

Abstract
Atrial fibrillation (AF) is the most common sustained clinical arrhythmia, with a lifetime incidence of up to 37%, and is a major contributor to population morbidity and mortality. Important components of AF management include control of cardiac rhythm, rate, and thromboembolic risk. In this narrative review article, we focus on rhythm-control therapy. The available therapies for cardiac rhythm control include antiarrhythmic drugs and catheter-based ablation procedures; both of these are presently neither optimally effective nor safe. In order to develop improved treatment options, it is necessary to use preclinical models, both to identify novel mechanism-based therapeutic targets and to test the effects of putative therapies before initiating clinical trials. Extensive research over the past 30 years has provided many insights into AF mechanisms that can be used to design new rhythm-maintenance approaches. However, it has proven very difficult to translate these mechanistic discoveries into clinically applicable safe and effective new therapies. The aim of this article is to explore the challenges that underlie this phenomenon. We begin by considering the basic problem of AF, including its clinical importance, the current therapeutic landscape, the drug development pipeline, and the notion of upstream therapy. We then discuss the currently available preclinical models of AF and their limitations, and move on to regulatory hurdles and considerations and then review industry concerns and strategies. Finally, we evaluate potential paths forward, attempting to derive insights from the developmental history of currently used approaches and suggesting possible paths for the future. While the introduction of successful conceptually innovative new treatments for AF control is proving extremely difficult, one significant breakthrough is likely to revolutionize both AF management and the therapeutic development landscape.
Keywords: Atrial fibrillation, Mechanisms, Personalized therapy, Antiarrhythmic drugs, Remodelling
Graphical Abstract

1. Introduction
Atrial fibrillation (AF) is a very common and important clinical problem.1 The other articles in this issue of Cardiovascular Research deal with recent advances in understanding AF, with the mechanistic factors that affect the likelihood of its occurrence and with efforts to apply basic research advances in specific fields to clinical applications. In this article, we aim to provide a general overview of the challenges involved in translating advances in the basic science of AF to clinical medicine. In particular, we discuss the clinical problem of AF and its therapeutics, the basic research models used to study underlying mechanisms and their therapeutic potential, the regulatory constraints and the industry landscape that determines the financial interest/viability of new therapy development. Consideration of these issues makes it clear why advancing AF therapeutics is such an important and challenging goal. We conclude with some ideas about how to approach some of these challenges in order to improve and accelerate the clinical translation of basic discoveries.
2. The problem
2.1 Nature of the problem
AF is the most common sustained arrhythmia, and its prevention and therapy remain a significant unmet medical need.1 AF is associated with substantial morbidity and mortality and is often difficult to adequately manage. While patients can be asymptomatic, AF is a frequent cause of hospitalizations, thromboembolic events, haemodynamic compromise, and heart failure (HF), along with symptoms such as fatigue, reduced exercise tolerance, dyspnoea, and syncope, which decrease the quality of life. AF-related stroke appears to be more medically severe than that due to non-AF aetiologies and the stroke risk in non-anticoagulated AF patients is increased ∼five-fold.2 AF is also associated with a two-fold increase in mortality,3 a three-fold increase in HF,3 and an increased risk of dementia. Occurring in ∼1% of patients <60 years of age, AF has a prevalence that increases in older individuals, affecting up to ∼12% of patients aged 75–84 years old; the overall lifetime risk is ∼ 37%.4 The costs associated with AF are large: the incremental associated cost in the USA was estimated at US $6–26 billion per year based on data from 2004 to 2006.5 A more recent estimate of the incremental annual economic burden of undiagnosed non-valvular AF is US $3.1 billion.6 Thus, AF is a common, clinically important and economically significant public health problem.
2.2 Current therapeutic landscape
AF is most often diagnosed in patients with underlying heart diseases, including hypertension, coronary artery disease, valvular diseases, and HF, and may occur concurrently with an exacerbation of the underlying cardiovascular condition. However, AF can also occur in patients without overt signs of heart disease or detectable structural remodelling. Varying aetiologies, genetic susceptibilities, and comorbidities contribute to substantial patient heterogeneity. Unlike life-threatening ventricular tachyarrhythmias, serious AF-induced haemodynamic instability is uncommon outside acute/intensive care settings. Severe hypotension, altered mental status, cardiac ischaemia, or decompensated HF are the most relevant indicators of such instability and typically prompt immediate (in most cases electrical) cardioversion.7–9
Detection or suspicion of AF in haemodynamically stable patients is generally followed by a thorough diagnostic assessment, including routine laboratory investigations (e.g. thyroid hormone status, electrolyte measurements, renal function analysis) as well as cardiac examinations like an electrocardiogram, echocardiography and Holter monitoring, or longer-term patch recordings to identify underlying cardiac diseases and determine the temporal pattern of AF. The treatment of any underlying cardiovascular risk factors and/or other precipitating contributors, like inflammation for post-operative AF, infection, trauma, sleep disordered breathing, obesity, excess alcohol consumption, hypertension, or cigarette smoking generally provides a first step in AF management. Moreover, concomitant heart disease directly affects therapeutic decision-making and the choice of drugs.
Figure 1 shows a simplified schema of the main steps in the management of AF. AF increases the risk of stroke or HF and consequently requires appropriate anticoagulation and control of ventricular rate in affected individuals. Beyond the prevention of any immediately life-threatening sequelae of this rhythm disorder, the consequences of restoring and maintaining sinus rhythm on mortality in AF-patients were assessed in two landmark trials comparing rate vs. rhythm control, the Atrial Fibrillation Follow-up Investigation of Rhythm Management10 and the Rate Control vs. Electrical Cardioversion for Persistent Atrial Fibrillation (RACE)11 studies. In both trials, no significant difference between the two treatment approaches could be demonstrated. The more recent Early Treatment of Atrial Fibrillation for Stroke Prevention Trial12 did show a reduction in significant cardiovascular outcomes with early institution of rhythm-control in AF-patients, perhaps because aggressive rhythm-control therapy was instituted early (before remodelling became advanced),13 reopening the debate and emphasizing the importance of improving the available pharmacological options for rhythm-control management. This outcome emphasizes the need for further study of the potential value of an early aggressive rhythm-control strategy and the need for improved pharmacological tools to help in achieving better rhythm control. This article will focus primarily on rhythm-control therapy, without delving into the details of aspects like anticoagulation and rate control.
Figure 1.
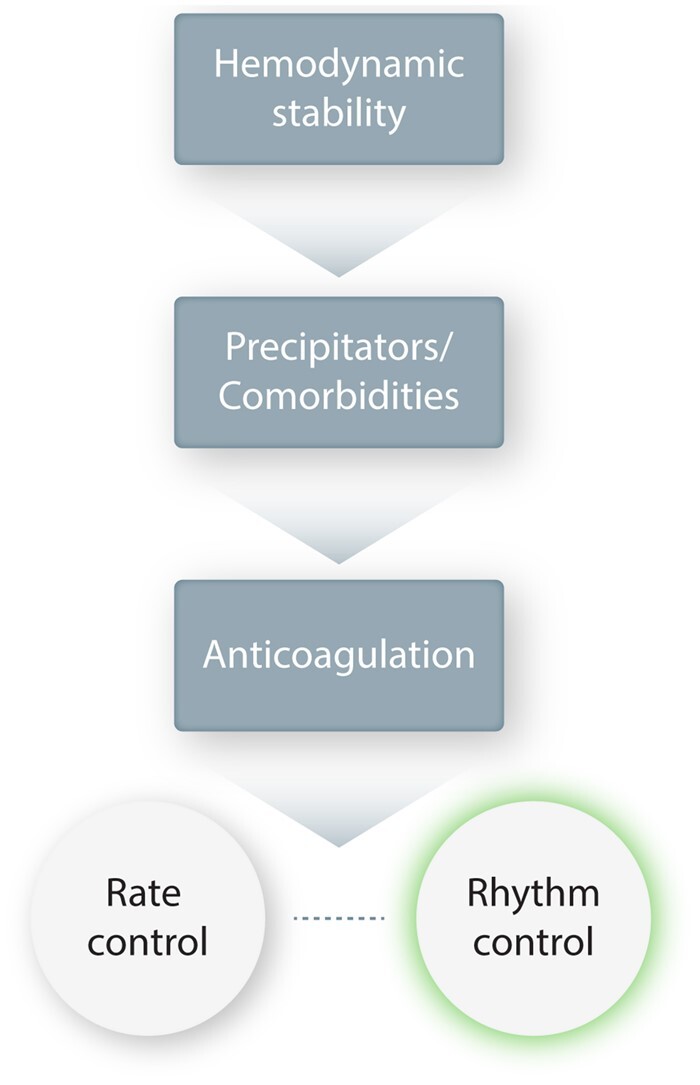
Overall AF treatment strategy schema. This is a simplified schema of the main steps in AF management. Haemodynamic stability must first be ensured, and any precipitators or comorbidities addressed. Anticoagulation is an essential early step in almost all patients to prevent thromboembolic complications. Rate control must be established, even in most patients for whom the primary approach will be rhythm control, since pharmacological rhythm control is not foolproof and rhythm-control drugs can cause a paradoxical acceleration in ventricular rate. The choice between rate control and rhythm control as the principal strategy is based on a variety of patient-specific considerations.
Control of the ventricular response rate is an important component of AF management to control symptoms, prevent ventricular dysfunction, and limit adverse atrial remodelling.14,15 The RACE-II trial unravelled a somewhat counterintuitive feature of AF therapy by suggesting a lack of benefit from a more strict (<80 beats/min at rest and <110 beats/min during moderate exercise) compared to lenient (<110 beats/min at rest) rate-control strategy.16 It must be noted that the treating physicians adjusted the treatment in each group according to their clinical judgement, and that while there was a 30 beat/min difference in the target heart-rate cutoffs for the two groups, the difference in mean heart rate was 17 beats/min at the end of dose titration and only 11 beats/min after 1 year in the study (patients were followed for 3 years). Much more needs to be learned to optimize rate-control management and tailor it to the individual patient.
