

Abstract
Increased levels of dysfunctional mitochondria within skeletal muscle are correlated with numerous age‐related physiopathological conditions. Improving our understanding of the links between mitochondrial function and muscle proteostasis, and the role played by individual genes and regulatory networks, is essential to develop treatments for these conditions. One potential player is the mitochondrial outer membrane protein Fis1, a crucial fission factor heavily involved in mitochondrial dynamics in yeast but with an unknown role in higher‐order organisms. By using Drosophila melanogaster as a model, we explored the effect of Fis1 mutations generated by transposon Minos‐mediated integration. Mutants exhibited a higher ratio of damaged mitochondria with age as well as elevated reactive oxygen species levels compared with controls. This caused an increase in oxidative stress, resulting in large accumulations of ubiquitinated proteins, accelerated muscle function decline, and mitochondrial myopathies in young mutant flies. Ectopic expression of Fis1 isoforms was sufficient to suppress this phenotype. Loss of Fis1 led to unbalanced mitochondrial proteostasis within fly muscle, decreasing both flight capabilities and lifespan. Fis1 thus clearly plays a role in fly mitochondrial dynamics. Further investigations into the detailed function of Fis1 are necessary for exploring how mitochondrial function correlates with muscle health during aging.
Keywords: aging, Drosophila melanogaster, Fis1, mitochondria
Ubiquitinated proteins and autophagic proteins accumulate within Drosophila melanogaster muscle fibers as they age. Here we generated Drosophila mutants for the gene Fis1, which influences mitochondrial dynamics, and observed similar phenotypes in these transgenic lines. The unbalanced proteostasis resulting from Fis1 mutation led to muscle fiber degeneration and reduced health spans. Fis1‐related mitochondrial quality control is an important factor in determining Drosophila muscle, and thus overall, health.
1. INTRODUCTION
The progressive decline in skeletal muscle strength and mass is a major cause of frailty during aging (Vainshtein et al., 2014). Studies have linked these declines to the dysregulation of proteostasis during aging (Labbadia & Morimoto, 2015). Although the process through which this change occurs remains debatable, redox‐related regulation pathways are considered the likely cause of age‐related changes in metabolism (Fernando et al., 2019). Mitochondria are the main source of cellular reactive oxygen species (ROS), which are by‐products of the electron transport chain (ETC; Ábrigo et al., 2018; Zorov et al., 2014). Compared with other tissues, skeletal muscle requires more adenosine triphosphate (ATP) for energy production and contains a larger number of mitochondria; mitochondria therefore heavily influence levels of oxidative stress in skeletal muscle (Ábrigo et al., 2018). Mitochondrial dysfunction can lead to increased ROS production, a hallmark of neurodegenerative and age‐related diseases such as Alzheimer's disease (AD; Wong‐Riley et al., 1997) and Parkinson's disease (Henchcliffe & Beal, 2008). Identifying the mechanisms that disrupt proteostasis and/or mitochondrial function in aging animals might thus lead to treatments that prevent disease incidence and delay age‐related illness.
Mitochondrial dynamics (fission/fusion) and mitophagy are two closely related mechanisms (Ashrafi & Schwarz, 2013; Twig & Shirihai, 2011) responsible for maintaining a healthy mitochondrial pool and eliminating damaged mitochondria, respectively (Lo Verso et al., 2014; Twig et al., 2008; Vainshtein et al., 2014). Mitochondrial fission and fusion processes are regulated by large guanosine triphosphatases (GTPases) of the dynamin family (Chan, 2012; Kraus & Ryan, 2017; Ni et al., 2015; Pernas & Scorrano, 2016; Tilokani et al., 2018; Westermann, 2010), which are highly conserved in yeast, flies, and mammals (Hales & Fuller, 1997; Hoppins et al., 2007; Liesa et al., 2009; Zhao et al., 2013). Mitochondrial fusion in mammals is mediated by the fusion proteins mitofusin (Mfn) 1, Mfn2, and optic atrophy 1 (OPA1); on the other hand, fission processes are mainly mediated by dynamin‐related protein 1 (Drp1; Kraus & Ryan, 2017; Ni et al., 2015; Pernas & Scorrano, 2016).
The first step of mitophagy‐mediated quality control is the segregation of healthy and damaged mitochondria, with damaged mitochondria possessing depolarized mitochondrial membrane potential (Ashrafi & Schwarz, 2013; Jin & Youle, 2012; Twig & Shirihai, 2011). Studies have reported that Mfn1 and Mfn2 are ubiquitinated during PINK1/parkin‐dependent mitophagy, resulting in the removal of damaged mitochondria in mammals and flies (Gegg et al., 2010; Geisler et al., 2010; Poole et al., 2010). As previously mentioned, Drp1, the mammalian homolog of the yeast Dnm1, is essential for mitophagy (Mao & Klionsky, 2013; Twig et al., 2008). As such, the core proteins that regulate mitochondrial dynamics also influence mitochondrial proteostasis; however, the interplay between mitochondrial dynamics and proteostasis during aging in muscle remains poorly understood.
Fis1, a crucial fission factor involved in the recruitment of Dnm1, is located on the mitochondrial outer membrane in yeast and may play a role in modulating mitochondrial dynamics (Mozdy et al., 2000; Shen et al., 2014). Although Fis1 is evolutionarily conserved from yeasts to humans, its direct participation in metazoan fission machinery remains debated (Hoppins et al., 2007; Okamoto & Shaw, 2005; Otera et al., 2010; Suzuki et al., 2003; Yu et al., 2019). Moreover, Fis1 knockout studies reported decreased mitophagy (Twig et al., 2008) and have suggested that Fis1 controls mitophagy through a regulated ER‐resident SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) protein (Xian et al., 2019). Consistent with this, Fis1 has been reported to fragment mitochondria and enhance the formation of mitochondria‐targeting autophagosomes (Gomes & Scorrano, 2008). Fis1 thus appears related to mitochondrial dynamics and mitophagy, although the mechanisms through which Fis1 regulates tissue‐specific mitochondrial dynamics and proteostasis in an age‐dependent manner are unknown.
Here, we used Drosophila melanogaster (D. melanogaster) as a model for studying Fis1‐mediated mitochondrial function and proteostasis in muscles. The transcription of Fis1 results in the production of at least six mRNA isoforms in D. melanogaster, denoted as C (698 bp), E (628 bp), A (630 bp), G (557 bp), D (560 bp), and F (704 bp) forms. After post‐translation modification, the isoforms translate into four Fis1 polypeptides: C (154 amino acids); E (148 amino acids); A and G (both with 98 amino acids); and D and F (both with 92 amino acids).
To simplify the nomenclature, we divided the isoforms into two groups, one for longer forms (more than 100 amino acids) and one for shorter forms (<100 amino acids). Next, we further subdivided each group according to isoform length. Both C and E forms were thus designated as long forms because they each contain more than 100 aa. However, because the C form (154 aa) is longer than the E form (148 aa), we denoted the C form as long form long (LL) and the E form as long form short (LS). Similarly, we designated the A and G forms as short form long (SL) and the D and F forms as short form short (SS).
By generating Fis1‐specific antibodies, we observed that the expression of the two longer forms increased with age. Loss of Fis1 led to mitochondrial enlargement and degeneration of mitochondrial inner membranes in both young and old flies. These Fis1‐lacking mitochondria exhibited multiple signatures of dysfunction, including decreased ETC complex protein levels, increased mitochondrial ROS leakage, and depolarized membrane potential. Moreover, by using Fis1 mutant flies, we found that the depletion of Fis1 dysregulated proteostasis and caused ubiquitinated protein accumulation within muscle fibers. Fis1 mutant flies also exhibited significantly reduced lifespans and mobility compared with controls. Our findings reveal that elevated ROS levels result in functional muscle decline, thus reducing proteostasis and ultimately leading to flight‐muscle degeneration. These results suggest that loss of Fis1 can lead to a decline in mitochondria function and accelerate muscle function decline.
2. MATERIALS AND METHODS
2.1. Drosophila rearing
Fly stocks were reared at 25 ± 1°C using commercial food (WellGenetics) on a 12‐h light/12‐h dark (LD 12:12) entrainment regime. All fly stocks were obtained from the Bloomington Drosophila Stock Center, Drosophila Genomics Resource Center, or were generated within our lab. Stock details are in key resource table (Table 1, Table 2).
TABLE 1.
