinkmalm A., An online nano-LC-ESI-FTICR-MS method for comprehensive characterization of endogenous fragments from amyloid β and amyloid precursor protein in human and cat cerebrospinal fluid. J. Mass Spectrom. 47, 591–603 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/25/eabg4855/DC1