In current clinical practice, the principal motivation for using antiarrhythmic drugs (AADs) is to reduce the symptom burden and improve quality of life in AF-patients.7–9 Symptoms like palpitations, dyspnoea, fatigue, light-headedness, or chest pain occur in many but not all patients. Their causal and temporal relation to AF episodes is complex and incompletely understood.17 Rhythm control with AADs and/or catheter ablation traditionally constitutes the last step in the treatment regime, restricted to patients in which symptoms are not adequately controlled by rate control alone. Importantly, because of its limited ability to prevent recurrence of AF, rhythm-control treatment does not obviate the need for anti-thrombotic or rate-control therapy in most patients.
The most commonly used AADs for rhythm control are listed in Figure 2. Based on the Singh/Vaughan-Williams classification, they belong to either class III, i.e. potassium channel blockers prolonging the effective refractory period, or class I, i.e. sodium channel blockers reducing excitability and therefore conduction velocity of the action potential, although many of them have actions of more than one class. Drug selection is guided to a significant extent by safety considerations. Many AADs have been associated with risks of bradyarrhythmias, excessive QT prolongation and torsades des pointes, or other types of ventricular tachyarrhythmias. Moreover, patients with underlying heart disease have considerably fewer options because of an increased risk of drug-induced arrhythmias (Figure 2A).7–9 The last significant FDA approval of an AAD in AF was dronedarone in 2009 (Figure 2B), which after going through a broad and ambitious development programme eventually fell somewhat short on efficacy and safety grounds and is not widely used today.18
Figure 2.

Most commonly used AF rhythm-control drugs (A) and a timeline showing their FDA approval year (B). In (A), the drugs are organized in alphabetical order. The detailed clinical decision-making approach for selection of these drugs is complex and not dealt with in this article. CAD, coronary artery disease; HF, heart failure.
The above information makes it clear that the available pharmacological toolbox for rhythm-control treatment is far from optimal. Catheter-based ablation techniques, most prominently pulmonary vein isolation, have emerged as an important alternative, but because of the very large size of the patient population, therapy with AADs still remains the cornerstone of AF treatment, with a clear unmet need for development of more efficient and safer AAD options. Of note, the rate of AAD prescription nearly tripled in the USA between 2004 and 2016.19 In addition, AADs play a significant role in many patients that undergo catheter ablation, whether while waiting for ablation, during the immediate post-ablation period (the ‘blanking period’) when transient recurrences are common and in cases where ablation is not sufficient and adjunctive AAD therapy is needed.
As for AAD therapy, the primary indication for catheter ablation is to reduce symptoms and improve quality of life, with ablation generally reserved for patients that have failed to respond to one or more AADs.7–9 The effectiveness of this intervention is greatest in patients with paroxysmal AF, followed by persistent AF, and even long-standing (>12 months) AF can be a therapeutic target.7–9 A steadily growing number of interventions and corresponding trials to assess sinus-rhythm maintenance with catheter ablation in AAD resistant and naïve patients have finally resulted in changed society guidelines7–9 that consider ablation as first-line treatment in selected AF-patients. Nevertheless, a recent meta-analysis demonstrated only incrementally improved success rates of catheter ablation for both paroxysmal and persistent AF between 2006 and 2016 (Figure 3), with present sinus-rhythm-maintenance rates averaging around 80% for paroxysmal AF and 65% for non-paroxysmal AF,20 suggesting that ablation is far from being a cure for AF. Moreover, outcome trials like CABANA have thus far been unable to show definitively a clear benefit of ablation vs. standard therapy.21 Part of the reason might be extensive crossovers from pharmacological to ablation therapy: both a treatment-received analysis and a per-protocol treatment comparison suggested all-cause mortality reductions with ablation therapy in CABANA.21 Although the Catheter Ablation vs. Standard Conventional Therapy in Patients with Left Ventricular Dysfunction and Atrial Fibrillation (CASTLE-AF) trial did suggest improved outcomes with a catheter-ablation rhythm-control strategy in HF patients,22 the subsequent Atrial Fibrillation Management in Congestive Heart Failure With Ablation (AMICA) study did not demonstrate similar improvement23 and the question remains open. Differences between the trials that might explain the discrepant outcomes include the number of patients per group (179/184 in CASTLE-AF vs. 68/72 in AMICA), the overall illness of the study populations (CASTLE-AF patients were less sick, with 30/35% in paroxysmal AF vs. 19/28% in AMICA, LV ejection fraction averaging 32/33% vs. 25/28%, respectively, class III or greater HF in 28/31% vs. 59/62%) and the follow-up duration (>3 years vs. 1 year, respectively).
Figure 3.

Evolution of success rates of catheter ablation of AF over time. Results shown are those obtained in a meta-analysis by Perino et al.20 (with permission from American Heart Journal and Elsevier). (A) Results for paroxysmal AF; (B) results for persistent AF. Regression lines suggest adjusted mean improvement rates (dashed lines) that were 1%/year for paroxysmal and 1.4%/year for persistent AF-ablations.
2.3 Drug development pipeline
With respect to pharmacological therapies, it is clear that despite their reasonably extensive continuing use, the currently available AADs have at most moderate efficacy and are associated with a risk of pro-arrhythmia, particularly in structurally remodelled heart. As shown in Figure 2, it has been over 10 years since the introduction of any new drug for AF treatment. Enthusiasm for AF drug development has been dampened by many factors, including rigorous regulatory requirements and a history of limited efficacy and well-recognized risk for cardiac and non-cardiac side effects. Figure 4 shows the current status of the AF development pipeline in the pharmaceutical industry and provides only limited hope for short-term progress, considering the notoriously low success rates for this indication. Pipeline assets offer limited innovation beyond already known motifs, including beta blockers, K+ channel blockers, modulators of cardiomyocyte Ca2+ handling, antioxidants, or parasympatholytics. The rationale and experimental basis for these specific targets goes beyond the scope of the present article—the interested reader is directed to recent detailed review articles.24,25 It is quite unclear whether the efforts in Figure 4 will be sufficient to produce an effective and widely usable new drug for AF.
Figure 4.

Present development pipeline for AF antiarrhythmic agents in the pharmaceutical industry. The agent being developed is shown at the left, followed by the originating entity, a description of the product and the present developmental stage. CaMKII, calcium/calmodulin dependent protein-kinase type II; IK,ACh, acetylcholine-gated potassium channel; Na+ channel, sodium channel; NOX4, NADPH oxidase type 4; RyR2, ryanodine receptor type 2; SK channel, small-conductance calcium-dependent potassium channel; UCLA, University of California at Los Angeles.
2.4 Upstream pharmacologic approaches
The idea that targeting development of the substrate leading to AF might be a more promising approach than attacking the final electrical end-product has been around for over 20 years.26 Upstream therapies, as such treatments have come to be called, aim to reduce or prevent structural and/or electrical remodelling that promotes AF and thus reduce the likelihood of the primary occurrence or progression of the arrhythmia. Structural remodelling results in fibrosis and other changes in the extracellular matrix, that promote atrial reentry.27 Electrical remodelling involves shortening of the atrial action potential duration and the atrial refractory period, also favouring reentry.27 In addition, abnormal cellular Ca2+-handling may lead to arrhythmogenic afterdepolarizations and spontaneous ectopic beats, while contributing to the progression of the arrhythmogenic substrate that initiates and maintains reentry.28
Approaches to upstream therapy that have been applied clinically include angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), aldosterone antagonists, beta blockers, mineralocorticoid inhibitors, statins, and polyunsaturated fatty acids.29–40 While there have been a number of disappointing trials, the available data point to the value of ACE inhibitors, ARBs and aldosterone inhibitors in preventing new-onset AF among patients with established stimuli to structural remodelling, like hypertension or HF associated with reduced ejection fraction.29–34,36 However, studies investigating ACE inhibitors and ARBs for secondary prevention are controversial and inconsistent, without a clear efficacy advantage.35 Although observational findings suggest potential value, the weight of available data suggest limited benefit, if any, for statins, beta blockers and polyunsaturated fatty acids in AF-prevention.35,37–40
3. Experimental development of new anti-AF approaches and challenges
3.1 Why are experimental models needed?
The development of new pharmacological therapies for AF, as for many other clinical conditions, is closely linked to an improved understanding of critical basic mechanisms.41 Experimental models are needed that mimic aspects of the complex clinical condition, in order to discover novel mechanisms that control the occurrence and progression of AF and to test specific mechanistic hypotheses. In addition, preclinical models are needed for the evaluation of candidate drugs and interventions for desired effects, ranging from the putative cellular or molecular mechanism of action to the desired beneficial effect on AF occurrence, maintenance, or progression.
3.2 What are the principal properties of the available preclinical models?
A variety of approaches and tools are available for the evaluation and discovery of tractable mechanisms at the organismal, organ, cellular, and molecular levels. Figure 5 summarizes the types of models that are used to investigate AF pathophysiology. Considerations within each model include the species to be studied and the manipulations used to create an arrhythmic substrate relevant to clinical AF.
Figure 5.

Summary of principal preclinical AF-models. Left: manipulations used to create experimental models of AF. Right: principal species in which various types of preclinical models have been created. CMs, cardiomyocytes; HF, heart failure; hIPSCs, human induced pluripotent stem cell; MI, myocardial infarction; NGF, nerve growth factor.
AF itself induces AF-promoting remodelling, largely through the effects of a very rapid atrial rate, often referred to as ‘AF begets AF’.42,43 Tachypacing models of the AF-substrate, which can involve very rapid regular atrial pacing or electrically induced AF, are limited to large animals in which tachycardia-pacemakers can be implanted, and have been predominantly created in dogs, pigs, and sheep. The development of the AF-substrate has a complex time-course.44,45 Electrical remodelling due to changes in ion-current function occurs within days and is primarily manifested as action potential/refractory period abbreviation, whereas structural remodelling consisting of extracellular matrix changes including atrial fibrosis develops much more slowly, over weeks to months.44,45 Rapid atrial activation is sufficient to cause both types of remodelling, but structural remodelling is enhanced when ventricular dysfunction occurs in response to poor ventricular rate control.15 Atrial tachycardia-remodelling provides insights into the mechanisms by which AF becomes progressively more resistant to therapy. A significant limitation of these models is that they provide no information about the mechanisms leading to the initial occurrence of AF, since AF has to start somehow before it can beget itself.