Detailed information of reagents
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Anti‐rabbit porin | Abcam, Cambridge, UK | #14734 |
| Anti‐mouse GAPDH | GeneTex, Hsinchu, Taiwan | #627408 |
| Anti‐mouse tubulin | GeneTex, Hsinchu, Taiwan | #628802 |
| Anti‐rabbit GFP | Abcam, Cambridge, UK | #6556 |
| Anti‐ mouse HA | Biolegend, USA | #901514 |
| Anti‐mouse HA | Merck Millipore | #05‐904 |
| Anti‐rabbit Fis1 | This paper | N/A |
| Anti‐rabbit ubiquitin | Cell Signaling Technology, Danvers, MA, USA | #3933S |
| Anti‐Atg8a | Millipore | ABC974‐1 |
| Anti‐Ref(2)p | Abcam | ab178440 |
| Anti‐NDUFS3 | Abcam | ab110246 |
| Anti‐chchd3 | A gift from Dr. Chao Tong | N/A |
| HRP‐conjugated goat anti‐rabbit IgG polyclonal antibody | GeneTex, Hsinchu, Taiwan | GTX213110‐02 |
| HRP‐conjugated goat anti‐mouse IgG polyclonal antibody | GeneTex, Hsinchu, Taiwan | GTX213110‐01 |
| Anti‐FK2 | Merck Millipore | #04‐263 |
| Anti‐rabbit 4‐HNE | Abcam, Cambridge, UK | #46545 |
| Anti‐mouse ATP5a | Abcam, Cambridge, UK | #14748 |
| Alex Flour 488 anti‐mouse | Thermo Fisher Scientific | Cat# A‐11029 |
| Alex Flour 647 anti‐rabbit | Thermo Fisher Scientific | Cat # A32733 |
| Chemicals, peptides, and recombinant proteins | ||
| VectaShield Antifade Mounting Medium | Vector Laboratories, Burlingame, CA, USA | # HV‐93952‐25 |
| Phalloidin | Sigma‐Aldrich, St. Louis, MO, USA | P1951 |
| Critical commercial assays | ||
| MitoSOX™ | Thermo Fisher Scientific | Cat# M36008 |
| MitoTracker® Green | Thermo Fisher Scientific | Cat# M7514 |
| JC‐1 Dye | Thermo Fisher Scientific | #T3168 |
| XCell™ SureLock™ Mini‐Cell | Thermo Fisher Scientific | #EI0001 |
| NativePAGE™ Running Buffer Kit | Thermo Fisher Scientific | #BN2007 |
| NativePAGE™ Sample Prep Kit | Thermo Fisher Scientific | #BN2008 |
| Colloidal Blue Staining Kit | Thermo Fisher Scientific | #LC6025 |
| NativeMark™ Unstained Protein Standard | Thermo Fisher Scientific | #LC0725 |
| ATP Bioluminescence Assay kit CLS II | Roche | #Cat 1699695 |
| Digitonin | Sigma‐Aldrich, St. Louis, MO, USA | #D141 |
| Experimental models: organisms/strains | ||
| Mef2‐GAL4 | Bloomington Drosophila Stock Center | #50742 |
| white 1118 | Bloomington Drosophila Stock Center | #3605 |
| Fis1 MI10520 | Bloomington Drosophila Stock Center | #55496 |
| gstD‐GFP | (Sykiotis et al., 2008) | N/A |
| gstD‐GFP‐Fis1mutant | N/A | N/A |
| UAS‐miR‐LL | N/A | N/A |
| UAS‐miR‐LL | N/A | N/A |
| UAS‐attB‐LL‐3xHA | N/A | N/A |
| UAS‐attB‐LS‐3xHA | N/A | N/A |
| Oligonucleotides | ||
| Rp49_Q‐PCR_F | N/A | TAAGCTGTCGCACAAATGGC |
| Rp49_Q‐PCR_R | N/A | GCACCAGGAACTTCTTGAAT |
| Fis1‐C/E_Q‐PCR‐F | N/A | ATCACGAACTTGAGTTGGATGGTG |
| Fis1‐C/E_Q‐PCR‐R | N/A | GTACCGACTTCGGACCAGAC |
| Fis1‐A/D_Q‐PCR‐F | N/A | TTGTATTTGCTAACAACATTTGCC |
| Fis1‐A/D_Q‐PCR‐R | N/A | AGAATGCGTACTTCTAAATCTTC |
| Fis1‐F/G_Q‐PCR‐F | N/A | TTCTTCTCAATTCTAATGACACAAT |
| Fis1‐F/G_Q‐PCR‐R | N/A | ATAGTCACGCCTTCCATCTG |
| nuc‐tub Q‐PCR F1 | N/A | TATAAGTAAAGGCAGCAGGGAGAC |
| nuc‐tub Q‐PCR R1 | N/A | ATCTGGGTACTCTTCCTCTCCATC |
| mt‐ND5 Q‐PCR F1 | N/A | GAAGTAAAGCTACATCCCCAATTCG |
| mt‐ND5 Q‐PCR R1 | N/A | GGTGAGATGGTTTAGGACTTGTTTC |
| mt‐CytB Q‐PCR‐F1 | N/A | TAGTGTTAATCATATTTGTCGAGACGTT |
| mt‐CytB Q‐PCR‐R1 | N/A | ATATGAACCGTAATAAATTCCTCGTCC |
| mt‐CoI Q‐PCR‐F1 | N/A | ATTAGGTGCTCCTGATATAGC |
| mt‐CoI Q‐PCR‐R1 | N/A | CAATTCCAGCGGATAGAGGTGG |
| Recombinant DNA | ||
| Fis1‐miR‐LL‐1 | This paper | GGCAGCTTACTTAAACTTAATCACAGCCTTTAATGTAACGGGAGAAATAGTAACGTCATAAGTTAATATACCATATC |
| Fis1‐miR‐LS‐2 | This paper | AATAATGATGTTAGGCACTTTAGGTACAACACGTTTTTAACCGACAATGTAGATATGGTATATTAACTTACATT |
| Mir6.1_5’EcoRI/BglII | This paper | GGCGAATTCCGCCAGATCTTTTAAAGTCCACAACTCATCAAGGAAAATGAAAGTCAAAGTTGGCAGCTTACTTAAACTTA |
| Mir6.1_3′NotI/BamHI | This paper | GGCCGCGGCCGCACGGATCCAAAACGGCATGGTTATTCGTGTGCCAAAAAAAAAAAAAATTAAATAATGATGTTAGGCAC |
| pUAST‐attB‐3xHA fusion Fis1‐CDS_F | This paper | AGGGAATTGGGAATTCATGGAGGATCTTTTAAACGAAGTTGTACCA |
| pUAST‐attB‐3xHA fusion Fis1‐LL‐CDS_R | This paper | CGTATGGGTACTCGAGTTTCTCCCGTTTTTGTTTATTTCTAGCCAT |
| pUAST‐attB‐3xHA fusion Fis1‐LS‐CDS_R | This paper | CGTATGGGTACTCGAGCTTTCTAGCCATAGCAATGCCAAGTCCTAA |
TABLE 2.
Abbreviation list
| Abbreviation | Name |
|---|---|
| 4HNE | 4‐Hydroxynonenal |
| AD | Alzheimer's disease |
| AGC | Automated gain control |
| Atg8a | Autophagy‐related protein 8a |
| ATP5a | ATP synthase alpha‐subunit |
| BN‐PAGE | Blue Native‐polyacrylamide gel electrophoresis |
| Chchd3 | Coiled‐Coil‐Helix‐Coiled‐Coil‐Helix Domain Containing 3 |
| Dnm1 | Dynamin 1 |
| Drp1 | Dynamin‐related protein 1 |
| DSM | Drosophila Schneider's Medium |
| ETC | Electron transport chain |
| FCCP | Carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone |
| Fis1 | Fission, mitochondrial 1 |
| Fis1‐LL | Fission, mitochondrial 1‐Long form long |
| Fis1‐LS | Fission, mitochondrial 1‐Long form short |
| Fis1‐SL | Fission, mitochondrial 1‐Short form long |
| Fis1‐SS | Fission, mitochondrial 1‐Short form short |
| GAPDH | Glyceraldehyde‐3‐phosphate dehydrogenase |
| gstD | Glutathione S transferase D1 |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LC‐MS/MS | Liquid chromatography‐tandem mass spectrometry |
| Marf | Mitochondrial associated regulatory factor |
| Mef2 | Myocyte enhancer factor‐2 |
| Mfn | Mitofusin |
| NDUFS3 | NADH:Ubiquinone Oxidoreductase Core Subunit S3 |
| OPA1 | Optic atrophy type 1 |
| OsO4 | Osmium tetroxide |
| PBS | Phosphate‐Buffered Saline |
| PBST | 1X Phosphate‐Buffered Saline containing 0.1% Triton X‐100 |
| PINK1 | PTEN‐induced kinase 1 |
| RCR | Respiratory control ratios |
| ROS | Reactive oxygen species |
| TPR | Tetratricopeptide repeat |
2.2. Lifespan assay
Control w 1118 and Fis1 mutant flies were collected within 24 h of eclosion and then maintained at 25°C in vials containing commercial fly food under an LD 12:12 cycle. Flies were provided fresh food every 3–4 days. Each vial contained 25 male or non‐virgin female flies, and different sexes and genotypes were housed separately. The sex ratio was maintained at 1:1 for experiments. Flies were counted at each transfer until all had died.
2.3. Behavioral assays
2.3.1. Climbing
Flies were maintained at 25°C in vials containing standard fly food. Each experimental repetition used 10–20 wild‐type or mutant flies. Flies were anesthetized using CO2 before being placed in vials at least 24 h prior to the start of each assay to reduce potential side effects. Each tube contained a maximum of 25 flies of the same sex.
Flies were then loaded into the first tube of a countercurrent apparatus without CO2. The flies were tapped down to the bottom of the tube and then allowed to climb into the top receiver tube for 30 s. Flies that climbed into the top tube were then forced into a second tube. This was repeated five times per assay. The distribution of flies between the tubes was counted each time to calculate the distribution ratio and performance index (PI). The tubes were scored from 0 to 5, left to right.
The distribution ratio was defined as:
The PI was defined as:
where i is the score of each tube.
2.3.2. Flight duration
Flies were fully anesthetized on ice and then gently placed dorsal side up on a cold Petri dish. The tip of a tether (copper wire) was gently dipped into wood glue before being placed onto the fly's head. The angle between the tether and the fly was approximately 30°, and care was taken to avoid disturbing the wing vibration. Fixed flies were then placed in a constant‐temperature environment (28°C) for 1 h in groups of 10 to allow for full recovery from the ice anesthesia. Next, fixed flies were set in the same orientation and puffed gently with air before immediately being filmed with a camera for 120 s. The total recorded flying time for each fly was estimated by counting the total time each individual spent during video playback. A minimum of 30 flies were tested per genotype in batches of 5–10.
2.4. Western blot
Protein was extracted from the thoraxes of 5–7‐day‐old male flies and was then homogenized in 1× RIPA buffer (20 mM Tris–HCl; pH 7.5, 150 mM NaCl; 1 mM Na2 EDTA; 1 mM EGTA; 1% NP‐40; 1% sodium deoxycholate; 2.5 mM sodium pyrophosphate; 1 mM Na3VO4; 1 mM β‐glycerophosphate), 10% protease inhibitor, and 4% phosphatase inhibitor. Samples were then centrifuged at 16,100 g for 10 min at 4°C and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (PAGE). Next, proteins were transferred to a PVDF membrane, which was probed with the following antibodies: anti‐GFP (1:10,000; Abcam), anti‐HA (1:10,000; Biolegend), anti‐Fis1 (1:10,000), anti‐GAPDH (1:10,000; GeneTex), anti‐tubulin (1:10,000; GeneTex), porin (1:10,000; Abcam), anti‐ubiquitin (1:10,000; Cell Signaling Technology), anti‐ATP5a (1:10,000; Abcam), anti‐NDUFS3 (1:10,000; Abcam), and anti‐Chchd3 (1:10,000; gifted by Dr. Chao Tong). Membranes were then incubated with mouse or rabbit peroxidase‐conjugated secondary antibody. Blots were detected using ECL Western Blotting Substrate (Merck Millipore), and images were analyzed using the ImageJ software package.
2.5. Adult muscle dissection and immunofluorescence
The thoracic muscle of male flies was dissected in phosphate‐buffered saline (PBS 1.37 M NaCl, 27 mM KCl, 4 mM Na2HPO4, 17.6 mM KH2PO4; pH 7.4). First, an incision was made between the fly's legs, and then, half of the thorax was washed in PBS buffer. The thoracic muscles were then fixed for 30 min in 4% paraformaldehyde (Sigma‐Aldrich)–1× PBS at room temperature (25°C). After fixation, the supernatant was removed and 0.1% PBST (0.1% triton in 1× PBS) was used to wash the muscle tissue three times for 10 min each time. Muscle tissue was blocked with 2% bovine serum albumin (BSA)/0.1% PBST for 2 h at room temperature. The solution was then removed, and the tissues were washed three times in 0.1% PBST for 10 min. The tissue was then incubated with the respective primary antibody, which was diluted using 2% BSA/0.1% PBST overnight at 4℃. Muscle was probed with the following antibodies: anti‐FK2 (Merck Millipore), anti‐HA (1:400; Abcam), and anti‐ATP5a (1:400; Abcam). The next day, the muscle tissue was washed three times for 10 min in 0.1% PBST. The secondary antibody and phalloidin (Sigma‐Aldrich) were then added to the muscle tissues, which were kept at room temperature (25°C) for 2–4 h. The Alexa Fluor 488 anti‐mouse (Thermo Fisher Scientific) and Alexa Fluor 647 anti‐rabbit (Thermo Fisher Scientific) secondary antibodies were used. Subsequent to this, the samples were washed three times in 0.1% PBST and mounted in VectaShield antifade mounting medium (Vector Laboratories). Images were captured using a laser confocal microscope (Leica SP5) with a hybrid detector (HyD). Images were analyzed and quantified using ImageJ.
2.6. JC‐1 staining
Adult flight muscles were dissected in Schneider's medium (DSM) and then incubated in 2 µM JC‐1 solution (diluted in DSM) for 45 min at room temperature. The muscles were then immediately mounted in DSM and imaged using the Leica SP5 laser confocal microscope. The intensity of fluorescent aggregates was quantified using ImageJ.
2.7. MitoSox staining
Adult flight muscles were dissected in DSM. Hemithoraxes were incubated in staining solution (5 μM MitoSox™ Red [M36008, Thermo Fisher Scientific] and 100 nM MitoTracker Green [M7514, Thermo Fisher Scientific]) for 12 min at room temperature. The hemithoraxes were washed twice with DSM for 30 s at room temperature. Samples were mounted in DSM solution. Slides were then imaged on the Leica SP5 laser confocal microscope using identical settings for each condition. MitoSox intensity was quantified using Image J.