LV dysfunction causes atrial fibrosis and thereby creates a clinically relevant substrate for AF-maintenance.46 AF-promoting LV-dysfunction can be induced by ventricular tachypacing,46 which is only feasible in larger animals, or by the induction of acute myocardial infarction in smaller animals like rodents. These models can be used to provide insights into the mechanisms that lead to fibrosis, a common and prominent component of the clinical AF-substrate.47 Limitations include the direct consequences of HF per se (like hypotension, neurohumoral changes, and acute atrial stretch) or of the intervention used to cause LV dysfunction, like the myocardial ischaemia associated with coronary artery occlusion.
The autonomic nervous system (ANS) plays an important role in determining AF initiation and maintenance, and ANS-manipulations can be used to create models of AF occurrence.48 Vagal-nerve stimulation produces a predictable and reversible substrate for AF-maintenance that can be used to screen antiarrhythmic-drug effectiveness for acute AF termination.49 Of note, the acetylcholine-gated potassium current IK,ACh, the main effector of vagal-nerve activity, develops agonist-independent constitutive IK,ACh activity mimicking persistent vagal-nerve function in atrial tachycardia-remodelled dog atria50 and in atria of patients with persistent AF,51 although the precise clinical role of this activity remains unclear. AF and various clinical AF-paradigms alter atrial autonomic innervation in ways that contribute to AF-promoting remodelling.48 Chronic alterations in autonomic function, like those induced by nerve growth-factor injection into sympathetic ganglia, create atrial profibrillatory changes that can result in spontaneous AF.48 Investigations in ANS-dependent animal models have been used to develop clinically applicable therapies that target the ANS.48
There is evidence for an important role of sterile inflammation in the development of the AF-substrate.52 Recent work shows that cardiomyocyte inflammatory signalling is enhanced in AF-patients and that activation of NLRP3-inflammasome signalling may be a common pathway involved in various clinical forms, including paroxysmal, persistent, and post-operative AF,53,54 as well as in conditions that promote AF (e.g. diabetes and obesity).55,56 Endurance exercise training in mice produces an inflammation-driven arrhythmogenic AF-substrate involving tumour necrosis factor-α activation.57 Thus, inflammation plays a key role in a number of animal models of AF, which can be used to explore novel interventions. The sterile pericarditis model typically involves substantial pericardial inflammation and is relevant to post-operative AF.58 Monocrotaline injection produces pulmonary hypertension and right-heart remodelling, with right-atrial dependent AF that shows enhanced inflammatory signalling.59 Inflammation resolution-promoting therapy prevents development of the AF-substrate in this model, showing the feasibility of acting on inflammation to prevent AF-related remodelling.60
Genetically engineered animals provide unmatched precision in identifying specific molecular mechanisms contributing to AF. Most of these are produced in mice, because of their well-characterized genetic structure and the ability to rapidly produce usable mouse lines bearing knock-in mutations, knockouts of specific genes (including inducible models to circumvent neonatal lethality and developmental effects), and animals with two or more simultaneous genetic alterations for complex hypothesis-testing.61 The recent development of atrial-targeted gene manipulation employing adeno-associated viruses and atrial-selective promoters allows for the alteration of atrial gene expression without confounding changes from ventricular effects.62 Limitations of these mouse models include small atrial size, which makes protein-expression analysis challenging, and aspects of electrophysiology not directly translatable to humans (like different ion-channel systems controlling repolarization, very rapid resting heart rates and possibly different AF-inducing and maintaining mechanisms). It is feasible to create genetically engineered AF-models in rats,63 but the longer production and breeding times make practical use more challenging. While rat atria are substantially larger than those of mice, significant electrophysiological differences from humans are present.
Atrial cardiomyocytes from AF-patients have been used extensively to study AF-related cellular electrophysiological and molecular abnormalities associated with AF.53,54,64 The results of such studies provide valuable information about clinical importance and relevance that is most powerful when used in conjunction with studies in animal models to address specific hypotheses and study cause–effect relationships.53,65 Limitations of studies in human tissues and cells relate primarily to the variability introduced by tissue procurement from patients: subjects of different ages, with various associated cardiac pathologies and different drug treatments. Careful control for these factors can deal with some of these concerns,54 but the associated extraneous sources of variance can never be completely eliminated even with careful propensity matching. Constant care is needed to ensure a reproducible yield of high-quality cardiomyocytes from the often very small atrial tissue-samples obtained from the operating room.
Induced pluripotent stem cells differentiated into cardiomyocytes (iPSC-CMs) are being used increasingly as models of heart disease.66 Protocols have been developed to preferentially drive iPSC differentiation towards an atrial cardiomyocyte phenotype, and such cells have been used to create 2-dimensional cell-sheets that show many atrial properties including the occurrence of arrhythmias with features of AF.67 However, caution is needed because these cells demonstrate many features of immature cell-types, including depolarized resting potentials with high levels of sodium-current inactivation and very slow conduction velocities (1–2 orders of magnitude slower than physiological).
3.3 What are the principal challenges to using experimental models?
Table 1 indicates some of the main challenges when using animal models for AF research. The first, and probably most important, is the clinical complexity of the condition being studied. Clinical AF varies enormously among different patient groups in its underlying pathophysiology and mechanisms. AF in a foetus in utero has very different mechanisms from familial AF in a 30 year-old, from paroxysmal AF in an otherwise healthy 50 year-old, from persistent AF in a 75 year-old patient with severe ischaemic cardiomyopathy and HF or from AF in a 45 year-old individual with thyrotoxicosis. These pathophysiological differences make it essential to be sure what questions are being asked about AF mechanisms, what target population is being considered for an intervention under development or what clinical condition is being mimicked. When it comes to AF, one size definitely does not fit all.
Table 1.
Limitations to clinical translation of results from current experimental models of AF
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A second important consideration is that all of the available experimental paradigms have limitations that must be considered. Rodent models do not realistically mimic the human electrophysiological phenotype in some important respects (e.g. basal heart rate and ion-currents governing repolarization) and certainly do not come close to human atrial dimensions, but large-animal models have other important drawbacks in terms of restricted availability of animals, larger purchase and handling costs and limited knowledge and manipulability of genetic make-up. Additional concerns include (i) the limited incorporation of clinically important risk factors like aging and comorbidities (e.g. hypertension and diabetes); (ii) the lack of models that manifest spontaneous AF, requiring AF induction to analyse markers of the AF-substrate like AF inducibility and duration; and (iii) the potential complicating effects of anaesthesia and invasive procedures required to obtain arrhythmia readouts. The human-cell models require great skill and sustained technical attention (with atrial-biopsy cellular/tissue studies), and are subject to contaminating effects by uncontrollable clinical variability. Studies with iPSC-CMs are limited by their often immature and poorly controllable phenotype, which tends to change over time in culture.
Extrapolating from animal systems to man is always challenging because of species differences, but also because the animal models are much more controlled and limited than the complex picture presented by patients. Patients are generally older, have multiple and varying comorbidities and take various drugs not usually present in animal models. In addition, each experimental system studied has technical limitations intrinsic to the methods used to develop and study it. Finally, the selected model system usually recapitulates only one or two aspects of the arrhythmogenic substrate, whereas in patients the AF-inducing and maintaining substrate results from the mixed effects of multiple comorbidities, risk factors, genetic determinants and medications, most of which act over much longer time periods than can be reproduced in experimental animal paradigms.
Overall, given the deficiencies of the different models available, it is important to be very clear on what one expects from a model, to what clinical paradigm(s) it is relevant and what the intrinsic limitations are of the information obtained. It is therefore key to use more than one model in order to obtain reliable insights, to replicate and to validate the findings.
4. Regulatory hurdles and considerations
Despite technological and methodological advances in catheter ablation, pharmacologic rhythm-control therapy remains an important component of the therapeutic strategy for many patients, particularly those who are highly symptomatic. In addition to their primary role, AADs are often needed as adjuncts to maintain sinus rhythm at various times after ablation. The regulatory threshold for approval of a drug to maintain sinus rhythm is challenging and has focused on endpoints that are clinically meaningful to patients, such as a reduction in symptoms (e.g. dyspnoea, fatigue, palpitations, and light-headedness), hospitalization, morbidity (e.g. new or worsening congestive HF, stroke, myocardial infarction, etc.), or mortality. The impact of drugs (as well as ablation) on symptoms does not necessarily correlate with sinus-rhythm maintenance, adding to the complexity of the regulatory pathway. Potential secondary outcome measures, which are generally not sufficient for a primary indication, include improved quality of life and exercise tolerance. For drugs with the potential to be proarrhythmic, the financial burden of development is complicated by the need for a cardiac outcome study, focused on cardiovascular mortality in higher risk patients, to demonstrate safety. Included in this category are agents whose mechanism of action is to modulate cardiac ion channels, with the potential to slow conduction and or prolong repolarization in the ventricles. Of course, the clinical benefit of any drug needs to be balanced against potential risks.
A drug that is safe and fully eliminates the recurrence of AF would be expected to have meaningful health benefits, but such a ‘magic bullet’ scenario is rather unlikely. Increases in the maintenance of sinus rhythm or prolongation of the time to first recurrence of AF alone (without improvement in other clinical endpoints) are only surrogate outcomes, which despite having been sufficient for approval of AADs in the past are no longer expected to constitute valid endpoints for the regulatory process. The reason for the insufficiency of such surrogate endpoints is that it is difficult to show that such an endpoint results in a meaningful health benefit to patients. An argument can also be made that a drug that converts persistent AF to a clinical picture in which patients go in and out of AF for significant periods of time might even increase the risk of thrombus dislodgment and embolic events.
The availability of multiple long-term ECG recording technologies, including implantable ECG recorders and ECG patches, has made it possible to study the effect of a drug on AF burden, and it may be possible to pursue this as a regulatory endpoint in certain situations. The focus would be to show that while AF is not obliterated, the episodes are likely insufficiently frequent and lasting to permit the formation of an atrial thrombus. Such a drug might potentially reduce the need for anticoagulation. Nevertheless, the patient population would need to be carefully defined and selected, since it is possible that such a drug would be less effective in patients with considerable electrical or structural remodelling. No drug has been approved using such a strategy, but it has potential value and would merit discussion with regulators.
5. Industry concerns and strategies
Despite the significant limitations of currently available AADs, AF pharmacotherapy has not seen any major innovation over the last three decades (Figure 2). Most of the advances in disease understanding documented in detail elsewhere28 have not yet been successfully translated to pharmacological application and/or clinical validation. Moreover, the research and development (R&D) pipeline analysis (Figure 4) reveals a quite sobering picture of the state of affairs of AF drug development across the industry, with only a few new molecular entities, most of which still lack crucial proof-of-concept in humans. It is also remarkable that ‘big pharma’ rarely shows up in the list. Does this reflect a generally low level of interest of major pharma companies to invest in R&D for AF drugs? There are many valid arguments to the contrary.
As already pointed out, the size of the problem related to the number of patients and AF-inflicted costs is enormous. The aging population and the constant increase in type 2 diabetes and obesity, two important AF risk factors, will further increase the unmet medical need. Non vitamin-K anticoagulants introduced for stroke prevention in AF-patients turned out to be a great success, not only for the patients (for whom devastating strokes could be avoided) and for the pharmaceutical industry, which produced blockbuster drugs, but also for society and healthcare systems, generating overall savings on the direct costs for stroke treatment and the enormous indirect costs associated with disability.68,69 But stroke is only one complication precipitated by AF. The majority of patients suffer from concomitant ischaemic heart disease and/or HF and these comorbid cardiovascular-risk patients are hospitalized twice as often compared to otherwise similar patient cohorts without AF. Irrespective of whether exacerbations of the underlying heart disease triggered by AF episodes (or vice versa) or symptomatic AF alone drives the patient to the emergency room, the number and costs of such hospitalizations—more than 550 000 hospitalizations per year (2010) in the USA,70 with concomitant AF adding another 70% to the costs5—provide the grounds to construct value propositions for AAD candidates. This approach obviously challenges the controversial notion of equivalence in outcomes between rhythm and rate control, an idea that has also been challenged by recent clinical data showing superiority of rhythm control in cardiac risk patients early in the natural history of AF (diagnosis within the last 12 months).12 Of note, only 20% of the patients assigned to early rhythm-control received AF-ablations; the rest were treated with AADs, clearly illustrating the great relevance of AAD approaches for rhythm-control strategies.
Poor efficacy and safety concerns associated with the currently used AADs provide ample opportunities for new-generation antiarrhythmic compounds with improved efficacy and safety profiles. It is important to note that the relatively rapid action of rhythm-control agents and readily accessible electrophysiological readouts in humans render antiarrhythmic efficacy (maintenance of sinus rhythm) and safety (signs of ventricular pro-arrhythmia) to be readily probed in smaller size studies of short duration before considering investing in costly outcome trials. Technical advances like the use of rhythm monitoring with smart-phone applications and patch-based remote recording devices are facilitating these types of approaches. Possible regulatory approaches to using AF burden as an approvable endpoint may also be helpful for highly efficacious agents. Today’s sparsely populated pipeline might partially reflect a well-functioning test cascade eliminating flawed candidates at early stages. Obviously, no candidate yet has been identified combining robust antiarrhythmic efficacy with the sufficient atrial specificity to prevent ventricular pro-arrhythmia risk, central nervous system or other unwanted target-related adverse effects.
It is conceivable that the efficacy of AADs—similar to ablation therapy—is limited to a large extent by the degree and anatomical complexity of atrial remodelling.71 This complex relationship is only partially captured by the current AF classification exclusively based on AF duration, which in itself cannot be defined precisely. While future clinical practice might further tolerate some degree of ‘trial-and-error’ to this end, especially for AADs with improved safety, there is a clear unmet need for clinical trials involving patients stratified according to ‘disease staging’ and ideally dominant AF mechanism.
Costs and risks are viewed significantly less favourably by many experts in industry when it comes to AF-prevention by targeting mechanisms acting to halt or potentially reverse the age- and heart disease-related remodelling processes that generate and contribute to the arrhythmogenic atrial substrate. The sample sizes and trial durations required to demonstrate efficacy substantially exceed those used for acute arrhythmia-treatment testing efficacy. Moreover, suitable dynamically responsive clinical surrogate markers (or ‘intermediate traits’) to indicate target engagement are needed to steer dose finding and de-risk the programme by generating initial relevant efficacy signals. At present, progression-specific markers are available for only a minority of the mechanisms linked to AF. Most importantly, despite being a central theme in AF research, atrial fibrosis remains a process difficult to monitor using biomarkers. The complexity of the issue is well-illustrated by the case of renin–angiotensin–aldosterone system (RAAS) inhibitors. While RAAS inhibitors appear to be of value for primary AF-prevention in studies of groups at high risk for fibrosis development (e.g. patients with substantial LV dysfunction post-myocardial infarction and those with severe hypertension),34 they have shown limited efficacy in secondary AF-prevention.35 The major challenge is that in the absence of reliable surrogate markers of fibrosis or fibrotic signalling, the only evidence for effectiveness is AF-prevention, which requires very large numbers of patients who are both at increased risk for developing a fibrotic AF-substrate and yet have not developed advanced atrial fibrosis. Thus, it is not surprising that the beneficial effects of ‘upstream therapy’ with RAAS blockers have been limited and difficult to demonstrate.72
Inflammation and inflammatory signalling are emerging as a second intensely studied driver mechanism, which combines the availability of relevant markers (e.g. interleukin-6 and high-sensitivity C-reactive protein), multiple candidate drugs, and a clinically relevant condition (post-operative AF) considered to be predominantly driven by inflammation of atrial tissue (target engagement).73 While this AF population might serve as a reasonable entry indication for anti-inflammatory drugs, the relevance of inflammatory signalling to the broader AF population is not well established. Nevertheless, recent evidence for a role of cardiomyocyte inflammatory signalling in varied forms of AF, including post-operative, paroxysmal, and persistent AF, points to potential therapeutic value of specific targeting of inflammatory signalling.53,54,74
Finally, patient heterogeneity with multiple different disease processes interacting at the individual patient level require considerably improved clinical classification schemes (‘disease staging’) to further personalize treatment and improve therapeutic efficacy.75 These considerations emphasize the need and relevance of further basic/translational and clinical research in AF to uncover novel and specific mechanisms for pharmacological intervention, while advancing our clinical tools to precisely define AF pathophysiology at the individual patient level. These efforts will benefit also from a constantly growing repertoire of therapeutic modalities, e.g. cell and gene therapies, potentially allowing for innovative interventions to tackle disease processes previously deemed ‘undruggable’.
6. Where do we go from here—possible paths forward
6.1 Consideration of basis for current therapies
In thinking about possible strategies to improve the development of new clinical approaches to treating AF, it may be instructive to evaluate the sources of therapies that are currently used. Figure 6 presents a variety of presently used rhythm-control interventions for AF, along with a statement of how they were developed.
Figure 6.

Principal currently used therapies for AF rhythm control (left) and their developmental origins (right). (A) Antiarrhythmic drugs. (B) Interventional methods.
Figure 6A summarizes the basis for the development of the AADs used to treat AF. Quinidine was first introduced for AF termination and sinus-rhythm maintenance about 100 years ago, as a safer and more effective analogue of quinine, which had been noted by Wenckebach to have effectiveness in AF conversion.76 Flecainide emerged as a result of chemical synthesis of a series of antiarrhythmic compounds,77 and was targeted for ventricular arrhythmia suppression.78 Its use as an AF-suppressing antiarrhythmic agent was subsequently noted empirically79 and created a challenge to prevailing mechanistic notions.80 Propafenone, like flecainide a class Ic AAD, was similarly first introduced as a drug to suppress ventricular ectopy,81 although its effectiveness in AF was observed early in its clinical development.82 Amiodarone was initially introduced as a vasodilator and anti-anginal drug, found to have selective action potential prolonging properties by Singh and Vaughan Williams83 and noted to be effective for a wide range of arrhythmias, including AF, by Rosenbaum.84 Dofetilide was identified in a search for novel molecules with class III properties,85 and soon noted to be of value in AF.86 Sotalol (initially known as MJ1999) was the first prototype class III agent as described in the classical Singh and Vaughan Williams’ article of 197287 and found to be highly effective in AF.88 Dronedarone was developed in a search for less toxic amiodarone congeners,89 but unlike most of the other agents was introduced primarily for the treatment of AF.90
Figure 6B summarizes the principal interventional approaches presently used for AF management and the basis for their development. The surgical maze procedure is the first approach to AF management targeted specifically to AF pathophysiology,91 has passed through many subsequent modifications92 and is probably still the most effective therapeutic modality for sinus-rhythm maintenance. Pulmonary vein-directed catheter procedures were initially described by Haissaguerre et al.93 based on empiric observations and remain the mainstay of catheter-based AF-ablation procedures.9 Linear catheter-ablation lesions are used for AF-ablation to mimic the therapeutic mechanisms of the maze procedure.94 Likely because of difficulties in successfully and safely creating linear lesions with complete bidirectional block by catheter ablation, the success of the maze procedure has yet to be replicated by catheter-based procedures. Based on experimental observations consistent with a role for a small number of discrete rotors in AF-maintenance,95 ablation procedures have been developed to target these rotors.96 Despite some initial success,96 procedures targeting cardiac rotors have overall been found to have limited efficacy to date,97 possibly related to observations showing that rotors tend to be multiple, short-lived, and spatially unstable.98
6.2 Challenges and possible paths forward
A few conclusions may be drawn from the history of AF therapy development. The first is that relatively few of the drugs in present use to treat AF were actually developed specifically to target AF. This fact relates to the compelling importance of ventricular tachyarrhythmias as a potentially lethal clinical target, notwithstanding the emerging great public health importance of AF. Nevertheless, there may be opportunities for the specific development of AF-targeting therapies. The single most effective treatment for AF, the surgical maze procedure, was targeted against the mechanisms underlying AF,83 providing some support for the promise of mechanistic targeting. On the other hand, the most effective catheter-based procedure for AF, pulmonary vein isolation, was an empirical development and the underlying mechanisms of pulmonary vein involvement in AF remain incompletely understood. Furthermore, the apparent failure of rotor ablation to date illustrates the challenges of developing new mechanism-based interventional AF therapies.