2.8. Mitochondrial respiration analysis
Mitochondrial respiration rates were measured using an Oxytherm System (Hansatech) with magnetic stirring, and thermostatic control was maintained at 25°C. Isolated mitochondria (1 mg/ml) from D. melanogaster were incubated in 120 mM KCl, 5 mM Pi‐Tris, 3 mM HEPES, 1 mM EGTA, and 1 mM MgCl2 (pH 7.2). Oxygen consumption was calculated according to the slope of the registered graph and plotted as Ng atoms: O2 × min−1 × mg−1. Respiratory control ratios (RCRs; adenosine diphosphate [ADP]‐stimulated respiration over basal respiration) and maximal respiration (FCCP‐stimulated respiration over basal respiration) were calculated (Von Stockum et al., 2019).
2.9. Plasmid construction
2.9.1. pUAST‐attB_miR‐Fis1‐LL or miR‐LS vector
After alignment analysis of all of the Fis1 alternative splicing mRNA sequences, each highly specific sequence with 22 contiguous nucleotides was identified for miR‐based RNAi design and located within the 3′ UTR region between the Fis1‐LL (Fis1‐C type) and Fis1‐LS (Fis1‐E type) isoform RNA transcripts (S1B). The functional stem–loop structure of the artificial mir‐based RNAi_Fis1‐LL and Fis1‐LS miRNAs was generated through the first primer set_ Fis1‐miR‐LL or Fis1‐miR‐LS primers in a polymerase chain reaction (PCR). The functional miRNAs were extended and flanking sequences added with restriction enzyme sites by the second primer set_Mir6.1_5′EcoRI/BglII and Mir6.1_3′NotI/BamHI primers to obtain precursor Fis1‐miRNA units (Chen et al., 2007). The restriction enzyme double‐digested EcoRI/NotI‐Fis1‐miRs were integrated into the EcoRI and NotI site region of the pUAST‐attB vector (GenBank: EF362409.1) to generate the pUAST‐attB_miR‐Fis1‐LL and pUAST‐attB_miR‐Fis1‐LS plasmids.
2.9.2. pUAST‐attB_Fis1‐3xHA vector
Fis1‐CDS PCR fragments without stop codons were generated through a PCR of the cDNA pools from the D. melanogaster w 1118 strain by using a pUAST‐attB_3xHA fusion Fis1‐CDS primer set. Agarose gel‐purified PCR products of Fis1‐CDS fragments were cloned into the EcoRI/XhoI sites of the pUAST‐attB vector with a C‐terminal‐fused 3xHA tag by using an In‐Fusion HD Cloning Kit (Clontech) to get the pUAST‐attB_Fis1‐3xHA plasmid.
2.9.3. Quantitative real‐time PCR
Total RNA was isolated from fly thoraxes with TRI reagent (Sigma). RNA quantity was evaluated using a microvolume spectrophotometer NanoDrop ND‐2000 (Thermo Fisher). After DNase treatment, first‐strand cDNA synthesis was performed using the SuperScript™ III First‐Strand Synthesis System (Invitrogen). The synthesized cDNA was amplified using a PCR in 10 μl reaction mixtures by using the ViiA 7 Real‐Time PCR System (Applied Biosystems) and the KAPA SYBR® FAST quantitative real‐time PCR (qPCR) Master Mix (2×) Kit (Sigma) with the following procedures: hold stage 95℃ for 20 s; PCR stage 40 cycles of 95℃ for 1 s and 60℃ for 20 s; and melt curve stage 95℃ for 15 s and 60℃ for 1 min. Measurements were taken using method quantification in triplicates, and relative RNA levels were normalized to rp49 levels.
2.10. Colorimetric assay of citrate synthase activity
In accordance with the procedure described by Ping Wei and colleagues (Wei et al., 2020), 10 fly thoraxes were homogenized with 105 μl of extraction buffer (20 mM HEPES [pH = 7.2], 1 mM EDTA, and 0.1% Triton X‐100), with a 5‐μl aliquot for protein content measurement. We added 400 μl of extraction buffer to each sample to increase the total volume to 500 μl and gently mixed. For each reaction, 1 μl of the sample was added to 150 μl of the freshly prepared reaction solution (20 mM Tris–HCl [pH 8.0], 0.1 mM DTNB, 0.3 mM acetyl CoA, and 1 mM oxaloacetic acid). The absorbance (OD) was measured at 412 nm every 10 s for 4 min at 25°C. The reaction rate was calculated using the following equation:
Citrate synthase activity was calculated as follows:
2.11. Hematoxylin and eosin staining
The segment between the thorax and abdomen of 4‐week‐old male flies was fixed in Carnoy's solution (60% ethanol, 30% chloroform, 10% glacial acetic acid) overnight at 4°C. Individual flies were held by their wings with tweezers and glued into a proper orientation on a Petri dish. Five to ten flies of the same genotype were used per test. The following day, samples were embedded in 3% agarose, transferred into 70% ethanol and sent to the pathology core lab of the National Health Research Institutes (NHRI) for hematoxylin and eosin (H&E) staining.
2.12. Mitochondria isolation
Mitochondria were purified using a mitochondrial isolation kit (Invitrogen) in accordance with the manufacturer's protocol. In total, 20–100 flies were washed twice with PBS. The PBS was then discarded, and the tissue was ground using a Dounce homogenizer in 400 μl of BSA/Reagent A solution. In addition, 400 μl of Reagent C solution was added, and the tube was inverted several times for mixing (it was not vortexed). Samples were then centrifuged at 700 g for 10 min at 4°C. Next, the pellet was discarded and the supernatant was transferred to a new tube. Subsequently, the supernatant cytosol was centrifuged at 12,000 g for 15 min at 4°C. After centrifugation, the mitochondrial pellet was isolated from the cytosol, and 500 µl of wash buffer was added to the pellet, which was then suspended by pipetting. Subsequently, the suspension was centrifuged at 12,000 g for 5 min and the supernatant was discarded. The mitochondrial pellet was maintained on ice for subsequent experiments.
2.13. Proteomic analysis
Proteomic analysis was performed as previously described (Cheung, et al., 2017). Nanoscale liquid chromatography coupled to tandem mass spectrometry (nano LC‐MS/MS) was performed using a mass spectrometer (LTQ‐Orbitrap XL; Thermo Fisher) equipped with a nanoACQUITY UPLC system (Waters). Next, peptide mixtures were loaded onto a 2 cm × 180 μm capillary trap column and then separated in a 75 μm ID × 25‐cm‐long C18 BEH nanoACQUITY column at a flow rate of 300 nl/min, where the mobile phases were A (0.1% formic acid [FA]) and B (0.1% FA/80% acetonitrile [ACN]). A linear gradient of 10%–40% B over 90 min and 40%–85% B over 10 min was used. Mass spectra from full survey scans were acquired on the Orbitrap (m/z 300–1500). The resolution of the instrument was set to 60,000 at 400 m/z with an automated gain control (AGC) value of 106. The 10 most intense precursor ions were selected from the MS scan for subsequent collision‐induced dissociation MS/MS scan in an ion trap (AGC target = 7000). For each biological sample, duplicate nano LC‐MS/MS analyses were performed. Two technical replicates were performed for each biological replicate.
2.14. ATP concentration measurement
Adult thoraxes were homogenized in water (10 µl/thorax) and centrifuged at 700 g. The supernatant was then carefully transferred to new Eppendorf tubes, and nine volumes of 100 mM Tris and 4 mM EDTA (pH 7.55) at boiling temperature were added. The mixture was incubated for 2 min at 100°C and kept on ice after heating. From each volume, 20 μl was obtained to detect the protein concentration by using a Bio‐Rad protein dye assay. At least 50 μl of the remaining samples was then transferred to a 96‐well microplate. Luciferase reagent was added to the samples (sample‐to‐luciferase reagent ratio of 1:1), and the measurement was taken using a microplate reader (425‐155, Hidex Chameleon). The blank was subtracted, and raw sample data were normalized to respective protein concentrations to calculate the relative ATP concentration.
2.15. Blue native gel
Pellets of mitochondria isolated from adult flies of the indicated genotypes were suspended at 10 mg/ml in 1× native PAGE sample buffer (Invitrogen) supplemented with protease inhibitor mixture (Sigma) and 2% (w/v) digitonin (Invitrogen). Samples were then immediately centrifuged at 100,000 g for 25 min at 4°C. The supernatant was transferred to a new tube, and the native PAGE 5% G‐250 sample additive (Invitrogen) was added. Samples were quickly loaded into a blue native polyacrylamide 3%–12% gradient gel (Invitrogen). After this step, electrophoresis was run in cathode buffer in darkness for 20 min at 150 V, and the condition was then changed to cathode buffer in the light and run for approximately 2 h at 250 V and 4°C. After electrophoresis, gels were fixed in 50% methanol +10% acetic acid for 20 min at room temperature, stained using a Colloidal Blue staining kit (Invitrogen) overnight at room temperature, and destained with deionized water.
2.16. Electron microscopy
Heads, abdomens, legs, and wings were excised from 1‐week‐old male flies. The thoraxes were then cut in half and fixed in 0.1 M cacodylate buffer (Sigma; pH 7.2) with 1% glutaraldehyde and 4% paraformaldehyde at 4°C overnight. Tissues were washed three times (15 min each) in 0.1 M cacodylate buffer at 4°C. Tissues were postfixed in OsO4/0.1 M cacodylate buffer (pH 7.2) at 4°C for 1 h and washed three times (15 min each) in 0.1 M cacodylate buffer at room temperature (25°C). After washing, the tissues were dehydrated twice in the following increasing concentrations of EtOH: 30%, 50%, and 70% for 10 min; 85% for 20 min; and then 95% and 100% for 20 min at room temperature. Tissues were then soaked twice in propylene oxide (10 min each) at room temperature. Tissue infiltration was conducted at room temperature by using a propylene oxide mixture containing epoxy resin 3:1, 1:1, and 1:3 for 8 h per concentration. Samples were then soaked in resin three times (8 h each) at room temperature and embedded in silicon rubber molds or in gelatin capsules. Finally, tissues were polymerized at 60°C for 24–48 h and sent to Academia Sinica for further sample processing.
2.17. Statistical analysis
At least three independent experiments were performed to obtain all results. Values are presented throughout as means ± standard deviations. Statistical data were processed using the GraphPad Prism software and Excel. Analyses utilized either a two‐tailed unpaired Student's t test (quantification and comparisons of Western blotting, blue native gel, ATP levels, complex IV activity, 4HNE, uniquitin, ATG8a, muscle vacuole area, and percentage of central myonuclei datasets), Chi‐squared test (quantification of mitochondrial size), Mann–Whitney U test (quantification of RCR), ANOVA/Bonferroni's multiple‐comparison test (flight duration experiments), or log‐rank Mantel–Cox test (lifespan experiments). The KEGG pathway enrichment analysis was performed using the ConsensusPathDB Web tool (http://ConsensusPathDB.org). p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****) were all considered as significant.