Efforts have been made to target atrial-specific ion channels like the ultra-rapid delayed rectifier K+-channel and IK,ACh.24 Neither of these targets have shown promise in initial clinical trials,99–101 illustrating the challenges in extrapolating from observations in experimental models and human tissue samples50,51,102 to clinical use. Novel ion-channel targeting drugs may still have promise. While the risk of pro-arrhythmia with Na+ and K+ channel blocking drugs are well recognized,103 it is interesting that such agents are still in wide clinical use for AF management.12 Harnessing advanced in silico approaches to help in designing AF-selective drug therapy104,105 and in silico/human pluripotent stem cell models to minimize proarrhythmic liability106,107 may allow for the development of improved ion-channel targeting AF therapies for clinical use. Late Na+-current and/or L-type Ca2+ current inhibition can reduce or eliminate torsades de pointes risk and may be a useful component of multiple-channel blocking drugs to reduce proarrhythmic propensities.
A wide range of potential molecular targets have been identified for prevention of AF-substrate development.28,41 A major limitation to their development is the lack of a clear development pathway. Interventions that prevent development of the AF-substrate must have acceptably low toxicity and clinical risk potential. Drugs that target pathways that can be beneficial for the primary disease in high-risk patients, like RAAS inhibitors in patients with HF or severe hypertension, have potential value,34 but in most cases will already be in common use for the primary pathology. For lower-risk patients, the toxicity/adverse effect potential would have to be extremely low. Drugs that promote the resolution of inflammation are thought to have very limited risk and might be potential candidates.60 Given the very high costs for the development of AADs, repurposing of existing drugs with anti-inflammatory actions (e.g. colchicine) might also constitute an alternative option for treatment targeting the atrial substrate that promotes AF. Classical anti-inflammatory agents have not emerged as practical because of limited efficacy and adverse effects,108,109 perhaps in part because they fail to target the specific forms of inflammation (e.g. preferably expressed in cardiomyocytes53,54) that appear to be important in more common forms of AF. The success of lifestyle interventions in controlling AF with substantial effectiveness over a practical timeframe110,111 points to the viability of substrate-prevention as a clinical approach to AF. Interestingly, there is an interaction between polygenic risk scores and lifestyle-factor associations with cardiovascular disease, arguing for the further investigation of novel approaches to guide and personalize AF interventions with the use of genetic information.112
7. Conclusions
It is clear that there is an important unmet need for improved therapeutic options for AF. There remains a paradox between the wide continuing use of electrically active AADs (with recognized limitations) and the limited ongoing efforts to develop better alternatives. Upstream therapy remains an attractive approach, but its clinical application presents both theoretical and practical obstacles. While the majority of AF treatments in present clinical use resulted from empirical development, new approaches based on arrhythmia mechanisms have yielded highly effective therapies in the past. Despite significant challenges, therapeutic innovation based on the translation of basic research findings to clinical applications remains a promising path towards the introduction of effective new treatment modalities for AF. While the introduction of successful, conceptually innovative approaches to AF control is a challenging objective, one significant breakthrough will clearly revolutionize both AF management and the therapeutic development landscape.
Conflict of interest: S.N. is a consultant for LQT Therapeutics. J.H. is an employee of Bayer AG, Germany. P.S. is Co-Founder, Board of Director member, and equity holder in Long QT Therapeutics, Consultant to Milestone Pharma, Acesion Pharma, Lilly, Cydan Pharma, MyoKardia, Pharmaron. J.H. has no disclosures. D.D. is member of the Scientific Advisory Boards of Omeicos Therapeutics GmbH and Acesion Pharma.
Funding
The authors’ work is supported by the Canadian Institutes of Health Research (Foundation Grant 148401 to S.N.) and the Heart and Stroke Foundation of Canada (16-12708 to S.N.), the Netherlands Organization for Scientific Research (ZonMW Vidi 09150171910029 to J.H.), the National Institutes of Health (R01-HL131517, R01-HL136389, and R01-HL089598 to D.D.), the European Union (large-scale network project MAESTRIA No. 965266 to D.D.), and the German Research Foundation (DFG, Do 769/4-1 to D.D.).
References
- 1. Du X, Dong J, Ma C. Is atrial fibrillation a preventable disease? J Am Coll Cardiol 2017;69:1968–1982. [DOI] [PubMed] [Google Scholar]
- 2. Kannel WB, Wolf PA, Benjamin EJ, Levy D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am J Cardiol 1998;82:2N–9N. [DOI] [PubMed] [Google Scholar]
- 3. Wang TJ, Larson MG, Levy D, Vasan RS, Leip EP, Wolf PA, D'Agostino RB, Murabito JM, Kannel WB, Benjamin EJ. Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the Framingham Heart Study. Circulation 2003;107:2920–2925. [DOI] [PubMed] [Google Scholar]
- 4. Geng M, Lin A, Nguyen TP. Revisiting antiarrhythmic drug therapy for atrial fibrillation: reviewing lessons learned and redefining therapeutic paradigms. Front Pharmacol 2020;11:581837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim MH, Johnston SS, Chu BC, Dalal MR, Schulman KL. Estimation of total incremental health care costs in patients with atrial fibrillation in the United States. Circ Cardiovasc Qual Outcomes 2011;4:313–320. [DOI] [PubMed] [Google Scholar]
- 6. Turakhia MP, Shafrin J, Bognar K, Goldman DP, Mendys PM, Abdulsattar Y, Wiederkehr D, Trocio J. Economic burden of undiagnosed nonvalvular atrial fibrillation in the United States. Am J Cardiol 2015;116:733–739. [DOI] [PubMed] [Google Scholar]
- 7. January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC Jr., Conti JB, Ellinor PT, Ezekowitz MD, Field ME, Murray KT, Sacco RL, Stevenson WG, Tchou PJ, Tracy CM, Yancy CW, Members AATF. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation 2014;130:2071–2104. [DOI] [PubMed] [Google Scholar]
- 8. Hindricks G, Potpara T, Dagres N, Arbelo E, Bax JJ, Blomstrom-Lundqvist C, Boriani G, Castella M, Dan GA, Dilaveris PE, Fauchier L, Filippatos G, Kalman JM, La Meir M, Lane DA, Lebeau JP, Lettino M, Lip GYH, Pinto FJ, Thomas GN, Valgimigli M, Van Gelder IC, Van Putte BP, Watkins CL, Group ESCSD. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association of Cardio-Thoracic Surgery (EACTS). Eur Heart J 2020;42:373–498. [DOI] [PubMed] [Google Scholar]
- 9. Andrade JG, Aguilar M, Atzema C, Bell A, Cairns JA, Cheung CC, Cox JL, Dorian P, Gladstone DJ, Healey JS, Khairy P, Leblanc K, McMurtry MS, Mitchell LB, Nair GM, Nattel S, Parkash R, Pilote L, Sandhu RK, Sarrazin JF, Sharma M, Skanes AC, Talajic M, Tsang TSM, Verma A, Verma S, Whitlock R, Wyse DG, Macle L, Members of the Secondary Panel. The 2020 Canadian Cardiovascular Society/Canadian Heart Rhythm Society Comprehensive Guidelines for the Management of Atrial Fibrillation. Can J Cardiol 2020;36:1847–1948. [DOI] [PubMed] [Google Scholar]
- 10. Wyse DG, Waldo AL, DiMarco JP, Domanski MJ, Rosenberg Y, Schron EB, Kellen JC, Greene HL, Mickel MC, Dalquist JE, Corley SD, Atrial Fibrillation Follow-up Investigation of Rhythm Management Investigators. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002;347:1825–1833. [DOI] [PubMed] [Google Scholar]
- 11. Van Gelder IC, Hagens VE, Bosker HA, Kingma JH, Kamp O, Kingma T, Said SA, Darmanata JI, Timmermans AJ, Tijssen JG, Crijns HJ, Rate Control versus Electrical Cardioversion for Persistent Atrial Fibrillation Study Group. A comparison of rate control and rhythm control in patients with recurrent persistent atrial fibrillation. N Engl J Med 2002;347:1834–1840. [DOI] [PubMed] [Google Scholar]
- 12. Kirchhof P, Camm AJ, Goette A, Brandes A, Eckardt L, Elvan A, Fetsch T, van Gelder IC, Haase D, Haegeli LM, Hamann F, Heidbuchel H, Hindricks G, Kautzner J, Kuck KH, Mont L, Ng GA, Rekosz J, Schoen N, Schotten U, Suling A, Taggeselle J, Themistoclakis S, Vettorazzi E, Vardas P, Wegscheider K, Willems S, Crijns H, Breithardt G, EAST-AFNET 4 Trial Investigators. Early rhythm-control therapy in patients with atrial fibrillation. N Engl J Med 2020;383:1305–1316. [DOI] [PubMed] [Google Scholar]
- 13. Nattel S, Guasch E, Savelieva I, Cosio FG, Valverde I, Halperin JL, Conroy JM, Al-Khatib SM, Hess PL, Kirchhof P, De Bono J, Lip GY, Banerjee A, Ruskin J, Blendea D, Camm AJ. Early management of atrial fibrillation to prevent cardiovascular complications. Eur Heart J 2014;35:1448–1456. [DOI] [PubMed] [Google Scholar]
- 14. Aguilar M, Nattel S. Clarity and controversy around rate control in AF, the orphan child in AF therapeutics. Int J Cardiol 2019;287:189–194. [DOI] [PubMed] [Google Scholar]
- 15. Guichard JB, Xiong F, Qi XY, L'Heureux N, Hiram R, Xiao J, Naud P, Tardif JC, Da Costa A, Nattel S. Role of atrial arrhythmia and ventricular response in atrial fibrillation induced atrial remodelling. Cardiovasc Res 2021;117:462–471. [DOI] [PubMed] [Google Scholar]
- 16. Van Gelder IC, Groenveld HF, Crijns HJ, Tuininga YS, Tijssen JG, Alings AM, Hillege HL, Bergsma-Kadijk JA, Cornel JH, Kamp O, Tukkie R, Bosker HA, Van Veldhuisen DJ, Van den Berg MP, RACE II Investigators. Lenient versus strict rate control in patients with atrial fibrillation. N Engl J Med 2010;362:1363–1373. [DOI] [PubMed] [Google Scholar]
- 17. Kranert M, Shchetynska-Marinova T, Berghoff T, Liebe V, Doesch C, Papavassiliu T, Custodis F, Akin I, Borggrefe M, Hohneck A. Arterial stiffness is associated with increased symptom burden in patients with atrial fibrillation. Can J Cardiol 2020;36:1949–1955. [DOI] [PubMed] [Google Scholar]
- 18. Dobrev D, Nattel S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet 2010;375:1212–1223. [DOI] [PubMed] [Google Scholar]
- 19. Markman TM, Geng Z, Epstein AE, Nazarian S, Deo R, Marchlinski FE, Groeneveld PW, Frankel DS. Trends in antiarrhythmic drug use among patients in the United States between 2004 and 2016. Circulation 2020;141:937–939. [DOI] [PubMed] [Google Scholar]
- 20. Perino AC, Leef GC, Cluckey A, Yunus FN, Askari M, Heidenreich PA, Narayan SM, Wang PJ, Turakhia MP. Secular trends in success rate of catheter ablation for atrial fibrillation: the SMASH-AF cohort. Am Heart J 2019;208:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Packer DL, Mark DB, Robb RA, Monahan KH, Bahnson TD, Poole JE, Noseworthy PA, Rosenberg YD, Jeffries N, Mitchell LB, Flaker GC, Pokushalov E, Romanov A, Bunch TJ, Noelker G, Ardashev A, Revishvili A, Wilber DJ, Cappato R, Kuck KH, Hindricks G, Davies DW, Kowey PR, Naccarelli GV, Reiffel JA, Piccini JP, Silverstein AP, Al-Khalidi HR, Lee KL, Investigators C, for the CABANA Investigators. Effect of catheter ablation vs antiarrhythmic drug therapy on mortality, stroke, bleeding, and cardiac arrest among patients with atrial fibrillation: the CABANA Randomized Clinical Trial. JAMA 2019;321:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marrouche NF, Brachmann J, Andresen D, Siebels J, Boersma L, Jordaens L, Merkely B, Pokushalov E, Sanders P, Proff J, Schunkert H, Christ H, Vogt J, Bansch D, CASTLE-AF Investigators. Catheter ablation for atrial fibrillation with heart failure. N Engl J Med 2018;378:417–427. [DOI] [PubMed] [Google Scholar]
- 23. Kuck KH, Merkely B, Zahn R, Arentz T, Seidl K, Schluter M, Tilz RR, Piorkowski C, Geller L, Kleemann T, Hindricks G. Catheter ablation versus best medical therapy in patients with persistent atrial fibrillation and congestive heart failure: the randomized AMICA trial. Circ Arrhythm Electrophysiol 2019;12:e007731. [DOI] [PubMed] [Google Scholar]
- 24. Dobrev D, Carlsson L, Nattel S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov 2012;11:275–291. [DOI] [PubMed] [Google Scholar]
- 25. Ang YS, Rajamani S, Haldar SM, Huser J. A new therapeutic framework for atrial fibrillation drug development. Circ Res 2020;127:184–201. [DOI] [PubMed] [Google Scholar]
- 26. Nattel S, Bourne G, Talajic M. Insights into mechanisms of antiarrhythmic drug action from experimental models of atrial fibrillation. J Cardiovasc Electrophysiol 1997;8:469–480. [DOI] [PubMed] [Google Scholar]
- 27. Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol 2008;1:62–73. [DOI] [PubMed] [Google Scholar]
- 28. Nattel S, Heijman J, Zhou L, Dobrev D. Molecular basis of atrial fibrillation pathophysiology and therapy: a translational perspective. Circ Res 2020;127:51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schneider MP, Hua TA, Bohm M, Wachtell K, Kjeldsen SE, Schmieder RE. Prevention of atrial fibrillation by renin-angiotensin system inhibition a meta-analysis. J Am Coll Cardiol 2010;55:2299–2307. [DOI] [PubMed] [Google Scholar]
- 30. Wachtell K, Lehto M, Gerdts E, Olsen MH, Hornestam B, Dahlof B, Ibsen H, Julius S, Kjeldsen SE, Lindholm LH, Nieminen MS, Devereux RB. Angiotensin II receptor blockade reduces new-onset atrial fibrillation and subsequent stroke compared to atenolol: the Losartan Intervention For End Point Reduction in Hypertension (LIFE) study. J Am Coll Cardiol 2005;45:712–719. [DOI] [PubMed] [Google Scholar]
- 31. Olsson LG, Swedberg K, Ducharme A, Granger CB, Michelson EL, McMurray JJ, Puu M, Yusuf S, Pfeffer MA, CHARM Investigators. Atrial fibrillation and risk of clinical events in chronic heart failure with and without left ventricular systolic dysfunction: results from the Candesartan in Heart failure-Assessment of Reduction in Mortality and morbidity (CHARM) program. J Am Coll Cardiol 2006;47:1997–2004. [DOI] [PubMed] [Google Scholar]
- 32. Okin PM, Wachtell K, Devereux RB, Harris KE, Jern S, Kjeldsen SE, Julius S, Lindholm LH, Nieminen MS, Edelman JM, Hille DA, Dahlof B. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA 2006;296:1242–1248. [DOI] [PubMed] [Google Scholar]
- 33. Marott SC, Nielsen SF, Benn M, Nordestgaard BG. Antihypertensive treatment and risk of atrial fibrillation: a nationwide study. Eur Heart J 2014;35:1205–1214. [DOI] [PubMed] [Google Scholar]
- 34. Savelieva I, Kakouros N, Kourliouros A, Camm AJ. Upstream therapies for management of atrial fibrillation: review of clinical evidence and implications for European Society of Cardiology guidelines. Part I: primary prevention. Europace 2011;13:308–328. [DOI] [PubMed] [Google Scholar]
- 35. Savelieva I, Kakouros N, Kourliouros A, Camm AJ. Upstream therapies for management of atrial fibrillation: review of clinical evidence and implications for European Society of Cardiology guidelines. Part II: secondary prevention. Europace 2011;13:610–625. [DOI] [PubMed] [Google Scholar]
- 36. Swedberg K, Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Shi H, Vincent J, Pitt B, EMPHASIS-HF Study Investigators. Eplerenone and atrial fibrillation in mild systolic heart failure: results from the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization And SurvIval Study in Heart Failure) study. J Am Coll Cardiol 2012;59:1598–1603. [DOI] [PubMed] [Google Scholar]
- 37. Valembois L, Audureau E, Takeda A, Jarzebowski W, Belmin J, Lafuente-Lafuente C. Antiarrhythmics for maintaining sinus rhythm after cardioversion of atrial fibrillation. Cochrane Database Syst Rev 2019; 9:CD005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rahimi K, Emberson J, McGale P, Majoni W, Merhi A, Asselbergs FW, Krane V, Macfarlane PW, PROSPER Executive. Effect of statins on atrial fibrillation: collaborative meta-analysis of published and unpublished evidence from randomised controlled trials. BMJ 2011;342:d1250. [DOI] [PubMed] [Google Scholar]
- 39. Zheng Z, Jayaram R, Jiang L, Emberson J, Zhao Y, Li Q, Du J, Guarguagli S, Hill M, Chen Z, Collins R, Casadei B. Perioperative rosuvastatin in cardiac surgery. N Engl J Med 2016;374:1744–1753. [DOI] [PubMed] [Google Scholar]
- 40. Kowey PR, Reiffel JA, Ellenbogen KA, Naccarelli GV, Pratt CM. Efficacy and safety of prescription omega-3 fatty acids for the prevention of recurrent symptomatic atrial fibrillation: a randomized controlled trial. JAMA 2010;304:2363–2372. [DOI] [PubMed] [Google Scholar]
- 41. Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D, Nattel S. The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovasc Res 2016;109:467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995;92:1954–1968. [DOI] [PubMed] [Google Scholar]
- 43. Wijffels MC, Kirchhof CJ, Dorland R, Power J, Allessie MA. Electrical remodeling due to atrial fibrillation in chronically instrumented conscious goats: roles of neurohumoral changes, ischemia, atrial stretch, and high rate of electrical activation. Circulation 1997;96:3710–3720. [DOI] [PubMed] [Google Scholar]
- 44. Martins RP, Kaur K, Hwang E, Ramirez RJ, Willis BC, Filgueiras-Rama D, Ennis SR, Takemoto Y, Ponce-Balbuena D, Zarzoso M, O'Connell RP, Musa H, Guerrero-Serna G, Avula UM, Swartz MF, Bhushal S, Deo M, Pandit SV, Berenfeld O, Jalife J. Dominant frequency increase rate predicts transition from paroxysmal to long-term persistent atrial fibrillation. Circulation 2014;129:1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tajiri K, Guichard JB, Qi X, Xiong F, Naud P, Tardif JC, Costa AD, Aonuma K, Nattel S. An N-/L-type calcium channel blocker, cilnidipine, suppresses autonomic, electrical, and structural remodelling associated with atrial fibrillation. Cardiovasc Res 2019;115:1975–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 1999;100:87–95. [DOI] [PubMed] [Google Scholar]