3. RESULTS
3.1. Fis1 long form expression increased in the thorax during aging
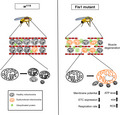
Mitochondrial Fis1 encodes the mitochondrial outer membrane protein Fis1. The Fis1 gene has been annotated with six transcripts and six polypeptides (four of which are unique) in D. melanogaster. In this study, we classified the four protein isoforms by the length of their amino acid sequence: short form short (SS), short form long (SL), long form short (LS), and long form long (LL; Figure 1a). All the polypeptides were found to include a single C‐terminal transmembrane domain with cytosolic α‐helixes at the N‐terminus. Unlike the short form group which only contained two α‐helixes, the long form group contained six α‐helixes, which consisted of two tetratricopeptide repeat (TPR) domains (Figure 1a).
FIGURE 1.
Fis1 LL levels increased with age. (a) Schematic of Fis1 protein isoform structures in a fly. The location of TPR motifs and transmembrane domains (TM) is indicated, and the black triangle indicates the location where the MiMIC cassette was inserted to generate the Fis1 mutant strain, Fis1 mutant. (b) qPCR analyses of Fis1 mRNA levels on weeks 1 and 4 and in w 1118 male flies. Each group was normalized by the total amount of Fis1 mRNA level of week‐old w 1118 males; n = 5 replicates with five flies per replicate. (c) Western blot detection and (d) quantification of Fis1 levels in thorax extracts from 4‐week‐old flies of w 1118, Fis1 mutant, mef2>UAS‐LL‐RNAi, and mef2>UAS‐LS‐RNAi. The two detected isoforms are indicated by arrows. The housekeeping protein GAPDH was used as an internal control, and the w 1118 and Fis1 mutant lines served as negative controls; n = 3 replicates with at least two flies per replicate, **p < 0.01; ***p < 0.001; unpaired t test. (e and f) Western blotting detection in flight‐muscle tissue revealed protein levels of endogenous Fis1 LL and LS (indicated by arrows) throughout the lifespan; n = 4 replicates with two flies per replicate, *p < 0.05; **p < 0.01; ***p < 0.001; unpaired t test
To identify the relationship between the expression of the six transcripts of Fis1 and muscle tissue aging, we examined mRNA levels within the thorax of 1‐, 4‐, and 7‐week‐old flies. qPCR analysis indicated that throughout the D. melanogaster lifespan, long form transcripts were more highly expressed than short form transcripts (Figures 1b and S1a). Long form transcript levels decreased between the first and fourth weeks, before slightly increasing between the fourth and seventh weeks (Figure 1b). To better understand which long forms were dominant at an endogenous protein level, we generated an antibody for Fis1 and examined w 1118 flies through Western blotting. By comparing controls with a MiMIC (Minos‐mediated integration cassette) insertion mutant strain (Fis1 MI10520, generated by Venken et al., 2011), we observed two protein isoforms (Figure 1c), which were identified as LL and LS through isoform‐specific transgenic targeting and knockdown by RNAi (Figures 1c,d and S1b). To further determine the age‐dependent changes in long form expression levels in D. melanogaster thoraxes, we examined specimens between 3 days and 7 weeks old. We found that LL expression levels increased with age, whereas LS expression levels remained constant throughout the aging process (Figure 1e,f).
Next, to determine the localization of LL and LS, we generated UAS‐Fis1‐LL and UAS‐Fis1‐LS lines with HA tags. Driven by muscle‐specific Gal4‐mef2, the two transgenic lines were dissected and the inner membrane marker, ATP5a, was discovered to be surrounded by an HA signal, indicating that both of these isoforms localized to the mitochondrial outer membrane (Figure S1c). To further confirm this distribution, we isolated mitochondria from wild‐type flies and examined protein expression through Western blotting. Although LL was localized only to mitochondria, LS was distributed in both the mitochondria and cytoplasm (Figure S1e).
These results suggest that Fis1 long forms are dominant throughout the Drosophila lifespan. The age‐dependent increase in LL suggests a link between Fis1 and aging.
3.2. Fis1 regulated mitochondrial morphology during aging
The decline in mitochondrial function is a hallmark of aging (López‐Otín et al., 2013). This decline is closely related to changes in the regulation of mitochondrial dynamics, and it may partially determine mitochondrial function (Chan, 2012; Schrepfer & Scorrano, 2016).
To investigate whether Fis1 mutation alters mitochondrial networks in an age‐dependent manner, we monitored mitochondrial morphology in the flight muscles of 1‐, 4‐, and 7‐week‐old Fis1 mutant flies through staining by using the inner membrane marker ATP5a. Loss of Fis1 in D. melanogaster led to enlarged mitochondria in young (1‐week‐old) flies compared with controls of the same age (Fisher's exact test; p = 0.0098; Figure 2a,d). Moreover, although mitochondria from control flies exhibited a progressive increase in size over time, we found no difference in mitochondrial size between 4‐week‐old mutant and control flies (p = 0.1196; Figure 2b,e), and by 7 weeks, mitochondria were significantly larger in controls than in Fis1 mutants (p = 0.0019; Figure 2c,f).
FIGURE 2.

Fis1 regulated mitochondrial morphology in muscle. (a–f) Immunostaining of indirect flight muscles from 1‐week‐old (a and a′), 4‐week‐old (b and b′), and 7‐week‐old (c and c′) w 1118 flies and 1‐week‐old (d and d′), 4‐week‐old (e and e′), and 7‐week‐old (f and f′) Fis1 mutant flies. Immunostaining revealed the mitochondrial inner membrane (green channel, anti‐ATP5a) and muscles (red channel, phalloidin/F‐actin); scale bar = 10 μm. (g) Left panel: quantification of mitochondrial area in muscle as shown in (a–b); n = 97, 106 (from left to right). **p < 0.01; Chi‐square. Central panel: quantification of mitochondrial area in muscle as shown in (c–d); n = 152, 169 from left to right. p = 0.1196, Chi‐square. Right panel: quantification of mitochondrial area in muscle as shown in (d–f); n = 134, 116 (from left to right); p = 0.0019, Chi‐square. (h–i) Representative electron micrographs of mitochondria in 1‐week‐old and 7‐week‐old w 1118 flight muscles; scale bar = 1 μm, p = 0.001. (j–k) Representative electron micrographs of mitochondria in 1‐week‐old and 7‐week‐old Fis1 mutant flight muscles; scale bar = 1 μm. p = 0.0198. (l) Quantification of mitochondria with incomplete inner membrane in (h–k); n = 3, every replicate contained one fly. *p < 0.05; **p < 0.01. p = 0.001, 0.0097, and 0.0198 from left to right comparison; unpaired Student's t test
Next, we examined the mitochondrial ultrastructure of young and aged Fis1 mutant flies through electron microscopy. We found that the percentage of mitochondria with incomplete cristae increased with age in w 1118 flies (Figure 2h,i,l). Fis1 mutant flies possessed larger numbers of impaired mitochondria early in life than did controls, and significantly more impaired mitochondria were observed in later life stages (Figure 2j–l).
These results suggest that loss of Fis1 increases the incidence of mitochondria with collapsed inner membranes. Because of the appearance of enlarged mitochondria and incomplete cristae, we examined the expression of proteins involved in mitochondrial biogenesis, fission, and fusion in Fis1 mutants. We investigated the expression of ATP5a (a subunit of ATP synthase), NDUFS3 (a subunit of complex I), porin (a mitochondrial outer membrane component for protein import), and ChChd3 (an inner membrane protein essential for maintaining cristae integrity). ATP5a, ChChd3, and porin exhibited no significant change, whereas NDUFS3 exhibited a significant reduction in expression compared with w1118 flies (Figure S2a,b; t test, p > 0.05 and p < 0.05, respectively). No significant changes in expression level of the fission‐induced protein Drpl or the fusion‐induced protein Marf in Fis1 mutants compared with w1118 flies were observed (Figure S3; t test, p > 0.05).
Collectively, the aforementioned data demonstrate that Fis1 regulates mitochondrial morphology without the involvement of mitochondrial dynamic proteins, and loss of Fis1 impairs the inner membrane structure.
3.3. Ablation of Fis1 impaired mitochondrial inner membrane integrity, leading to defects in mitochondrial function
Mitochondrial dynamics regulate mitochondrial quality and are closely related to mitochondrial function (Lo Verso et al., 2014; Twig et al., 2008; Vainshtein et al., 2014).
According to our analyses, loss of Fis1 increased the number of mitochondria with incomplete cristae (Figure 2j,k), suggesting a decline in the function of these mitochondria. The main function of mitochondria is to produce ATP through oxidative phosphorylation. To produce ATP, electrons are transferred within the ETC to generate a proton gradient for ATP production. Thus, the integrity of the ETC is crucial for mitochondrial function.
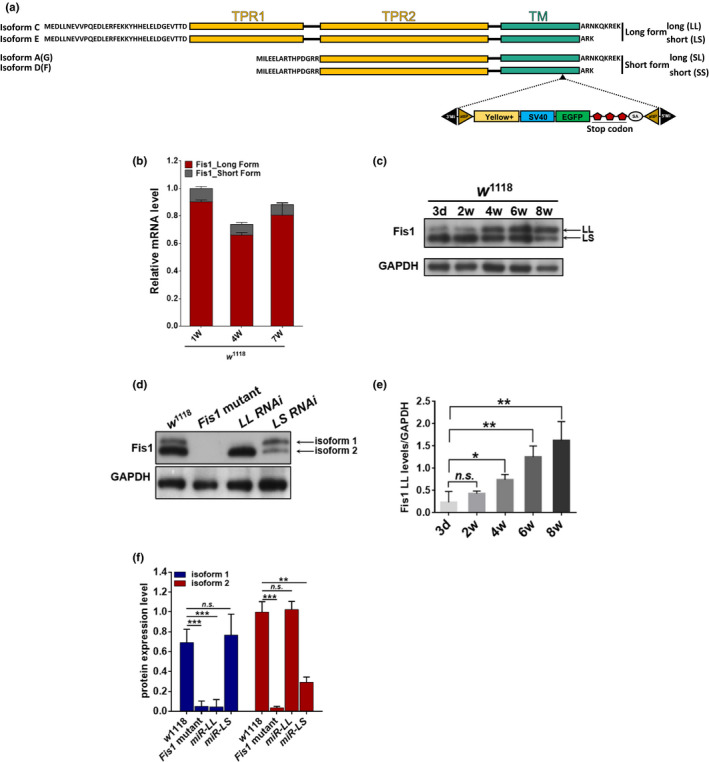
We examined mitochondrial respiration by measuring both ADP‐stimulated (RCR) and FCCP‐stimulated (maximal) respiration. Loss of Fis1 resulted in a lower RCR and maximal respiration activity (Figure 3a,b). This result supports the hypothesis that loss of Fis1 diminishes respiration chain activity, which may be caused by damage to ETC complexes.
FIGURE 3.
Ablation of Fis1 impaired mitochondrial inner membrane integrity and led to functional defects in mitochondria. (a–b) RCRs (ADP‐stimulated respiration over basal respiration) and maximal respiration (FCCP‐stimulated over basal respiration) calculations in 6‐week‐old w1118 and Fis1 mutant flies;*p < 0.05, **p < 0.01; Mann–Whitney U test. (c) KEGG pathway enrichment analysis of significantly differentially expressed proteins (fold‐change >1.5 or <0.67). Enriched terms are presented with the number of hit proteins (green) or p values (pink), respectively. The heatmap depicts the normalized intensities of proteins involved in oxidative phosphorylation function; n = 3, every replicate contained at least 100 thoraxes. (d) Heatmap depicting hierarchical mitochondrial proteomics of w 1118 (left) and Fis1 mutant flies (right). Red and blue represent the protein intensity from high to low; p values are shown as indicated. (e) BN gel of mitochondrial extraction of thoraxes from 4‐week‐old w 1118 and Fis1 mutant flies. Each replicate contained at least 100 thoraxes. (f) Quantification of complex IV level as shown in (e); n = 3, ****p < 0.0001; Student's t test. (h) Activity of citrate synthase in 2‐week‐old w1118 and Fis1 mutant flies; n = 3; each replicate contained ten flies; *p < 0.05; Student's t test. (h) Activity of complex IV was examined in 2‐week‐old w1118 and Fis1 mutant flies; n = 3; each replicate contained ten flies; *p < 0.05; Student's t test. (i) Immunostaining of indirect flight muscle stained with JC‐1 probe. JC‐1 monomers (green) proportionally accumulated in mitochondria according to the mitochondrial membrane potential. JC‐1 dye tended to aggregate in healthy mitochondria and form JC‐1 aggregates (red). Fis1 mutant flies had decreased JC‐1 aggregates (red) compared with wild‐type flies during aging. Scale bar: 10 μm. (j) Quantification of JC‐1 aggregate in muscles as shown in (i); n = 4, each replicate contained two flies. *p < 0.05 **p < 0.01; ***p < 0.001; unpaired t test
To examine protein levels and the integrity of ETC complexes, we isolated mitochondria specifically from the thorax and subjected the digested proteins to LC‐MS/MS (Figure S4). Among all 13 proteins with upregulated or downregulated expression in Fis1 mutant flies, those related to oxidative phosphorylation were the most abundant (Figure 3c). All the oxidative phosphorylation‐related proteins were downregulated and contained four subunits of complex IV (Figure 3d). To further determine the content of ETC complexes, we isolated mitochondria from fly thoraxes and examined them using blue native PAGE (BN‐PAGE). Compared with w 1118 flies, complex IV from the Fis1 mutant flies appeared to decrease (Figure 3e, quantified in 3f; unpaired t test, p < 0.0001). The BN‐PAGE results confirmed the decreased expression level of complex IV observed in proteomic data. Because ETC complexes are responsible for producing energy, we tested ATP concentration at different ages and found that Fis1 depletion resulted in decreased ATP concentration throughout the fly lifespan (unpaired t test; p < 0.0001 [1w], p = 0.0053 [4w], p = 0.0169 [7w]; Figure 3g). We also examined the activity of complex IV, which is a crucial catalytic enzyme in the TCA cycle. Fis1 mutant flies had lower complex IV activity compared with w 1118 flies, indicating that the TCA cycle is impaired in Fis1 mutants (Figure 3h). Furthermore, we tested the mitochondrial membrane potential by using JC‐1 and observed a reduced potential in Fis1 mutant flight muscles (Figure 3i, quantified in 3j; p = 0.0004 [1w], p = 0.0023 [7w]). Thus, Fis1 loss results in the ablation of mitochondrial function in muscle tissues.
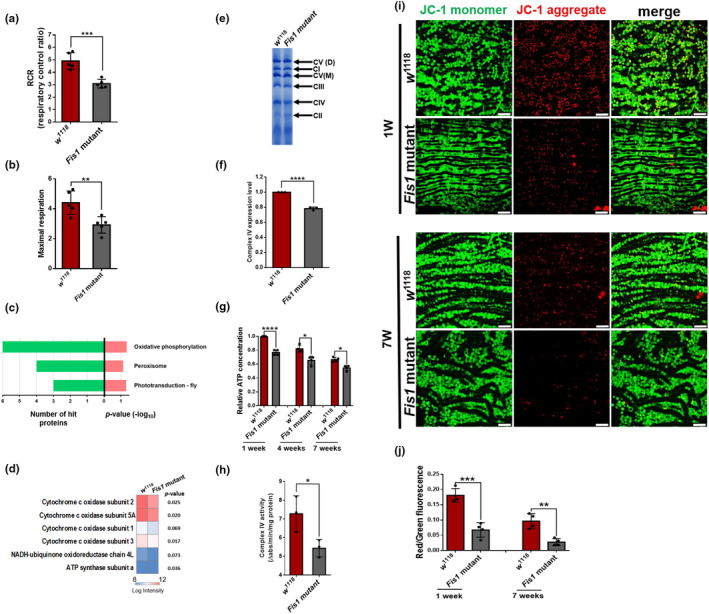
3.4. Depletion of Fis1 increases ROS production
Mitochondria are the main source of ROS (Ábrigo et al., 2018; Balaban et al., 2005; Cadenas & Davies, 2000; Starkov & Fiskum, 2003). A study reported that the mutation of complex IV led to ROS leakage in aged mice (Reichart et al., 2019). We assumed that a lack of Fis1 would increase ROS levels in Fis1 mutant flies; therefore, we expressed the ROS reporter gstD with a GFP tag in the fly thorax (Sykiotis & Bohmann, 2008; Figure 4a), which allowed us to monitor the basal level of GFP in flies. In adult flies carrying the gstD–GFP reporter, GFP activity can be used to monitor the production of ROS at basal conditions after subtracting the baseline GFP signal from the abdomen (Figure 4b). By using Western blotting, we found that aged flies exhibited an increase in GFP levels. In addition, Fis1 mutant flies produced a greater amount of ROS than did wild‐type flies at all ages tested (Figure 4c), suggesting that the unusually high level of ROS resulted from loss of Fis1. Next, to confirm whether the elevated ROS originated from mitochondria, we stained 7‐week‐old flight muscles with mitochondrial ROS indicator (mitoSOX) and found significantly increased mitochondrial ROS in Fis1 mutant flies (Figure 4d, quantified in 4e; p < 0.001). Excess ROS provokes the oxidation of phospholipids in mitochondrial membranes and forms lipid peroxidative products (Yin et al., 2011). Among these products, 4‐hydroxy‐2‐nonenals (4‐HNE) is one of the most bioactive and representative lipid alkenals (Benedetti et al., 1980). When stained using the 4‐HNE antibody, Fis1 mutant flies also exhibited increased 4‐HNE signals within muscle mitochondria, indicating increased oxidative damage (Figure 4f,g; quantification in 4g; p < 0.0001). Specifically, by using mef2>Fis1‐LL RNAi and ef2>Fis1‐LS RNAi flies, we verified that the knockdown of Fis1‐LL or Fis1‐LS resulted in increased oxidative damage in the muscles of mutant flies (Figure 4f–i, quantified in 4j). We also found that the ectopic expression of Fis1‐LL or Fis1‐LS suppressed the upregulation of 4‐HNE in Fis1 mutants (Figure S5d–g). Collectively, loss of both long forms of Fis1 increased ROS levels and the amount of oxidative lipids in the thoraxes of flies, which consequently increased oxidative damage.
FIGURE 4.
Depletion of Fis1 increases ROS production and oxidative damages. (a) Structure of the gstD–GFP transgene reporter. GFP is produced when gstD is induced by ROS. (b) Expression of the transgenic reporter construct gstD–GFP was visualized in 1‐, 4‐, and 7‐week‐old flies (from left to right) through fluorescence stereomicroscopy. Transcription of the gstD enhancer was potently induced in the gut and in other tissues using oxidants. (c) Western blot analysis of GFP expression levels in thoracic tissues of homozygous gstD–GFP reporter alone or gstD–GFP–Fis1 recombined reporter upon various ages. Each group of GFP intensity was compared with the GFP intensity of 3‐day‐old gstD–GFP reporter. The relative intensity was calculated and labeled. (d) Staining of indirect flight muscles from 7‐week‐old w 1118 and Fis1 mutant males, showing mitochondria (green channel, Mitotracker green staining) and levels of superoxide radicals (red channel, staining with MitoSOX reagent); scale bar = 10 μm. (e) Quantified ratio of mitoSOX signal to mitochondrial area as depicted in (d); unpaired t test, n = 4, each replicate contained two flies. **p < 0.01; ***p < 0.001. (f–i) Immunostaining of indirect flight muscles from 4‐week‐old Mef2 control, Fis1 mutant, mef2>miR‐LL, and mef2>miR‐LS flies, showing 4‐HNE aggregates (green channel) and phalloidin (red channel); scale bar = 10 μm. (j) Quantification of accumulated 4‐HNE aggregates in muscle as depicted in (f–i). The area of aggregated 4‐HNE was normalized by the total mitochondria area. Unpaired t test, n = 3, each replicate contained two flies. ***p < 0.001; unpaired t test
3.5. Depletion of Fis1 leads to remarkable ubiquitinated protein aggregation during aging
Accumulated ubiquitinated protein in muscle is representative of aging, and oxidative stress is known to play a role in regulating proteostasis (Fernando et al., 2019).
However, the role of increased ROS in disrupting proteolytic homeostasis in muscle remains unknown. In this study, we examined whether mitochondrial ROS, which is caused by Fis1 loss, causes an imbalance in proteostasis in muscle.
We first examined the accumulation of ubiquitinated protein within flight muscles through immunofluorescence microscopy and found a predominant accumulation of ubiquitinated protein in week‐old Fis1 mutant flies compared with w 1118 flies (Figure 5a,d and a′,d′). Furthermore, Fis1 loss not only caused premature ubiquitin accumulation but also highly accelerated the accumulation (Figure 5a–f, quantified in 5g). These results were also confirmed by Western blotting (Figure 5h), suggesting that Fis1 participates in the protein degradation system throughout the fly lifespan and in the accumulation of ubiquitinated protein in early age. Therefore, loss of Fis1 accelerated the accumulation of ubiquitinated proteins in muscle during aging.
FIGURE 5.