- 47. Marrouche NF, Wilber D, Hindricks G, Jais P, Akoum N, Marchlinski F, Kholmovski E, Burgon N, Hu N, Mont L, Deneke T, Duytschaever M, Neumann T, Mansour M, Mahnkopf C, Herweg B, Daoud E, Wissner E, Bansmann P, Brachmann J. Association of atrial tissue fibrosis identified by delayed enhancement MRI and atrial fibrillation catheter ablation: the DECAAF study. JAMA 2014;311:498–506. [DOI] [PubMed] [Google Scholar]
- 48. Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res 2014;114:1500–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nattel S, Liu L, St-Georges D. Effects of the novel antiarrhythmic agent azimilide on experimental atrial fibrillation and atrial electrophysiologic properties. Cardiovasc Res 1998;37:627–635. [DOI] [PubMed] [Google Scholar]
- 50. Cha TJ, Ehrlich JR, Chartier D, Qi XY, Xiao L, Nattel S. Kir3-based inward rectifier potassium current: potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation 2006;113:1730–1737. [DOI] [PubMed] [Google Scholar]
- 51. Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M, Ravens U. The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation 2005;112:3697–3706. [DOI] [PubMed] [Google Scholar]
- 52. Harada M, Van Wagoner DR, Nattel S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ J 2015;79:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yao C, Veleva T, Scott L Jr, Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha ID, Ghezelbash S, Reynolds CL, Shen YH, LeMaire SA, Schmitz W, Muller FU, El-Armouche A, Tony Eissa N, Beeton C, Nattel S, Wehrens XHT, Dobrev D, Li N. Enhanced cardiomyocyte NLRP3 inflammasome signaling promotes atrial fibrillation. Circulation 2018;138:2227–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, Wang Q, Abu-Taha IH, Gorka M, Kunzel S, El-Armouche A, Reichenspurner H, Kamler M, Nikolaev V, Ravens U, Li N, Nattel S, Wehrens XHT, Dobrev D. Atrial myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation. Circ Res 2020;127:1036–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fender AC, Kleeschulte S, Stolte S, Leineweber K, Kamler M, Bode J, Li N, Dobrev D. Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res Cardiol 2020;115:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Scott L Jr, Fender AC, Saljic A, Li L, Chen X, Wang X, Linz D, Lang J, Hohl M, Twomey D, Pham TT, Diaz-Lankenau R, Chelu MG, Kamler M, Entman ML, Taffet GE, Sanders P, Dobrev D, Li N. NLRP3 inflammasome is a key driver of Obesity-Induced atrial arrhythmias. Cardiovasc Res 2021; doi:10.1093/cvr/cvab1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aschar-Sobbi R, Izaddoustdar F, Korogyi AS, Wang Q, Farman GP, Yang F, Yang W, Dorian D, Simpson JA, Tuomi JM, Jones DL, Nanthakumar K, Cox B, Wehrens XH, Dorian P, Backx PH. Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNFalpha. Nat Commun 2015;6:6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ishii Y, Schuessler RB, Gaynor SL, Yamada K, Fu AS, Boineau JP, Damiano RJ Jr. Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation. Circulation 2005;111:2881–2888. [DOI] [PubMed] [Google Scholar]
- 59. Hiram R, Naud P, Xiong F, Al-u’datt D, Algalarrondo V, Sirois MG, Tanguay J-F, Tardif J-C, Nattel S. Right atrial mechanisms of atrial fibrillation in a rat model of right heart disease. J Am Coll Cardiol 2019;74:1332–1347. [DOI] [PubMed] [Google Scholar]
- 60. Hiram R, Xiong F, Naud P, Xiao J, Sirois M, Tanguay JF, Tardif JC, Nattel S. The inflammation-resolution promoting molecule resolvin-D1 prevents atrial proarrhythmic remodeling in experimental right heart disease. Cardiovasc Res 2020;doi:10.1093/cvr/cvaa1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dobrev D, Wehrens XHT. Mouse models of cardiac arrhythmias. Circ Res 2018;123:332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ni L, Scott L Jr, Campbell HM, Pan X, Alsina KM, Reynolds J, Philippen LE, Hulsurkar M, Lagor WR, Li N, Wehrens XHT. Atrial-specific gene delivery using an adeno-associated viral vector. Circ Res 2019;124:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Peng W, Li M, Li H, Tang K, Zhuang J, Zhang J, Xiao J, Jiang H, Li D, Yu Y, Sham PC, Nattel S, Xu Y. Dysfunction of myosin light-chain 4 (MYL4) leads to heritable atrial cardiomyopathy with electrical, contractile, and structural components: evidence from genetically-engineered rats. J Am Heart Assoc 2017;6:e007030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dobrev D, Ravens U. Remodeling of cardiomyocyte ion channels in human atrial fibrillation. Basic Res Cardiol 2003;98:137–148. [DOI] [PubMed] [Google Scholar]
- 65. Moreira LM, Takawale A, Hulsurkar M, Menassa DA, Antanaviciute A, Lahiri SK, Mehta N, Evans N, Psarros C, Robinson P, Sparrow AJ, Gillis MA, Ashley N, Naud P, Barallobre-Barreiro J, Theofilatos K, Lee A, Norris M, Clarke MV, Russell PK, Casadei B, Bhattacharya S, Zajac JD, Davey RA, Sirois M, Mead A, Simmons A, Mayr M, Sayeed R, Krasopoulos G, Redwood C, Channon KM, Tardif JC, Wehrens XHT, Nattel S, Reilly S. Paracrine signalling by cardiac calcitonin controls atrial fibrogenesis and arrhythmia. Nature 2020;587:460–465. [DOI] [PubMed] [Google Scholar]
- 66. Parrotta EI, Lucchino V, Scaramuzzino L, Scalise S, Cuda G. Modeling cardiac disease mechanisms using induced pluripotent stem cell-derived cardiomyocytes: progress, promises and challenges. IJMS 2020;21:4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Laksman Z, Wauchop M, Lin E, Protze S, Lee J, Yang W, Izaddoustdar F, Shafaattalab S, Gepstein L, Tibbits GF, Keller G, Backx PH. Modeling atrial fibrillation using human embryonic stem cell-derived atrial tissue. Sci Rep 2017;7:5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Seiffge DJ, Meinel TR. The real prize of direct oral anticoagulant blockbuster. Heart 2021;107:8–9. [DOI] [PubMed] [Google Scholar]
- 69. Orlowski A, Gale CP, Ashton R, Petrungaro B, Slater R, Nadarajah R, Cowan JC, Buck J, Smith W, Wu J. Clinical and budget impacts of changes in oral anticoagulation prescribing for atrial fibrillation. Heart 2021;107:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Patel NJ, Deshmukh A, Pant S, Singh V, Patel N, Arora S, Shah N, Chothani A, Savani GT, Mehta K, Parikh V, Rathod A, Badheka AO, Lafferty J, Kowalski M, Mehta JL, Mitrani RD, Viles-Gonzalez JF, Paydak H. Contemporary trends of hospitalization for atrial fibrillation in the United States, 2000 through 2010: implications for healthcare planning. Circulation 2014;129:2371–2379. [DOI] [PubMed] [Google Scholar]
- 71. Sohns C, Marrouche NF. Atrial fibrillation and cardiac fibrosis. Eur Heart J 2020;41:1123–1131. [DOI] [PubMed] [Google Scholar]
- 72. Zakeri R, Van Wagoner DR, Calkins H, Wong T, Ross HM, Heist EK, Meyer TE, Kowey PR, Mentz RJ, Cleland JG, Pitt B, Zannad F, Linde C. The burden of proof: the current state of atrial fibrillation prevention and treatment trials. Heart Rhythm 2017;14:763–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dobrev D, Aguilar M, Heijman J, Guichard JB, Nattel S. Postoperative atrial fibrillation: mechanisms, manifestations and management. Nat Rev Cardiol 2019;16:417–436. [DOI] [PubMed] [Google Scholar]
- 74. Scott L Jr, Li N, Dobrev D. Role of inflammatory signaling in atrial fibrillation. Int J Cardiol 2019;287:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fabritz L, Guasch E, Antoniades C, Bardinet I, Benninger G, Betts TR, Brand E, Breithardt G, Bucklar-Suchankova G, Camm AJ, Cartlidge D, Casadei B, Chua WW, Crijns HJ, Deeks J, Hatem S, Hidden-Lucet F, Kaab S, Maniadakis N, Martin S, Mont L, Reinecke H, Sinner MF, Schotten U, Southwood T, Stoll M, Vardas P, Wakili R, West A, Ziegler A, Kirchhof P. Expert consensus document: defining the major health modifiers causing atrial fibrillation: a roadmap to underpin personalized prevention and treatment. Nat Rev Cardiol 2016;13:230–237. [DOI] [PubMed] [Google Scholar]
- 76. Frey W. Weitere erfahrungen mit chinidin bei absoluter Herzunregelmässigkeit. Berliner Klin Wochenschr 1918;55:849–853. [Google Scholar]
- 77. Banitt EH, Bronn WR, Coyne WE, Schmid JR. Antiarrhythmics. 2. Synthesis and antiarrhythmic activity of N-(piperidylalkyl)trifluoroethoxybenzamides. J Med Chem 1977;20:821–826. [DOI] [PubMed] [Google Scholar]
- 78. Somani P. Antiarrhythmic effects of flecainide. Clin Pharmacol Ther 1980;27:464–470. [DOI] [PubMed] [Google Scholar]
- 79. Goy JJ, Maendly R, Grbic M, Finci L, Sigwart U. Cardioversion with flecainide in patients with atrial fibrillation of recent onset. Eur J Clin Pharmacol 1985;27:737–738. [DOI] [PubMed] [Google Scholar]