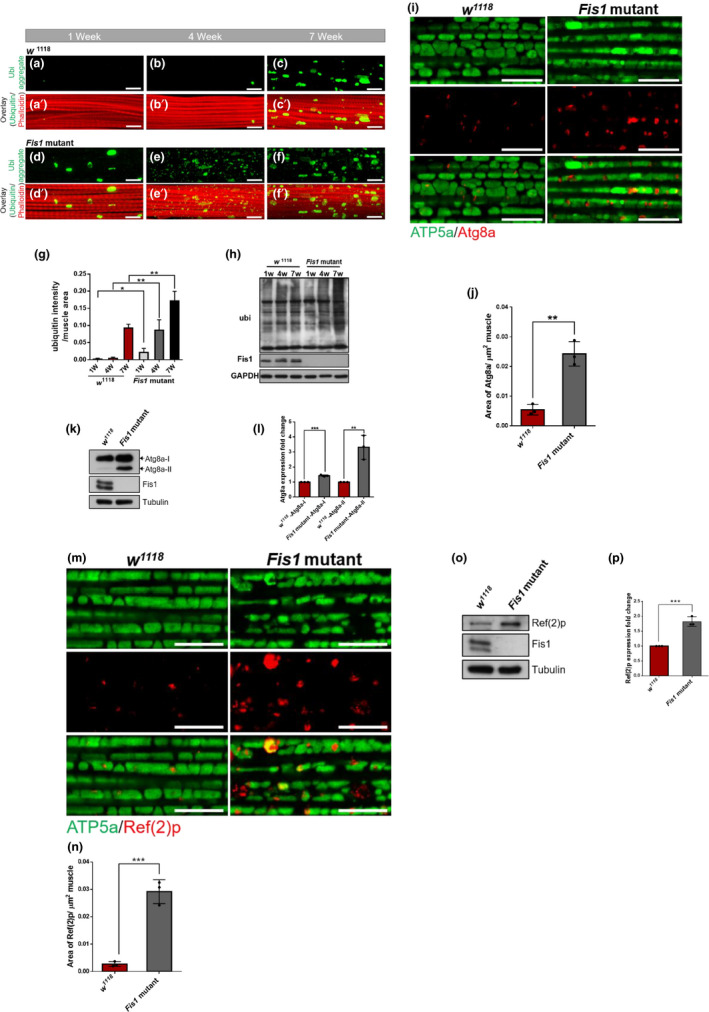
Protein degradation pathways were impaired with Fis1 inhibition in adult muscles. (a–c′) Immunostaining of indirect flight muscles from 1‐week‐old (a and a′), 4‐week‐old (b and b′), and 7‐week‐old (c and c′) w 1118 flies, showing polyubiquitinated aggregates (green channel, anti‐FK2) and muscles (red channel, phalloidin/F‐actin); scale bar = 10 μm. (d–f′) Immunostaining of indirect flight muscles from 1‐week‐old (d and d′), 4‐week‐old (e and e′), and 7‐week‐old (f and f′) Fis1 mutant flies, showing polyubiquitinated aggregates (green channel, anti‐FK2) and muscles (red channel, phalloidin/F‐actin); scale bar = 10 μm. (g) Quantification of polyubiquitin aggregates in muscle as depicted in (a–f); n = 3, each replicate contained two flies. *p < 0.05; **p < 0.01; unpaired t test. (h) Western blot detection and densitometry of total ubiquitin‐conjugated proteins between w 1118 and Fis1 mutant flies between 1 and 7 weeks old. (i) Immunostaining of indirect flight muscles from 2‐week‐old w 1118 and Fis1 mutant flies, showing mitochondria (green channel, anti‐ATP5a) and Atg8a aggregates (red channel, anti‐Atg8a); scale bar = 10 μm. (j) Quantification of Atg8a aggregates as depicted in (i); n = 3, each replicate contained two flies. **p < 0.01; unpaired t test. (k) Atg8a‐II was accumulated in 2‐week‐old Fis1 mutant. (l) Quantification of Atg8a‐II as depicted in (K); n = 3, each replicate contained two flies. **p < 0.01; unpaired t test. (m) Immunostaining of indirect flight muscles from 2‐week‐old w 1118 and Fis1 mutant flies, showing mitochondria (green channel, anti‐ATP5a) and Ref(2)p aggregates (red channel, anti‐Ref(2)p); scale bar = 10 μm. (n) Quantification of Ref(2)p aggregates as shown in (m); n = 3, each replicate contained two flies. ***p < 0.001; unpaired t test. (o) Ref(2)p was accumulated in 2‐week‐old Fis1 mutants. (p) Quantification of Ref(2)p as shown in (O); n = 3, each replicate contained two flies. ***p < 0.001; unpaired t test
Studies have described how the muscle‐specific inhibited mitochondrial fission protein Drp1 leads to a reduction of mitochondrial function and elevation of impaired autophagy markers in mammal muscle, which results in an atrophy phenotype (Dulac et al., 2020; Favaro et al., 2019).
To further clarify whether a lack of Fis1 impaired autophagy within the muscle cells in flies, we probed muscles with the steady‐state autophagy marker Atg8a to visualize the endogenous autophagosome. We found that 2‐week‐old Fis1 mutant flies accumulated Atg8a protein (Figure 5i, quantified in 5j). The increased aggregation of Atg8a could also be observed in Western blotting (Figure 5k, quantified in 5l). Furthermore, similar to the accumulation of ubiquitinated protein, the level of aggregated Ref(2)p in 2‐week‐old mutant flies significantly increased compared with w 1118 flies (Figure 5m, quantified in 5n). The increased aggregation of Ref(2)p could also be observed in Western blotting (Figure 5o, quantified in 5p). These data indicate that loss of Fis1 impairs the proteostasis mechanism in fly muscles from a young age.
3.6. Fis1 deletion causes myopathy in indirect flight muscle and led to exercise intolerance
Changes in proteostasis can accelerate aging (Labbadia & Morimoto, 2015). Without an appropriate clearance system for damaged proteins, increased cytotoxicity may occur and the lifespan may be shortened (Kevei & Hoppe, 2014).
We found that Fis1 mutant flies exhibited a reduced median lifespan (37 days) by almost one‐third compared with wild‐type flies (55 days; Figure 6a; p < 0.0001). Similarly, the use of mef2 gal4 to drive UAS‐Fis1 LL or LS RNAi targeting led to a shortened lifespan (Figure S6; p < 0.0001). Because age‐related diseases are often accompanied by impaired mobility, we tested the climbing ability and flight maintenance capability of both young and old flies. Both tests demonstrated that flies lacking Fis1 exhibited significantly decreased mobility at all ages, which worsened with age (Figure 6b,c).
FIGURE 6.
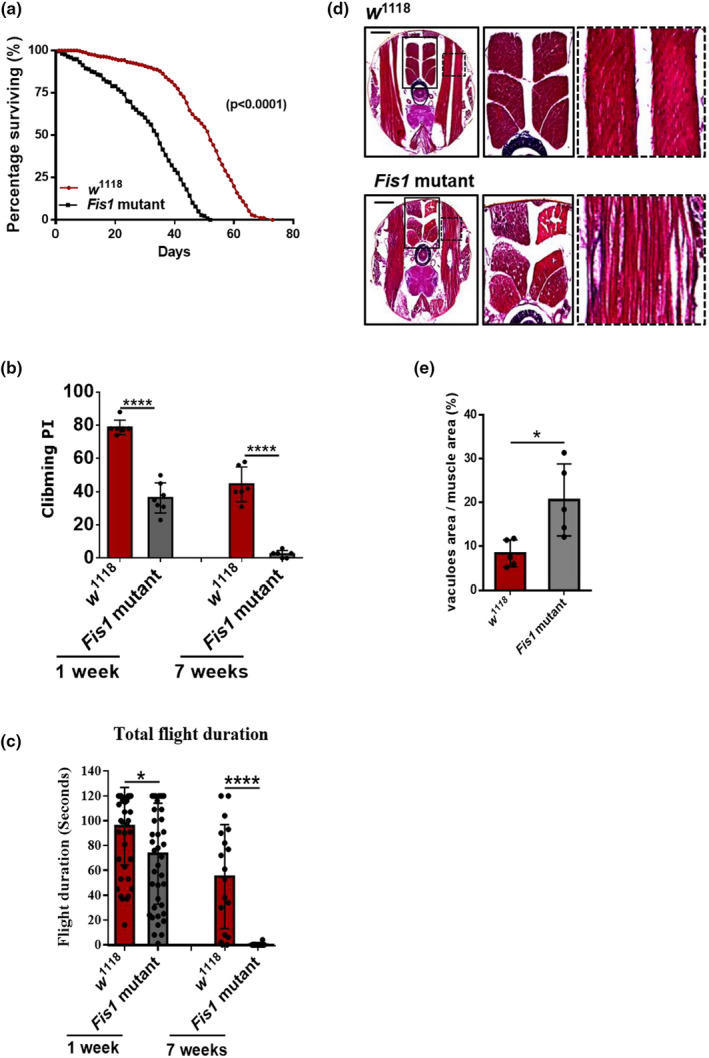
Fis1 deletion caused myopathy in indirect flight muscles and led to exercise intolerance. (a) The survival curve of w 1118 and Fis1 mutants indicates that Fis1 deletion resulted in lethality after eclosion. w 1118 flies, n = 347; Fis1 mutants, n = 219. ****p < 0.0001; log‐rank (Mantel–Cox) test. (b) Compounded climbing PI of 1 and 7‐week‐old w 1118 and mutant flies. The PIs of Fis1 mutants were analyzed and compared with those of Fis1 w 1118 flies; n = 6, each replicate contained at least 90 flies. ****p < 0.0001; Student's t test. (c) Total flight duration was significantly reduced in animals with knockdown of Fis1 compared with controls at 1 and 7 weeks old. From left to right, n = 47, 42, 18, 20. *p < 0.05; ****p < 0.0001; ANOVA/Bonferroni's multiple‐comparisons test. (d) H&E staining of flight muscle revealed that 4‐week‐old Fis1 mutant flies lost muscle fiber integrity compared with controls. (e) Quantified ratio of vacuole area to muscle area as shown in (d); n = 5, *p < 0.05; Student's t test
In neurodegenerative conditions such as AD and Parkinson's disease, proteostasis dysfunction leads to the aberrant accumulation of proteins, causing neuron death (Sweeney et al. 2017). This disease process can also be observed in muscles, leading to the degeneration of muscle cells (Fernando et al., 2019). The evidence of accumulated ubiquitin within muscles and declined mobility led us to investigate the possibility of muscle degeneration in Fis1 mutant flies. To test whether these mobility defects resulted from muscle degeneration, we performed H&E staining on Fis1 mutant flies and found severe muscle degeneration; specifically, the impaired indirect flight muscles were packed in a disorderly manner and vascular areas were visible (Figure 6d, quantified in 6e).
In addition, the number of centrally nucleated myofibers has been used as a marker to reflect previous cycles of muscle degeneration and regeneration (Dulac et al., 2020; Favaro et al., 2019; Sakellariou et al., 2011). Drp1 mutants exhibited accelerated muscle degeneration with the appearance of centrally localized myonuclei (Dulac et al., 2020; Favaro et al., 2019). To further examine Fis1‐induced muscle degeneration, we examined the location of myonuclei in Fis1 mutants.
By using phalloidin to indicate the muscle area and DAPI as a nucleus marker, we found a higher ratio of centrally located myonuclei in the aged control flies compared with the young control flies (Figure S7a,b, quantified in e). However, aged Fis1 mutant flies had a higher ratio of centrally nucleated myofibers than did the aged control flies (Figure S7c,d, quantified in e), indicating that loss of Fis1 accelerates muscle degeneration. Together, these findings indicate that loss of Fis1 causes an imbalance in mitochondrial proteostasis within muscle, affecting the health and lifespan of Drosophila.
4. DISCUSSION
Results from numerous model systems have suggested that aging is linked to alterations in the regulation of mitochondrial dynamics (Sharma et al., 2019). However, the underlying mechanisms that modulate these changes and the causal relationships between altered mitochondrial dynamics and age‐related health decline remain unclear. In this study, we addressed whether Fis1, an outer membrane fission factor in yeast, could modulate tissue and organismal aging in Drosophila. We first discovered that the mRNA of long form Fis1 transcripts was dominant throughout the Drosophila lifespan. In addition, we found that the protein expression levels of LL increased with age, whereas equivalent levels of LS had a mild effect.
We further found that loss of Fis1‐LL or Fis1‐LS in muscle resulted in elevated ROS levels (Figure 4f–j), and their ectopic expression led to the suppression of oxidative stress (Figure S5). These results suggest that both Fis1‐LL and Fis1‐LS have similar functions in influencing the production of ROS and oxidative lipids. Although LL and LS are similar in protein sequence and structure, their C‐terminal tails differ slightly; therefore, LL possesses greater similarity to human Fis1. Another study reported that the transmembrane domain, which is located at the C‐terminus, is crucial for human Fis1 to function as a regulator of mitochondrial dynamics (Yu et al., 2019). The differences in C‐terminal structure between LL and LS may guide them to different locations as well as cause them to have different functions.
Muscle proteomic results in humans indicated that the expression of the mitochondrial fission component dynamin 2 increases with age (Murgia et al., 2017). Fis1 and S‐nitrosylated Drp1 (SNO‐Drp1) significantly increased in brain tissues and skin fibroblasts in patients with AD (Wang et al., 2012).
Of the two major isoforms of Fis1 in D. melanogaster, LL possesses a greater similarity to human Fis1. The increased LL levels suggest that LL has potential for use as a biomarker of aging or as a therapeutic target against aging or age‐related diseases. Future work can focus on whether LL possesses specific functions in aging and whether these functions differ among other isoforms.
Inducing mitochondrial fission is known to rescue deterioration in PINK1 and Parkin mutant flies (Deng et al., 2008; Yang et al., 2008), supporting the hypothesis that mitochondrial fission can facilitate mitophagy (Mao & Klionsky, 2013; Twig et al., 2008). However, little is known about the relationships among mitochondrial dynamics, mitophagy, and aging. Fis1 is involved in recruiting Dnm1p (the yeast Drp1 homolog) to mitochondria through interaction with Mdv1p and Caf4p, and it is involved in promoting mitochondrial fission processes in yeast (Hoppins et al., 2007). Given the absence of Mdv1p and Caf4p in metazoans, the actual role of Fis1 in the mitochondrial fission process remains unclear (Hoppins et al., 2007; Okamoto & Shaw, 2005). Some studies have suggested that Fis1 participates in fission processes (James et al., 2003; Shen et al., 2014; Stojanovski et al., 2004) or inhibits fusion factors (Yu et al., 2019), whereas others have suggested that Fis1 is dispensable to fission processes or has only mild effects on mitochondrial morphology when mutated or overexpressed (Shen et al., 2014; Suzuki et al., 2003).
In this study, we examined the mitochondrial morphology of Fis1 mutant flies at different ages (1, 4, and 7 weeks) and found that loss of Fis1 led to enlarged mitochondria at young (1 week) and old (7 weeks) ages. However, the mutation of Fis1 had a minor effect on mitochondrial morphology at 4 weeks.
Loss of Fis1 increased the number of enlarged mitochondria with incomplete cristae during aging, suggesting that Fis1 regulates mitochondrial morphology in an age‐dependent manner. Furthermore, mitochondrial size and Fis1‐LL protein expression increased with age, and the ectopic expression of Fis1‐LL not only induced smaller mitochondria (Figure S1c,d) but also reduced oxidative damage (Figure S5a–c). We propose that Fis1 may have a protective function during aging; however, the increased expression of Fis1‐LL with age is insufficient for fully suppressing age‐related mitochondrial impairment. We believe that Fis1‐LL plays a role in maintaining proteostasis and preventing the overenlargement of mitochondria; therefore, loss of Fis1 should cause mitochondrial enlargement and incomplete cristae during aging (Figure 2). However, further investigations are required to clarify the detailed mechanism.
Mitochondrial dynamics (fission/fusion) and mitophagy are closely related mechanisms for maintaining a healthy mitochondrial pool and eliminating damaged mitochondria (Ashrafi & Schwarz, 2013; Lo Verso et al., 2014; Twig et al., 2008; Twig & Shirihai, 2011; Vainshtein et al., 2014). In this study, we found that Fis1 plays a protective role in proteostasis. Fis1 mutation compromised the integrity of the mitochondrial inner membrane in young flies and was accompanied by defects in complex IV. A study suggested that the mutation of complex IV in a mouse model reduced lifespans with increased ROS production as well as downregulation of Fis1 (Reichart et al., 2019). However, elevated ROS levels in aged w 1118 flies were also observed; this observation is crucial. Oxidative stress is defined as an imbalance between the production of ROS and the antioxidant defense systems (Luceri et al., 2018). The accumulating ROS attack DNA, induce DNA damage, and disrupt the antioxidant defense systems (Peng et al., 2014), causing age‐related aggregation of ROS in control strains. Our study suggests that loss of Fis1 produces an imbalance in oxidative stress through defects in complex IV, which potentially mediates mitochondrial ROS production.
Furthermore, defects within the ETC not only increase ROS levels but also downregulate ATP concentration. Our findings indicate that mitochondria in Fis1 mutant flies exhibit reduced membrane potentials, suggesting a decline in mitochondrial function, which occurs early in the aging process. Because this cumulative damage is toxic to cells, it is crucial to clear dysfunctional mitochondria. Fis1 was reported to regulate PINK1‐independent mitophagy in humans (Xian et al., 2019), and mutation of Fis1 was found to induce abnormal mitophagy in a mouse model (Zhang et al., 2019). We observed a dramatic accumulation of ubiquitinated protein aggregates in the muscles of young Fis1 mutant flies. These aggregates continuously accumulated with age, and eventually mutant muscle fibers were almost completely filled with protein aggregates. This accumulation is a striking phenotype of aging. Furthermore, an endogenous autophagic marker, Atg8a, aggregated during early life stages. Fis1 may thus be a key protein linking mitochondrial function and mitophagy during aging.
Several AD animal models and AD patient brains have exhibited evidence of reduced ATP concentration, diminished complex IV activity, and enhanced oxidative stress compared with controls (Hauptmann et al., 2009; Reddy, 2009; Xie et al., 2013). Fis1 has also been reported to be a potential biomarker of AD because its expression is increased in the peripheral blood lymphocytes of patients with AD (Wang et al., 2012). Moreover, increased levels of Drp1–Fis1 interaction, which promotes mitochondrial fission, have been discovered in fibroblasts derived from patients with AD (Joshi et al., 2018). A study also reported that the C9ORF72 gene, the mutation of which causes amyotrophic lateral sclerosis, has a strong and lethal genetic interaction with Fis1 (Chai et al., 2020). These studies implicate Fis1 as a causal factor of the aforementioned diseases. Finally, our findings reveal how loss of Fis1 can lead to a decline in mitochondria and eventual acceleration of the aging process.
CONFLICT OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
T.‐T.L., P.‐L.C., J.‐C.L., S.V.S., E.Z, and C.‐H.C. conceived and designed experiments; J.‐C.L. designed and assembled plasmids; T.‐T.L., P.‐L.C., J.‐C.L., Y.‐W.C., and S.V.S. performed experiments; T.‐T.L., P.‐L.C., Y.‐W.C., H.‐F.J., M.P.S., and C.‐H.C. analyzed data; and T.‐T.L., P.‐L.C., J.‐C.L., M.P.S., Y.‐W.C., J.‐M.Y., H.‐D.W., and C.‐H.C. wrote the paper.
Supporting information
Fig S1‐S7
ACKNOWLEDGMENTS
We would also like to thank Uni‐edit (www.uni‐edit.net) for editing and proofreading this manuscript. This study was supported by the NHRI (National Health Research Institutes, Taiwan; grant no. NHRI‐08A1‐MRGP12‐035; awarded to C.‐H.C.). Fly lines were supplied by the Bloomington Stock Center, the FlyORF Stock Center, or the Taiwan Fly Core. T.E.M. was supported by the IMB, Academia Sinica.
Lee and Chen equal contribution.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Ábrigo, J. , Elorza, A. A. , Riedel, C. A. , Vilos, C. , Simon, F. , Cabrera, D. , Estrada, L. , & Cabello‐Verrugio, C. (2018). Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxidative Medicine and Cellular Longevity, 2018, 2063179. 10.1155/2018/2063179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi, G. , & Schwarz, T. L. (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death & Differentiation, 20(1), 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban, R. S. , Nemoto, S. , & Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell, 120(4), 483–495. [DOI] [PubMed] [Google Scholar]
- Benedetti, A. , Comporti, M. , & Esterbauer, H. (1980). Identification of 4‐hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochimica Et Biophysica Acta (BBA) – Lipids and Lipid Metabolism, 620(2), 281–296. [DOI] [PubMed] [Google Scholar]
- Cadenas, E. , & Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biology and Medicine, 29(3–4), 222–230. [DOI] [PubMed] [Google Scholar]
- Chai, N. , Haney, M. S. , Couthouis, J. , Morgens, D. W. , Benjamin, A. , Wu, K. , Ousey, J. , Fang, S. , Finer, S. , Bassik, M. C. , & Gitler, A. D. (2020). Genome‐wide synthetic lethal CRISPR screen identifies FIS1 as a genetic interactor of ALS‐linked C9ORF72. Brain Research, 1728, 146601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, D. C. (2012). Fusion and fission: Interlinked processes critical for mitochondrial health. Annual Review of Genetics, 46, 265–287. [DOI] [PubMed] [Google Scholar]
- Chen, C. H. , Huang, H. , Ward, C. M. , Su, J. T. , Schaeffer, L. V. , Guo, M. , & Hay, B. A. (2007). A synthetic maternal‐effect selfish genetic element drives population replacement in Drosophila. Science, 316(5824), 597–600. [DOI] [PubMed] [Google Scholar]
- Cheung, C. H. Y. , Hsu, C. L. , Chen, K. P. , Chong, S. T. , Wu, C. H. , Huang, H. C. , & Juan, H. F. (2017). MCM2‐regulated functional networks in lung cancer by multi‐dimensional proteomic approach. Scientific Reports, 7(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, H. , Dodson, M. W. , Huang, H. , & Guo, M. (2008). The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proceedings of the National Academy of Sciences, 105(38), 14503–14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulac, M. , Leduc‐Gaudet, J.‐P. , Reynaud, O. , Ayoub, M.‐B. , Guérin, A. , Finkelchtein, M. , Hussain, S. N. A. , & Gouspillou, G. (2020). Drp1 knockdown induces severe muscle atrophy and remodelling, mitochondrial dysfunction, autophagy impairment and denervation. The Journal of Physiology, 598(17), 3691–3710. [DOI] [PubMed] [Google Scholar]
- Favaro, G. , Romanello, V. , Varanita, T. , Andrea Desbats, M. , Morbidoni, V. , Tezze, C. , Albiero, M. , Canato, M. , Gherardi, G. , De Stefani, D. , Mammucari, C. , Blaauw, B. , Boncompagni, S. , Protasi, F. , Reggiani, C. , Scorrano, L. , Salviati, L. , & Sandri, M. (2019). DRP1‐mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nature Communications, 10(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando, R. , Drescher, C. , Nowotny, K. , Grune, T. , & Castro, J. P. (2019). Impaired proteostasis during skeletal muscle aging. Free Radical Biology and Medicine, 132, 58–66. [DOI] [PubMed] [Google Scholar]
- Gegg, M. E. , Cooper, J. M. , Chau, K. Y. , Rojo, M. , Schapira, A. H. , & Taanman, J. W. (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin‐dependent manner upon induction of mitophagy. Human Molecular Genetics, 19(24), 4861–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler, S. , Holmström, K. M. , Skujat, D. , Fiesel, F. C. , Rothfuss, O. C. , Kahle, P. J. , & Springer, W. (2010). PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biology, 12(2), 119–131. [DOI] [PubMed] [Google Scholar]
- Gomes, L. C. , & Scorrano, L. (2008). High levels of Fis1, a pro‐fission mitochondrial protein, trigger autophagy. Biochimica et Biophysica Acta (BBA)‐Bioenergetics, 1777(7–8), 860–866. [DOI] [PubMed] [Google Scholar]
- Hales, K. G. , & Fuller, M. T. (1997). Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell, 90(1), 121–129. [DOI] [PubMed] [Google Scholar]
- Hauptmann, S. , Scherping, I. , Dröse, S. , Brandt, U. , Schulz, K. L. , Jendrach, M. , Leuner, K. , Eckert, A. , & Müller, W. E. (2009). Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiology of Aging, 30(10), 1574–1586. [DOI] [PubMed] [Google Scholar]
- Henchcliffe, C. , & Beal, M. F. (2008). Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nature Clinical Practice Neurology, 4(11), 600–609. [DOI] [PubMed] [Google Scholar]