- 80. Nattel S. Antiarrhythmic drug classifications. A critical appraisal of their history, present status, and clinical relevance. Drugs 1991;41:672–701. [DOI] [PubMed] [Google Scholar]
- 81. Seipel L, Breithardt G. Propafenone–a new antiarrhythmic drug. Eur Heart J 1980;1:309–313. [DOI] [PubMed] [Google Scholar]
- 82. Beck OA, Lehmann HU, Hochrein H. [Propafenon and lidoflazine in chronic atrial fibrillation and flutter (author's transl)]. Dtsch Med Wochenschr 1978;103:1068–1072. [DOI] [PubMed] [Google Scholar]
- 83. Singh BN, Vaughan Williams EM. The effect of amiodarone, a new anti-anginal drug, on cardiac muscle. Br J Pharmacol 1970;39:657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rosenbaum MB, Chiale PA, Halpern MS, Nau GJ, Przybylski J, Levi RJ, Lazzari JO, Elizari MV. Clinical efficacy of amiodarone as an antiarrhythmic agent. Am J Cardiol 1976;38:934–944. [DOI] [PubMed] [Google Scholar]
- 85. Cross PE, Arrowsmith JE, Thomas GN, Gwilt M, Burges RA, Higgins AJ. Selective class III antiarrhythmic agents. 1 Bis(arylalkyl)amines. J Med Chem 1990;33:1151–1155. [DOI] [PubMed] [Google Scholar]
- 86. Suttorp MJ, Polak PE, van 't Hof A, Rasmussen HS, Dunselman PH, Kingma JH. Efficacy and safety of a new selective class III antiarrhythmic agent dofetilide in paroxysmal atrial fibrillation or atrial flutter. Am J Cardiol 1992;69:417–419. [DOI] [PubMed] [Google Scholar]
- 87. Singh BN, Vaughan Williams EM. A third class of anti-arrhythmic action. Effects on atrial and ventricular intracellular potentials, and other pharmacological actions on cardiac muscle, of MJ 1999 and AH 3474. Br J Pharmacol 1970;39:675–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Singh BN, Deedwania P, Nademanee K, Ward A, Sorkin EM. Sotalol. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use. Drugs 1987;34:311–349. [DOI] [PubMed] [Google Scholar]
- 89. Manning A, Thisse V, Hodeige D, Richard J, Heyndrickx JP, Chatelain P. SR 33589, a new amiodarone-like antiarrhythmic agent: electrophysiological effects in anesthetized dogs. J Cardiovasc Pharmacol 1995;25:252–261. [DOI] [PubMed] [Google Scholar]
- 90. Hohnloser SH, Crijns HJ, van Eickels M, Gaudin C, Page RL, Torp-Pedersen C, Connolly SJ, ATHENA Investigators. Effect of dronedarone on cardiovascular events in atrial fibrillation. N Engl J Med 2009;360:668–678. [DOI] [PubMed] [Google Scholar]
- 91. Cox JL, Schuessler RB, D'Agostino HJ Jr, Stone CM, Chang BC, Cain ME, Corr PB, Boineau JP. The surgical treatment of atrial fibrillation. III. Development of a definitive surgical procedure. J Thorac Cardiovasc Surg 1991;101:569–583. [PubMed] [Google Scholar]
- 92. Cox JL, Churyla A, Malaisrie SC, Kruse J, Pham DT, Kislitsina ON, McCarthy PM. When is a Maze Procedure a Maze Procedure? Can J Cardiol 2018;34:1482–1491. [DOI] [PubMed] [Google Scholar]
- 93. Haïssaguerre M, Jaïs P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Métayer P, Clémenty J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med 1998;339:659–666. [DOI] [PubMed] [Google Scholar]
- 94. Haíssaguerre M, Jaís P, Shah DC, Gencel L, Pradeau V, Garrigues S, Chouairi S, Hocini M, Métayer P, Roudaut R, Clémenty J. Right and left atrial radiofrequency catheter therapy of paroxysmal atrial fibrillation. J Cardiovasc Electrophysiol 1996;7:1132–1144. [DOI] [PubMed] [Google Scholar]
- 95. Jalife J, Berenfeld O, Skanes A, Mandapati R. Mechanisms of atrial fibrillation: mother rotors or multiple daughter wavelets, or both? J Cardiovasc Electrophysiol 1998;9:S2–S12. [PubMed] [Google Scholar]
- 96. Narayan SM, Krummen DE, Shivkumar K, Clopton P, Rappel WJ, Miller JM. Treatment of atrial fibrillation by the ablation of localized sources: CONFIRM (Conventional Ablation for Atrial Fibrillation With or Without Focal Impulse and Rotor Modulation) trial. J Am Coll Cardiol 2012;60:628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tilz RR, Lenz C, Sommer P, Roza MS, Sarver AE, Williams CG, Heeger C, Hindricks G, Vogler J, Eitel C. Focal impulse and rotor modulation ablation vs. pulmonary vein isolation for the treatment of paroxysmal atrial fibrillation: results from the FIRMAP AF study. Europace 2020;10.1093/europace/euaa1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Haissaguerre M, Hocini M, Denis A, Shah AJ, Komatsu Y, Yamashita S, Daly M, Amraoui S, Zellerhoff S, Picat MQ, Quotb A, Jesel L, Lim H, Ploux S, Bordachar P, Attuel G, Meillet V, Ritter P, Derval N, Sacher F, Bernus O, Cochet H, Jais P, Dubois R. Driver domains in persistent atrial fibrillation. Circulation 2014;130:530–538. [DOI] [PubMed] [Google Scholar]
- 99. Walfridsson H, Anfinsen OG, Berggren A, Frison L, Jensen S, Linhardt G, Nordkam AC, Sundqvist M, Carlsson L. Is the acetylcholine-regulated inwardly rectifying potassium current a viable antiarrhythmic target? Translational discrepancies of AZD2927 and A7071 in dogs and humans. Europace 2015;17:473–482. [DOI] [PubMed] [Google Scholar]
- 100. Shunmugam SR, Sugihara C, Freemantle N, Round P, Furniss S, Sulke N. A double-blind, randomised, placebo-controlled, cross-over study assessing the use of XEN-D0103 in patients with paroxysmal atrial fibrillation and implanted pacemakers allowing continuous beat-to-beat monitoring of drug efficacy. J Interv Card Electrophysiol 2018;51:191–197. [DOI] [PubMed] [Google Scholar]
- 101. Camm AJ, Dorian P, Hohnloser SH, Kowey PR, Tyl B, Ni Y, Vandzhura V, Maison-Blanche P, de Melis M, Sanders P. A randomized, double-blind, placebo-controlled trial assessing the efficacy of S66913 in patients with paroxysmal atrial fibrillation. Eur Heart J Cardiovasc Pharmacother 2019;5:21–28. [DOI] [PubMed] [Google Scholar]
- 102. Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res 1993;73:1061–1076. [DOI] [PubMed] [Google Scholar]
- 103. Huang H, Pugsley MK, Fermini B, Curtis MJ, Koerner J, Accardi M, Authier S. Cardiac voltage-gated ion channels in safety pharmacology: review of the landscape leading to the CiPA initiative. J Pharmacol Toxicol Methods 2017;87:11–23. [DOI] [PubMed] [Google Scholar]
- 104. Aguilar-Shardonofsky M, Vigmond EJ, Nattel S, Comtois P. In silico optimization of atrial fibrillation-selective sodium channel blocker pharmacodynamics. Biophys J 2012;102:951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Aguilar M, Xiong F, Qi XY, Comtois P, Nattel S. Potassium channel blockade enhances atrial fibrillation-selective antiarrhythmic effects of optimized state-dependent sodium channel blockade. Circulation 2015;132:2203–2211. [DOI] [PubMed] [Google Scholar]
- 106. Raphel F, De Korte T, Lombardi D, Braam S, Gerbeau JF. A greedy classifier optimization strategy to assess ion channel blocking activity and pro-arrhythmia in hiPSC-cardiomyocytes. PLoS Comput Biol 2020;16:e1008203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Margara F, Wang ZJ, Levrero-Florencio F, Santiago A, Vazquez M, Bueno-Orovio A, Rodriguez B. In-silico human electro-mechanical ventricular modelling and simulation for drug-induced pro-arrhythmia and inotropic risk assessment. Prog Biophys Mol Biol 2021;159:58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. van Osch D, Dieleman JM, van Dijk D, Jacob KA, Kluin J, Doevendans PA, Nathoe HM, group DEfCSs, group DEfCSDs. Dexamethasone for the prevention of postoperative atrial fibrillation. Int J Cardiol 2015;182:431–437. [DOI] [PubMed] [Google Scholar]
- 109. Chokesuwattanaskul R, Chiengthong K, Thongprayoon C, Lertjitbanjong P, Bathini T, Ungprasert P, Cato LD, Mao MA, Cheungpasitporn W. Nonsteroidal anti-inflammatory drugs and incidence of atrial fibrillation: a meta-analysis. QJM 2020;113:79–85. [DOI] [PubMed] [Google Scholar]
- 110. Abed HS, Wittert GA, Leong DP, Shirazi MG, Bahrami B, Middeldorp ME, Lorimer MF, Lau DH, Antic NA, Brooks AG, Abhayaratna WP, Kalman JM, Sanders P. Effect of weight reduction and cardiometabolic risk factor management on symptom burden and severity in patients with atrial fibrillation: a randomized clinical trial. JAMA 2013;310:2050–2060. [DOI] [PubMed] [Google Scholar]
- 111. Chung MK, Eckhardt LL, Chen LY, Ahmed HM, Gopinathannair R, Joglar JA, Noseworthy PA, Pack QR, Sanders P, Trulock KM, American Heart Association Electrocardiography and Arrhythmias Committee and Exercise, Cardiac Rehabilitation, and Secondary Prevention Committee of the Council on Clinical Cardiology; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Council on Lifestyle and Cardiometabolic Health. Lifestyle and risk factor modification for reduction of atrial fibrillation: a scientific statement from the American Heart Association. Circulation 2020;141:e750–e772. [DOI] [PubMed] [Google Scholar]
- 112. Ye Y, Chen X, Han J, Jiang W, Natarajan P, Xhao H. Interactions between enhanced polygenic risk scores and cardiovascular disease, diabetes mellitus and lipid levels. Circ Genom Precis Med 2021;14:e003128. [DOI] [PMC free article] [PubMed] [Google Scholar]