- Hoppins, S. , Lackner, L. , & Nunnari, J. (2007). The machines that divide and fuse mitochondria. Annual Review of Biochemistry, 76, 751–780. [DOI] [PubMed] [Google Scholar]
- James, D. I. , Parone, P. A. , Mattenberger, Y. , & Martinou, J. C. (2003). hFis1, a novel component of the mammalian mitochondrial fission machinery. Journal of Biological Chemistry, 278(38), 36373–36379. [DOI] [PubMed] [Google Scholar]
- Jin, S. M. , & Youle, R. J. (2012). PINK1‐and Parkin‐mediated mitophagy at a glance. Journal of Cell Science, 125(4), 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi, A. U. , Saw, N. L. , Shamloo, M. , & Mochly‐Rosen, D. (2018). Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer's disease. Oncotarget, 9(5), 6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevei É., Hoppe T. (2014). Ubiquitin sets the timer: impacts on aging and longevity. Nature Structural & Molecular Biology, 21(4), 290–292. 10.1038/nsmb.2806 [DOI] [PubMed] [Google Scholar]
- Kraus, F. , & Ryan, M. T. (2017). The constriction and scission machineries involved in mitochondrial fission. Journal of Cell Science, 130(18), 2953–2960. [DOI] [PubMed] [Google Scholar]
- Labbadia, J. , & Morimoto, R. I. (2015). The biology of proteostasis in aging and disease. Annual Review of Biochemistry, 84, 435–464. 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa, M. , Palacín, M. , & Zorzano, A. (2009). Mitochondrial dynamics in mammalian health and disease. Physiological Reviews, 89(3), 799–845. [DOI] [PubMed] [Google Scholar]
- Lo Verso, F. , Carnio, S. , Vainshtein, A. , & Sandri, M. (2014). Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy, 10(11), 1883–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Otín, C. , Blasco, M. A. , Partridge, L. , Serrano, M. , & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luceri, C. , Bigagli, E. , Femia, A. P. , Caderni, G. , Giovannelli, L. , & Lodovici, M. (2018). Aging related changes in circulating reactive oxygen species (ROS) and protein carbonyls are indicative of liver oxidative injury. Toxicology Reports, 5, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao, K. , & Klionsky, D. J. (2013). Mitochondrial fission facilitates mitophagy in Saccharomyces cerevisiae. Autophagy, 9(11), 1900–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdy, A. D. , McCaffery, J. M. , & Shaw, J. M. (2000). Dnm1p GTPase‐mediated mitochondrial fission is a multi‐step process requiring the novel integral membrane component Fis1p. The Journal of Cell Biology, 151(2), 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia, M. , Toniolo, L. , Nagaraj, N. , Ciciliot, S. , Vindigni, V. , Schiaffino, S. , Reggiani, C. , & Mann, M. (2017). Single muscle fiber proteomics reveals fiber‐type‐specific features of human muscle aging. Cell Reports, 19(11), 2396–2409. [DOI] [PubMed] [Google Scholar]
- Ni, H. M. , Williams, J. A. , & Ding, W. X. (2015). Mitochondrial dynamics and mitochondrial quality control. Redox Biology, 4, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, K. , & Shaw, J. M. (2005). Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annual Review of Genetics, 39, 503–536. [DOI] [PubMed] [Google Scholar]
- Otera, H. , Wang, C. , Cleland, M. M. , Setoguchi, K. , Yokota, S. , Youle, R. J. , & Mihara, K. (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. Journal of Cell Biology, 191(6), 1141–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, C. , Wang, X. , Chen, J. , Jiao, R. , Wang, L. , Li, Y. M. , Zuo, Y. , Liu, Y. , Lei, L. , Ma, K. Y. , Huang, Y. , & Chen, Z.‐Y. (2014). Biology of ageing and role of dietary antioxidants. BioMed Research International, 2014, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas, L. , & Scorrano, L. (2016). Mito‐morphosis: Mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annual Review of Physiology, 78, 505–531. [DOI] [PubMed] [Google Scholar]
- Poole, A. C. , Thomas, R. E. , Yu, S. , Vincow, E. S. , & Pallanck, L. (2010). The mitochondrial fusion‐promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One, 5(4), e10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, P. H. (2009). Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease. Experimental Neurology, 218(2), 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichart, G. , Mayer, J. , Zehm, C. , Kirschstein, T. , Tokay, T. , Lange, F. , Baltrusch, S. , Tiedge, M. , Fuellen, G. , Ibrahim, S. , & Köhling, R. (2019). Mitochondrial complex IV mutation increases reactive oxygen species production and reduces lifespan in aged mice. Acta Physiologica, 225(4), e13214. [DOI] [PubMed] [Google Scholar]
- Sakellariou, G. K. , Pye, D. , Vasilaki, A. , Zibrik, L. , Palomero, J. , Kabayo, T. , McArdle, F. , Van Remmen, H. , Richardson, A. , Tidball, J. G. , McArdle, A. , & Jackson, M. J. (2011). Role of superoxide‐nitric oxide interactions in the accelerated age‐related loss of muscle mass in mice lacking Cu, Zn superoxide dismutase. Aging Cell, 10(5), 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrepfer, E. , & Scorrano, L. (2016). Mitofusins, from mitochondria to metabolism. Molecular Cell, 61(5), 683–694. [DOI] [PubMed] [Google Scholar]
- Sharma, A. , Smith, H. J. , Yao, P. , & Mair, W. B. (2019). Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Reports, 20(12), e48395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Q. , Yamano, K. , Head, B. P. , Kawajiri, S. , Cheung, J. T. M. , Wang, C. , Cho, J.‐H. , Hattori, N. , Youle, R. J. , & van der Bliek, A. M. (2014). Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Molecular Biology of the Cell, 25(1), 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov, A. A. , & Fiskum, G. (2003). Regulation of brain mitochondrial H2O2 production by membrane potential and NAD (P) H redox state. Journal of Neurochemistry, 86(5), 1101–1107. [DOI] [PubMed] [Google Scholar]
- Stojanovski, D. , Koutsopoulos, O. S. , Okamoto, K. , & Ryan, M. T. (2004). Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. Journal of Cell Science, 117(7), 1201–1210. [DOI] [PubMed] [Google Scholar]
- Suzuki, M. , Jeong, S. Y. , Karbowski, M. , Youle, R. J. , & Tjandra, N. (2003). The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR‐like helix bundle. Journal of Molecular Biology, 334(3), 445–458. [DOI] [PubMed] [Google Scholar]
- Sweeney P., Park H., Baumann M., Dunlop J., Frydman J., Kopito R., McCampbell A., Leblanc G., Venkateswaran A., Nurmi A., Hodgson R. (2017). Protein misfolding in neurodegenerative diseases: implications and strategies. Translational Neurodegeneration, 6(1), 10.1186/s40035-017-0077-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykiotis, G. P. , & Bohmann, D. (2008). Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Developmental Cell, 14(1), 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilokani L., Nagashima S., Paupe V., Prudent J. (2018). Mitochondrial dynamics: overview of molecular mechanisms. Essays in Biochemistry, 62(3), 341–360. 10.1042/ebc20170104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig, G. , Elorza, A. , Molina, A. J. A. , Mohamed, H. , Wikstrom, J. D. , Walzer, G. , Stiles, L. , Haigh, S. E. , Katz, S. , Las, G. , Alroy, J. , Wu, M. , Py, B. F. , Yuan, J. , Deeney, J. T. , Corkey, B. E. , & Shirihai, O. S. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. The EMBO Journal, 27(2), 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig, G. , & Shirihai, O. S. (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxidants & Redox Signaling, 14(10), 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein, A. , Grumati, P. , Sandri, M. , & Bonaldo, P. (2014). Skeletal muscle, autophagy, and physical activity: The ménage à trois of metabolic regulation in health and disease. Journal of Molecular Medicine (Berlin, Germany), 92(2), 127–137. 10.1007/s00109-013-1096-z. [DOI] [PubMed] [Google Scholar]
- Venken, K. J. T. , Schulze, K. L. , Haelterman, N. A. , Pan, H. , He, Y. , Evans‐Holm, M. , Carlson, J. W. , Levis, R. W. , Spradling, A. C. , Hoskins, R. A. , & Bellen, H. J. (2011). MiMIC: A highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nature Methods, 8(9), 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Stockum, S. , Sanchez‐Martinez, A. , Corrà, S. , Chakraborty, J. , Marchesan, E. , Locatello, L. , Da Rè, C. , Cusumano, P. , Caicci, F. , Ferrari, V. , Costa, R. , Bubacco, L. , Rasotto, M. B. , Szabo, I. , Whitworth, A. J. , Scorrano, L. , & Ziviani, E. (2019). Inhibition of the deubiquitinase USP8 corrects a Drosophila PINK1 model of mitochondria dysfunction. Life Science Alliance, 2(2), e201900392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Song, J. , Tan, M. , Albers, K. M. , & Jia, J. (2012). Mitochondrial fission proteins in peripheral blood lymphocytes are potential biomarkers for Alzheimer’s disease. European Journal of Neurology, 19(7), 1015–1022. [DOI] [PubMed] [Google Scholar]
- Wei, P. , Liu, Q. , Xue, W. , & Wang, J. (2020). A colorimetric assay of citrate synthase activity in Drosophila melanogaster . Journal of Visualized Experiments: JOVE, (155). 10.3791/59454 [DOI] [PubMed] [Google Scholar]
- Westermann, B. (2010). Mitochondrial fusion and fission in cell life and death. Nature Reviews Molecular Cell Biology, 11(12), 872–884. [DOI] [PubMed] [Google Scholar]
- Wong‐Riley, M. , Antuono, P. , Ho, K.‐C. , Egan, R. , Hevner, R. , Liebl, W. , Huang, Z. , Rachel, R. , & Jones, J. (1997). Cytochrome oxidase in Alzheimer's disease: Biochemical, histochemical, and immunohistochemical analyses of the visual and other systems. Vision Research, 37(24), 3593–3608. [DOI] [PubMed] [Google Scholar]
- Xian, H. , Yang, Q. , Xiao, L. , Shen, H. M. , & Liou, Y. C. (2019). STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nature Communications, 10(1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, H. , Guan, J. , Borrelli, L. A. , Xu, J. , Serrano‐Pozo, A. , & Bacskai, B. J. (2013). Mitochondrial alterations near amyloid plaques in an Alzheimer's disease mouse model. Journal of Neuroscience, 33(43), 17042–17051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Ouyang, Y. , Yang, L. , Beal, M. F. , McQuibban, A. , Vogel, H. , & Lu, B. (2008). Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proceedings of the National Academy of Sciences, 105(19), 7070–7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, H. , Xu, L. , & Porter, N. A. (2011). Free radical lipid peroxidation: Mechanisms and analysis. Chemical Reviews, 111(10), 5944–5972. [DOI] [PubMed] [Google Scholar]
- Yu, R. , Jin, S. B. , Lendahl, U. , Nistér, M. , & Zhao, J. (2019). Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. The EMBO Journal, 38(8), e99748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Sliter, D. A. , Bleck, C. K. , & Ding, S. (2019). Fis1 deficiencies differentially affect mitochondrial quality in skeletal muscle. Mitochondrion, 49, 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, J. , Lendahl, U. , & Nistér, M. (2013). Regulation of mitochondrial dynamics: Convergences and divergences between yeast and vertebrates. Cellular and Molecular Life Sciences, 70(6), 951–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov, D. B. , Juhaszova, M. , & Sollott, S. J. (2014). Mitochondrial reactive oxygen species (ROS) and ROS‐induced ROS release. Physiological Reviews, 94(3), 909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.